

Abstract
After the release of bilateral ureteral obstruction (BUO), postobstructive diuresis from an impaired urine concentration mechanism is associated with reduced aquaporin 2 (AQP2) abundance in the inner medullary collecting duct (IMCD). However, the underlying molecular mechanism of this AQP2 reduction is incompletely understood. To elucidate the mechanisms responsible for this phenomenon, we studied molecular changes in IMCDs isolated from rats with 4-h BUO or sham operation at the early onset of AQP2 downregulation using mass spectrometry-based proteomic analysis. Two-hundred fifteen proteins had significant changes in abundances, with 65% of them downregulated in the IMCD of 4-h BUO rats compared with sham rats. Bioinformatic analysis revealed that significantly changed proteins were associated with functional Gene Ontology terms, including “cell-cell adhesion,” “cell-cell adherens junction,” “mitochondrial inner membrane,” “endoplasmic reticulum chaperone complex,” and the KEGG pathway of glycolysis/gluconeogenesis. Targeted liquid chromatography-tandem mass spectrometry or immunoblot analysis confirmed the changes in 19 proteins representative of each predominant cluster, including AQP2. Electron microscopy demonstrated disrupted tight junctions, disorganized adherens junctions, swollen mitochondria, enlargement of the endoplasmic reticulum lumen, and numerous autophagosomes/lysosomes in the IMCD of rats with 4-h BUO. AQP2 and seven proteins chosen as representative of the significantly altered clusters had a significant increase in immunofluorescence-based colocalization with autophagosomes/lysosomes. Immunogold electron microscopy confirmed colocalization of AQP2 with the autophagosome marker microtubule-associated protein 1A/1B-light chain 3 and the lysosomal marker cathepsin D in IMCD cells of rats with 4-h BUO. We conclude that enhanced autophagic degradation of AQP2 and other critical proteins, as well as endoplasmic reticulum stress in the IMCD, are initiated shortly after BUO.
Keywords: aquaporin 2, autophagy, bilateral ureteral obstruction, endoplasmic reticulum, inner medullary collecting duct, proteomics
INTRODUCTION
Urinary tract obstruction is a common urological condition observed in both children and adults. Congenital urinary tract obstruction leading to obstructive uropathy is the second most common cause of end-stage renal disease in children (1). Several solid organ malignancies (e.g., bladder, cervix, and prostate cancers) can induce obstructive urine flow from both kidneys, leading to acute kidney injury (4). Urinary tract obstruction alters renal blood flow and impairs glomerular filtration and tubular function (2). In humans, massive water diuresis and salt wasting, symptoms of postobstructive diuresis, have been observed after the release of bladder outlet obstruction caused by prostrate carcinoma (55). Likewise, a 24-h bilateral ureteral obstruction (BUO) rat model has shown similar effects immediately after the release of obstruction, and postobstructive diuresis persisted for 7 days (14). Various animal studies have investigated several of the underlying molecular mechanisms of postobstructive diuresis. For example, in 24-h unilateral urinary tract obstruction (UUO) and BUO models, decreased levels of water channel proteins [aquaporin (AQP)2, AQP3, and AQP4], located in the collecting duct (CD) (1, 14, 21), and urea transporters (UT-A1, UT-A2, and UT-B) (24), located in the inner medullary CD (IMCD), are involved in the development and maintenance of urinary concentration defects after obstruction and the release of obstruction. In addition, 24-h UUO- and BUO-induced reductions in major Na+ channels likely contribute to salt wasting during postobstructive diuresis (25–27).
The mechanisms responsible for the alterations in various transport proteins are beginning to be unraveled. In a 48-h BUO model, arginine vasopressin, a major modulator of AQPs and urea transporters, not only fails to increase but in fact decreases levels of AQPs (AQP2, AQP3, and AQP4), the type 2 vasopressin receptor (V2R), cAMP, and adenylyl cyclase type VI in the renal medulla (21). PGE2, which antagonizes arginine vasopressin effects on water permeability and Na+ transport in the CD, and the major enzyme responsible for its production (46), cyclooxygenase 2 (COX-2), are increased in the inner medulla (IM) of rats at 6 and 12 h after BUO, respectively (39). A previous study (36) in COX-2 knockout mice has confirmed a role of COX-2-derived prostaglandins in postobstructive diuresis. The renin-angiotensin system is also involved in BUO pathology, as angiotensin II type 1 (AT1) receptor blockage attenuated V2R, AQP2, and phospho-Ser256 AQP2 reductions 24 h after BUO (18). Despite these inroads, studies focusing on the initial mechanisms of altered regulation of AQP2 are limited. A time course study of BUO showed that reduced V2R levels observed at 12-h BUO are not responsible for the reduction of AQP2 mRNA detected at 2-h BUO (49). Furthermore, in a 12-h BUO model, AQP2 and phospho-Ser261 AQP2 colocalized with early endosomal and lysosomal markers, indicating a potential role of the lysosomal pathway in AQP2 degradation (49).
Recently, our group has demonstrated that autophagic degradation of AQP2 in the IMCD is responsible for the development of a nephrogenic diabetes insipidus (NDI) phenotype in rats with hypokalemia (20) and hypercalcemia (19). Autophagy is a conserved biological process that plays a crucial role in cellular homeostasis by removal of damaged organelles, protein aggregates, and other macromolecules and subsequent transport of these components to the lysosome for degradation followed by reuse of some metabolites (23). As a prosurvival mechanism, autophagy is elevated in response to various cellular stresses, e.g., oxidative stress and endoplasmic reticulum (ER) stress. Autophagy eliminating damaged mitochondria (mitophagy) is necessary to maintain quality control of mitochondria and prevents apoptosis since damaged mitochondria produce reactive oxygen species (ROS) and release proapoptotic factors (42). Autophagy protects cells from apoptosis; however, with prolonged stress, apoptosis may overwhelm the protective effect of autophagy (13). The mechanism by which hypokalemia and hypercalcemia stimulate autophagy in the IMCD remains unclear. Alteration of extracellular K+ and Ca2+ levels could change mitochondrial K+ and Ca2+ levels, resulting in decreased ATP production (41, 52). Given the evidence of gross mitochondrial damage in the IMCD of hypokalemic rats and downregulation of mitochondrial membrane proteins in the IMCD of hypercalcemic rats, we speculated that mitochondrial damage might activate autophagy and mitophagy in both NDI models (19, 20).
ER stress is known to induce autophagy in the kidney (5). In eukaryotic cells, the ER is responsible for protein synthesis, folding, and degradation, known as ER proteostasis, to maintain protein quality control. Various stresses, including oxidative stress, cause accumulation of unfolded and/or misfolded proteins in the ER, inducing disruption of ER proteostasis (ER stress). ER stress activates the three major signal transducers that induce the unfolded protein response (UPR) pathway (45). The UPR pathway initiates signaling events, including expression of chaperones, attenuation of protein translation, and degradation of unfolded proteins, to avoid irreversible cellular damage. In addition, the UPR pathway also regulates ER contacts with other organelles and the plasma membrane, with ER stress altering their functions (44, 45). The UPR pathway responds to ER stress in a binary fashion, depending on severity (17). Under mild stress, the adaptive branch of the UPR pathway promotes cell survival; however, under severe or prolonged stress, the apoptotic UPR pathway is emphasized, followed by cell death (17). In both cases, ER stress induces swollen mitochondria, autophagy, and mitophagy (28). Indeed, elevation of markers for oxidative stress, ER stress, and ER-associated apoptotic proteins are observed in whole kidney tissue after 4 h of UUO, followed by an elevation of apoptosis markers (Bax/Bcl2 ratio and caspase-3) 4 h later (58). Recently, we showed that the COOH terminus of heat shock complex 70-interacting protein, which has a protective role in the UPR during ER stress, interacts with AQP2 and facilitates its degradation (56).
The present study aimed to delineate the early mechanisms of AQP2 downregulation in response to BUO. Proteomics were initially used to investigate protein changes in the IMCD at the early onset of AQP2 downregulation in a 4-h BUO model. Subsequent immunofluorescence and electron microscopy imaging demonstrated enlargement of the ER lumen, swollen mitochondria, and mitophagy, which were associated with increased autophagic degradation of AQP2 and other critical protein targets in the IMCD. Together, our data show a key role of autophagy in the pathology of BUO.
MATERIALS AND METHODS
Brief descriptions of the experimental procedures are provided. For complete details, see the Supplemental Materials and Methods (available online at https://doi.org/10.6084/m9.figshare.10000406.v1).
