

Abstract
Regulation of axon guidance and pruning of inappropriate synapses by class 3 semaphorins are key to the development of neural circuits. Collapsin response mediator protein 2 (CRMP2) has been shown to regulate axon guidance by mediating semaphorin 3A (Sema3A) signaling; however, nothing is known about its role in synapse pruning. Here, using newly generated crmp2 −/− mice we demonstrate that CRMP2 has a moderate effect on Sema3A‐dependent axon guidance in vivo, and its deficiency leads to a mild defect in axon guidance in peripheral nerves and the corpus callosum. Surprisingly, crmp2 −/− mice display prominent defects in stereotyped axon pruning in hippocampus and visual cortex and altered dendritic spine remodeling, which is consistent with impaired Sema3F signaling and with models of autism spectrum disorder (ASD). We demonstrate that CRMP2 mediates Sema3F signaling in primary neurons and that crmp2 −/− mice display ASD‐related social behavior changes in the early postnatal period as well as in adults. Together, we demonstrate that CRMP2 mediates Sema3F‐dependent synapse pruning and its dysfunction shares histological and behavioral features of ASD.
Keywords: axon guidance, collapsin response mediator protein 2, dendritic spines, semaphorins, synapse pruning
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Neuroscience
CRMP2 mediates not only Sema3A‐dependent axon guidance, but also Sema3F‐dependent stereotyped axon pruning and pruning of dendritic spines. Crmp2−/− mice display behavioral defects related to autism spectrum disorder.

Introduction
The pattern of axonal connections is established during pre‐ and postnatal development by a cascade of multiple events. In embryogenesis, axonal growth cones are guided to their targets and multiple axon branches are formed. Since both correct and incorrect projections are formed, the embryonic brain connectome is only transient and the inaccurate connections are eliminated (pruned) in the early postnatal development 1. Defects in development and maturation of brain circuits have been linked to several neurodevelopmental disorders including autism spectrum disorder (ASD), schizophrenia, or epilepsy 2, 3, 4.
Generally, two types of pruning are recognized: (i) small‐scale axon pruning, regulated by neural activity or trophic support and (ii) large‐scale stereotyped axon pruning, which is genetically predetermined 1, 3. Stereotyped pruning can be further histologically divided into degeneration‐like 5 and retraction‐like 6, which has been linked to secreted semaphorins and their coreceptors, e.g., plexin‐A4 and plexin‐A3 6, 7. Intracellular mediators that transmit signals from plexins in axon pruning are not completely understood. One of the key molecule downstream of semaphorin 3A (Sema3A) signaling that directly interacts with cytoskeleton components is collapsin response mediator protein 2 (CRMP2) 8, 9, 10. In its non‐phosphorylated state, CRMP2 binds to tubulin dimers and promotes their polymerization 11. However, upon phosphorylation, it dissociates from microtubules promoting growth cone collapse 12. Two splice variants of Crmp2 have been found that differ at the N‐terminus—CRMP2A and CRMP2B 13, 14. CRMP2B is phosphorylated by cyclin‐dependent kinase 5 (CDK5) at Ser522, by GSK‐3β at Thr509, Thr514, and Ser518, and by Rho kinase at Thr555 12, 15, 16. We have recently shown that CRMP2A is phosphorylated at N‐terminal Ser27 by Cdk5, which promotes its interaction and stabilization by prolyl isomerase Pin1 14. Recently, CRMP2 deficiency in conditional knockout mice has been linked to schizophrenia due to changes in dendritic morphology (decreased spine number in CA1 neurons and layer 5 cortical neurons), behavioral changes (hyperactivity and social behavior impairment), and prepulse inhibition (PPI) deficit 17, 18. In addition to schizophrenia, deregulation of CRMP2 has been in humans associated with autism spectrum disorder (ASD), mood disorders, epilepsy, or Alzheimer's disease 19, 20, 21, 22, 23, 24, (SFARI Gene database). Among these, ASD and schizophrenia are of special interest since their symptomatic proximity but distinct pathogenesis. The exact role of CRMP2 in the development of these conditions has so far been elusive. Although schizophrenia and ASD share some behavioral characteristics (e.g., decreased cognitive functions, impaired social skills, repetitive behavior), they differ in the timing of their onset (early childhood for ASD, late adolescence for schizophrenia) 25 and the nature of the underlying neuronal connectivity disorder. Whereas hypoconnectivity (lower number of dendrites, dendritic spines, general decrease of white matter) is often present in schizophrenia, ASD has been associated with local hyperconnectivity caused by either increased synapse formation or their incomplete pruning 26. Because CRMP2 is downstream of semaphorin signaling, which controls axonal pruning 6, and it has been linked to both schizophrenia and ASD, it is one of the prime candidates to regulate this process. However, the in vivo analysis of both conditional and full CRMP2 knockout mice has to date mainly focused on the dendritic phenotype and associated behavioral aspects. In addition, the role of CRMP2 in class 3 semaphorin signaling (other than Sema3A), which has been previously linked to defects in pruning and ASD (particularly Sema3F), is so far not known.
In order to characterize the function of CRMP2 in axon growth, guidance, and pruning and its role in class 3 semaphorin signaling in vivo, we generated CRMP2 full knockout mice (crmp2 −/−). We show that CRMP2 participates in regulation of axon guidance in both central and peripheral nervous systems. In peripheral nervous system, deficiency of CRMP2 leads to mild overgrowth and increased branching of ophthalmic branch of trigeminal nerve and other selected peripheral nerves. In the central nervous system, we detected defects in postnatal callosal axon growth and guidance. Both of these systems are regulated by Sema3A, which was previously shown to induce CRMP2 phosphorylation and signaling in vitro. Indeed, we confirm that primary motor and DRG neurons isolated from crmp2 −/− mice have defects mediating Sema3A signaling.
Importantly, we show that CRMP2 is essential also for synaptic refinement as crmp2 −/− mice demonstrate defective stereotyped pruning of axons arising from hippocampus and visual cortex and inadequate elimination of dendritic spines in dentate gyrus (DG). Pruning in both of these systems is dependent on Sema3F rather than Sema3A, and its defect is in accord with ASD rather than schizophrenia‐like phenotype. In agreement with this hypothesis, we show that CRMP2 is essential for Sema3F‐induced axon retraction and dendritic spine remodeling in primary hippocampal cultures and that crmp2 −/− mice suffer from ultrasonic vocalizations defect in early postnatal stages as well as social behavioral changes in adults linked to ASD.
In summary, we provide evidence that in addition to its role in Sema3A‐dependent axon guidance, CRMP2 is a key mediator of Sema3F‐dependent axon pruning and dendritic spine remodeling. Our data highlight the importance of CRMP2 in neural circuit formation and refinement in vivo and demonstrate that its deficiency leads to defects in neural development associated with neurodevelopmental disorders, in particular ASD and schizophrenia.
Results
Crmp2 deficiency leads to axonal growth defects in peripheral nerves
We used TALEN (transcription activator‐like effector nucleases) mutagenesis to delete the second and third exons of the Crmp2 locus leading to a knockout of both CRMP2 protein isoforms CRMP2A and CRMP2B (Figs 1A and B, and EV1A and B). The knockout mice were viable and fertile, and the size of their brains was similar to the WT littermates (Fig EV1C). In agreement with Ref. 17, we observed ventriculomegaly in the homozygous mice (Fig EV1D and E).
Figure 1. CRMP2 deficiency leads to axonal growth defects in peripheral nerves.

-
AGeneration of crmp2 knockout mice by TALEN mutagenesis. Alternative first exons are highlighted in black and red.
-
BCRMP2 expression in adult cortex (layers VI–IV), corpus callosum (CC), and hippocampal area CA1. CRMP2 (green) is expressed throughout the cortex and hippocampus, and CRMP2A (red) is particularly strongly expressed in a subset of callosal axons. Nuclei are counterstained with Hoechst 33342. Scale bars: 50 μm.
-
CWhole‐mount immunohistochemistry with anti‐neurofilaments antibody for visualization of peripheral nerves. In crmp2 −/− embryos, the growth of ophthalmic branch of the trigeminal nerve is increased and axons are defasciculated (first and second rows, arrowheads). Similarly, we detected increased growth and branching of lateral branches of spinal nerves in crmp2 −/− embryos (third row, arrowheads). Trigeminal nerve (V) and its branches (Op—ophthalmic, Mx—maxillar, Md—mandibular) are indicated. Scale bars: 500 μm.
-
D, EQuantification of areas innervated by ophthalmic branches of trigeminal ganglion and spinal nerve, normalized to WT littermates (each dot depicts one embryo). Area of ophthalmic branch in crmp2 −/− was increased by 66% (P < 0.05, n = 10, 3 litters). For spinal nerves, total area in knockouts was increased by 45% (P < 0.05, WT n = 8, crmp2 −/− n = 7, 3 litters). Mean ± SD, *P < 0.05, t‐test.
-
FGrowth of motor neurons from E11.5–E12.5 WT and crmp2 −/− spinal cord explants in microfluidic chambers. Excerpts from 14‐h time‐lapse imaging are shown. Upon application of Sema3A into distal compartment, WT axons tend to stop or slow down their growth unlike crmp2 −/− axons. Purple lines highlight the growth path of individual axons in a distal chamber. Scale bars: 100 μm. See also Movie EV1.
-
GQuantification of the fraction of growing (> 50 μm) axons in one imaging field (WT: control 76.8 ± 5.7%, Sema3A 57.7 ± 10.6%, P < 0.001, crmp2 −/−: control 75.4 ± 10.4%, Sema3A 75.4 ± 6.7%, P = 0.99), n = 3 experiments per genotype, 7–8 explants per condition. Mean ± SD. ***P < 0.001, 2‐way ANOVA with Bonferroni's multiple comparison test.
Figure EV1. Related to Fig 1. Analysis of the brain morphology of crmp2 −/− mice.

- CRMP2 (A + B isoforms, arrows) and CRMP2A (arrowhead) isoform immunostaining of coronal brain sections from adult WT and crmp2 −/− mice. Both isoforms are missing in crmp2 −/− mice. Yellow rectangles depict the area shown in Fig 1B. CRMP2B is present throughout the cortex, and hippocampal CA1 and CA3 regions. CRMP2A is localized mostly in callosal axons (CC), mossy fibers (mf), and inner molecular layer (mo) of dentate gyrus. Scale bars: 100 μm.
- Western blot analysis of brain lysates of crmp2 +/+, +/− and −/− mice.
- Upper view of WT and crmp2 −/− brains showing similar brain size. Scale bars: 5 mm.
- Three 250‐μm coronal sections from n = 3 WT and crmp2 −/− mice demonstrate enlarged ventricles in crmp2 −/− mice. Scale bars: 5 mm.
- Quantification of the ventricle areas normalized to total slice area (ventricle/slice index, WT 0.011 ± 0.001, crmp2 −/− 0.027 ± 0.003, P < 0.001, n = 3 animals/genotype). Mean ± SD, ***P < 0.001, t‐test.
- Analysis of cortical neuron migration (WT/crmp2 +/− n = 6 embryos, crmp2 −/− n = 4 embryos). Both WTs and heterozygous (Het) embryos were used as controls. Embryos were electroporated with GFP expressing plasmid at E14.5 and analyzed at E17.5. Quantification of percentages of GFP‐positive neurons in different layers is shown. CP—cortical plate, IVZ—interventricular zone, VZ—ventricular zone. Scale bars: 100 μm, mean ± SD, t‐test.
CRMP2 has been shown to mediate Sema3A signaling and regulate axon guidance in vitro 8. To test whether CRMP2 deficiency leads to altered axon growth also in vivo, we first analyzed the development of peripheral nerves in E10.5–E12.5 embryos using whole‐mount immunohistochemistry (Fig EV2A) as their development is regulated by Sema3A 27, 28, 29, 30. We quantified changes in neuron growth by measuring the area innervated by axons from each given nerve and branching by counting of the total number of branches. The growth and branching of ophthalmic branch of the trigeminal nerve were significantly increased in crmp2 −/− mice at E10.5–E12.5 and similarly increased was the sprouting of the lateral branches of the spinal nerves. (Axon projections from dorsal root ganglions (DRGs) to the spinal cord were not changed in crmp2 −/− mice) (Figs 1C–E, and EV2B and C). Since CRMP2 deficiency in vivo led to increased, rather than reduced axon growth, our data indicate that the growth‐promoting function of CRMP2 can be compensated in vivo at least in the tested neurons, but that its function as a mediator of repulsive axon guidance signals is unique and cannot be fully rescued by other proteins. Since the growth of both ophthalmic branch and the lateral branches of the spinal nerves is negatively regulated by Sema3A 28, our data support the function of CRMP2 as a mediator of Sema3A signaling.
