

Abstract
Purpose
Maturity-onset diabetes of the young type 3 (MODY 3) is a consequence of heterozygous germline mutations in HNF1A, and a subtype of hepatocellular adenoma (HCA) is caused by biallelic somatic HNF1A mutations; rare HCA may be related to MODY 3. This study aimed to investigate the cosegregation of HNF1A mutations with diabetes and HCA in two families.
Methods
Two patients suffering from HCA and diabetes were screened for HNF1A germline and somatic mutations using direct sequence analysis and methylation-specific multiplex-ligation-dependent probe amplification (MS-MLPA) assay. Further, we screened eight relatives in the two independent families for diabetes, HCA and HNF1A variants. Additionally, we reviewed the literature concerning the phenotypes of MODY 3 and HCA at the background of HNF1A mutations.
Results
Here we reported two families (a total of six relatives) with two missense germline mutations of HNF1A identified initially using direct sequence analysis (c.686G>A in family A and c.526 + 1G>A in family B). Somatic deletion of the second allele of HNF1A was found in liver tumor tissues in both probands who were diagnosed with HCA. There are a total of ten cases of both MODY 3 and HCA phenotypes reported in the literature to date; incomplete penetrance for HCA was observed, and all the patients with HCA developed diabetes. The onset of diabetes and HCA was highly variable, the treatment of diabetes varied from diet to insulin, and the clinical expression of HCA ranged from silent to hemorrhage. Further, the severity of diabetes mellitus was not related to the occurrence of HCA.
Conclusions
This study describes the association of HCA and MODY 3 at the background of HNF1A mutations and highlights the importance of screening for HCA in MODY 3 families to avoid the possibility of severe complications. Further, the current study indicated that there may be a special mutational spectrum of HNF1A correlated with HCA in MODY 3 families.
Keywords: HNF1A, MODY 3, Hepatocellular adenoma
Introduction
Maturity-onset diabetes of the young (MODY) is an autosomal dominantly inherited type of diabetes that results from heterozygous mutations in various transcription factors acting in the development and function of pancreatic beta cells [1]. MODY 3 is caused by inactivating germline heterozygous mutations in the hepatocyte nuclear factor 1a (HNF1A) gene, which accounts for nearly 2–5% of insulin independent diabetes and 30–65% of MODY and is characterized by positive glycosuria, microvascular complications tendency, and sulfonylurea priority [2]. HNF1A is mainly expressed in pancreatic beta cells, the intestine, and the liver [1], and plays an important role in the cellular function to regulate glycolipid metabolism. Mutations in HNF1A lead to decreased insulin secretion, and progressive damage of beta cells and thus cause the onset of diabetes. Hepatocellular adenoma (HCA), which is known as a liver tumor characterized as being rare and benign, is widely believed to be related to oral contraception [3], and usually manifests as a single tumor, however, when several adenomas are discovered in one patient, this HCA is termed liver adenomatosis [4]. Regardless of solitary or multiple, HCAs can be complicated by ache, hemorrhage, and increased risk of hepatocellular carcinoma. HCAs can be classified into molecular subtypes; ~30% of HCAs are associated with the inactivation of HNF1A, and 90% of the cases are somatic mutations [5]. Thus, we hypothesized the existence of a relationship between HNF1A mutations and phenotypes of both MODY 3 and HCA, sometimes with the two pathologies appearing in the same individuals [6–9]. To date, there have been four reports, and a total of ten cases showing the connections between the two phenotypes in subjects with HNF1A mutations.
We herein report the cosegregation phenotypes of HCA and diabetes in two unrelated Chinese MODY 3 families with germline and somatic HNF1A mutations, and reviewed the literature concerning phenotypes of MODY 3 and HCA.
Subjects and methods
Subjects
The probands from two families exhibited early-onset noninsulin-dependent diabetes as well as HCA. HCA was discovered by noninvasive radiological imaging, and confirmed by histological evaluation (Fig. 4), which was performed from surgical specimens. The diagnosis of diabetes was based on the diagnostic criteria of the American Diabetes Association [10]. Screening for HCA was based on liver ultrasonography and/or computed tomography (CT).