Experimental Animals
All experiments were approved by the Animal Care and Use Committee of Thammasat University. To avoid the possible effects of the female reproductive cycle, (e.g., estrogen and progesterone altering Na+ reabsorption), the present study was performed with male Sprague-Dawley rats weighing 180–200 g. Rats had free access to water and to a standard rodent diet. Rats were randomly assigned to the following two groups for each protocol: 4 h of BUO and sham operation (sham) control. Rats were anesthetized with isoflurane and placed on a heating pad to maintain body temperature at 37°C during surgery. Through a midabdominal incision, both upper parts of the ureter were ligated using 3-0 silk in four rats. Rats in the sham control group were prepared in parallel with the 4-h BUO group. Rats were allowed to recover after surgery in metabolic cages. We collected urine dripping from the urethra of sham control rats for osmolality measurement. Four hours after the operation, rats were decapitated using a guillotine while conscious to avoid stress-induced arginine vasopressin secretion that might alter the IMCD protein abundance.
Protocols
Protocol 1.
Eight rats were allocated to the 4-h BUO (n = 4) and sham (n = 4) groups. IMs were isolated, and the IMCD was prepared for a nontargeted proteomic study. Three independent experiments were performed.
Protocol 2.
Eight rats were allocated to the 4-h BUO (n = 4) and sham (n = 4) groups. IMs were isolated followed by IMCD isolation for a targeted proteomic study. Three independent experiments were performed.
Protocol 3.
Twelve rats were allocated to the 4-h BUO (n = 6) and sham (n = 6) group. IMs were isolated for immunoblot analysis. Trunk blood was collected for serum urea, creatinine, Na+, and K+ analyses. Control rats stayed in metabolic cages after surgery to collect urine for urine volume until euthanization. Urine dripping from the urethra of sham control rats was collected for osmolality measurement. For 4-h BUO rats, urine was aspirated from their pelvises for osmolality measurement.
Protocol 4.
Six rats were allocated to the 4-h BUO (n = 3) and sham (n = 3) groups. The left kidneys were harvested for immunofluorescence, and the right kidneys were harvested for electron microscopy. Two independent experiments were performed.
Protocol 5.
Six rats were allocated to the 4-h BUO (n = 3) and sham (n = 3) groups. IMs were dissected for immunogold electron microscopy. Two independent experiments were performed.
Protocol 6.
Twenty-eight rats were allocated to the 10-h BUO grouph (n = 7) versus the sham group for 10 h (n = 7) and to the 24-h BUO group (n = 7) versus the sham group for 24 h (n = 7). IMs from the right kidneys were dissected for immunoblot analysis, and the left kidneys were harvested for electron microscopy. Rats were put in metabolic cages after surgery until euthanization to collect urine for volume. Urine dripping from the urethra of sham control rats was collected for osmolality measurement. Urine was aspirated from the pelvis of BUO rats for osmolality measurement.
IMCD and Peptide Preparation
The IMCD was prepared from the IM according to Stokes et al. (50) with modifications. In brief, kidney IMs were digested by an incubation at 37°C for 70–90 min in digestion solution. The resulting suspension was centrifuged at 70 g for 30 s to harvest the IMCD-enriched fraction. Pellets from rats in each group were pooled and lysed in 8 M urea, 50 mM Tris·HCl, and 75 mM NaCl containing protease inhibitors (Roche, Mannheim, Germany). Protein samples were sonicated and centrifuged at 14,000 g for 10 min at 4°C, and supernatants were collected. Protein concentration was determined using the BCA protein assay (Pierce, ThermoFisher Scientific). A total of 200 µg protein from each group was reduced, alkylated, and quenched followed by trypsin digestion, as previously described (19). The peptides were then quantified by a Quantitative Fluorometric Peptide Assay (Pierce).
Dimethyl Labeling and Peptide Fractionation
A total of 100 µg peptides from each group was subjected to in-solution stable isotope dimethyl labeling (3). In brief, peptides from the control group were labeled with regular formaldehyde (CH2O; Sigma-Aldrich) and sodium cyanoborohydride (NaBH3CN; Sigma-Aldrich), which generated a mass increase of 28.0313 Da per primary amine on a peptide (light labeling). Heavy labeling peptides from the BUO group were generated by combining deuterated [13C]formaldehyde (13CD2O) with sodium cyanoborodeuteride (NaBD3CN; Cambridge Isotope Laboratories), increasing a mass of 36.0757 Da per primary amine. The two differentially labeled samples were mixed and fractionated into five fractions using a high-pH, reversed-phase peptide fractionation kit per the manufacturer’s protocol (Pierce, ThermoFisher Scientific).
Liquid Chromatography-Tandem Mass Spectrometry Analysis and Database Searching
Fractionated samples were submitted to nano-liquid chromatography EASY-nLC 1000 (ThermoFisher Scientific) coupled with Q-exactive plus mass spectrometer (Q Exactive Plus Hybrid Quadrupole-Orbitrap, ThermoFisher Scientific). The mass spectrometry (MS) methods included a full MS scan followed by 10 data-dependent MS2 scans. MS2 spectra were searched and analyzed using Maxquant software (version 1.6.0.13) based on a database from Uniprot Rattus norvegicus (12,353 proteins, February 2018). Labeled peptide analysis and quantification were performed using Maxquant (7) and Perseus software (54). The median peptide abundance of all peptides identified for a given protein was used to infer the protein abundance change. Values are presented as means ± SE of log2 of BUO-to-sham ratios for each protein across all three biological replicates. All proteins that passed criteria two-tailed t tests against log2(1) (P < 0.05) were considered significantly changed. The DAVID bioinformatics tool (http://david.abcc.ncifcrf.gov/) (16) was used to identify clusters of proteins based on Gene Ontology (GO) terms. A modified Fischer’s exact test was used to assess the level of enrichment.
Parallel Reaction Monitoring
We performed three independent experiments. Each experiment contained eight rats, which were randomly assigned into the following two groups: 4-h BUO (n = 4) and sham (n = 4). IMCDs from rats in each group were pooled. Ten micrograms of peptides from each group were analyzed using a Q-Exactive orbitrap MS (Thermo Scientific). The target peptides were selected based on prior nontargeted proteomic data. Peak areas for each peptide were extracted using Skyline software (31). Values are presented as means of log2 of BUO-to-sham ratios for each protein across all three biological replicates. The quantification was based on the sum of the areas under the curve (AUCs) of the selected peptides and the ratio between light and heavy isotope peptide fragments. All proteins that passed criteria two-tailed t tests against log2(1) (P < 0.05) were considered significantly changed.
Immunoblot Analysis
Independent experiments were performed for immunoblot analysis. Twelve rats were allocated to the 4-h BUO (n = 6) and sham (n = 6) groups. The kidneys were homogenized in Laemmli sample buffer containing protease inhibitor. The total protein concentration of the homogenate was measured using the BCA protein assay. Loading gels with Coomassie blue stain were performed to adjust the final protein levels in each sample before the immunoblot analysis. Anti-AQP2 (15) and anti-phospho-Ser256 AQP2 (37) were kindly provided by Dr. Mark Knepper (National Institutes of Health). A full list of the primary and secondary antibodies is provided in the Supplemental Materials and Methods. Electrophoresis and immunoblot analysis were performed as previously described (43). Images were developed and quantified using near-infrared fluorescence (Li-Cor Odyssey).
Immunofluorescence Microscopy
Six rats were allocated to the 4-h BUO (n = 3) and sham (n = 3) groups. The left kidneys were harvested for immunofluorescence, and the right kidneys were harvested for electron microscopy. Two independent experiments were performed. The immunofluorescence labeling of kidney sections was performed as previously described (20). Primary antibodies and secondary antibodies are provided online in the Supplemental Materials and Methods. Fluorescence images were acquired using an Axio Observer Z1 microscope in conjunction with ApoTome2 (Carl Zeiss, Jena, Germany). The degree of fluorescence colocalization was analyzed by Pearson correlation coefficient, and P < 0.05 was considered significant.
Electron Microscopy
Fresh renal IMs from three sham and three BUO rats were minced into 1-mm3 cubes and fixed in a standard fixative followed by dehydration, as previously described (20). The tissues were embedded in epoxy resin. Ultrathin sections were mounted on naked copper grids and then examined under a transmission electron microscope (FEI Tecnai G2 TWIN 200 kV). Two independent experiments were performed.
Immunogold Electron Microscopy
All procedures have been previously described in detail (12). Six rats were allocated to the 4-h BUO (n = 3) and sham (n = 3) groups. The kidneys were removed, and IMs were dissected. A list of primary antibodies is provided online in the Supplemental Materials and Methods. Primary antibodies were visualized using various combinations (see figures) and sequential application of donkey anti-rabbit IgG, goat anti-rabbit IgG, or donkey anti-goat IgG conjugated to various sizes of colloidal gold particles (BioCell Research Laboratories). Sections were examined with a FEI Morgagni electron microscope. Two independent experiments were performed.
Statistics
Value are presented as means ± SE. Individual sample sizes (n) are indicated in the figures. Statistical comparisons between two groups were made by a Student’s unpaired t test. P values of <0.05 were considered significant.