Figure EV2. Related to Fig 1. Peripheral nerve growth and motor neuron cultures in microfluidic chambers.

- WT and crmp2 −/− E11.5 embryos stained with antibody against neurofilaments. All peripheral nerves are present in crmp2 −/−. III—oculomotor, IV—trochlear, V—trigeminal, VII—facial, VIII—vestibulocochlear, IX—glossopharyngeal, X—vagal, and XI—accessory nerve. Op—ophthalmic branch, Mx—maxillar branch, and Md—mandibular branch of the trigeminal nerve.
- Whole‐mount immunolabeling of embryos at E12.5 (WT n = 4, crmp2 −/− n = 5, 2 litters), note the increased branching of the Op branch in crmp2 −/−. Scale bars: 500 μm.
- Quantification of the number of the Op branches (WT 67 ± 4.5, crmp2 −/− 86 ± 10.3, P < 0.05), mean ± SD are shown, *P < 0.05, t‐test.
- Increased levels of CRMP4 and CRMP1 in crmp2 −/− brain lysates.
- Adjacent coronal slices of E12.5 embryonic head sections stained with anti‐neurofilaments and anti‐CRMP4 antibodies to confirm the presence of CRMP4 in peripheral nerves. Arrows indicate the same nerves in different sections. Scale bars: 200 μm.
- Adjacent coronal sections of E12.5 embryonic heads stained with anti‐neurofilaments and anti‐CRMP2/2A antibodies to confirm the presence of CRMP2 isoforms in peripheral nerves. Arrows indicate the same nerves in different sections. Scale bars: 100 μm.
- Growth cone collapse assay revealed decreased reactivity to Sema3A in DRGs isolated from crmp2 −/− embryos. DRGs (n ≥ 3 explants per genotype and condition) were stimulated with different Sema3A concentrations for 30 min, fixed, and stained for actin (green) and β3‐tubulin (red). Scale bars: 100 μm, mean ± SD, ***P < 0.001, 2‐way ANOVA with Bonferroni's multiple comparison test.
- Experimental design of motor neuron cultures in microfluidic chambers using spinal cord explants. Dashed squares in the second row show areas that were scanned and analyzed.
To directly demonstrate that CRMP2 deficiency interferes with Sema3A signaling in peripheral neurons isolated from crmp2 −/− mice, we took an advantage of microfluidic chamber approach for extraaxonal environment manipulation. In this chamber, cell bodies and axons are separated in proximal or distal compartment, respectively 31. We prepared spinal cord explants from WT and crmp2 −/− E11.5–E12.5 embryos and cultured them for 4–5 days until motor neuron axons crossed to the distal compartment (Fig EV2H). Then, we stained axons with Alexa 647‐conjugated cholera toxin subunit B and analyzed their growth by live imaging. Without Sema3A addition, the growth of WT and crmp2 −/− explants and their axons were comparable indicating again that the axon growth‐promoting function of CRMP2 is redundant, at least in the tested neurons. When we added Sema3A (5 nM) to the distal (axonal) compartment, we observed decrease in the number of growing WT axons, as expected (P < 0.001, Fig 1F and G, Movie EV1). However, in crmp2 −/− explants, Sema3A had no significant effect on the number of growing axons (P = 0.99, Fig 2F and G, Movie EV1). In addition, we analyzed Sema3A‐induced growth cone collapse in E11.5–E12.5 isolated DRGs. Neurons were treated with Sema3A for 30 min, then fixed, and stained for actin and β3‐tubulin. In crmp2 −/− neurons, responsiveness to Sema3A was significantly decreased (Fig EV2G) which is in accord with the previously published data 8, 32. Thus, our data demonstrate that CRMP2 mediates Sema3A signaling in vitro and participates in guidance of multiple peripheral nerves in Sema3A‐regulated regions in vivo.
Figure 2. CRMP2 regulates postnatal development of corpus callosum.

- CRMP2 deficiency leads to callosal hypoplasia. Shortening of corpus callosum (arrowheads) is apparent in both sagittal (first row) and coronal sections (second row) of adult brains. Scale bars: 1 mm.
- Quantification of callosal length in 30‐day‐old mice (P30, n = 3, crmp2 −/− is 80.8 ± 5% of WT, P = 0.003) and adult mice (n = 3, crmp2 −/− is 80.5 ± 3% of WT, P = 0.002), mean ± SD, *P < 0.05, t‐test.
- Labeling of adult corpus callosum with anti‐neurofilaments antibody in sagittal sections. Outline depicts missing posterior part of the tract in crmp2 −/− mice. Caudal part of the corpus callosum in WTs is located dorsally above the habenular commissure (hbc), while in crmp2 −/− mice, callosum terminates rostrally before reaching the hbc (arrows). PC indicates posterior commissure. Scale bars: 500 μm.
- The growth of GFP‐labeled callosal axons in the contralateral cortex at P6 (embryos were electroporated at E15.5). Note the disorganized paths of crmp2 −/− axons. Scale bars: 200 μm.
- Schematic drawing of the callosal axon path and axonal tortuosity calculation. Pyramidal neurons in layer II/III project their axons into the contralateral cortex [rectangle depicts the area displayed in (D)]. Axonal tortuosity quantification as shown in (F), tortuosity = 1 if a = b.
- Quantification of tortuosity of axons (WT n = 6 pups, crmp2 −/− n = 3 pups) upon their exit from the callosal tract (WT 1.016 ± 0.003 vs. crmp2 −/− 1.044 ± 0.012, P < 0.001), mean ± SD, ***P < 0.001, t‐test.
- DiI‐labeled callosal axons from P9 oblique brain sections (see the Appendix Fig S1) and their reconstruction in Neurolucida 360 (WT n = 6, crmp2 −/− n = 9). Scale bars: 200 μm.
- Polar histograms of callosal axons reconstructed in (G). Note the broader range of axon growth angles in crmp2 −/− mice.
- Left: schematic representation of polar histogram analysis by clustering the traced axons (WT n = 6 pups, crmp2 −/− n = 9 pups) into three groups based on the growth angles. Right: proportion of axons growing in selected clusters (WT, 0° to ±20°: 67.3 ± 10.3%; ±20° to ±40°: 21.2 ± 5.3%; ±40° to ±60°: 6.22 ± 3.2%; crmp2 −/−, 58.5 ± 5%, P = 0.047, 27.7 ± 4.3%, P = 0.022, 8.64 ± 2.25%, P = 0.11), mean ± SD, *P < 0.05, t‐test.
CRMP2 regulates anatomy and axon guidance in corpus callosum
In addition to the peripheral nerves, we analyzed the effect of CRMP2 deficiency on axonal growth also in the central nervous system. We detected anatomical changes in the largest axonal bundle of the brain, the corpus callosum, in crmp2 −/− mice. The length of corpus callosum was significantly reduced in juvenile (−19.2%, P < 0.05) as well as adult (−19.5%, P < 0.05) knockout mice as can be appreciated in both sagittal and coronal sections (Fig 2A and B). More detailed labeling of the tract with anti‐neurofilaments antibody showed that the posterior part of the corpus callosum (splenium) is markedly hypoplastic in crmp2 −/− mice, ending rostrally to the habenular commissure, while the WT splenium is longer, located just above the habenular commissure (Fig 2C).
Callosal axons have been shown to be guided by Sema3A 33, 34. Thus, we analyzed whether dysgenesis of the corpus callosum we detected in crmp2 −/− mice is also accompanied by defects in callosal axon guidance. We in utero electroporated WT and crmp2 −/− embryonal cortices with pCAGGS‐EGFP vector at E15.5, which results in labeling of cortical layer 2/3 (i.e., mainly callosal‐projecting neurons). The brains were collected at postnatal day 6 (P6), fixed in 4% PFA, and cut coronally to trace callosal axons in a hemisphere contralateral to the electroporation site (Fig 2D and E). At this stage, axons from somatosensory cortex enter the contralateral cortex in WT 35. Most of the WT axons showed well organized, parallel growth. In contrast, crmp2 −/− axons often failed to grow in an organized, parallel way upon leaving the main callosal tract, their distribution in the cortex seemed uneven. We tracked the electroporated axons after leaving callosal bundle at the contralateral site and quantified their tortuosity (i.e., the ratio of real length of the segment vs. distance of the first and last point of the segment, Fig 2E). We found that tortuosity was significantly higher in crmp2 −/− in comparison with WT (P < 0.001, Fig 2F).
Importantly, in some coronal sections of crmp2 −/− mice we detected deregulated growth of callosal axons even in the midline. Together with the reduced length of corpus callosum in crmp2 −/− mice in the rostro‐caudal axis we detected (Fig 2A–C), it was suggested that CRMP2 deficiency may alter the rostro‐caudal guidance of callosal axons in the midline. To test this hypothesis, we traced the callosal axons by injecting DiI into the fixed P9 and adult somatosensory cortices and analyzed organization and fasciculation of the traced axons in the midline (see Materials and Methods). We found that at P9, crmp2 −/− axons were significantly more distorted as seen upon plotting to polar histograms or fan‐in diagrams (interval (0°, ±20°): P < 0.05, interval (±20°, ±40°): P = 0.02, Fig 2H–I, Appendix Fig S1A and B).
Together, our results show that CRMP2 regulates growth and guidance of selected axons in both peripheral (PNS) and central nervous system (CNS). In addition, since CRMP2 deficiency in crmp2 −/− mice leads to enhanced, rather than reduced axon growth, our results suggest that in the analyzed PNS and CNS regions, the mediation of the repulsive axon guidance signals by CRMP2 takes precedence over its axon growth‐promoting function.
CRMP2 mediates Sema3F‐driven, but not Sema3A‐driven axon pruning
CRMP2 has been associated with neurodevelopmental disorders like schizophrenia and ASD characterized by altered brain connectivity 36 and defects in postnatal synaptic refinement through axon and dendrite pruning 26, 37. Importantly, in many regions Sema3F seems to play a more important role in pruning than Sema3A 6, 7. Therefore, we asked whether CRMP2 regulates axon pruning and if, in this function, it mediates Sema3A or rather Sema3F signaling. For this, we analyzed two developing axonal systems showing either Sema3F‐mediated pruning (the infrapyramidal bundle, IPB; 6) or Sema3A‐mediated pruning (the hippocamposeptal bundle; 6). First, we analyzed stereotyped pruning of infrapyramidal bundle (IPB) of hippocampal mossy fibers taking place between P20 and P40 and regulated by Sema3F and its receptor complex (Nrp2/PlxnA3) 6. At P14 (i.e., before pruning), calbindin immunostaining revealed the presence of IPB in both WT and crmp2 −/− mice (Fig 3A and B). Synapses were formed in both IPB and the main bundles as revealed by VGluT1 staining (Fig 3A and B). In 7‐week‐old animals, however, when pruning was complete in WT, the IPB remained present in crmp2 −/− mice, and their IPB index (IPB length/main bundle length) was significantly higher than in WT (P < 0.001, Fig 3C–G). The same pattern of IPB pruning was detected also in sagittal sections (Fig EV3A). To determine the maturity of IPB synapses, we stained adult coronal sections with antibodies against VgluT2 or VgluT1, which are both expressed in developing mossy fibers, but in adult hippocampus only VgluT1 is present 38. We found VgluT1 in the main bundles of WTs and crmp2 −/− mice, as well as in the unpruned IPBs of crmp2 −/− (Fig 3E and F). We did not detect the immature VgluT2 signal (Fig 3C and D). Thus, the IPB axons formed during postnatal development persist in crmp2 −/− mice into adulthood and form mature synapses. These results demonstrate that IPB pruning is defective in crmp2 −/− mice (as is the case in Sema3F −/−, Nrp2 −/−, and PlxnA3 −/− mice) 6.
Figure 3. Infrapyramidal bundle fails to prune in crmp2 −/− mice.