Fig. 4.
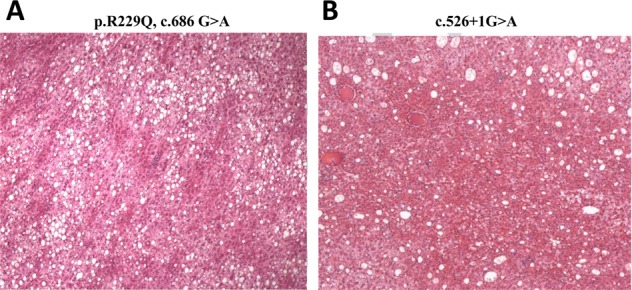
Histological characteristics of the hepatocellular adenoma. a proband 1: CD34 (part+), CK19 (−), CK7 (scattered+), Ki-67 (index 5%), β-catenin (membrane+), AFP (−), GPC-3 (−), and reticulum fiber (+). b proband 2: GPC-3 (−), CK7 (−), CEA (−), IMP3 (−), Ki-67 (index 2%), CD34 (vessel+), CAM5.2 (+), AFP (−), Hepatocyte (+), β-catenin (membrane+), and CK19 (−)
DNA extraction
Peripheral blood samples (4 ml) were obtained from the two MODY probands and the relatives available from the two families. Genomic DNA was extracted from whole blood using a QIAamp DNA Mini Kit (Qiagen China Co., Ltd., Shanghai, China) according to the manufacturer’s recommendations [11].
Liver tumor tissue samples collected from two probands with proven HCA by surgical biopsy were frozen immediately in liquid nitrogen and stored at −80 °C. Tumor DNA was extracted using a salting-out procedure. Informed consent was obtained from all individual participants and/or parents/guardians included in the study.
Germline HNF1A mutation analysis
The ten exons, the exon–intron boundaries, and the promoter region of HNF1A were screened for mutations in the two probands by direct sequencing of PCR products as previously described [11]. The NCBI BLAST database was used to identify variants by aligning with reference sequences NM_000545.6 (HNF1A). The direct diagnosis of the mutation identified in the probands were then offered to relatives independently of their clinical status.
Somatic HNF1A mutation analysis
The direct sequencing of PCR products and the methylation-specific multiplex-ligation-dependent probe amplification (MS-MLPA) assay were performed in tumor DNA. MS-MLPA was performed with SALSA MLPA Probemix P241-E1 MODY Mix 1 (MRC-Holland, Amsterdam, The Netherlands) in Beijing Novocardio Biotechnology Co., Ltd. The P241-E1 MODY Mix 1 Probemix contains 52 MLPA probes with amplification products between 130 and 500 nt. It contains probes for the HNF4A, GCK, HNF1A, and HNF1B genes and is therefore specific for MODY 1, 2, 3, and 5. For the HNF4A gene, 12 probes are included, furthermore 11 for the GCK gene, 11 for the HNF1A gene, and 10 for the HNF1B gene are included. In addition, eight reference probes are included in this probe mix. The identity of the genes detected by the reference probes is available online (www.mlpa.com). The validation experiment should result in a standard deviation <0.10 for all probes over the experiment (0.08 for proband 1, 0.05 for proband 2). The threshold for chromosomal abnormalities was established as follows: the lower limit was 0.7 for deletion and the upper limit was 1.33 for duplication. Abnormal values were plotted outside the threshold line. Probes for HNF1A are listed in Supplementary Table 1.
Literature review
The literature search occurred in March 2019. We systematically identified all potentially relevant articles from the following three electronic databases: MEDLINE, PubMed and Web of Science. Search terms about diabetes such as “maturity-onset diabetes of the young (MODY)” and “HNF1A-MODY”, and liver neoplasms, such as “liver tumor”, “hepatocellular adenoma” and “liver neoplasms”, were used in various combinations and permutations across the databases. Language restriction (English) was applied. There were a total of four papers that included ten patients that met the requirements [6–9] (Table 2).
Table 2.