RESULTS
Four Hours of BUO Induced Renal Impairment and Expression of AQP2
Four hours of BUO induced significant increases in plasma urea (17.7 ± 0.4 vs. 33.1 ± 0.4 mg/dL, P < 0.01) and creatinine (0.3 ± 0.07 vs. 0.8 ± 0.2 mg/dL, P < 0.01) and a significant decrease in urine osmolality compared with sham (1,662.7 ± 33.7 vs. 552.7 ± 14.1 mosmol·kg−1·H2O−1, P < 0.001) (see Supplemental Table S1; all Supplemental Data for this article are provided online at https://doi.org/10.6084/m9.figshare.10271465.v1). The urine volume of the sham group is provided online in Supplemental Table S1.
In IM homogenates, BUO significantly decreased the abundance of phospho-Ser256 AQP2 (vasopressin-regulated site) by 65.8% (P < 0.001) and total AQP2 by 42.2% (P < 0.01; Fig. 1A). The reduction in phospho-Ser256 AQP2 was significantly greater than the reduction in total AQP2 (P < 0.001; see Supplemental Table S2).
Fig. 1.
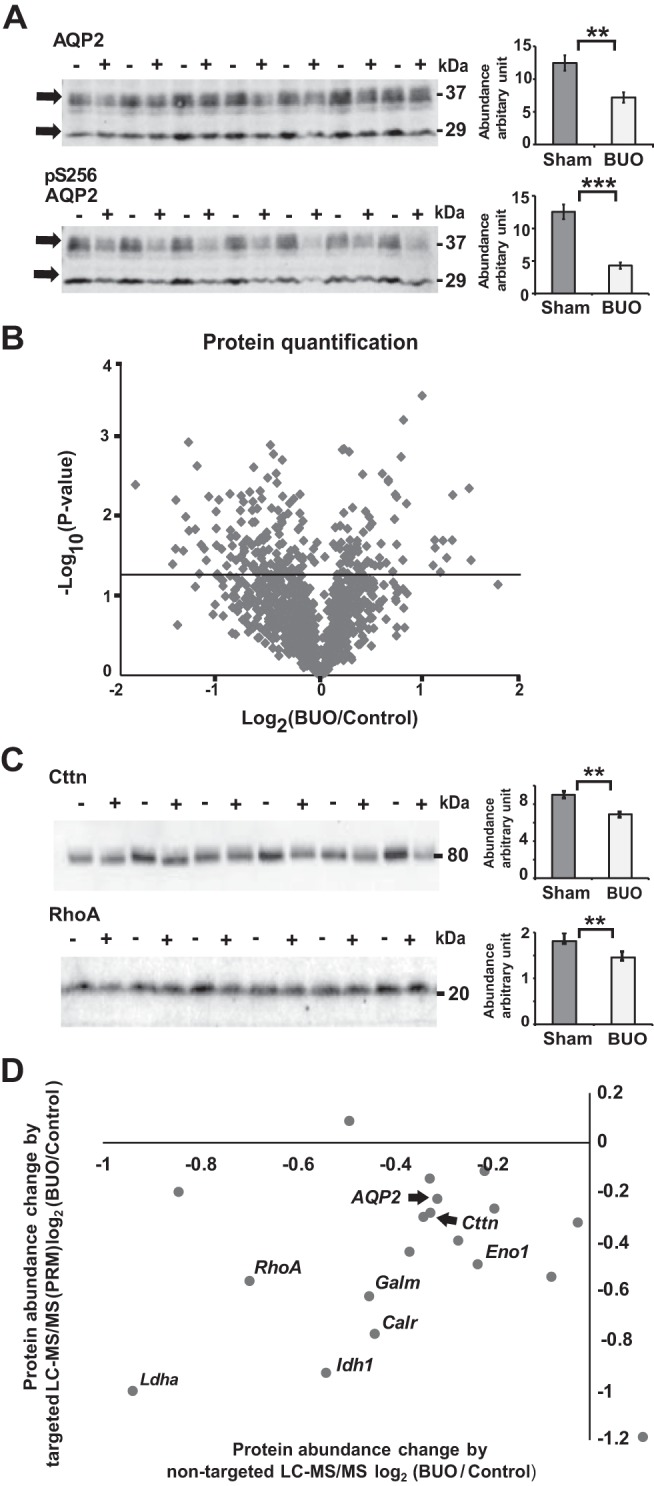
Immunoblot and proteomic analyses of the inner medulla and inner medullary collecting duct (IMCD) from 4-h bilateral ureteral obstruction (BUO) and sham rats and data verification. A: densitometry analysis revealed significant downregulation of total aquaporin 2 (AQP2) and phospho-Ser256 (pS256) AQP2 by 42% (**P < 0.005) and 65% (***P < 0.001) in 4-h BUO (+; n = 6) compared with sham (−; n = 6) rats, respectively (unpaired t tests). B: proteomic analysis of IMCD proteins from 4-h BUO and sham rats from 3 independent experiments. The volcano plot demonstrates the average log2 protein abundance ratio of BUO to sham groups for 1,046 proteins identified in all 3 biological replicates (x-axis) versus the significance, expressed as –log10 (P value, y-axis). The horizontal solid line indicates a P value of 0.05 [−log (P value) of 1.3]. A total of 215 proteins showed significant changes in abundance in 4-h BUO IMCDs compared with sham IMCDs (unpaired t tests, P < 0.05, above the solid line). C: immunoblot analysis of inner medulla samples from sham (−; n = 6) and 4-h BUO (+; n = 6) rats probed with RhoA and cortactin (Cttn) antibodies. Densitometry analysis revealed 20% downregulation of RhoA (unpaired t tests, **P < 0.01) and 23% downregulation of Cttn in 4-h BUO compared with sham rats. D: the x-axis represents the mean of the log2 value of the protein abundance ratio for paired 4-h BUO and sham rats [two-tailed t test against log2 (1), P < 0.05] resulting from nontargeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis from 3 independent experiments. The y-axis represents the mean of the log2 value of the protein abundance ratio of the same proteins verified by targeted LC-MS/MS from 3 independent experiments [two-tailed t test against log2 (1), P < 0.05]. See text for definitions of abbreviations.
Identification of Global Molecular Changes in the IMCD of 4-h BUO Rats by Proteomics and Bioinformatics
Nontargeted liquid chromotography-tandem MS (LC-MS/MS) analysis identified a total of 16,681 unique peptides, corresponding to 2,311 proteins. Figure 1B shows a volcano plot of the average log2 protein abundance ratio of BUO to sham for 1,046 proteins identified in all 3 biological replicates (x-axis) versus the significance. A total of 215 proteins changed significantly in the IMCD of BUO rats compared with sham rats (see Supplemental Data available online at https://doi.org/10.6084/m9.figshare.9731345.v1). Of these, 140 proteins were significantly decreased in abundance, whereas 75 proteins were significantly increased. The complete list of protein quantifications along with associated GO terms are provided in a publicly accessible online database (https://hpcwebapps.cit.nih.gov/ESBL/Database/BUO/). The DAVID bioinformatics resource was used to further investigate the classes of proteins that exhibited a significant change. Highly represented annotations were grouped into clusters. The most significantly enriched clusters included proteins with the functional GO terms “cell-cell adhesion,” “cell-cell adherens junction,” “mitochondrial inner membrane,” and “endoplasmic reticulum chaperone complex.” KEGG pathway analysis revealed significant “glycolysis/gluconeogenesis” enrichment in the IMCD of BUO compared with sham rats (Table 1).
Table 1.
Gene Ontology annotation among 216 significantly changed proteins that relate to kidney histopathology
| Proteins | |
|---|---|
| Biological process | |
| Cell-cell adhesion | RhoA*, Bzw2*, Capza1, Cast*, Ccs, Hsp90ab1*, Hspa5*, Idh1*, Kif5b*, Ldha*, Pdlim5, Pfn1*, Ppme1, Prdx6*, Sept9*, Stk24, Tagln2*, Tmpo*, Ywhaz* |
| Cellular components | |
| Cell-cell adherens junction | Afdn, Anxa1*, Bzw2*, Capza1, Cast*, Ccs, Dlg1, Hsp90ab1*, Hspa5*, Idh1*, Jup, Kif5b*, Ldha*, Pdlim5, Pfn1*, Ppme1, Prdx6*, Sept9*, Stk24*, Tagln2*, Tmpo*, Ywhaz* |
| Mitochondrial inner membrane | Aldh3a2*, Atp5d, Ckmt1, Coq9, Cycs*, Eci1, Hadh*, Letm1*, Lgals3*, Pc, Rps3*, Sdhb, Slc25a11*, Tomm22*, Uqcrh, Vdac2* |
| Endoplasmic reticulum chaperone complex | Calr*, Dnajb11*, Hspa5*, Hyou1*, Pdia4*, Ppib*, Rcn2* |
| KEGG pathway | |
| Glycolysis/gluconeogenesis | Aldh3a2*, Galm*, Ldha*, Akr1a1*, Pfkl*, Aldh1a3*, Eno1*, Aldoa |
Significantly decreased proteins observed by nontargeted liquid chromatography-tandem mass spectrometry [two-tailed against log2(1) (P < 0.05)].