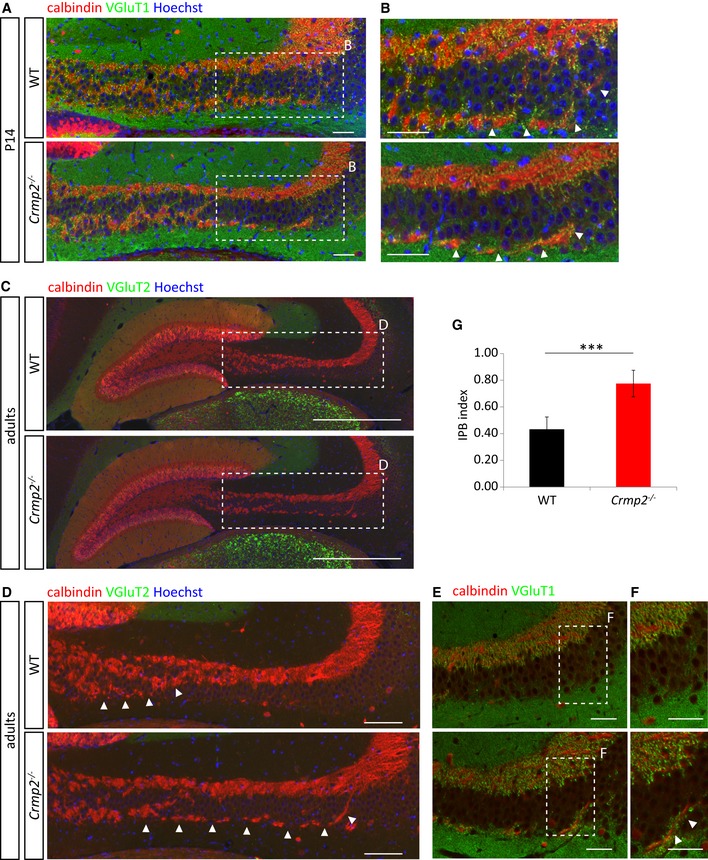
-
A, B(A) Coronal sections of P14 brains stained for calbindin and VgluT1. Infrapyramidal bundle (IPB) progresses into hippocampal CA3 region in both WT and crmp2 −/− mice. Nuclei are counterstained with Hoechst 33342. (B) Details from depicted regions in (A), arrowheads show the course of IPB. Scale bars: 100 μm.
-
C, D(C) Staining for calbindin shows unpruned infrapyramidal bundle in adult crmp2 −/− mice. No VGluT2 signal in hippocampal CA3 region is present in WT and crmp2 −/− suggesting mature synapses. Scale bars: 500 μm. (D) Details from depicted regions in (C), arrowheads show the course of IPB. Scale bars: 100 μm.
-
E, FStaining for calbindin and VgluT1 shows mature synapses in the main bundle in both genotypes and in the IPB in crmp2 −/− mice (arrowheads). Scale bars: 100 μm.
-
GQuantification of IPB index (length of the IPB vs. main bundle, WT 0.43 ± 0.05, crmp2 −/− 0.78 ± 0.05, P < 0.001) counted from adult coronal sections (WT n = 4 mice, crmp2 −/− n = 5 mice). Mean ± SD, ***P < 0.001, t‐test.
Figure EV3. Related to Fig 3. Axon pruning in crmp2 −/− mice is altered in Sema3F‐related but not Sema3A‐related regions.
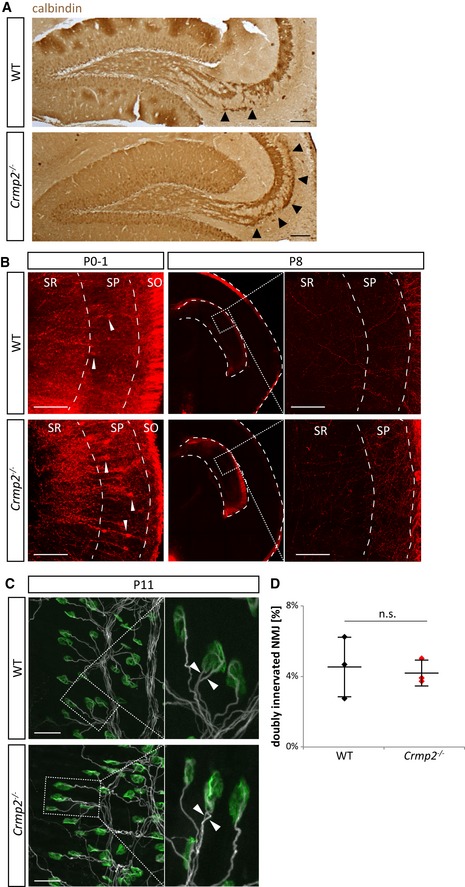
- Calbindin immunostaining of sagittal brain sections shows defects in pruning similar to those seen in coronal sections of WT and crmp2 −/− mice in Fig 3. Arrowheads indicate the infrapyramidal bundle. Scale bars: 100 μm.
- Hippocamposeptal axons are pruned in both WT and crmp2 −/− mice (n = 3). At P0–1, retrogradely DiI‐labeled CA1 pyramidal neurons showed strong signal in both WTs and crmp2 −/− indicating the presence of CA1 projections into the medial septum. (Arrowheads indicate retrogradely labeled cell bodies.) Conversely, at P8, no retrogradely labeled CA1 cell bodies were detected in CA1 granular layer in either WT or crmp2 −/− mice. SR, SP, and SO indicate stratum radiatum, stratum pyramidale, and stratum oriens, respectively. Scale bars: 100 μm.
- Maximum intensity projections of P11 triangularis sterni muscle NMJs of WT and crmp2 −/− mice (n = 3). Axons were stained with βIII‐tubulin antibody (white) and postsynaptic acetylcholine receptors with α‐bungarotoxin (green). Boxed areas are shown on the right at higher magnification with examples of doubly innervated NMJs where two axons are competing for synaptic innervation (arrowheads). Scale bars: 50 μm.
- Quantification of the number of doubly innervated NMJs revealed no significant difference (WT 4.5 ± 1.8%, cmrp2 −/− 4.2 ± 0.7%, P > 0.99, n = 3 pups/genotype). Mean ± SD, Mann–Whitney test.
We next tested whether stereotyped pruning of a different group of axons arising from hippocampus—the hippocamposeptal axons of CA1 pyramidal neurons—is also affected in crmp2 −/− mice. CA1 neurons send their axons into medial and lateral septum at P0–1. However, at P8, only the axons projecting to the lateral septum persist, while the ones projecting into the medial septum are pruned 39. In this system, the pruning is mediated by Sema3A and not Sema3F 6. Our analysis of the development of the CA1 hippocamposeptal axons in crmp2 −/− mouse brains by retrograde DiI tracing (Fig EV3B) showed no change in these brains as compared to WT brains at P0 or P8, indicating that the Sema3A‐mediated pruning of hippocamposeptal axons is not affected in crmp2 −/− mice.
To further support the role of CRMP2 as a mediator of Sema3F‐driven pruning, we analyzed pruning of corticospinal axons of visual cortex neurons that have been previously shown to be dependent on Sema3F signaling. In the early developmental stages, these neurons send their projections not only to the superior colliculus but also to two inappropriate targets, i.e., the inferior colliculus (IC) and the spinal cord. During the third postnatal week, inappropriate axons are eliminated through a pruning process regulated by Sema3F 7. We analyzed the development of the visual cortex projection by the means of DiI anterograde tracing (Fig 4A and B). At P9, before the pruning period, there was no significant difference between WT and crmp2 −/− in the VP index (fluorescence intensity of corticospinal axons after vs. before the branch point). In adult WT mice, though, the inappropriate corticospinal axons were largely pruned [VP index significantly dropped (P < 0.01, Fig 4C)], while in crmp2 −/− mice, they were still largely present [the VP indexes were not significantly different between P9 and adult (P = 0.24); Fig 4C]. These data demonstrate that CRMP2 participates in postnatal refinement of corticospinal visual axons and is consistent with its role in mediating Sema3F signaling.
Figure 4. Defective pruning of corticospinal visual axons in crmp2 −/− mice.

- Upper row: DiI tracing of the visual cortex axons at P9 (before pruning), sagittal sections. Branching point of the tract is shown. Lower row: visual cortex axons in adult mice after pruning period. Note significantly reduced number of axons continuing into pyramidal tract in WT (dashed arrow) (arrows—2 branches of corticospinal visual axons). Maximum projections are shown. Scale bars: 200 μm.
- Schematic drawing of DiI injection and axon tracing. Axons that initially enter pyramidal tract fail to prune in crmp2 −/− mice (red line).
- Quantification of VP (visual pruning) index (fluorescence intensity of pyramidal axons after vs. before the branch point). In WT animals after the pruning period, only a minor part of axons descends toward the pyramidal tract (WT: P9 0.65 ± 0.09, adults 0.17 ± 0.04, P = 0.006). However, in crmp2 −/− mice, corticospinal axons are still largely present, and their VP indexes are not significantly different between adult and P9 stages (crmp2 −/−: P9 0.54 ± 0.18, adults 0.62 ± 0.27, P = 0.24). Mean ± SD, **P < 0.01, 2‐way ANOVA with Bonferroni's multiple comparison test. P9: n = 3 pups/genotype, adults: n = 5 mice/genotype.
While, in development, both CNS and PNS projections are refined by pruning, we did not detect changes in the pruning of the peripheral neuromuscular junctions (NMJs) in crmp2 −/− mice using trigonum sterni muscle [the number of double‐innervated NMJs was similar in WT and mutant mice at P11 (P > 0.99, Fig EV3C and D)].
Finally, we asked whether Sema3F‐dependent axonal guidance is also mediated by CRMP2. Axon guidance is affected in two specific axonal bundles in Sema3F −/− mice 40—namely the anterior commissure (AC) and the retroflex fascicle. Immunostaining of P6 and P14 brains with anti‐neurofilaments antibody revealed that both structures are present in crmp2 −/− mice (Fig EV4A and D). However, temporal limb of the AC appeared to be hypoplastic. We therefore analyzed morphology of AC in the horizontal sections (Fig EV4B). In knockouts, the diameter of temporal limb was indeed significantly thinner and the olfactory limb defasciculated (Fig EV4B and C). The data thus indicate that CRMP2 also contributes to Sema3F‐driven axon guidance.
Figure EV4. Related to Fig 4. Growth of anterior commissure and retroflex fascicle in crmp2 −/− mice.

- At P14, anterior commissure (AC) is present in both WT and crmp2 −/− mice. Coronal sections labeled with antibody against neurofilaments show reduced temporal limb extension. Scale bars: 1 mm.
- AC hypoplasia in crmp2 −/− mice. Adult brains (n = 3) were sliced horizontally (250 μm sections) to reveal both frontal and temporal limbs of an AC. Temporal limb in knockouts is thinner (arrowheads, right magnification box), and frontal limb shows various degree of defasciculation. Midline thickness (arrowheads, left magnification box) was similar in both WT and knockouts. Scale bars: 5 mm (left), 1 mm (details on the right).
- Quantification of the diameter of AC in the temporal limb (crmp2 −/− is 70 ± 3% of WT, P = 0.0056) and in the midline (crmp2 −/− is 88 ± 1% of WT, P = 0.12, n = 3 mice/genotype). Mean ± SD, *P < 0.05, t‐test.
- Retroflex fascicle (fr) and mammillothalamic tract (mt) are normally formed in crmp2 −/− mice. Coronal sections at the level of posterior commissure (pc) are shown. Scale bars: 1 mm.
In conclusion, we found significant differences between WT and crmp2 −/− mice in stereotyped pruning in regions controlled by Sema3F (visual cortex axons, IPB). In contrast, we found no differences in regions controlled by Sema3A (i.e., hippocamposeptal axons) suggesting that CRMP2 mediates Sema3F‐driven, but not Sema3A‐driven axon pruning.
CRMP2 mediates Sema3F‐dependent axon retraction in vitro
In order to directly show that CRMP2 is necessary for Sema3F‐triggered axon pruning, but dispensable for Sema3A‐dependent pruning, we tested pruning in an in vitro system. We prepared dissociated hippocampal cultures from WT and crmp2 −/− E16.5 embryos and transfected them with EGFP at DIV1 (1 day in vitro). At DIV7, we added Sema3A or Sema3F into the cultures and analyzed the axon behavior in fluorescence microscope by time‐lapse imaging. DIV7, which is an early stage of synapse formation 41, was chosen to facilitate analysis of neurons in still less complex connectivity patterns (Fig 5A and B, [Link], [Link]). We analyzed only stable axon terminals that did not show any movement in 1‐h period prior to addition of the guidance cues (45% of all labeled axon terminals). In control conditions in both wild‐type and knockout, a small number of the stable axons were spontaneously retracting (WT 13%, KO 20%, P = 0.03, Fig 5C). After addition of either Sema3A or Sema3F into WT culture, we observed a threefold increase in axon retractions (32% for Sema3A, P < 0.001, 41% for Sema3F, P < 0.001, Fig 5C). However, in crmp2 −/−, this increase was detectable only after Sema3A (30%, P < 0.05) and not after Sema3F (22%, P > 0.99). These data demonstrate, that in primary neuron cultures undergoing synaptogenesis, CRMP2 is essential to mediate Sema3F but not Sema3A signaling. This is in agreement with our in vivo findings that stereotyped pruning in crmp2 −/− is affected in Sema3F‐controlled, but not in Sema3A‐controled regions.