Clinical characteristics of patients presenting with hepatocellular adenoma and diabetes mellitus and carrying the HNF1A mutations
| Diabetes | Hepatocellular adenoma | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | BMI (kg/m2) | Age at onset (y) | Current treatment | HbA1c (%) | Age at diagnosis (y) | Contraceptive use (y) | Clinical manifestations at diagnosis | Liver feature imaging at diagnosis | Duration of follow-up (y) | Evolution | Treatment | |
| Family 1c. 686G>A, p. Arg229Gln | ||||||||||||
| IIa | Mno detection | 24.3 | 52 | Sulfonylurea | 7.6 | / | / | / | / | / | / | / |
| IIIa (Proband 1) | Mdeletion | 22.2 | 26 | Sulfonylurea | 5.8 | 31 | No | None, US discovery | Solitary nodule 6 cm | 1.0 | Stable | Liver resection |
| Familyc.526+1G>A | ||||||||||||
| IIb | Fno detection | 20.2 | 47 | Sulfonylurea | / | / | / | / | / | / | / | / |
| IIIb | Fno detection | 19.2 | 23 | Diet | 6.2 | / | / | / | / | / | / | / |
| IIIc | Mno detection | 23.1 | 36 | Sulfonylurea | / | / | / | / | / | / | / | |
| IVa (Proband 2) | Mdeletion | 20.2 | 13 | Diet | 5.7 | 17 | No | None, US discovery | Multiple nodules including a 5 cm adenoma | 1.5 | Progressive growth of nodules (3 cm) | Liver resection |
| [6] P291fs (872_873insC) | Fno detection | 32 | Insulin | / | 17 | No | Hepatomegaly | Multiple nodules including a 19 cm adenoma | 22 | Progressive growth of nodules (1–7 cm); intrahepatic bleeding | Right lobeotomy, then liver transplantation | |
| Fno detection | 24 | Insulin | / | 51 | No | None, US discovery | Multiple nodules (0.5–2.5 cm) | 7 | Stable | None | ||
| Fno detection | 15 | Metformin | / | 16 | 1.0 | Ip bleeding of a 15 cm nodule | Massive liver adenomatosis (0.5–15 cm) | / | Death (16 y) | |||
| MF277_H279del | 15 | Insulin | / | 15 | / | None, US discovery | Multiple nodules (1–4 cm) | 5 | Stable | Liver resection | ||
| Fdeletion | 15 | Nutritional advice | / | 14 | No | Hepatomegaly | Multiple nodules including a 12-cm adenoma | 3 | Stable | Liver resection | ||
| Fno detection | 25 | Nutritional advice (GD on insulin) | / | 29 | No | None, US discovery | Multiple nodules (0.3–1 cm) | <1.0 | Stable | None | ||
| [7]c.1340 C>T (p.P447L) | Fno detection | 17 | Insulin | / | 55 | 10 | None, US discovery | Multiple nodules (7–16 cm) | 1.0 | Stable | Liver resection | |
| [8] ex2–3del | Mno detection | 10 | Sulponylurea | / | 31 | / | Rupture of a 10.5 cm liver mass | Multiple nodules (≤10.5 cm) | / | Stable | Left hepatectomy | |
| Fno detection | 7 | / | / | 56 | / | None, US discovery | Solitary nodule (1.5 cm) | / | Stable | Liver resection | ||
| [9]c.130dup,pLeu44fs | Fno detection | 13 | Insulin | 7.7 | 16 | / | None, MRI discovery | Multiple nodules (5–8 cm) | / | / | / | |
F Female, BMI body mass index, GD gestational diabetes, M male, y years old, US ultrasonography, / not available
Superscript in the first column of the table represents germline mutations; Superscript in the second column of the table represents somatic mutations
Results
MODY expression
The familial history of diabetes and the age at diagnosis of diabetes in both families suggested that the two probands may be diagnosed as MODY (Fig. 1; Table 1). The screening of HNF1A was therefore initiated in the two probands (Fig. 2). A heterozygous mutation (c. 686G>A, p. Arg229Gln) in HNF1A was found in proband 1. The mutation caused a change in the protein from Arg to Gln at p. Arg229, which is located in exon 3 of HNF1A. The mutation was subsequently searched for in other available family members in family 1, and was also found in the father. A heterozygous mutation (c.526 + 1G>A) in HNF1A was found in proband 2, and in other three relatives in family 2, and the mutation cosegregated with the clinical phenotypes of MODY 3 within the pedigree. The mutation is in the first nucleotide of intron 2 in HNF1A, and this variant is a splicing mutation that may affect protein function and cause diabetes. Neither of the two mutations were found in the ExAC database [12].