Data Verification by Targeted LC-MS/MS and Immunoblot Analysis
To confirm the prior nontargeted LC-MS/MS analysis, we performed three new experiments to generate samples for immunoblot and targeted LC-MS/MS analyses. Immunoblot analysis (Fig. 1C) confirmed significant downregulation of RhoA and cortactin (Cttn), which play important roles in cell-cell adhesion (10). Consistent with the nontargeted LC-MS/MS analysis, targeted LC-MS/MS demonstrated downregulation of AQP2 plus proteins representative of cell adhesion [RhoA, Cttn, heat shock protein 90α family class B member 1 (Hsp90ab1), transgelin-2 (Tagln2), and 14-3-3 protein-ζ/δ (Ywhaz)], the mitochondria inner membrane [isocitrate dehydrogenase 1 (Idh1), voltage-dependent anion-selective channel protein 2 (Vdac2), 40S ribosomal protein S3a (Rps3a), hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit-β (Hadhb), and lactate dehydrogenase A (Ldha)], the ER chaperone complex [calreticulin (Calr), hypoxia upregulated protein 1 (Hyou1), and peptidylprolyl isomerase B (Ppib)], and the KEGG pathway of glycolysis/gluconeogenesis [heat shock 70-kDa protein 5 (Hspa5), galactose mutarotase (Galm), enolase 1 (Eno1), calpastatin (Cast), aldehyde dehydrogenase 3 family member A2 (Aldh3a2), and aldehyde dehydrogenase 1 family member A3 (Aldh1a3)] (Fig. 1D and Table 2).
Table 2.
A list of 20 selected proteins with the log2 value of protein abundance ratio paired BUO and sham rats by nontargeted and targeted LC-MS/MS analysis
| Log2 (BUO/Sham) |
|||
|---|---|---|---|
| Protein Group (Protein Name) | Gene Symbol | Selected reaction monitoring | Nontargeted LC-MS/MS |
| Cell-cell adhesion | |||
| Aquaporin 2 | AQP2 | −0.34* | −0.35* |
| Transforming protein RhoA | RhoA | −0.62* | −0.59* |
| Src substrate cortactin | Cttn | −0.35* | −0.39* |
| Heat shock protein-90β | HSP90ab1 | −0.31* | −0.47* |
| Transgelin-2 | Tgaln2 | −0.26* | −0.38* |
| 14-3-3 protein-ζ/δ | Ywhaz | −0.17* | −0.57* |
| Mitochondria | |||
| Isocitrate dehydrogenase [NADP] cytoplasmic | Idh1 | −0.50* | −0.85* |
| Voltage-dependent anion-selective channel protein 2 | Vdac2 | −0.13* | −0.42* |
| 40S ribosomal protein S3a | Rps3A | −0.33* | −0.14* |
| Trifunctional enzyme subunit-β, mitochondrial | Hadhb | −0.50* | 0.09* |
| l-lactate dehydrogenase A chain | Ldha | −0.79* | −0.90* |
| Endoplasmic reticulum chaperone complex | |||
| Calreticulin | Calr | −0.43* | −0.74* |
| Hypoxia upregulated protein 1 | Hyou1 | −0.38* | −0.5* |
| Peptidyl-prolyl cis-trans isomerase B | Ppib | −0.72* | −0.33* |
| Glycolysis/gluconeogenesis | |||
| 78-kDa glucose-regulated protein | Hspa5 | −0.36* | −0.40* |
| Aldose 1-epimerase | Galm | −0.44* | −0.63* |
| α-Enolase | Eno1 | −0.28* | −0.54* |
| Calpastatin | Cast | −0.27* | −0.27* |
| Aldehyde dehydrogenase family 3 member A2 | Aldh3a2 | −0.02* | −1.04* |
| Aldehyde dehydrogenase family 1 member A3 | Aldh1a3 | −1.57 | −0.91* |
Values are averages of log2 [bilateral ureteral obstruction (BUO)/sham]. LC-MS/MS, liquid chromatography-tandem mass spectrometry.
Statistically significant by two-tailed against log2(1) (P < 0.05).
Estimated Half-Lives of Downregulated Proteins
We cross-referenced mouse CD protein half-lives (47) with the downregulated proteins identified in the IMCD of rats with 4-h BUO. The majority of the protein half-lives were longer than 4 h (see Supplemental Fig. S1, available online at https://doi.org/10.6084/m9.figshare.10000286.v1). Given the expected lag between BUO onset (stimulation), reduction of mRNA levels, and reduced protein synthesis (30), these data indicated that increased protein degradation accounts for much of the decreased proteins after BUO. This result aligns with previous work suggesting that protein degradation/stabilization accounts for much of the altered proteome upon ER stress (6).
Structural Abnormalities of the IMCD in BUO
Electron microscopy demonstrated dilated tubular lumens, rupture of the apical membrane, and clear cytoplasm of IMCD cells in BUO rats (Fig. 2, A and B). Electron microscopy highlighted tight-junction disruption, disorganized adherens junctions, and many lysosomes adjacent to ruptured apical membranes in BUO IMCD cells (Fig. 2, C and D). In every BUO IMCD cell examined, there were significantly increased numbers of double-membrane autophagosomes and single-membrane autolysosomes containing electron-dense materials (i.e., 5–8 autophagosomes and 20–30 autolysosomes per cell compared with only 1 autophagosome without autolysosome in control rats, P < 0.001). Autolysosomes were also located adjacent to the adherens junction in the BUO IMCD (Fig. 2, E and F). In addition, enlarged mitochondria were observed in BUO IMCD cells relative to sham IMCD cells (Fig. 2, G and H). Interestingly, we observed generalized enlargement of the ER lumen in BUO IMCD cells compared with the ER in sham IMCD cells (Fig. 2, I and J).
Fig. 2.

Structural abnormalities in inner medullary collecting duct (IMCD) cells of 4-h bilateral ureteral obstruction (BUO) rats. A: compared with sham IMCDs (n = 3), electron micrography showed clear cytoplasm of tubular cells (arrows) and dilated tubular lumens (*) in IMCD cells of 4-h BUO rats (n = 3). B: magnified image (scale bar = 2 µm). C: electron micrograph of the intercellular junction of sham IMCD cells demonstrating a thin tight junction (TJ) and light, delicate adherens junction (AJ). D: a disrupted apical membrane (black arrow) surrounded by endosomes (white arrowhead), disrupted tight junction, disorganized AJ (black arrowheads), and clear area of cytoplasm (**) were observed in 4-h BUO IMCD cells. E and F: electron micrographs demonstrating single-membrane autophagolysosomes (white arrowheads) containing electron-dense membrane structures and double-membrane autophagosomes (black arrowheads) (E) and autolysosomes (white arrowheads) located adjacent to the AJ in 4-h BUO IMCD cells (F). G: normal mitochondria (MT) showing sharp cristae in IMCD cells of sham rats. H: large and swollen mitochondria (arrow) were observed in 4-h BUO IMCD cells. I: arrowheads indicating a normal endoplasmic reticulum (ER) in IMCD cells of sham rats (arrows). J: enlargement of the ER lumen in 4-h BUO IMCD cells (arrowheads). Scale bar = 100 nm. K and L: in BUO IMCD cells, microtubule-associated protein 1A/1B-light chain 3 (LC3)-positive membrane-bound autophagosomes (arrows) containing AQP2 (arrowheads) (K) and cathepsin D-positive membrane-bound autolysosomes (10-nm gold particles; arrows) containing AQP2 (15-nm gold particles; arrowheads) (L) were frequently observed. Two independent experiments were performed.
Colocalization of Downregulated Proteins With Autophagosomes/Lysosomes
To assess a potential role of autophagy in mediating the reduction in AQP2 and select proteins in BUO IMCDs, immunofluorescence colocalization experiments were performed using antibodies specific for proteins associated with the autophagosome [microtubule-associated protein 1A/1B-light chain 3 (LC3) or autophagy related 12 (ATG12)] and lysosome [lysosomal associated membrane protein 1 (Lamp1)]. A high degree of colocalization of AQP2 with LC3 and Lamp1 was detected in BUO IMCDs (Fig. 3, A–C). Double-labeling immunogold electron microscopy confirmed colocalization of AQP2 with both LC3 (autophagosome marker) and cathepsin D (lysosomal marker; Fig. 2, K and L). Furthermore, AQP2 was colocalized with various other downregulated proteins in a punctate pattern, including RhoA and Cttn (Fig. 4), and representative proteins of cell-cell adhesion [catenin-β1 (Ctnnb1) and integrin-β1 (Itgb1)], cell-cell adherens junction [vinculin (Vcl) and erm], and mitochondrial inner membrane [ATP synthase F1 subunit-α (ATP5a)] (Fig. 5 and Supplemental Fig. S2). No colocalization of AQP2 with these proteins was observed in IMCDs of sham rats. These findings support that the process of autophagy is involved in downregulation of these groups of proteins in BUO.
Fig. 3.