Figure 5. CRMP2 mediates Sema3F signaling in primary neurons.
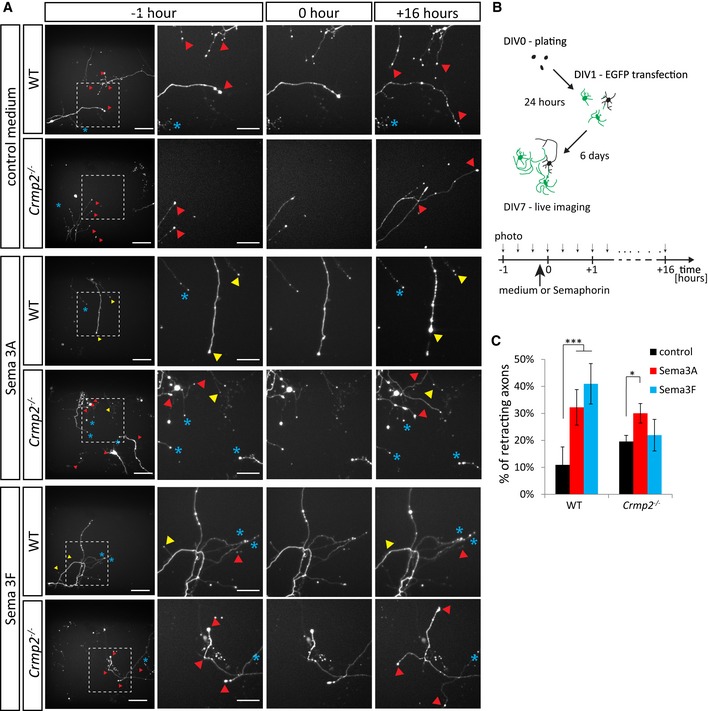
- Time‐lapse imaging of DIV7 cultured hippocampal neurons before and after semaphorin stimulation. Upper panel: axon growth without semaphorin stimulation. Middle panel: stimulation with Sema3A (0 h) (1 nM, n = 669 axons for WT, 761 axons for knockout) causes retraction of both WT and crmp2 −/− neurons. Lower panel: stimulation with Sema3F (5 nM, n = 602 axons for WT, 955 axons for knockout) causes axon retraction in WT, but not in crmp2 −/− neurons. Red triangles depict growing axons, yellow retracting axons, and blue asterisks indicate steady non‐growing axons. See also [Link], [Link]. Scale bars: 100 μm (whole image field) and 50 μm (magnified).
- Schematic drawing of the experimental setup.
- Quantification of retracting axons (number of retracting vs. steady axons, three experiments). WT: control 13.4 ± 5%, Sema3A 31.2 ± 6% (P < 0.001), Sema3F 36.9 ± 8.7% (P < 0.001); crmp2 −/−: control 19.8 ± 1.8%, Sema3A 28.5 ± 4.4% (P < 0.05), Sema3F 22.4 ± 4.1% (P > 0.99), mean ± SD are shown. ***P < 0.001, *P < 0.05, 2‐way ANOVA with Bonferroni's multiple comparison test.
CRMP2 regulates dendritic spine remodeling in hippocampal granule cells
Besides triggering axon pruning, Sema3F regulates also the development of some classes of dendritic spines (e.g., spines of dentate gyrus (DG) granule cells) 42. In contrast, Sema3A/Nrp1 signaling seems to be dispensable for dendritic spine morphogenesis 42, 43. In order to test whether CRMP2 participates also in spine development/morphogenesis, we DiOlistically labeled DG neurons and analyzed their dendritic spines. Surprisingly, we found significantly increased spine density in crmp2 −/− adult DG granule cells compared to WTs (Fig 6B and C, P < 0.001). This phenotype was similar to that found in Sema3F −/− and Nrp2 −/− 42. Next, we analyzed branching of DG granule cell dendrites. Sholl analysis revealed no differences between WT and crmp2 −/− mice (Fig 6D and E, P > 0.99), which is again in line with the phenotype of Sema3F −/− mice 42.
Figure 6. CRMP2 regulates dendritic spine refinement in dentate gyrus granule cells.
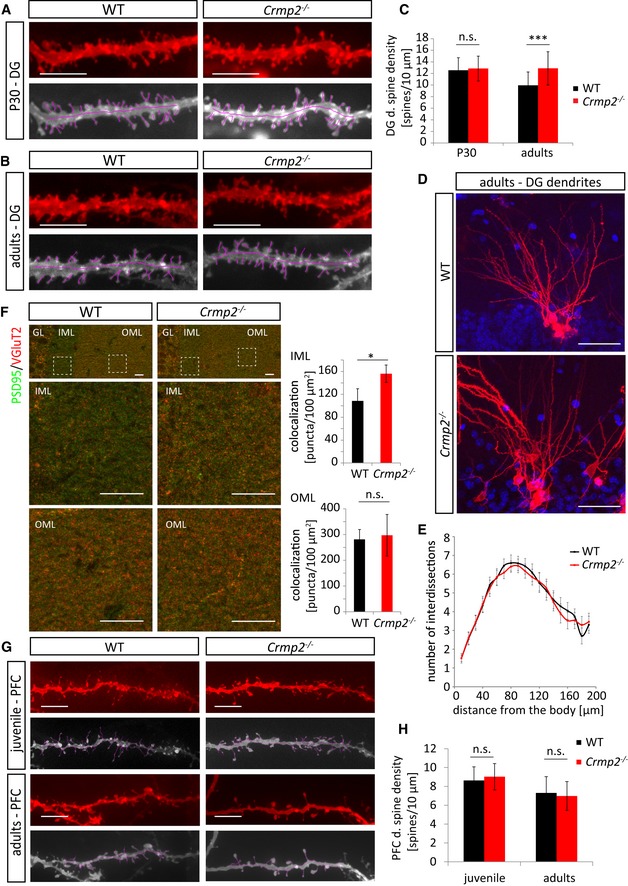
-
A, BDendritic spine density in DiOlistically labeled DG granule cells is similar in WT and crmp2 −/− mice at P30. In adults, however, spine density in crmp2 −/− granule cells is increased comparing to WT. Scale bars: 5 μm.
-
CQuantification of dendritic spine density in DG granule cells in the inner molecular layer (50–100 μm away from the soma). WT, P30: 12.5 ± 2.2 spines/10 μm, adults: 9.95 ± 2.3; crmp2 −/−, P30: 12.86 ± 2.1 (P > 0.99), adults: 12.88 ± 2.9 (P < 0.001), mean ± SD, ***P < 0.001, 2‐way ANOVA with Bonferroni correction. P30: 3 animals/genotype, WT = 61 dendrites, knockout = 64 dendrites; adults: 3 animals/genotype, WT = 41 dendrites, knockout = 37 dendrites.
-
D, EAnalysis of branching of DiOlistically labeled DG granule cell dendrites in adult WT and crmp2 −/− mice (n = 3 animals, ≥ 25 dendrites). Quantification of granule cell branching by Sholl analysis showed no significant differences (P > 0.99). Scale bars: 50 μm, mean ± SEM, 2‐way ANOVA with Bonferroni correction.
-
FDefects in synapse elimination in the inner molecular layer revealed by double immunostaining with PSD95 and VGluT2 antibodies (n = 5 mice/genotype). GL indicates granule cell layer, and IML/OML indicate inner/outer molecular layer, respectively. Density of colocalized PSD95/VGLuT2 puncta was counted. IML: WT 109 ± 21, crmp2 −/− 156 ± 15 (P = 0.004). OML: WT 281 ± 38, crmp2 −/− 297 ± 80 (P = 0.7). Scale bars: 10 μm, mean ± SD, *P < 0.05, t‐test.
-
GSpine density in DiOlistically labeled prefrontal cortex (PFC) is similar in WT and crmp2 −/− mice in both juvenile and adult mice. Scale bars: 5 μm.
-
HQuantification of dendritic spine density of PFC pyramidal neurons, basal dendrites (WT, juvenile: 8.63 ± 1.44 spines/10 μm, adults: 7.3 ± 1.4; crmp2 −/−, juvenile: 9.05 ± 1.73, P = 0.97, adults: 6.98 ± 1.53, P > 0.99), mean ± SD, 2‐way ANOVA with Bonferroni's multiple comparison test. P25: n = 3, ≥ 50 dendrites; adults: n = 3, ≥ 50 dendrites.
Higher spine density could be a result of either increased generation of new spines or defective pruning of spines or both. Considering the axon pruning defects we found in Sema3F‐regulated areas in crmp2 −/− mice (see above), and considering that Sema3F promotes loss of spines in vitro 42, we hypothesized that Sema3F regulates dendritic spine pruning through CRMP2. To test this hypothesis, we labeled and counted DG dendritic spine density in P30 (adolescent) mice when dendritic spines are virtually all formed and the pruning process starts 44, 45. We found no differences in dendritic spine density between WTs and mutants at P30 (Fig 6A and C, P > 0.99). This indicates that the formation of spines is unaltered in crmp2 −/− mice and that it is the process of dendritic spine pruning that is defective in these mice. To support this hypothesis, we tested whether the number of excitatory synapses is also increased in DG of adult crmp2 −/− mice using PSD95 and VGluT2 post‐ and pre‐synaptic markers, respectively. We found that in crmp2 −/− mice, the density of colocalized PSD95/VGluT2 puncta was indeed increased in the inner part of the molecular layer (Fig 6F), which is in agreement with DG synaptic pruning deficit. As DG spine morphogenesis is regulated through Sema3F 42, our findings indicate that CRMP2 mediates Sema3F‐dependent synapse pruning.
Finally, we analyzed the role of CRMP2 in Sema3F‐induced dendritic spine remodeling in vitro. We prepared hippocampal cultures, transfected neurons with GFP at DIV14, and analyzed the same dendritic segments at DIV21 and DIV25 (Fig EV5B). We detected significantly decreased spine density at DIV25 in WT neurons consistent with ongoing spine remodeling, but not in crmp2 −/− neurons (Fig EV5C). Importantly, treatment of WT DIV25 neurons with Sema3F induced elimination of their spines, but no significant effect was detected in CRMP2‐deficient neurons (Fig EV5D and E). These observations are in accord with our in vivo findings and further support the role of CRMP2 as mediator of Sema3F‐driven spine remodeling.
Figure EV5. Related to Fig 6. Additional dendritic spine defects revealed in vitro and in vivo .

-
AAnalysis of 3D‐reconstructed spine head diameters in DG revealed increased proportion of spines with enlarged heads at P30, but no changes in adults. Scale bars: 5 μm, ***P < 0.001, Kolmogorov–Smirnov test.
-
B, CAnalysis of dendritic spine remodeling in vitro. E18.5 hippocampal neurons were transfected with GFP (at DIV 14), and the same dendrite segments were scanned at DIV 21 and DIV 25. Spine density decreased in WT neurons (DIV21 vs. DIV 25: 9.6 ± 2.2 spines/10 μm vs. 8.4 ± 2.4 spines/10 μm, P = 0.003, n = 21 neurons) but not in knockout neurons (DIV21 vs. DIV 25: 10.3 ± 2.3 spines/10 μm vs. 9.9 ± 2.6 spines/10 μm, P = 0.25, n = 37 neurons). Scale bars: 5 μm, mean ± SD, *P < 0.05, paired t‐test.
-
D, EAt DIV 25, neurons were photographed, treated with 5 nM Sema3F or control Fc for 3 h, and photographed again. CRMP2 deficiency prevents Sema3F‐induced spine elimination. WT neurons: Fc treatment (before vs. after) 7.8 ± 2 vs. 8.6 ± 1.8 spines/10 μm (P = 0.07, n = 10 neurons), Sema3F treatment (before vs. after) 9.2 ± 3.2 vs. 8.4 ± 2 spines/10 μm (P = 0.01, n = 15 neurons). Crmp2 −/− neurons: Fc treatment (before vs. after) 9 ± 2.5 vs. 9.4 ± 2.9 spines/10 μm (P = 0.04, 16 neurons), Sema3F treatment (before vs. after) 10.9 ± 2.2 vs. 11.2 ± 2.4 spines/10 μm (P = 0.2, 41 neurons). Scale bars: 5 μm, mean ± SD, *P < 0.05, paired t‐test.
-
F, GAnalysis of DiOlistically labeled dendritic spine density in hippocampal CA1 region, secondary apical dendrites (n = 3 mice per genotype, 36 dendrites) (WT 15.2 ± 2.8 spines/10 μm, crmp2 −/− 13.3 ± 3.4 spines/10 μm, P = 0.02). Scale bars: 5 μm, *P < 0.05, t‐test.