Fig. 1.
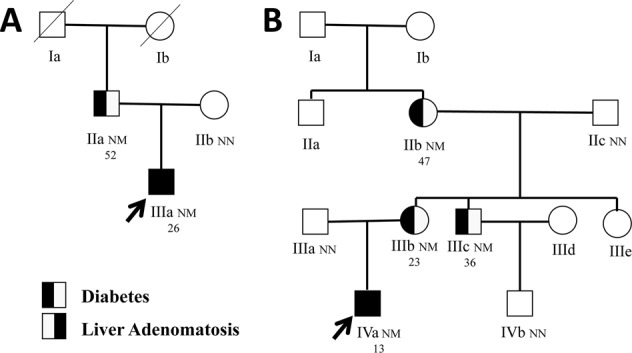
Pedigree of families of proband 1 (a) and proband 2 (b). Squares represent male family members, while circles represent female family members. The black symbol on the left represents individuals with diabetes, the black symbol on the right represents individuals with hepatocellular adenoma, and the blank symbols represent normal individuals. Arrows indicate probands in the families (a, IIIa; b, IVa). Variant carrier status is presented as N: Normal allele and M: Mutation. The sequence data displayed heterozygous mutations in HNF1A (c.686G>A, p.R229Q in proband 1; c.526 + 1G>A in proband 2). The second line under the symbols corresponds to the onset age of diabetes
Table 1.
Laboratory investigation
| Laboratory (serum) | Proband 1 | Proband 2 | Normal range |
|---|---|---|---|
| Sex | M | M | / |
| Age (year) | 32 | 19 | / |
| Birthweight (kg) | 3 | 3.7 | / |
| Height (cm) | 175 | 186 | / |
| Weight (kg) | 68 | 70 | / |
| Body mass index (kg/m2) | 22.2 | 20.2 | / |
| Systolic blood pressure (mmHg) | 120 | 110 | / |
| Diastolic blood pressure (mmHg) | 80 | 75 | / |
| Fasting glucose (mmol/L) | 7.5 | 4.9 | 3.9–6.1 |
| 2 h-postprandial glucose (mmol/L) | 9.1 | 11.3 | / |
| Fasting C-peptide (ng/ml) | 0.83 | 0.72 | 0.9–4.2 |
| 2 h-postprandial C-peptide (ng/ml) | 4.3 | 3.75 | / |
| Glycated albumin (%) | 17.3 | 13.4 | 10.8–17.1 |
| Glycated hemoglobin (%) | 5.8 | 5.7 | 4.5–6.3 |
| Current therapy | Sulfonylurea | Diet | / |
| Inheritance | Father | Mother | / |
| Total cholesterol (mmol/L) | 4.04 | 3.7 | 2.85–5.7 |
| Triglyceride (mmol/L) | 0.8 | 0.54 | 0.45–1.7 |
| HDL-C (mmol/L) | 1.14 | 1.14 | 0.93–1.81 |
| LDL-C (mmol/L) | 2.28 | 2.19 | <3.37 |
| Hs-CRP (mg/L) | 0.16 | 0.06 | 0–3.0 |
| Alanine transaminase (U/L) | 30 | 19 | 9–50 |
| Aspartate aminotransferase (U/L) | 27 | 19 | 15–40 |
| Total bilirubin (umol/L) | 18 | 17.2 | 5.1–22.2 |
| Direct bilirubin (umol/L) | 6.8 | 7.4 | 0–6.8 |
| GGT (U/L) | 11 | 17 | 10–60 |
| Albumin (g/L) | 47 | 50 | 35–52 |
| Creatinine (umol/L) | 84 | 81 | 59–104 |
| Uric acid (umol/L) | 192 | 349 | 210–416 |
| AFP (ng/ml) | 1.4 | 1.9 | 0–20 |
| CEA (ng/ml) | 0.76 | 0.37 | 0–5 |
| CA199 (U/ml) | 4.8 | 6.3 | 0–34 |
| Routine urine | |||
| GLU | NEG | NEG | NEG |
| PRO | NEG | NEG | NEG |
| KET | NEG | NEG | NEG |
| ACR (mg/g Cr) | 3 | 4 | 0–30 |
HDL-C high-density lipoprotein cholesterol, LDL-C low-density lipoprotein cholesterol, HOMA-IR homeostasis model assessment of insulin resistance, hsCRP high sensitivity C reactive protein, AFP alpha-fetoprotein, CEA carcinoembryonic antigen
Fig. 2.