Colocalization of aquaporin 2 (AQP2) with autophagy markers in inner medullary collecting duct (IMCD) cells of 4-h bilateral ureteral obstruction (BUO) and sham (S) rats. A: inner medulla sections of sham (n = 3) and 4-h BUO (n = 3) were triple labeled against AQP2 (red), lysosomal associated membrane protein 1 (Lamp1; pink), and microtubule-associated protein 1A/1B-light chain 3 (LC3; green). Insets: magnified views of the areas where significant colocalizations were observed as a group of puncta that were not seen in control sections. Scale bars = 4 µm. B: representative scatterplots demonstrating the high degree of colocalization between AQP2 and LC3 (Pearson correlation coefficient, r2 = 0.90, P < 0.05) or Lamp1 (r2 = 0.67, P < 0.05) in IMCD cells of a rat with 4-h BUO. C: bar graph summarizing Pearson correlation coefficients of AQP2 and LC3 or Lamp1 from 30 colocalized puncta. Two independent experiments were performed.
Fig. 4.

Colocalization of cell-cell adhesion proteins with autophagy markers in the inner medullary collecting duct of 4-h bilateral ureteral obstruction (BUO) rats. Inner medulla sections of sham (S; n = 3) and 4-h BUO (n = 3) rats were triple labeled against aquaporin-2 (AQP2; red), lysosomal associated membrane protein 1 (Lamp1; pink), and downregulated proteins [RhoA and cortactin (Cttn)]. Insets: magnified views of the areas where significant colocalization was observed as a group of puncta. These colocalized puncta were not observed in control sections. Scale bars = 4 µm. Scatterplots demonstrate the degree of colocalization between RhoA and Lamp1 (Pearson correlation coefficient, r2 = 0.81, P < 0.05), Cttn, and Lamp1 (r2 = 0.94, P < 0.05). Two independent experiments were performed.
Fig. 5.

Colocalization of proteins representative of the cell-cell adherens junction and mitochondrial inner membrane with autophagy markers in the inner medullary collecting duct (IMCD) of 4-h bilateral ureteral obstruction (BUO) rats. Inner medulla sections of sham (S; n = 3) and 4-h BUO (n = 3) rats were triple labeled against aquaporin 2 (AQP2; red), lysosomal associated membrane protein 1 (Lamp1; pink), and proteins representative of cell-cell adherens junctions [catenin-β1 (Ctnnb1) and vinculin (Vcl); green]. The IMCD section was labeled with proteins associated with the mitochondrial inner membrane [ATP synthase F1 subunit-α (ATP5a); pink] and autophagosome [autophagy related 12 (ATG12; green) and AQP2 (red)]. Insets: magnified views of the areas where significant colocalization was observed as a group of puncta. These colocalized puncta were not observed in control sections. Scale bars = 4 µm. Scatterplots demonstrate the degree of colocalization between Ctnnb1 and Lamp1 (Pearson correlation coefficient, r2 = 0.89, P < 0.05), Vcl, and Lamp1 (r2 = 0.86, P < 0.05) as well as ATP5a and ATG12 (r2 = 0.80, P < 0.05). Two independent experiments were performed.
Apoptosis Was Induced in the IM at 4 h of BUO
To investigate whether apoptosis occurs in IMCD cells after longer periods of BUO, we performed 10- and 24-h BUO models and studied morphology under electron microscopy as well as immunoblot analysis for the key apoptotic markers AQP2, RhoA, and Cttn. Rats with BUO were unable to concentrate urine compared with sham rats (Supplemental Table S3). Their IMCD cells became condensed and severely deformed after 10-h BUO (Fig. 6A). Compared with the 4-h BUO condition, we observed disruption of the adherens junctions, swelling and rupture of mitochondrial membranes, enlargement of ER lumens, rupture of the nuclear membrane, and chromatin condensation, indicating the initiation of the apoptosis pathway in IMCD cells after 10-h BUO (Fig. 6, B–D). Even more extreme effects were seen at 24-h BUO, with high levels of cell separation, distortion, and shrinkage, disappearance of the epithelial membrane, and appearance of lipid droplets as well as condensation of nuclear chromatin (pyknosis) and rupture of the nuclear membrane, all of which are indicative of apoptosis (Fig. 6, E–H). As expected, significant downregulation of AQP2 and RhoA, a cell-cell adhesion representative, was observed in the IM after 10- and 24-h BUO (Fig. 7, A–E). Interestingly, cleaved poly(ADP-ribose) polymerase (PARP), a marker of DNA breakage and late apoptosis (53), was significantly upregulated in the IM as early as 4 h after BUO, with continued upregulation at 10 h after BUO (Fig. 7, F–H); however, the initial marker of apoptosis cleaved caspase-9 was not significantly upregulated (Fig. 8, A–B). Such an expression pattern, combined with oxidative stress, autophagy, and the absence of apoptotic bodies, is indicative of caspase-free regulated cell death (53). In agreement with IMCD morphology, cleaved PARP was still significantly upregulated, along with cleaved caspase-9, cleaved caspase-6 and AQP2 downregulation, at 24-h BUO compared with sham (Figs. 7C and 8, C and F).
Fig. 6.

Structural abnormalities in inner medullary collecting duct (IMCD) cells of 10- and 24-h bilateral ureteral obstruction (BUO) rats. A: electron micrograph showing the condensed cytoplasm, severely deformed IMCD cells, and collapsed tubular lumens (*) in 10-h BUO rats (n = 3). Scale bar = 1 µm. B–D: IMCD cells of 10-h BUO rats (n = 3) contained disrupted adherens junctions (AJ; black arrowhead; B), enlargement of the endoplasmic reticulum (ER) lumen (white arrowheads; C), and slightly enlarged mitochondria (MT) with loss of membranes (black arrowhead; D). Scale bars = 100 nm. E–H: as for 24-h BUO rats (n = 3), IMCD cells contained many lipid droplets (black arrowheads) in a shrunken cytoplasm and pyknotic nuclei (NU; white arrowhead). Scale bars = 1 µm. IMCD cells demonstrated rupture of nuclei (white arrowhead; E), rupture of epithelium tubular lumens (black arrowheads; F), enlargement of the ER lumen and loss of the ER membrane (white arrowhead; G), and loss of the mitochondrial membrane and cristae (black arrowhead; H). Scale bars = 100 nm. Two independent experiments were performed.
Fig. 7.
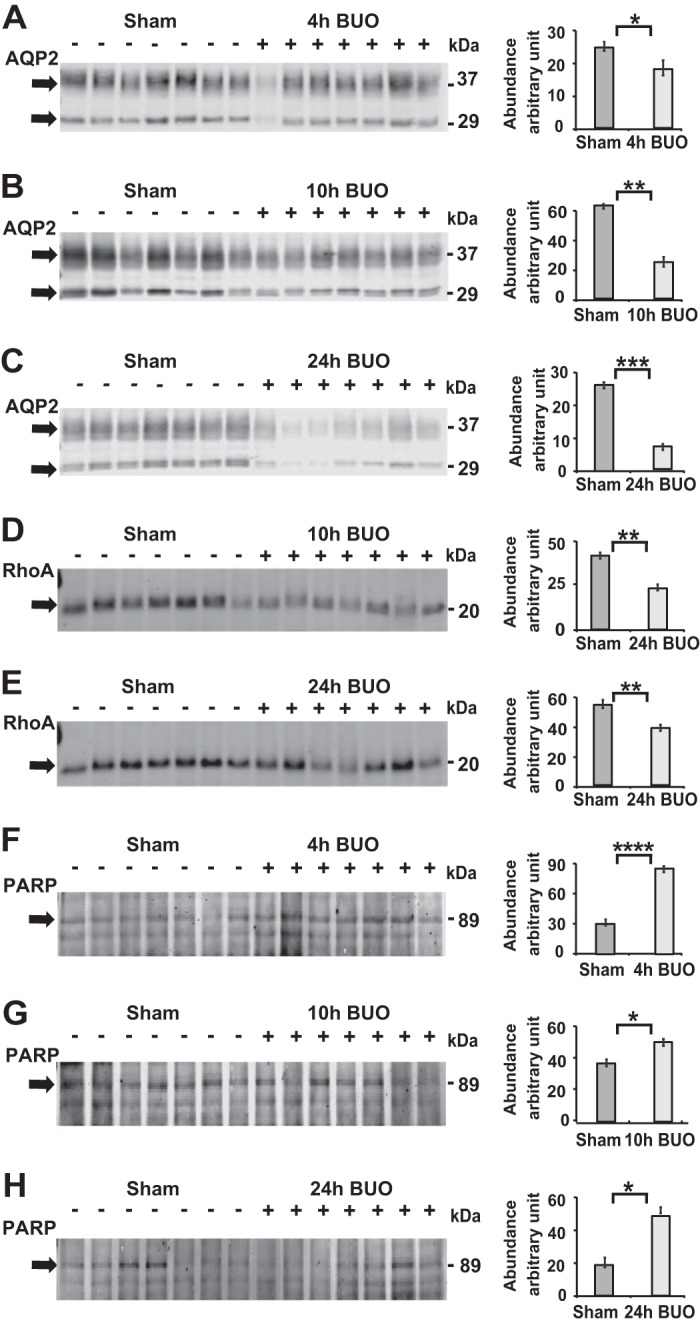
Downregulation of aquaporin-2 (AQP2) and RhoA and upregulation of cleaved poly(ADP-ribose) polymerase (PARP) in the inner medulla of 10- and 24-h bilateral ureteral obstruction (BUO) rats. A–C: representative immunoblots from whole inner medullas of 4-h BUO (+, n = 7) versus sham (−, n = 7), 10-h BUO (+, n = 7) versus sham (−, n = 7), and 24-h BUO (+, n = 7) versus sham (−, n = 7) rats demonstrated downregulations of AQP2 by 27%, 61%, and 73% after 4, 10, and 24 h, respectively, of BUO compared with sham. D and E: significantly downregulated RhoA by 55% and 77% was observed in the inner medullary collecting duct (IMCD) of 10- and 24-h BUO, respectively, compared with sham rats. F–H: significant upregulation of cleaved PARP by 182%, 157%, and 166% after 4, 10, and 24 h of BUO, respectively, compared with sham rats (unpaired t tests: *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001).