Defects in the distribution of dendritic spines due to aberrant synapse refinement are one of the key features of both ASD and schizophrenia. Generally, dendritic spine number in ASD patients is higher than in control subjects or variable in different regions, while in schizophrenia patients, it is lower 36. This applies particularly to some brain regions, e.g., prefrontal cortex (PFC), where excessive spine elimination has been associated with pathogenesis of schizophrenia 46. Since some phenotypical aspects of conditional crmp2 −/− mice like impaired sensorimotor gating have been related to schizophrenia 17, we asked whether CRMP2 deficiency also leads to spine overpruning and reduced spine density in PFC. At P25, PFC spine density was similar in both WT and mutants (Fig 6F and G, P = 0.97) and similar to our findings in the DG. Importantly, unlike in DG, we did not detect any significant difference in PFC spine density between adult WT and crmp2 −/− mice (Fig 6G and H, P > 0.99), which is not consistent with schizophrenia‐like phenotype.
Crmp2 −/− mice display juvenile sociability defects, memory impairment, and decreased anxiety
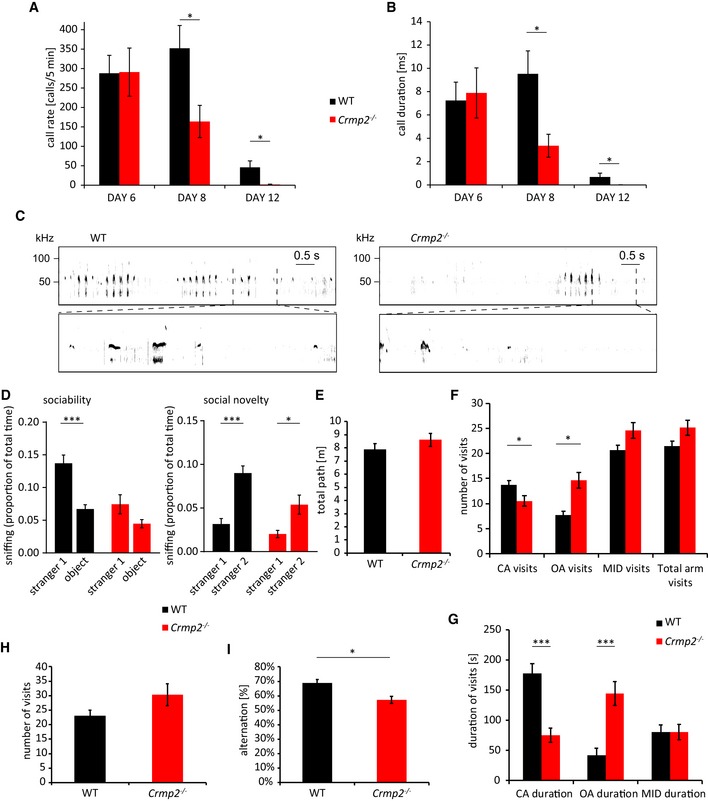
While schizophrenia shares some behavioral symptoms with autism (e.g., cognitive and social deficits), it differs in the onset of the disease, as ASD manifests typically in 3‐year‐old children while schizophrenia in the adolescence at the earliest 25, 37. This suggests that any changes underlying ASD must be present already very early in postnatal development. To assess this hypothesis, we wanted to see whether CRMP2 deficiency results in juvenile behavioral changes. To this effect, we analyzed ultrasonic vocalization (USV) in P6, P8, and P12 WT and crmp2 −/− pups as a measure of their sociability 47. We recorded USVs of WT and mutant pups in 5‐min intervals after isolation from their mothers and found that at P6 the number of calls was similar between both groups (P = 0.73), but decreased significantly in the mutants at P8 (P = 0.015; Fig 7A–C). At P12, the mutants were almost completely silent (P = 0.002); in fact, only two mutant pups from 13 vocalized at all (10/14 in WT). The duration of the individual calls was also significantly shorter in crmp2 −/− pups in both P8 (P = 0.011) and P12 (P = 0.003) (Fig 7B). This early onset social behavior defects have been described in numerous mouse models of ASD with dendritic spine pathology 26, some of which also showed dendritic spine pathology later in adults 48, 49, 50, 51. To broaden the analysis of crmp2 −/− social abnormalities also to the adult animals, we performed a three‐chamber sociability test. We found disruption of social preference in knockouts since they spent similar time exploring both stranger mice and neutral object, unlike WT, that preferred the mice (WT P < 0.001, crmp2 −/− P = 0.07, Fig 7D, Appendix Fig S2A). On the other hand, social novelty seems to be preserved in crmp2 −/− mice, although less expressed than in WT animals (WT P < 0.001, crmp2 −/− P < 0.05, Fig 7D, Appendix Fig S2B).
Figure 7. Behavioral analysis of crmp2 −/− mice.
-
A, BUltrasonic vocalization was measured at P6, P8, and P12 (WT n = 14 pups, crmp2 −/− n = 13 pups). In crmp2 −/− mice, there is a significant decrease in the rate and duration of calls at P8 (call rate, WT 352 ± 58/5 min, crmp2 −/− 164 ± 41/5 min, P = 0.015; call duration, WT 9.52 ± 2 ms, crmp2 −/− 3.36 ± 1 ms, P = 0.011) and P12 (call rate, WT 46 ± 16/5 min crmp2 −/− 1.54 ± 1.24/5 min, P = 0.002; call duration, WT 0.68 ± 0.35 ms, crmp2 −/− 0.007 ± 0.006 ms, P = 0.002). Mean ± SEM, *P < 0.05, t‐test (P6, P8), Mann–Whitney test (P12).
-
CRepresentative sonograms of the P8 mice.
-
D3‐chamber test (WT n = 11 mice, crmp2 −/− n = 13 mice). In sociability phase, WT mice spent significantly more time with a social partner (stranger 1), unlike knockouts [WT—stranger 1: 0.14 ± 0.01, object: 0.07 ± 0.007 (P = 0.0001); crmp2 −/−—stranger 1: 0.07 ± 0.01, object: 0.04 ± 0.006 (P = 0.07)]. In social novelty phase, when an object was substituted with a second social partner (stranger 2), both WTs and knockouts preferred novel mice to known mice [WT—stranger 1: 0.03 ± 0.006, stranger 2: 0.09 ± 0.008 (P < 0.0001); crmp2 −/−—stranger 1: 0.02 ± 0.004, stranger 2: 0.05 ± 0.01 (P = 0.02)]. Mean ± SEM, *P < 0.05, ***P < 0.001, t‐test.
-
E–GElevated plus maze test (n = 10 mice/genotype). (E) Total distance walked is similar in WT and crmp2 −/− (WT 7.9 ± 0.45 m, crmp2 −/− 8.6 ± 0.5 m, P = 0.3). (F) Frequency and (G) duration of open arm (OA) visits are increased in crmp2 −/− mice suggesting decreased anxiety. CA denotes closed arms, MID denotes the transition zone between arms, and total arm visits represent a sum of visits in all four arms. (CA frequency: WT 13.7 ± 0.9/5 min, crmp2 −/− 10.5 ± 1.1/5 min, P = 0.04; OA frequency: WT 7.7 ± 0.8/5 min, crmp2 −/− 14.6 ± 1.7/5 min, P = 0.002; MID frequency: WT 20.6 ± 1/5 min, crmp2 −/− 24.6 ± 1.6/5 min, P = 0.06; total arm visits: WT 21.4 ± 1/5 min, crmp2 −/− 25.1 ± 1.5/5 min, P = 0.07; CA duration: WT 177 ± 17 s, crmp2 −/− 75.3 ± 12.6 s, P < 0.001; OA duration: WT 42 ± 12.6 s, crmp2 −/− 144.4 ± 21 s, P < 0.001; MID duration: WT 80.26 ± 11.5 s, crmp2 −/− 80.17 ± 12.6 s, P = 0.99), mean ± SEM, *P < 0.05, ***P < 0.001, t‐test.
-
H, IY‐maze test (WT n = 9 mice, crmp2 −/− n = 8 mice). Decreased alternations between arms of the maze indicate impaired working memory (number of visits, WT: 23.1 ± 1.9, crmp2 −/− 30.4 ± 3.8, P = 0.12; alternations, WT 68.8 ± 2.5%, crmp2 −/− 57.3 ± 2.5%, P = 0.006), mean ± SEM, *P < 0.05, t‐test.
We next asked whether hippocampus‐dependent memory functions are affected in crmp2 −/− mice as they exhibit aberrant inputs from DG into CA3 (unpruned mossy fibers) and increased spine density in DG granule cells (input from entorhinal cortex). We tested working memory using Y‐maze (a three‐arm maze) where WT and mutants showed comparable level of exploratory activity (Fig 7H). However, the ratio of spontaneous arm alternation was significantly lower in crmp2 −/− mice (Fig 7I, P = 0.0058) indicating a working memory impairment 17. In contrast, long‐term memory and general behavioral flexibility seem not to be affected in crmp2 −/− mice as revealed by active place avoidance on a rotating arena test (Appendix Fig S2C).
Similar to other CRMP2‐deficient mouse models 17, 52, we detected anxiety impairment in crmp2 −/− mice using elevated plus maze. In the task, the knockouts also demonstrated increased activity during exploration of the maze (Fig 7E), spent more time in the open arms of the maze (P < 0.001), and visited them more often (P = 0.0016) than their WT counterparts (Fig 7F and G) suggesting decreased anxiety, or perhaps a more general lack of adequate response to potentially dangerous situations. This phenotype may reflect the hippocampal phenotype of crmp2 −/− mice as lesions in particularly ventral hippocampus have been shown to result in similar decreased anxiety in mouse models 53.
Discussion
CRMP2 has been long considered an important regulator of semaphorin 3A‐mediated axon guidance during embryonic development. Its expression, though, is high even in the early postnatal neurons, but its role in the postnatal development and adult neurons has so far been elusive. In the present study, we demonstrated that CRMP2 is not only mediator of Sema3A signaling regulating axon guidance in embryonic development, but importantly, that it plays a central role in the postnatal refinement of the nervous system. By generating new crmp2 −/− mice and analyzing their phenotype, we first showed that CRMP2 deficiency in vivo leads to axon guidance defects in CNS and PNS that could be attributed to changes of Sema3A signaling. Strikingly, we demonstrated that CRMP2 mediates also Sema3F signaling and that CRMP2 deficiency disrupts early postnatal Sema3F‐mediated axon and dendritic spine refinement in multiple areas of the CNS and in hippocampal neuron cultures. Changes in Sema3F signaling pathway have been considered a risk factor in the pathogenesis of ASD. In accord with that, we showed that crmp2 −/− mice suffer from altered pruning and both early postnatal and adult social interaction defects previously linked to autism. Together, our in vivo and in vitro data demonstrated a novel function of CRMP2 in postnatal fine‐tuning of the nervous system by Sema3F and showed that its deficiency in mice leads to neurodevelopmental defects associated with pathogenesis of the ASD in human.
Regulation of axon and dendritic pruning by Sema3F and CRMP2
Initial growth of axons and dendrites during embryonic and early postnatal period results in an embryonic template that must be later refined to generate a functional healthy nervous system 54. Stereotyped refinement of nervous system was uncovered in several regions: (i) Infrapyramidal bundle (IPB) axons of hippocampal mossy fibers are retracted between P14 and P30 in mice 55; (ii) excess of layer 2/3 callosal axons of motor, sensory, and visual cortex is refined until P30 33; and (iii) corticospinal axons from layer 5 visual cortex are eliminated between P9 and P25 7. In addition, density of dendritic spines in various brain regions in mice and humans peaks between childhood and adolescence 44, 45 and its significant portion is eliminated until adulthood. Finally, pruning also occurs in peripheral nervous system—originally polyneuronal innervation of muscle fibers is later in development refined so that each muscle fiber is innervated by only one motor axon 56, 57.
Knockout mouse studies identified extracellular cues and receptors that mediates pruning in rodents. These include Sema3A and its receptor complex (Nrp1/PlxnA4), and Sema3F and its receptors (Nrp2/PlxnA3), ephrin‐B3–EphB2 reverse signaling, and C4 component of complement 6, 58, 59. Intracellular pathways that translate extracellular signal to cytoskeleton are however poorly understood. Here, we identified CRMP2 as a novel mediator of pruning in rodent brain triggered by Sema3F.