The sequencing chromatogram and position of the mutation in HNF1A gene. Arrows indicate the changed nucleotide
The clinical manifestations of diabetes in the two families were extraordinarily variable, the age of onset ranged from 13 to 52, and the treatment varied from diet to sulfonylurea. Further, they had BMI ranges from 19.2 to 24.3 kg/m2. Laboratory testing was only notable for elevated fasting glucose (7.5 mmol/L), 2 h-postprandial glucose (9.1 mmol/L) and glycated albumin (17.3%) in proband 1 and for elevated 2 h-postprandial glucose (11.3 mmol/L) in proband 2.
Hepatocellular adenoma (HCA)
HCA was detected in the two probands but was not in other relatives diagnosed with MODY 3. In both probands, patients did not have abdominal symptoms or liver enzyme disturbances, and therefore HCA was diagnosed through ultrasonography screening. The tumor markers (CA199, CEA and AFP) were normal in both probands. Computed tomography (CT) scans were performed in the two probands (Fig. 3). In proband 1, a round soft tissue density shadow with a clear boundary (60 mm × 57 mm × 48 mm) was identified, located next to the posterior segment of the right hepatic lobe and the inferior vena cava and protruded outside of the liver (the CT value was ~52 HU). In proband 2, an irregular low-density lesion (47 mm × 42 mm) in the right lobe of the liver (CT value was ~48 HU) was found.
Fig. 3.
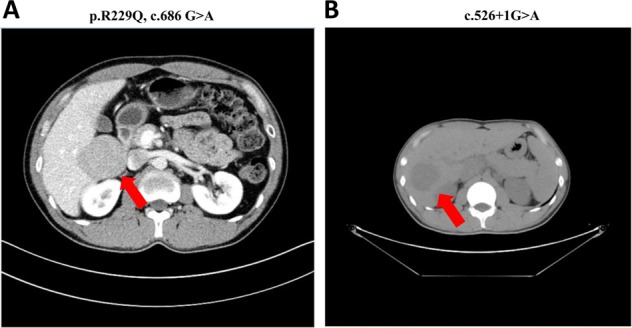
Computed tomography (CT) scans of the two probands. CT scans taken on the preoperative day (a: proband 1; b: proband 2). The size of the tumors of proband 1 was 60 mm × 57 mm × 48 mm (52 HU), and that of proband 2 was 47 mm × 42 mm (48 HU) (arrowhead)
The two probands underwent hepatectomy and recovered uneventfully. HCA was confirmed by histology in both patients. Immunostains for AFP, glypican-3, and CK19 were both negative (Fig. 4). One-year follow-up was available in the two patients (Table 2). In proband 2, progressive growth of hepatic nodules was observed after a 1-year period of follow-up while no progression was observed in proband 1.
Biallelic inactivation of HNF1A
The two probands were investigated for somatic alterations of HNF1A in their liver-cell tumors. Sanger sequencing found no abnormalities; thus, MS-MLPA was performed to search for the variants in HNF1A. A heterozygous deletion mutation in exons 1–10 of the HNF1A gene was found in both patients (Fig. 5).
Fig. 5.