Fig. 8.
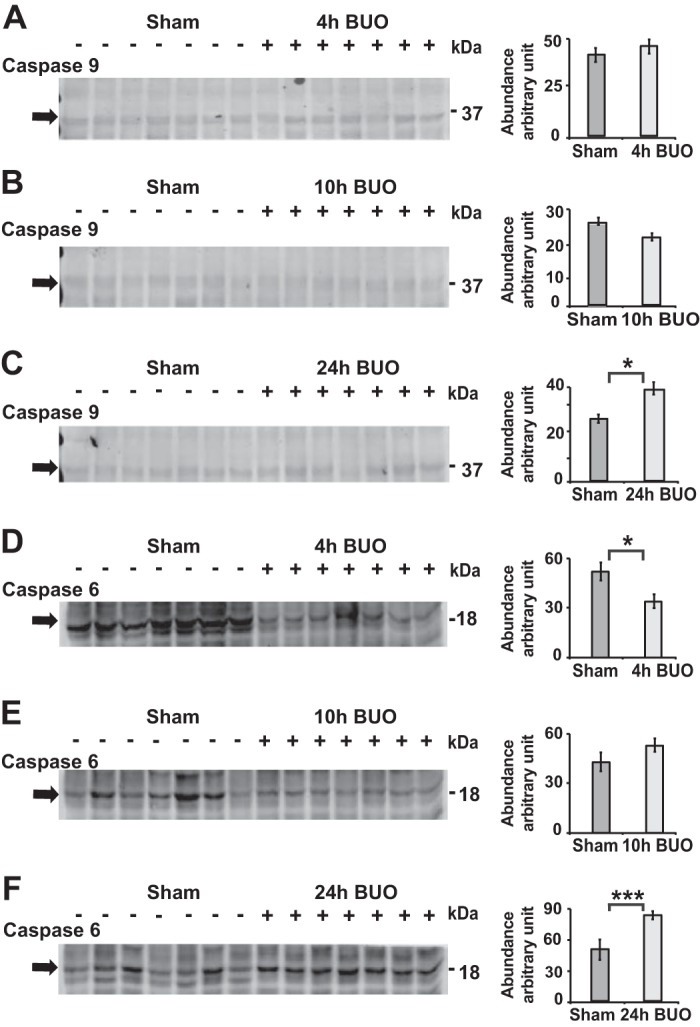
Upregulation of cleaved caspase-6 and cleaved caspase-9 in the inner medulla of 24-h bilateral ureteral obstruction (BUO) rats. A and B: representative immunoblots from whole inner medullas of 4-h BUO (+, n = 7) versus sham (−, n = 7) and 10-h BUO (+, n = 7) versus sham (−, n = 7) rats demonstrated that cleaved caspase-9 abundance did not significant differ from sham rats. D and E: cleaved caspase-6 was significant downregulated after 4-h BUO by 34% (unpaired t tests, *P < 0.05; D) but did not significant change compared with sham rats after 10-h BUO (E). C and F: densitometry analysis revealed upregulations of cleaved caspase-6 and cleaved caspase-9 by 48% and 49%, respectively, in 24-h BUO (+, n = 7) compared with sham (−, n = 7) rats (unpaired t tests, *P < 0.05, ***P < 0.005).
DISCUSSION
Urinary tract obstruction impairs renal blood flow, glomerular filtration, and tubular function. It has been known for more than six decades that pronounced urinary water and Na+ loss are postobstruction features (55). However, the initial molecular mechanisms of postobstructive diuresis remain incompletely understood. Although several studies have examined BUO at 2–48 h, they have focused on a limited number of target molecules in the kidney (18, 21, 36, 38, 39, 49). Because a previous study (49) of BUO observed decreased phosphorylation of AQP2 at 6 h postobstruction followed by a total AQP2 protein reduction at 12 h after BUO, we focused on the initial pathogenesis of obstructive diuresis by investigating proteomic changes that occur at 4 h of BUO. Our use of an unbiased systems approach allowed us to generate the most detailed view to date of the early effects of BUO.
Our proteomic analysis pointed toward the initial molecular mechanisms underlying BUO pathogenesis to be increased degradation of proteins. Supporting this, after 4 h of BUO, AQP2 in IMCD cells was sequestered in autophagosomes/lysosomes. The urea transporter UT-A1 is also expressed exclusively in IMCD cells and is essential for urine concentration (11). Although we did not identify any UT-A1 peptides in our proteomic analysis, we observed that UT-A1 colocalized with AQP2 and lysosomal markers in the kidney of BUO rats (Supplemental Fig. S3). Because decreased abundance of AQP2 would be sufficient to produce the urinary concentrating defect in BUO, it is likely that autophagy is the mechanism for AQP2 downregulation and subsequent development of postobstructive diuresis. However, to formally demonstrate that autophagy leads directly to AQP2 degradation, it would be necessary to show that inhibition of autophagy prevents AQP2 degradation. The use of a renal-collecting, tubule-specific, autophagy-related gene knockout mouse model would be required to assess this.
Autophagy is considered to be a cytoprotective response to maintain kidney tubule homeostasis under various stresses. This is demonstrated in many renal tubular injury models, including a progressive renal fibrosis model of 3-day UUO (22). In addition, a previous study (9) demonstrated that autophagy suppressed renal tubulointerstitial fibrosis in a 7-day UUO model using LC3−/− and beclin 1 heterozygous mice. More recently, a study by Nam et al. (33) demonstrated that autophagy attenuates tubulointerstitial fibrosis in a 7-day UUO model using renal distal, tubule-specific, autophagy related gene 7 knockout mice. However, the BUO model induced significantly higher intrarenal hydrostatic pressure than a UUO model at 24 h of obstruction (57). This level of severity may result in activation of an apoptotic response versus a prosurvival response, as demonstrated by upregulated PARP in the IM at 4 h of BUO in this study. An in vitro proteomic study in which rat proximal tubular cells were periodically stretched over 24 h as an obstruction mimic showed similar activation of a proapoptotic response in addition to downregulation of cytoskeletal proteins and tight and adherens junction proteins (40).
Mitophagy was also observed in acute kidney injury at 48 h, 7 days after UUO (29, 51), and at the early onset of hypokalemia-induced NDI (20). Here, downregulation of 16 mitochondrial inner membrane proteins, 8 proteins of which are involved in glycolysis/gluconeogenesis in the IMCD of 4-h BUO rats, supports a similar role of mitophagy in BUO. With respect to the IMCD, AQP2 cycling between the cytosol and apical plasma membrane and regulation of AQP2 phosphorylation at multiple sites are likely energy-consuming events. Therefore, the reduction in AQP2 as a common and early response to hypokalemia-induced NDI (20) and hypercalcemia-induced NDI (19) and BUO, being instigated by autophagy to minimize energy-consuming functions, seems viable.
Autophagic responses can regulate epithelial junctions to maintain cellular homeostasis (34). Our observation that autophagic degradation of cell-cell adhesion proteins is common between hypokalemia and hypercalcemia-induced NDI; also, BUO is intriguing and supports an interplay of autophagy and epithelial junctions, e.g., tight junctions, gap junctions, and adherens junctions, during pathological conditions, e.g., starvation (34). However, this interplay may manifest differently depending on the different types and severities of insults. For example, under nutrient starvation, autophagy enhances intestinal tight junction barrier function and reduces the permeability of ions and small-sized solutes by degradation of pore-forming claudin-2 (35).