Sema3F binds preferentially to Nrp2/PlxnA3 receptor complex and is important regulator of neural development. Deficiency of Sema3F, Nrp2, or PlxnA3 results in defects in stereotyped pruning of hippocampal infrapyramidal bundle, distribution of DG dendritic spines, or anterior commissures, 42, 55. Moreover, PlxnA3/A4 −/− mice display defects in pruning of visual axons. Similarly, in our crmp2 −/− mice, we found all: IPB pruning defect, alteration of DG spine density (Figs 3 and 6), and defects in pruning of visual cortex axons (Fig 4). Moreover, our in vitro assays (Figs 5 and EV5) showed Sema3F is unable to induce axon and dendritic spine retraction in hippocampal neurons isolated from crmp2 −/− embryos. In addition to Sema3F, IPB is also regulated by ephrin‐B3 reverse signaling. It is possible to speculate that CRMP2, and in particular CRMP2A isoform, which is expressed in mossy fibers, conveys signaling of both Sema3F and ephrin‐B3. Further studies will assess this hypothesis in more detail.
Sema3A has been shown to orchestrate pruning of, e.g., CA1 hippocamposeptal or callosal axons 3, 33. As CRMP2 has originally been identified as a mediator of Sema3A 8, we hypothesized that CRMP2 is involved also in Sema3A‐triggered pruning. However, we did not find any evidence that would support this hypothesis. We did not detect DiI‐positive CA1 neuron bodies by retrograde tracing from medial septum after the pruning period in crmp2 −/− pups 6 (Fig EV3B). In callosal axons, we found significant differences in axon guidance between WT and crmp2 −/− mice in P6–P9 mice (Fig 2), but the significance was lost in adult mice (Appendix Fig S1C–E), which could be due to the presence of effective Sema3A‐dependent axon pruning. In line with these in vivo data, we found that Sema3A (but not Sema3F) was able to partially induce axon retraction of the stalling crmp2 −/− axons in 1‐week‐old hippocampal neuron cultures (Fig 6). Relatively high variability in this experiment could be due to different sensitivity of individual synapses to semaphorin stimulation.
Importantly, we also did not find any defect in the pruning of neuromuscular junctions (NMJs) in crmp2 −/− mice (Fig EV3C and D). At the end of the embryonic development, each synapse is innervated by up to 10 axon branches of different motor units 56. During the first two postnatal weeks, all except one terminal branch are pruned back establishing singly innervated NMJs 60. While the exact molecular cascade regulating motor axon pruning is not known, Sema3A seems to play a role in the process as its receptor, Nrp1, is expressed in pre‐synaptic axon terminals 61. Moreover, Sema3A secreted from Schwann cells participates in NMJ remodeling 62. Sema3F signaling has so far not been linked to motor axon pruning.
Dendritic spine density changes dynamically during childhood and adolescence. In mice, spine density peaks around 1 month and then decreases to reach stable levels around 2 months 45. It has been shown that distribution of dendritic spines is regulated by class 3 semaphorins 42. Previous in vitro experiments showed that Sema3F, but not Sema3A, decreases PSD‐95‐positive puncta in dissociated DG neurons 42. Accordingly, Sema3F‐treated cortical neurons displayed decrease in apical dendrite spine density 42. Adult Sema3F −/−, Nrp2 −/−, and PlxnA3 −/− mice show increased spine density in several brain regions, in particular DG dendrites 42, 63. Crmp2 −/− mice partially mimic this phenotype as we also found increased spine density in adult, but not in P30 DG granule cells (Fig 6). Interestingly, in Sema3F −/− mice, increased DG spine density is detectable already during spine generation (P21) and is largely retained into adulthood, while in WT, they are subsequently pruned 42. In crmp2 −/− mice, we did not find increased DG spine density in the pre‐pruning period (P30, Fig 6A), but similar to Sema3F −/− mice, we did find defects in DG spine pruning. Before pruning, spines also tended to be relatively enlarged in crmp2 −/− mice (Fig EV5A). Similar observations were found in Sema3F‐ and Nrp2‐deficient mice using electron microscopy 42. Aberrant spine size could reflect disruption in actin dynamic. In vitro analysis of dendritic spine remodeling further supports our in vivo findings (Fig EV5). Together, these data suggest that while Sema3F signaling regulates both spine generation and pruning, CRMP2 contributes mainly to spine pruning.
CRMP2 in axonal growth in vivo
As demonstrated in knockout lines of Sema3A and its downstream targets (Sema3A −/−, Nrp1 −/−, and PlxnA4 −/− mice), Sema3A signaling is an essential regulator of the development of rodent trigeminal nerve, facial nerve, DRGs projection, olfactory bulb, hippocampal formation, and corpus callosum 27, 28, 33, 34, 64, 65, 66, 67. Surprisingly, although Sema3A or Nrp1 deletion causes strong overgrowth of some peripheral nerves (e.g., trigeminal and spinal axons) 27, 28, we found only a mild overgrowth and increased branching of these axons upon deletion of its downstream mediator CRMP2 in crmp2 −/− mice (Figs 1 and EV2). This could be due to a partial rescue of Sema3A signaling in these neurons by other CRMP family members as mentioned before. Indeed, we and others 52 found that full CRMP2 deficiency in vivo is associated with increase of CRMP1 and CRMP4 levels (Fig EV2). CRMP1 and CRMP4 are sequentially and functionally close to CRMP2, and their expression pattern partially overlaps 68, 69. Moreover, CRMP4 (Fig EV2) and CRMP1 70 are expressed in peripheral nerves suggesting that their elevation could rescue the reduced axon growth caused by acute CRMP2 deficiency. Increased expression of CRMP1 or CRMP4 could partially compensate for CRMP2 deficiency not only in Sema3A‐dependent axon guidance but also in pruning, as we did not detect significant defects in Sema3A‐dependent axon pruning in crmp2 −/− neurons in vivo or in vitro (Figs 5 and EV3).
Previous in vitro studies demonstrated axon growth‐promoting effect of CRMP2, while we did not detect any axon growth reduction in crmp2 −/− mice. This may be because the role of CRMP2 in regulation of axon growth, neuron polarization, and migration has so far been studied in vitro using an acute knockdown of CRMP2 rather than in vivo in full knockout mice, which we used in our experiments and where the CRMP2 deficiency may be better compensated by other genes (e.g., CRMP1 or CRMP4).
Electroporation studies of Sema3A −/− or Nrp1 floxed/floxed brains demonstrated their role in the development of corpus callosum, with mispositioned axons in callosal midline and axonal mistargeting in contralateral cortex at P8 33. Defects in axon pruning of this region were also suggested 33. Using the in utero electroporation and DiI tracing in crmp2 −/− mice, we also found defective guidance of callosal axons in the contralateral cortex and their altered orientation in the midline in the rostro‐caudal axis (Fig 2).
Notably, Sema3F signaling is essential also for guidance of specific cranial nerves and was related to the development of limbic system and anterior commissure 40, 71. From these, we only detected partial malformation of AC in crmp2 −/− mice (AC is missing in Sema3F −/− or Nrp2 −/− mice) suggesting CRMP2 may participate also in Sema3F‐mediated axon guidance (Fig EV4).
Previous in vitro and in vivo experiments suggested that CRMP2 also regulates neuronal migration 72. However, using the in utero electroporation, we found no significant changes in neuron distribution in the developing WT and mutant cortical plates at E17.5 (Fig EV1F). This likely reflects different experimental paradigms used in the studies (somatic knockdown vs. full knockout) 72.
CRMP2 involvement in pathogenesis of neurodevelopmental disorders
Deregulation of CRMP2 has been linked to several neurodevelopmental disorders (SFARI Gene database, https://gene.sfari.org/database/human-gene/DPYSL2). Recently published analysis of conditional brain‐specific 17 and full crmp2 knockout mice 52 showed multiple behavioral defects associated with CRMP2 deficiency. Notably, conditional knockout mice revealed hyperactivity and prepulsed inhibition (PPI) deficit together with social behavior impairment. PPI is a test for evaluating sensorimotor gating—the phenomenon that is often altered in schizophrenia patients. In addition, clozapine (an antipsychotic drug) treatment was capable to reduce hyperactivity in conditional crmp2 −/− mice. Furthermore, morphological analysis showed increased volume of brain ventricles and impaired dendritic development in hippocampal CA1 and DG neurons, which is associated with schizophrenia, but also other neurodevelopmental disorders 17. Importantly, the analysis of the full and conditional CRMP2 knockout mice revealed also their significant differences. In particular, while PPI was reduced in the conditional mice and in the full crmp2 −/− mice, it was not significantly different to WT 17, 52. This suggests that even a minor difference in the spatio‐temporal inactivation of CRMP2 during development can have a major impact on the development and severity of the resulting neurodevelopmental defects. In the full CRMP2 knockout mice, we generated, we found several phenotypical defects present in the published conditional and full CRMP2 knockout mice (e.g., ventriculomegaly, spine density changes in DG, working memory defects, or hyperactivity) 17, 73. We also found brain sizes comparable in both WTs and crmp2 −/− mice, similar as reported in the CRMP2 knockout mice 17, 73, although we detected a non‐significant tendency for a thinner cortex in the knockout mice (not shown) in agreement with the hypoplastic corpus callosum and anterior commissure. Changes in interhemispheric connectivity have recently been linked to ASD and schizophrenia through CYFIP1, a CRMP2 binding partner 74, 75. Importantly, we demonstrate that CRMP2 knockout leads to defects in axonal pruning and dendritic spine remodeling compatible with ASD rather than schizophrenia 25 (Figs 3, 4 and 6). Similar to other ASD mouse models 49, also in crmp2 −/− mice the dendritic spine pruning deficiency is not present in all brain regions [e.g., we did not detect it in the prefrontal cortex (Fig 6F and G) or CA1 neurons (Fig EV5F and G)]. This may reflect specific spatio‐temporal combinations of expression of CRMP2 (and its isoforms) and semaphorins in different brain regions (of note, there is a strong expression of CRMP2A isoform specifically in the inner molecular layer of DG (Fig EV1A), where we detected the pruning deficiency). The detail role of CRMP2 isoforms in axon pruning will be analyzed in future studies.
Morphological changes were accompanied by altered social communication in early postnatal (P8 and P12) mutants and decreased sociability in adults (Fig 7) further corroborating the role of CRMP2 in the pathogenesis of ASD. Defects in early postnatal USVs followed by dendritic spine pathology have been previously observed in several mouse models of ASD 49, 50, 51. The connection between early postnatal altered sociability and impairment of stereotyped axon pruning is much less clear. Nevertheless, defects in stereotyped axon pruning have been associated with social interaction deficit and ASD in humans and mice, e.g., through functional variants and knockouts of Otx‐1 gene 76, 77.
Importantly, Sema3F signaling has been also implicated in the pathogenesis of ASD. Sema3F‐ or NRP2‐deficient mice show both behavioral and neuropathological aspects of ASD 78, and Sema3F interacts with multiple ASD‐related genes, e.g., fragile X mental retardation protein or MECP2 79. Thus, by linking Sema3F and CRMP2 signaling and comparing the histological as well as behavioral effects of their deficiency, our data strongly implicate that the Sema3F‐CRMP2 signaling plays an important role in ASD pathogenesis. Since previously CRMP2 has been linked with pathogenesis of schizophrenia, it may serve as a molecular link connecting class 3 semaphorin signaling defects to both ASD and schizophrenia.
Materials and Methods
Animals
All animal studies were ethically reviewed and performed in accordance with European directive 2010/63/EU and were approved by the Czech Central Commission for Animal Welfare. Mice, all in C57BL6/N background, were housed and handled according to the institutional committee guidelines with free access to food and water. Unless stated otherwise, adult mice used for experiments were 12–16 weeks old. See Appendix Table S1 for number of animals used in experiments.