Results of the MS-MLPA in the region of HNF1A in proband 1 (a) and proband 2 (b). The methylation-specific multiplex-ligation-dependent probe amplification (MS-MLPA) assay was performed with SALSA MLPA Probemix P241-E1 MODY Mix 1 (MRC-Holland, Amsterdam, The Netherlands). The P241-E1 MODY Mix 1 Probemix contains 52 MLPA probes with amplification products between 130 and 500 nt. It contains probes for the HNF4A, GCK, HNF1A, and HNF1B genes and is therefore specific for MODY 1, 2, 3, and 5. For the HNF4A gene, 12 probes are included, furthermore 11 for the GCK gene, 11 for the HNF1A gene, and 10 for the HNF1B gene are included. In addition, eight reference probes are included in this probe mix. The identity of the genes detected by the reference probes is available online (www.mlpa.com). The validation experiment should result in a standard deviation <0.10 for all probes over the experiment (0.08 for proband 1, 0.05 for proband 2). The threshold for chromosomal abnormalities was established as follows: the lower limit was 0.7 for deletion and the upper limit was 1.33 for duplication. Abnormal values were plotted outside the threshold line. Heterozygous deletion mutation in exons 1–10 of HNF1A gene was found in both proband 1 and proband 2
Discussion
We herein describe the occurrence of HCA in MODY 3 patients from two Chinese families. HNF1A gene alterations were identified as heterozygous germline mutations in peripheral blood cells and as biallelic inactivated variants in liver adenoma cells in two affected patients. To further evaluate the connection of HCA and MODY 3, we reviewed the relevant literature published in English to date [6–9].
HNF1A is a transcription factor that is expressed in a series of tissues, including the pancreas, liver, and digestive tract. However, the role of HNF1A in liver function and proliferation has not yet been elucidated. Studies in mice have reported that HNF1A inactivation can result in liver enlargement and hepatic dysfunction [13], and the expression of HNF1A decreased in preneoplastic nodules[14]; thus, HNF1A was believed to play an important role in cellular growth in liver nodules. In addition, microRNA-122 (miR-122) is the most abundant miRNA in the liver, and its deregulation is associated with hepatocellular carcinoma metastasis. Interestingly, miR-122 is under the transcriptional control of HNF1A. These results further indicate the strong correlation between HNF1A and liver adenomas.
HNF1A mutations are found in ~30% of HCAs; however, most of these patients do not suffer from MODY 3. The mutations are biallelic in most cases of HCA, which is consistent with the role of HNF1A as a tumor suppressor [5]. Exons 2–3 of HNF1A contain the majority of the DNA binding domain, and mutations in these positions may result in a dominant-negative effect on HNF1A function, thus leading to the high penetrance of HCA in carriers of those specific mutations. In the current study, we found biallelic mutations in the DNA binding domain of HNF1A in the tumor tissues of the two probands. However, this phenomenon was only observed in two patients suffering from both HCA and MODY 3 in the literature [8]. Further research on the molecular mechanism may be expected to explain this appearance. In addition, germline missense mutations in HNF1A may occur outside the dimerization domain in patients with MODY 3; therefore, the encoded proteins may dimerize with the wild-type protein but disrupt transcriptional function [15]. Consistent with the results, the two germline mutations found in our study were located beyond the dimerization domain, and have been reported as disease-causing variants in former studies [16, 17]. The missense mutation c.686G>A changes the residues in the homeodomain region of the protein that are highly conserved in human, rat, mouse, hamster, chicken and salmon [16]. The splicing site mutation c.526+1G>A occurrs within the DNA-binding domain, and is conserved across the rat, mouse, hamster, chicken, xenopus, and salmon [17]. These findings raise the possibility that a particular loss of function mutational spectrum in HNF1A may be associated with the development of HCA.