The enlargement of the ER lumen and swollen mitochondria identified in IMCD cells after 4-h BUO are unique to the BUO model relative to other models we have studied with NDI (19, 20). These distinct findings may represent more severe IMCD stress in BUO than the other models. Under moderate stress, ER chaperones involved in the adaptive UPR pathway are upregulated (28); however, these proteins are downregulated under severe stress via the apoptotic UPR pathway (28). Our results are in parallel with the latter case, with significant reductions in four of seven ER chaperones [Hyou1, Hspa5, DnaJ heat shock protein family member B11 (Dnajb11), and Calr] involved in the UPR pathway. Hyou1 and Hspa5 have been previously shown to respond to mild and severe stress in renal tubules (28). The UPR pathway also regulates morphological changes to the ER, which may subsequently affect mitochondrial structure and function via altered mitochondria-associated ER membrane proteins, e.g., mitofusin-2 and protein kinase R-like kinase (PERK) (32). Disruption of adherens junctions was also demonstrated in a model of ER stress via chemical induction and, ultimately, apoptosis in HK-2 cells (8). This suggests an interplay between ER stress and autophagy responses on cell-cell adhesion molecules. Although the signaling pathway inducing autophagic AQP2 degradation after 4-h BUO remains unknown, we observed evidence of ER stress concurrent with swollen mitochondria and mitophagy in the IMCD. Therefore, well-established signaling pathways (e.g., PKR-like ER kinase) involved in cross-talk of ER stress-mediated autophagy (48) may play major roles in AQP2 degradation in this model. Meanwhile, the COOH terminus of heat shock complex 70-interacting protein interacting with AQP2 (56) may facilitate AQP2 degradation via lysosomal and/or ER-associated protein degradation pathways during ER stress.
In summary, the present study used an unbiased systems biology approach to generate a broad picture of the altered protein landscape within IMCD cells shortly after the onset of BUO. The results demonstrate an interplay between the ER, mitochondria, cell-cell junction proteins, autophagy, and mitophagy for maintaining cellular homeostasis under the stress imposed by BUO. It appears that although autophagy and the adaptive UPR pathway are enhanced during BUO to protect IMCD cells, the stress of BUO is ultimately severe enough to initiate the apoptotic UPR pathway and apoptosis is initiated.
GRANTS
This work was supported by Research Scholar Grants RMU 5380032 and TRG 5880224 from the Thailand Research Fund and Thammasat University. The proteomic study is supported by the Center of Excellent in Systems Biology, Research affairs, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand. The immunogold electron microscopic study is supported by the Danish Medical Research Council and the Lundbeck Foundation. T. Pisitkun is supported by the Chulalongkorn Academic Advancement into its 2nd Century project.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.P. and S.K. conceived and designed research; P.S., N.C., R.A.F., and S.K. performed experiments; P.S., C.B., K.C., N.C., R.A.F., T.P., and S.K. analyzed data; T.P, R.A.F., and S.K. interpreted results of experiments; S.K. prepared figures; S.K. drafted manuscript; K.G.H., R.A.F., and S.K. edited and revised manuscript; R.A.F., T.P., and S.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Mark A. Knepper, Dr. Chung-Lin Chou, and Dr. David M. Payne for advice. We thank Christina Schmidt for assistance with electron microscopy. The animal experiments were performed at the laboratory animal center at Thammasat University.
REFERENCES
- 1.EMMES Corporation North American Pediatric Renal Transplant Cooperative Study (NAPRTCS): Annual Report. Rockville, MD: EMMES Corporation, 2008. [Google Scholar]
- 2.Bander SJ, Buerkert JE, Martin D, Klahr S. Long-term effects of 24-hr unilateral ureteral obstruction on renal function in the rat. Kidney Int 28: 614–620, 1985. doi: 10.1038/ki.1985.173. [DOI] [PubMed] [Google Scholar]
- 3.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4: 484–494, 2009. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- 4.Campbell GA, Hu D, Okusa MD. Acute kidney injury in the cancer patient. Adv Chronic Kidney Dis 21: 64–71, 2014. doi: 10.1053/j.ackd.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Chandrika BB, Yang C, Ou Y, Feng X, Muhoza D, Holmes AF, Theus S, Deshmukh S, Haun RS, Kaushal GP. Endoplasmic reticulum stress-induced autophagy provides cytoprotection from chemical hypoxia and oxidant injury and ameliorates renal ischemia-reperfusion injury. PLoS One 10: e0140025, 2015. doi: 10.1371/journal.pone.0140025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng Z, Teo G, Krueger S, Rock TM, Koh HW, Choi H, Vogel C. Differential dynamics of the mammalian mRNA and protein expression response to misfolding stress. Mol Syst Biol 12: 855, 2016. doi: 10.15252/msb.20156423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372, 2008. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 8.Dickhout JG Chalal J, Matthews A, Carlisle RE, Tat V, Naiel S. Endoplasmic reticulum stress causes renal epithelial cell disjunction. J Pharmacol Rep 4: 62–68, 2016. [Google Scholar]
- 9.Ding Y, Kim S, Lee SY, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 25: 2835–2846, 2014. doi: 10.1681/ASN.2013101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Sayegh TY, Arora PD, Fan L, Laschinger CA, Greer PA, McCulloch CA, Kapus A. Phosphorylation of N-cadherin-associated cortactin by Fer kinase regulates N-cadherin mobility and intercellular adhesion strength. Mol Biol Cell 16: 5514–5527, 2005. doi: 10.1091/mbc.e05-05-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fenton RA, Chou CL, Stewart GS, Smith CP, Knepper MA. Urinary concentrating defect in mice with selective deletion of phloretin-sensitive urea transporters in the renal collecting duct. Proc Natl Acad Sci USA 101: 7469–7474, 2004. doi: 10.1073/pnas.0401704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fenton RA, Moeller HB, Hoffert JD, Yu MJ, Nielsen S, Knepper MA. Acute regulation of aquaporin-2 phosphorylation at Ser-264 by vasopressin. Proc Natl Acad Sci USA 105: 3134–3139, 2008. doi: 10.1073/pnas.0712338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fougeray S, Pallet N. Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat Rev Nephrol 11: 34–45, 2015. doi: 10.1038/nrneph.2014.201. [DOI] [PubMed] [Google Scholar]
- 14.Frøkiaer J, Marples D, Knepper MA, Nielsen S. Bilateral ureteral obstruction downregulates expression of vasopressin-sensitive AQP-2 water channel in rat kidney. Am J Physiol Renal Physiol 270: F657–F668, 1996. doi: 10.1152/ajprenal.1996.270.4.F657. [DOI] [PubMed] [Google Scholar]
- 15.Hoffert JD, Fenton RA, Moeller HB, Simons B, Tchapyjnikov D, McDill BW, Yu MJ, Pisitkun T, Chen F, Knepper MA. Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J Biol Chem 283: 24617–24627, 2008. doi: 10.1074/jbc.M803074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57, 2009. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 17.Inagi R, Ishimoto Y, Nangaku M. Proteostasis in endoplasmic reticulum–new mechanisms in kidney disease. Nat Rev Nephrol 10: 369–378, 2014. doi: 10.1038/nrneph.2014.67. [DOI] [PubMed] [Google Scholar]
- 18.Jensen AM, Bae EH, Fenton RA, Nørregaard R, Nielsen S, Kim SW, Frøkiaer J. Angiotensin II regulates V2 receptor and pAQP2 during ureteral obstruction. Am J Physiol Renal Physiol 296: F127–F134, 2009. doi: 10.1152/ajprenal.90479.2008. [DOI] [PubMed] [Google Scholar]
- 19.Khositseth S, Charngkaew K, Boonkrai C, Somparn P, Uawithya P, Chomanee N, Payne DM, Fenton RA, Pisitkun T. Hypercalcemia induces targeted autophagic degradation of aquaporin-2 at the onset of nephrogenic diabetes insipidus. Kidney Int 91: 1070–1087, 2017. doi: 10.1016/j.kint.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Khositseth S, Uawithya P, Somparn P, Charngkaew K, Thippamom N, Hoffert JD, Saeed F, Michael Payne D, Chen SH, Fenton RA, Pisitkun T. Autophagic degradation of aquaporin-2 is an early event in hypokalemia-induced nephrogenic diabetes insipidus. Sci Rep 5: 18311, 2015. doi: 10.1038/srep18311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SW, Cho SH, Oh BS, Yeum CH, Choi KC, Ahn KY, Lee J. Diminished renal expression of aquaporin water channels in rats with experimental bilateral ureteral obstruction. J Am Soc Nephrol 12: 2019–2028, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Kim WY, Nam SA, Song HC, Ko JS, Park SH, Kim HL, Choi EJ, Kim YS, Kim J, Kim YK. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology (Carlton) 17: 148–159, 2012. doi: 10.1111/j.1440-1797.2011.01541.x. [DOI] [PubMed] [Google Scholar]