Crmp2 −/− mice generation
We used TALEN mutagenesis (transcription activator‐like effector nucleases) to generate crmp2 −/− mice. Two TALEN pairs targeting sequences 185–150 bp 5′ of crmp2 exon 2 and 183–218 bp 3′ of exon 3 (Fig 1A) were designed using TAL Effector Nucleotide Targeter 2.0 (https://tale-nt.cac.cornell.edu/) 80, 81, assembled using the Golden Gate Cloning system 80, and cloned into the ELD‐KKR backbone plasmid. DNA‐binding domains of TALENs specific for the desired target sites within the crmp2 locus consisted of following repeats: HD‐HD‐NN‐HD‐HD‐HD‐NG‐NI‐NN‐HD‐NG‐NN‐NN‐NI‐NG‐HD‐NG (5′ TALEN‐crmp2‐ex1), NN‐HD‐NI NI‐NG‐HD‐HD‐NG‐HD‐NG‐NN‐NG‐HD‐NG‐HD‐NG‐NG (3′ TALEN‐crmp2‐ex1), HD‐HD‐NI‐NI‐NN‐NI‐NN‐NG‐HD‐NI‐HD‐NG‐NN‐NI‐NN‐HD‐NG‐NG (5′ TALEN‐crmp2‐ex2), and NN‐HD‐NI‐HD‐NI‐NG‐NG‐HD‐NG‐NI‐HD‐HD‐NI‐NI‐NG‐NN‐NG (3′ TALEN‐crmp2‐ex2). All TALEN plasmids were used for production of TALEN encoding mRNA as described previously 82. TALEN mRNAs (with total RNA concentration of 40 ng/μl) were microinjected into C57BL6/N‐derived zygotes. Genomic DNA isolated from tail biopsies of newborn mice was screened by PCR for deletion of exons 1 and 2 (3,673 bp) (primers F1: 5′‐ATATCCCACGATTCTGACCAATCA‐3′ and R1: 5′‐CCAAATAACTGCAGTGTAGCCTAT‐3′), and deletion was confirmed by locus sequencing and mice used as founders of crmp2 −/− line. The mouse line was genotyped by PCR using locus‐specific primers: R1: ACTTACCGTGATGCGTGGAA, F1: TCACCCTCCCGGGACGAT, and R2: TCTACCAATGTTACAACACAGA.
Antibodies, cell dyes, and plasmids
Primary antibodies used in this study are as follows: mouse anti‐CRMP2 and hamster anti‐CRMP1 (WAKO, IHC 1:200, WB 1:5,000), rabbit anti‐TUC4 (CRMP4, Millipore, IHC 1:400, WB 1:5,000), rabbit affinity purified anti‐CRMP2A (IHC 1:75) 14, rabbit anti‐CRMP2A (WB, 1:30,000) 14, mouse anti‐neurofilaments (2H3 antigen, DSHB Iowa, 1:150), rabbit anti‐MAP2 (Abcam, 1:300), rabbit anti‐calbindin (Swant, 1:600), mouse anti‐VgluT2 and anti‐VGluT1 (Millipore, 1:400), goat anti‐PSD95 (Millipore, 1:200), mouse anti‐tau (Abcam, 1:500), mouse anti‐βIII‐Tubulin conjugated to Alexa 488 (BioLegend, 1:200), mouse anti‐actin (Sigma, 1:500), and rabbit anti‐βIII‐Tubulin (Sigma, 1:500). Secondary fluorescent antibodies were conjugated with various Alexa Fluor dyes: anti‐mouse (Alexa 488 or 594), anti‐rabbit (Alexa 594), and anti‐goat (Alexa 488), diluted 1:400. Hoechst 33342 (1 μg/ml) was used to counterstain cell nuclei. In some cases, ABC kit (Vector Laboratories) was used for detection. For whole‐mount immunostaining, secondary anti‐mouse antibody conjugated with HRP was used (1:1,000). For Western blots, secondary anti‐mouse, anti‐rabbit, or anti‐hamster conjugated with HRP was used (1:10,000). Cholera toxin subunit B conjugated to Alexa 647, DiI, and DiO was purchased from Thermo Fisher. α‐Bungarotoxin conjugated to Alexa 594 was purchased from Invitrogen (50 μg/ml; 1:50). EGFP was cloned into pCAGGS vector.
Histology, immunohistochemistry, and biochemistry
Mice were perfused transcardially with PBS and ice‐cold 4% paraformaldehyde (PFA) in PBS. Brains were isolated and postfixed overnight at 4°C in 4% PFA/PBS. Subsequently, brains were washed in PBS and processed as described previously 14, 83. Seven‐μm‐thick paraffin sections were created. Immunohistochemistry was done as described previously 14. Sections were deparaffinated as follows: 2 × 10 min 100% xylene, 2 × 10 min 100% ethanol, 3 min 90% EtOH, 3 min 70% EtOH, 3 min 50% EtOH, and PBS. Antigen retrieval was performed in some cases using citrate‐based antigen retrieval solution (Vector) diluted 1:100 in water. Slices were blocked in 1% BSA/0.2% Tween/PBS (PBST) and incubated with primary antibodies overnight at 4°C. Next, slices were washed 3 × 5 min in PBST and incubated with secondary antibodies conjugated with Alexa Fluors, 2 h at RT. Then, slices were washed 3 × 5 min in PBST and mounted in Mowiol with Hoechst (1:1,000). For bright‐field microscopy, slices were pretreated 15 min in 3% H2O2 prior to blocking and ABC kit (Vector) was used according to the manufacturer's instructions. HRP activity was detected with 0.05% DAB. Subsequently, slices were dehydrated in ethanol–xylene and embedded into Eukitt. Brain protein isolation, SDS–PAGE, and Western blotting were done as described previously 14.
Whole‐mount immunohistochemistry
Whole‐mount immunohistochemistry was performed as described previously 14 with some modifications. E10.5–E12.5 embryos were isolated from time‐pregnant mothers. WTs and knockouts from the same litter were compared. Embryos were fixed in 4% PFA/PBS overnight, 4°C. Next day, they were washed with PBS and unmasked in 1:100 diluted antigen retrieval solution (Vector). Embryos were then washed again in PBS for 10 min (RT) followed 30 min in Dent fixative (20% DMSO/methanol) at 4°C. Subsequently, all samples were bleached overnight at 4°C in 5% H2O2/20% DMSO/methanol. Next, embryos were blocked overnight in 10% FBS/20% DMSO/PBS at 4°C and then incubated in anti‐neurofilaments antibody clone 2H3 for 4 days (dilution 1:100 in blocking solution) followed by secondary HRP‐conjugated antibody (1:1,000) for 24 h. Then, embryos were washed in 20% DMSO/PBS and incubated in 0.6% Tween/PBS overnight. Finally, samples were washed in PBS and incubated in 0.05% DAB for 2 h. H2O2 was then used as a substrate. Labeled embryos were washed in PBS and cleared in ascending glycerol concentration (20, 40, 60, and 80%). They were stored in 80% glycerol at 4°C. Images were captured using Nikon SMZ18 stereomicroscope and post‐processed in Helicon focus to create sharp images. Surface area occupied by a given nerve was measured in ImageJ. Axons were traced in Neurolucida 360.
DiOlistics and DiI tracing
We used DiOlistic approach using Gene Gun helium‐powered system from Bio‐Rad. Bullets were prepared as described 84, and tubing was coated with 10 mg/ml polyvinylpyrollidone (PVP). We mixed 100 mg Tungsten beads and 2.5 mg DiI or DiO (dissolved in CH2Cl2). After CH2Cl2 evaporation, resulted powder was transferred into aluminum‐wrapped falcon tube and 3 ml H2O was added. Solution was sonicated at 4°C until no clumps were visible (30–45 min). Then, solution was sucked into tubing in prep station, beads were able to settle down, and water was removed. Tubing was rotated 1 h during continuous drying with nitrogen (2–3 l/min). Finally, 1.3‐cm bullets were cut from tubing and stored in 4°C with silica gel beads to prevent rehydration.
Slices for DiOlistics were prepared as follows: Mice were perfused with 20 ml 4% PFA/PBS, and brains were isolated and postfixed 30 min in 4% PFA/PBS. Then, brains were washed 1–3 h in PBS and 20 min in 15% sucrose/PBS following another 20 min in 30% sucrose/PBS. 250 μm coronal slices were prepared using vibratome Leica 2000S. Prior to shooting, slices were treated 5 min in 15% sucrose and 5 min in 30% sucrose. Dye was carried using pressure 120 Psi and modified filter as described 84. After shooting, slices were washed 3× in PBS quickly and dye was let to diffuse 40 min at 4°C. Slices were then mounted onto glass slice in 0.5% n‐propyl gallate/90% glycerol/PBS (NPG) and imaged by CARV II/Nikon Ti‐E spinning disk.
For carbocyanine dye tracing, mice were perfused transcardially with PBS and fixed with 4% PFA/PBS. Next, either small DiI crystal was placed or 0.1 μl of DiI solution was injected into target area. We used 2.5 mg/100 μl concentration, and DiI was dissolved in DMSO. Slices were prepared in vibratome and scanned by Leica TCS SP8. Details are as follows.
Corpus callosum tracing
DiI solution was injected into superficial layers of cortex. Brains were maintained in 4% PFA/PBS in 37°C for 3–4 weeks. Then, brains were cut in oblique (horizontal + 20°) direction (Appendix Fig S1A), 150‐μm‐thick sections were prepared. Slices with traced axons were mounted onto glass slide in NPG mounting solution.
Hippocamposeptal axon tracing
After fixation, brains were trimmed to expose septum. DiI crystals were inserted into the medial septum and brains were maintained in 4% PFA/PBS for 1–2 weeks. 100 μm (P0–1) or 150 μm (P8) coronal slices were prepared. We found a strong labeling of septohippocampal projections (e.g., axons arising from the septum entering the hippocampus) and also subicular neurons projecting to the medial septum in both WTs and knockouts. We screened for retrogradely labeled CA1 neuron bodies in the hippocampus, whose presence at P8 indicates incomplete pruning.
Visual corticospinal axon tracing
DiI solution was injected into primary visual area. After 2–3 weeks, brains were cut sagitally to 150–180‐μm slices that were mounted in NPG with Hoechst. The tracing pattern was compared with data from Allen brain atlas connectivity studies to ensure that we targeted correct area. We observed two axon branches in the diencephalon: first branch growing into pyramidal tract (corticospinal axons) and second to superior colliculus (collicular axons). To quantify the axonal growth into the pyramidal tract, we compared fluorescence intensity of corticospinal axons vs. intensity of axons before branching. We refer to this ratio as visual pruning index (VP index), with its lower values indicating the presence of refinement.
In utero electroporations
In utero electroporations were done as previously described 85. Briefly, pregnant mice were anaesthetized by 2.5% isoflurane. Anesthesia was maintained by 2% isoflurane. We injected pCAGGS‐EGFP plasmid (3 μg/μl) into ventricles of E14.5 embryos (migration assay, analysis at E17.5) or E15.5 embryos (callosal axons, analyzed at P6). Electroporation was carried out by small paddle electrodes (35 V, five pulses, 950‐ms interval) to target sensory cortex. For migration assays, embryos were harvested at E17.5, and brains were fixed in 4% PFA/PBS, sliced (150‐μm vibratome sections), counterstained with Hoechst, and scanned by Leica TCS SP8. In this case only, both WTs and crmp2 +/− embryos were used as controls. Callosal axons were analyzed at P6. After birth, pups were nurtured by a foster mother. At P6, mice were sacrificed, and brains were fixed in 4% PFA/PBS, sliced (150 μm vibratome sections), and analyzed by CARV II/Nikon Ti‐E spinning disk.
Semaphorin assays and live imaging
Mouse E16.5 hippocampal neurons were prepared and cultured as described 14. Briefly, pregnant mice were sacrificed, and embryos isolated and decapitated in cold HBSS with 10 mM Hepes. Hippocampi were isolated, moved to Neurobasal medium (Neurobasal (Gibco) with 2.5% Β27 supplement (Gibco), 2.5 mM glutamine, and 1% penicillin/streptomycin solution), and triturated. Subsequently, solution was strained through a 40‐μm strainer (Biologix) and spun down (300 g, 2 min, 4°C). The supernatant was removed, and the remaining cells were resuspended in a fresh Neurobasal medium and counted. Neurons were plated into 24‐glass bottom plates (100,000 cells/well) coated with laminin (1 μg/ml) and poly‐d‐lysine (50 μg/ml) and cultured in Neurobasal medium that was refreshed every 2–3 days. Neurons were transfected with pCAGGS‐EGFP using Lipofectamine 2000 (Invitrogen), 24 h after plating (axon retraction assay) or at DIV14 (dendritic spine analysis). Mouse semaphorin 3A and 3F (carrier free) or control Fc were purchased from R&D systems and were diluted to 1 mg/ml stock concentration. For axon retraction assay at DIV 7, medium volume was adjusted to 300 μl in each well. Neurons were photographed three times (with 20 min gaps), and then, 50 μl of fresh medium with Fc or Sema3A (final concentration 1 nM) or Sema3F (final concentration 5 nM) was added. Subsequently, neurons were imaged every 20 min for 16 h by Leica DMI6000 equipped with a heating box and CO2 atmosphere. For analysis of dendritic spines, the same segments were photographed at DIV21 and DIV25. At DIV25, neurons were stimulated with Sema3F (5 nM) for 3 h and photographed again.