To date, there are a total of four case reports describing ten patients with MODY 3 and HCA [6–9]; the exact prevalence of HCA in MODY 3 patients is still unknown. Our team consecutively recruited MODY patients in the outpatient clinic of Endocrinology at Peking Union Medical College Hospital, Beijing, China; between January 2014 and December 2018 [18], there were a total of 25 patients from 11 families identified as having MODY 3, among which only two patients were identified with HCA (8%), and this result reflects the incomplete penetrance of liver tumors in MODY 3 individuals. Despite a large tumor burden, our patients did not have abdominal symptoms or complaints, and there were no significant liver function abnormalities. These results highlighted the importance of abdominal ultrasound for MODY 3 patients. Furthermore, by reviewing the features of the 12 patients from our study and previous case reports, we found that the clinical expression of HCA was highly variable, from silent to hemorrhage, and the course of HCA was either stable or associated with complications that required surgery. In addition, the patients demonstrated a wide range of ages at diagnosis, from adolescence to late onset after age 50. Notably, most of the patients received well-controlled glucose with HbA1c 5.7–7.7%; thus, there is no evidence that the liver phenotype worsened the severity of diabetes. In contrast with solitary liver adenoma, where nearly 90% of female patients have a history of using contraceptives [3], there was no apparent environmental factor found in our patients and in the publicly reported cases retrieved from the literature with phenotypes of MODY 3 and HCA.
In summary, our study enabled us to evaluate the cosegregation of HCA and MODY 3 with loss of function mutations of HNF1A, and advised systematic screening of HCA through noninvasive liver imaging in MODY 3 pedigrees. In the future, additional studies may help to establish the prevalence of HCA in MODY 3 patients and the potential mechanism beneath this connection.
Supplementary information
Acknowledgements
The authors thank the participants for their continuing participation in this research effort.
Funding
This work was supported by the grants from National Key R&D Program of China (2017YFC1309603), National Key Research and Development Program of China (2016YFA0101002), National Natural Science Foundation of China (Nos. 81170736, 81570715, 81870579), and Medical Epigenetics Research Center, Chinese Academy of Medical Sciences (2017PT31036, 2018PT31021).
Author contributions
J.L.F. analyzed the data and wrote the manuscript; T.W. and X.Z. contributed to data collection; X.H.X. contributed to acquisition and interpretation of the data, and revised the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Peking Union Medical College Hospital Ethics Committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The reference number is B137 and date of approval is 2015–6–29.
Informed consent
Informed consent was obtained from all individual participants and/or parents/guardians included in the study.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1007/s12020-019-02138-x) contains supplementary material, which is available to authorized users.
References
- 1.Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, Southam L, Cox RD, Lathrop GM, Boriraj VV, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3) Nature. 1996;384:455–458. doi: 10.1038/384455a0. [DOI] [PubMed] [Google Scholar]
- 2.R. Naylor, A. Knight Johnson, D. del Gaudio: Maturity-Onset Diabetes of the Young Overview. In: GeneReviews((R)). (Edited by.) Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., Amemiya A. Seattle (WA): University of Washington, Seattle University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved (1993) [PubMed]
- 3.Flejou JF, Barge J, Menu Y, Degott C, Bismuth H, Potet F, Benhamou JP. Liver adenomatosis. An entity distinct from liver adenoma? Gastroenterology. 1985;89:1132–1138. doi: 10.1016/0016-5085(85)90220-3. [DOI] [PubMed] [Google Scholar]