- 23.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 132: 27–42, 2008. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li C, Klein JD, Wang W, Knepper MA, Nielsen S, Sands JM, Frøkiaer J. Altered expression of urea transporters in response to ureteral obstruction. Am J Physiol Renal Physiol 286: F1154–F1162, 2004. doi: 10.1152/ajprenal.00453.2003. [DOI] [PubMed] [Google Scholar]
- 25.Li C, Wang W, Kwon TH, Knepper MA, Nielsen S, Frøkiaer J. Altered expression of major renal Na transporters in rats with bilateral ureteral obstruction and release of obstruction. Am J Physiol Renal Physiol 285: F889–F901, 2003. doi: 10.1152/ajprenal.00170.2003. [DOI] [PubMed] [Google Scholar]
- 26.Li C, Wang W, Kwon TH, Knepper MA, Nielsen S, Frøkiaer J. Altered expression of major renal Na transporters in rats with unilateral ureteral obstruction. Am J Physiol Renal Physiol 284: F155–F166, 2003. doi: 10.1152/ajprenal.00272.2002. [DOI] [PubMed] [Google Scholar]
- 27.Li C, Wang W, Norregaard R, Knepper MA, Nielsen S, Frøkiaer J. Altered expression of epithelial sodium channel in rats with bilateral or unilateral ureteral obstruction. Am J Physiol Renal Physiol 293: F333–F341, 2007. doi: 10.1152/ajprenal.00372.2006. [DOI] [PubMed] [Google Scholar]
- 28.Lindenmeyer MT, Rastaldi MP, Ikehata M, Neusser MA, Kretzler M, Cohen CD, Schlöndorff D. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol 19: 2225–2236, 2008. doi: 10.1681/ASN.2007121313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu M, Sun Y, Xu M, Yu X, Zhang Y, Huang S, Ding G, Zhang A, Jia Z. Role of mitochondrial oxidative stress in modulating the expressions of aquaporins in obstructive kidney disease. Am J Physiol Renal Physiol 314: F658–F666, 2018. doi: 10.1152/ajprenal.00234.2017. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Beyer A, Aebersold R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165: 535–550, 2016. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 31.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966–968, 2010. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muñoz JP, Ivanova S, Sánchez-Wandelmer J, Martínez-Cristóbal P, Noguera E, Sancho A, Díaz-Ramos A, Hernández-Alvarez MI, Sebastián D, Mauvezin C, Palacín M, Zorzano A. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J 32: 2348–2361, 2013. doi: 10.1038/emboj.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nam SA, Kim WY, Kim JW, Park SH, Kim HL, Lee MS, Komatsu M, Ha H, Lim JH, Park CW, Yang CW, Kim J, Kim YK. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis 10: 78, 2019. doi: 10.1038/s41419-019-1356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nighot P, Ma T. Role of autophagy in the regulation of epithelial cell junctions. Tissue Barriers 4: e1171284, 2016. doi: 10.1080/21688370.2016.1171284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nighot PK, Hu CA, Ma TY. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J Biol Chem 290: 7234–7246, 2015. doi: 10.1074/jbc.M114.597492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilsson L, Madsen K, Topcu SO, Jensen BL, Frøkiær J, Nørregaard R. Disruption of cyclooxygenase-2 prevents downregulation of cortical AQP2 and AQP3 in response to bilateral ureteral obstruction in the mouse. Am J Physiol Renal Physiol 302: F1430–F1439, 2012. doi: 10.1152/ajprenal.00682.2011. [DOI] [PubMed] [Google Scholar]
- 37.Nishimoto G, Zelenina M, Li D, Yasui M, Aperia A, Nielsen S, Nairn AC. Arginine vasopressin stimulates phosphorylation of aquaporin-2 in rat renal tissue. Am J Physiol 276: F254–F259, 1999. doi: 10.1152/ajprenal.1999.276.2.F254. [DOI] [PubMed] [Google Scholar]
- 38.Nørregaard R, Jensen BL, Li C, Wang W, Knepper MA, Nielsen S, Frøkiaer J. COX-2 inhibition prevents downregulation of key renal water and sodium transport proteins in response to bilateral ureteral obstruction. Am J Physiol Renal Physiol 289: F322–F333, 2005. doi: 10.1152/ajprenal.00061.2005. [DOI] [PubMed] [Google Scholar]
- 39.Nørregaard R, Jensen BL, Topcu SO, Wang G, Schweer H, Nielsen S, Frøkiaer J. Urinary tract obstruction induces transient accumulation of COX-2-derived prostanoids in kidney tissue. Am J Physiol Regul Integr Comp Physiol 298: R1017–R1025, 2010. doi: 10.1152/ajpregu.00336.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orton DJ, Doucette AA, Maksym GN, MacLellan DL. Proteomic analysis of rat proximal tubule cells following stretch-induced apoptosis in an in vitro model of kidney obstruction. J Proteomics 100: 125–135, 2014. doi: 10.1016/j.jprot.2013.11.025. [DOI] [PubMed] [Google Scholar]
- 41.Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De Stefani D, Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53: 726–737, 2014. doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28: R170–R185, 2018. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pisitkun T, Jacob V, Schleicher SM, Chou CL, Yu MJ, Knepper MA. Akt and ERK1/2 pathways are components of the vasopressin signaling network in rat native IMCD. Am J Physiol Renal Physiol 295: F1030–F1043, 2008. doi: 10.1152/ajprenal.90339.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saheki Y, De Camilli P. Endoplasmic reticulum-plasma membrane contact sites. Annu Rev Biochem 86: 659–684, 2017. doi: 10.1146/annurev-biochem-061516-044932. [DOI] [PubMed] [Google Scholar]
- 45.Saito A, Imaizumi K. Unfolded protein response-dependent communication and contact among endoplasmic reticulum, mitochondria, and plasma membrane. Int J Mol Sci 19: 3215, 2018. doi: 10.3390/ijms19103215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakairi Y, Jacobson HR, Noland TD, Breyer MD. Luminal prostaglandin E receptors regulate salt and water transport in rabbit cortical collecting duct. Am J Physiol Renal Physiol 269: F257–F265, 1995. doi: 10.1152/ajprenal.1995.269.2.F257. [DOI] [PubMed] [Google Scholar]
- 47.Sandoval PC, Slentz DH, Pisitkun T, Saeed F, Hoffert JD, Knepper MA. Proteome-wide measurement of protein half-lives and translation rates in vasopressin-sensitive collecting duct cells. J Am Soc Nephrol 24: 1793–1805, 2013. doi: 10.1681/ASN.2013030279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song S, Tan J, Miao Y, Zhang Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J Cell Physiol 233: 3867–3874, 2018. doi: 10.1002/jcp.26137. [DOI] [PubMed] [Google Scholar]
- 49.Stødkilde L, Nørregaard R, Fenton RA, Wang G, Knepper MA, Frøkiær J. Bilateral ureteral obstruction induces early downregulation and redistribution of AQP2 and phosphorylated AQP2. Am J Physiol Renal Physiol 301: F226–F235, 2011. doi: 10.1152/ajprenal.00664.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stokes JB, Grupp C, Kinne RK. Purification of rat papillary collecting duct cells: functional and metabolic assessment. Am J Physiol Renal Physiol 253: F251–F262, 1987. doi: 10.1152/ajprenal.1987.253.2.F251. [DOI] [PubMed] [Google Scholar]
- 51.Sun Y, Zhang Y, Zhu Y, Zhang A, Huang S, Yin X, Ding G, Liu M, Jia Z. Inhibition of mitochondrial complex-1 restores the downregulation of aquaporins in obstructive nephropathy. Am J Physiol Renal Physiol 311: F777–F786, 2016. doi: 10.1152/ajprenal.00215.2015. [DOI] [PubMed] [Google Scholar]
- 52.Szabò I, Leanza L, Gulbins E, Zoratti M. Physiology of potassium channels in the inner membrane of mitochondria. Pflugers Arch 463: 231–246, 2012. doi: 10.1007/s00424-011-1058-7. [DOI] [PubMed] [Google Scholar]
- 53.Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res 29: 347–364, 2019. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740, 2016. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 55.Witte MH, Short FA, Hollander W Jr. Massive polyuria and natruresis following relief of urinary tract obstruction. Am J Med 37: 320–326, 1964. doi: 10.1016/0002-9343(64)90015-4. [DOI] [PubMed] [Google Scholar]
- 56.Wu Q, Moeller HB, Stevens DA, Sanchez-Hodge R, Childers G, Kortenoeven MLA, Cheng L, Rosenbaek LL, Rubel C, Patterson C, Pisitkun T, Schisler JC, Fenton RA. CHIP regulates aquaporin-2 quality control and body water homeostasis. J Am Soc Nephrol 29: 936–948, 2018. doi: 10.1681/ASN.2017050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yarger WE, Aynedjian HS, Bank N. A micropuncture study of postobstructive diuresis in the rat. J Clin Invest 51: 625–637, 1972. doi: 10.1172/JCI106852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeh CH, Chiang HS, Lai TY, Chien CT. Unilateral ureteral obstruction evokes renal tubular apoptosis via the enhanced oxidative stress and endoplasmic reticulum stress in the rat. Neurourol Urodyn 30: 472–479, 2011. doi: 10.1002/nau.20855. [DOI] [PubMed] [Google Scholar]