- 4.Chiche L, Dao T, Salame E, Galais MP, Bouvard N, Schmutz G, Rousselot P, Bioulac-Sage P, Segol P, Gignoux M. Liver adenomatosis: reappraisal, diagnosis, and surgical management: eight new cases and review of the literature. Ann. Surg. 2000;231:74–81. doi: 10.1097/00000658-200001000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bluteau O, Jeannot E, Bioulac-Sage P, Marques JM, Blanc JF, Bui H, Beaudoin JC, Franco D, Balabaud C, Laurent-Puig P, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat. Genet. 2002;32:312–315. doi: 10.1038/ng1001. [DOI] [PubMed] [Google Scholar]
- 6.Reznik Y, Dao T, Coutant R, Chiche L, Jeannot E, Clauin S, Rousselot P, Fabre M, Oberti F, Fatome A, et al. Hepatocyte nuclear factor-1 alpha gene inactivation: cosegregation between liver adenomatosis and diabetes phenotypes in two maturity-onset diabetes of the young (MODY)3 families. J. Clin. Endocrinol. Metab. 2004;89:1476–1480. doi: 10.1210/jc.2003-031552. [DOI] [PubMed] [Google Scholar]
- 7.Cantu S, Krier J, Hashemi N. Hepatocyte nuclear factor 1alpha mutation-associated MODY-3 and familial liver adenomatosis. J. Clin. Gastroenterol. 2016;50:181–182. doi: 10.1097/MCG.0000000000000461. [DOI] [PubMed] [Google Scholar]
- 8.Willson JS, Godwin TD, Wiggins GA, Guilford PJ, McCall JL. Primary hepatocellular neoplasms in a MODY3 family with a novel HNF1A germline mutation. J. Hepatol. 2013;59:904–907. doi: 10.1016/j.jhep.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 9.Harryvan TJ, Tushuizen ME, Young A. Patient with diabetes and liver tumors. Gastroenterology. 2018;155:25–26. doi: 10.1053/j.gastro.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 10.American Diabetes Association, Diabetes Advocacy: Standards of Medical Care in Diabetes-2019. Diabetes Care. 42, S182–S183 (2019) [DOI] [PubMed]
- 11.Wang Xiaojing, Li Wei, Ma Liangkun, Ping Fan, Liu Juntao, Wu Xueyan, Mao Jiangfeng, Wang Xi, Nie Min. Variants in MODY genes associated with maternal lipids profiles in second trimester of pregnancy. The Journal of Gene Medicine. 2017;19(6-7):e2962. doi: 10.1002/jgm.2962. [DOI] [PubMed] [Google Scholar]
- 12.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pontoglio M, Barra J, Hadchouel M, Doyen A, Kress C, Bach JP, Babinet C, Yaniv M. Hepatocyte nuclear factor 1 inactivation results in hepatic dysfunction, phenylketonuria, and renal Fanconi syndrome. Cell. 1996;84:575–585. doi: 10.1016/S0092-8674(00)81033-8. [DOI] [PubMed] [Google Scholar]
- 14.Flodby P, Liao DZ, Blanck A, Xanthopoulos KG, Hallstrom IP. Expression of the liver-enriched transcription factors C/EBP alpha, C/EBP beta, HNF-1, and HNF-4 in preneoplastic nodules and hepatocellular carcinoma in rat liver. Mol. Carcinog. 1995;12:103–109. doi: 10.1002/mc.2940120207. [DOI] [PubMed] [Google Scholar]
- 15.Jeannot E, Mellottee L, Bioulac-Sage P, Balabaud C, Scoazec JY, Tran Van Nhieu J, Bacq Y, Michalak S, Buob D, Laurent-Puig P, et al. Spectrum of HNF1A somatic mutations in hepatocellular adenoma differs from that in patients with MODY3 and suggests genotoxic damage. Diabetes. 2010;59:1836–1844. doi: 10.2337/db09-1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaisaki PJ, Menzel S, Lindner T, Oda N, Rjasanowski I, Sahm J, Meincke G, Schulze J, Schmechel H, Petzold C, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in MODY and early-onset NIDDM: evidence for a mutational hotspot in exon 4. Diabetes. 1997;46:528–535. doi: 10.2337/diab.46.3.528. [DOI] [PubMed] [Google Scholar]
- 17.Frayling TM, Evans JC, Bulman MP, Pearson E, Allen L, Owen K, Bingham C, Hannemann M, Shepherd M, Ellard S, et al. beta-cell genes and diabetes: molecular and clinical characterization of mutations in transcription factors. Diabetes. 2001;50(Suppl 1):S94–S100. doi: 10.2337/diabetes.50.2007.S94. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Wang T, Yu M, Zhang H, Ping F, Zhang Q, Xu J, Feng K, Xiao X. Screening of HNF1A andHNF4A mutation and clinical phenotype analysis in a large cohort of Chinese patients with maturity-onsetdiabetes of the young. Acta Diabetol. 2019;56:281–288. doi: 10.1007/s00592-018-1232-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.