Microfluidic chambers and DRG collapse assay
Microfluidic chambers were prepared as described before 31, 86. E11.5–E12.5 spinal cord explants were dissected in cold HBSS and placed into laminin (3 μg/ml)‐ and poly‐l‐ornithine (1.5 μg/ml)‐coated proximal well. After 3–4 days, axons entered the distal compartment. Then, explants were labeled by Alexa 647‐conjugated cholera toxin subunit B. 5 nM Sema3A of control Fc was applied distally, and axons were photographed by Leica DMI6000 microscope every 10 min during 14‐h interval. Axons growing at least 50 μm were analyzed. Growth cone collapse was analyzed in DRG explants isolated at E11.5–E12.5. DRGs were plated on coverslips coated with laminin (1 μg/ml) and poly‐D‐lysine (50 μg/ml) and cultured in Neurobasal medium supplemented with NGF (R&D systems, 25 ng/ml). The day after plating, explants were stimulated with various Sema3A concentrations for 30 min, fixed, and stained with antibodies against actin (to label growth cones) and β‐3 tubulin (to label axons). DRGs were imaged by Nikon spinning disk.
Analysis of developmental motor axon pruning
WT and crmp2 −/− pups (both sexes) were sacrificed on P11, and the thorax was excised as previously described 87, 88, 89, 90 and fixed in 4% PFA in 0.1 M phosphate buffer (PB) for 1 h on ice. The triangularis sterni muscle was dissected and incubated overnight (4°C) in anti‐βIII‐tubulin antibody conjugated to Alexa 488 (BioLegend 801203; 1:200)‐ and Alexa 594‐conjugated α‐bungarotoxin (Invitrogen B13423; 50 μg/ml; 1:50) in blocking solution (5% BSA, 0.5% Triton X‐100 in 0.1 M PB). Muscles were then washed in 0.1 M PB and mounted in Vectashield (Vector Laboratories). Z‐stacks were recorded using a confocal microscope (FV1000, Olympus) equipped with a 20×/0.8 N.A. oil‐immersion objective and analyzed for the percentage of doubly innervated NMJs using Fiji (cell counter plugin) 91.
Behavioral tests
Ultrasonic vocalizations were recorded from P6, P8, and P12 pups (WT n = 14, crmp2 −/− n = 13). Each pup was taken from its home cage, put into Styrofoam box, and recorded for 5 min with microphone (Dodotronic Ultramic 250K, Italy) placed at the top of the box. Audacity software (freely available) was used for recordings with sampling frequency set to 250 kHz. The vocalizations were analyzed automatically using Avisoft‐SASLab Pro; however, the automatic analysis was checked and manually corrected if necessary. Main parameters measured were number of vocalizations and its total length.
Sociability and social novelty preference were tested in three‐chambered apparatus (54 × 20 × 33 cm) made from clear plexiglass. The chambers were divided by transparent walls with squared openings (5 × 5 cm) and sliding doors. Each mouse (WT n = 11, crmp2 −/− n = 13) was first placed in the middle compartment for 10 min. After the habituation, unknown male mice (stranger 1) were enclosed in a little wire cage and placed in either left or right compartment. Black plug (4.5 cm in diameter) was used as an object and placed in the opposite compartment inside identical wire cage. The position of stranger mouse and object was counterbalanced between trials, and stranger mice were previously habituated to the cage. Sliding doors were then opened, and the test mouse was allowed to freely explore the apparatus for 10 min. After end of this part of experiment, the object was removed and another unknown mouse (stranger 2) was put inside the same chamber. The test mouse was then allowed to explore all chambers for another 5 min. The behavior was recorded by a camera placed above the apparatus. Time spent in each chamber and time spent sniffing the wire cages were analyzed manually in BORIS software.
The elevated plus maze (EPM) is a cross‐shaped maze elevated 40 cm above the floor level, with all four arms (30 cm long, 5 cm wide, 16 cm high) accessible from the central platform. Two opposite arms were enclosed by opaque walls (closed arms), while the other two were without walls (open arms). The test assesses spontaneous and anxiety‐like behaviors of the animals. The closed arms are perceived as safer by the animals, as they are darker, more protected, and without risk of falling. Anxious animals are expected to spend most of the time in the closed arms, while less anxious and more explorative individuals explore the open arms more often. The behavior of the animals during each 5‐min session was recorded by a web camera placed above the apparatus. Locomotor activity (total distance) and time spent in different compartments were analyzed offline by digital tracking software (EthoVision, Noldus). Numbers of animals: n = 10 for both WT and knockouts, aged 31–44 weeks.
Spontaneous alternation was tested in three‐armed maze (Y‐maze), with each arm 35 cm long, 6 cm wide, and 18 cm high. The mice were left free to explore the empty apparatus for 8 min. Between trials, the apparatus was cleaned by ethanol and then wiped clean and dry to erase any scent marks. Number of arm visits was counted, indicating the exploratory activity (a visit was counted if the mouse placed all paws into the arm). Spontaneous alternation was measured as the ratio of actual triads (three different arms entered in three subsequent visits) to potential triads (theoretical maximum performance). In the Y‐maze task, 17 male mice (9 WT, 8 KO), aged 31–44 weeks, were tested.
The active place avoidance task, or AAPA 92, 93 (for review, see Ref. 94), is a rodent cognitive task testing hippocampal functions, including spatial navigation, cognitive coordination, and flexibility. As a dry‐arena task, it is more suitable for mice than the Morris water maze, as mice are worse swimmers than rats and tend to be overtly stressed by water immersion 95. We used a carousel maze (circular arena 56 cm in diameter) with electrified grid floor, surrounded by a transparent plexiglass wall, rotating at approximately 1 rotation per minute. The apparatus was located in a dimly lit room with abundant extramaze cues, with and additional, highly contrast cue card in close proximity (1.5 m) of the arena. A computer‐based tracking system (Tracker, Biosignal Group, USA) recorded the positions of the mouse and the arena at a sampling rate of 25 Hz. A 60‐deg unmarked to‐be‐avoided sector was defined in the coordinate frame of the room by tracking software. Each entrance into the sector was punished by mild electric foot shocks (scrambled; 100 Hz alternating current; 40–80 V) delivered by the tracking system into the grid floor. Each shock lasted 0.5 s and was repeated after 0.9 s if the mouse failed to escape the to‐be‐avoided sector in time. Intensity of the shock was individualized for each mouse (0.2–0.4 mA), to ensure escape reaction while avoiding excessive pain. The training schedule consisted of five acquisition sessions and four reversal sessions, where the sector position was changed by 180°. Two 10‐min sessions were scheduled for each experimental day, separated by approximately 3 h of rest in the home cage. The arena rotated while the sector remained fixed in the reference frame of the room; therefore, the mice had to move actively away from the sector in the direction opposite to arena rotation; otherwise, they would be carried into the sector. For successful avoidance, the animal had to separate the distant room‐frame cues, which could be used to locate and avoid the sector, from the irrelevant, arena‐frame cues. Selection of the correct spatial cues and achievement of the correct behavioral strategy requires segregation of spatial frames, a skill that is considered equivalent of human cognitive coordination 96. Furthermore, cognitive flexibility was tested in reversal sessions, to adjust the behavioral response to reversed sector location. The trajectory of the mice was analyzed offline using the custom‐made and freely available Carousel Maze Manager 4.0 97. Total distance walked, a measure of locomotor activity of the mouse, and the number of entrances, a measure of its ability to avoid entering the to‐be‐avoided sector, were used as the most important output parameters. Active place avoidance performance was evaluated by two consecutive RM‐ANOVA analyses, with sessions taken as repeated measures, and genotype as a between‐subject factor. In this task, 21 male mice (11 KO, 10 WT), 12–14 weeks old, were tested.
Light microscopy
Five types of microscopes were used in this study: (i) Leica TCS SP8 confocal equipped with 405‐nm laser (Hoechst), 488‐nm laser (DiO, EGFP excitation), 552‐nm laser (DiI excitation), 10×/0.3 dry, and 25×/0.75 immerse‐oil objectives (Figs 2G, 4, 6D, F, EV1F, EV3B, and EV5B, D, Appendix Fig S1C). (ii) Inverted fluorescent microscope Leica DMI6000 equipped with 20×/0.4 dry objective, 37°C incubator, and CO2 chamber for live imaging (Figs 1F and 5). (iii) CARV II/Nikon Ti‐E spinning disk equipped with 20×/0.5 dry, 40×/1.3, and 100×/1.4 immerse‐oil objectives (Figs 1B, 2C, D, 3, 6A, B, F, EV1A, F, EV2G, EV3A, EV4A, D, and EV5A, F). (iv) Olympus FV1000 confocal microscope equipped with 20×/0.8 objective (Fig EV3C). (v) Nikon SMZ18 stereomicroscope for microdissections and whole‐mount analysis equipped with 1× and 2× SHR Plan Apo objectives (Figs 1C, 2A, EV1C, D, EV2A, B, E, F, and EV4B). Microscopy images from Figs 1, 2, 3, 4 and EV1, EV3, and EV4 are compositions of more fields automatically generated by Leica LAS X software or NIS elements software. Maximum projections were generated in ImageJ.
Image processing and statistics
We used Neurolucida 360 software to reconstruct callosal axons from both in utero electroporation and DiI‐tracing experiments. In case of oblique sections, we picked only midline regions (approximately 100 μm from midline to each side) and traced axons semi‐automatically using AutoNeuron algorithm. To display overall axon growth direction, polar histograms and fan‐in diagrams were generated using Neurolucida explorer. Polar histograms show total axon length in a specific degree range. We counted lengths in each 20° of histogram. Fan‐in diagrams represent the same in another graphical view when all traced axons rise from a single point. Dendrites and dendritic spines were analyzed either with Neurolucida360 or with NeuronJ plugin in ImageJ. Spine head diameter was measured after 3D reconstruction of spines in Neurolucida 360. Images from whole‐mount preparations were processed in Helicon focus to create sharp projections. Growth of peripheral nerves and all data from time‐lapse imaging series were analyzed in ImageJ. Neuron migration was analyzed in ImageJ using cell counter plugin. Measurements of axon pruning from immunohistochemistry and DiI‐tracing experiments were done in ImageJ. In all figures, different channels of image series were combined in pseudo‐color using the “screen” function in Adobe Photoshop and adjusted to enhance low‐intensity objects. GraphPad Prism was used for statistical analysis. For comparison of two independent groups, unpaired t‐test was used unless stated otherwise. Multiple comparisons were performed using two‐way ANOVA. Data from behavioral experiments were analyzed by unpaired t‐test (P6, P8 vocalization, sociability, Y‐maze, EPM), ANOVA (AAPA), or Mann–Whitney test (P12 vocalization). All data are presented as means ± SD, unless stated otherwise.
Author contributions
Study design: MB, JZ; data acquisition and analysis: JZ, MB, RW, KJ, MJ, RM, TP, BP, MW, MSB, MK, PK, XZ; supervision: MB, EP, AS, RS, TM, GA‐B; writing—original draft: JZ, MB, MSB; writing—review and editing: JZ, MB, GA‐B, TM, RM, AS, TP, MSB; project administration: MB.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Review Process File
Acknowledgements
The work of MB, JZ, MJ, TP, BP, and AS was supported by Czech Health Research Council grant no. NV18‐04‐00085. MB, RW, BP, JZ, and MK were supported by Czech Science Foundation grant no. 16‐15915S. The work of JZ, RW, and BP was supported by Grant Agency of the Charles University grant nos. 682217, 524218, and 1062216, respectively. MJ, TP, and AS were supported by Czech Science Foundation grant no. 19‐03016S and Czech Health Research Council grant no 17‐30833A. MW, MSB, and TM were supported by the ERC (FP/2007‐2013; ERC Grant Agreement No. 616791), the German‐Israeli Foundation, SFB 870, DFG grant CRC870‐A11, and the Munich Cluster for Systems Neurology (SyNergy; EXC2145). RS was supported by RVO 68378050 by Academy of Sciences of the Czech Republic. Czech Centre for Phenogenomics infrastructure, used during the project, was supported by grants LM2015040, CZ.1.05/2.1.00/19.0395, and CZ.1.05/1.1.00/02.0109 funded by the Ministry of Education, Youth and Sports and the European Regional Development Fund. We are grateful to Dr. Jan Krůšek from the Department of Cellular Neurophysiology, Institute of Physiology, CAS, Prague, for his help with preparation of glass micropipettes. We acknowledge the Microscopy Centre—Light Microscopy Core Facility, IMG ASCR, Prague, Czech Republic, supported by MEYS (LM2015062), OPPK (CZ.2.16/3.1.00/21547) and (LO1419), and Light Microscopy Core Facility, IPHYS ASCR, Prague, Czech Republic, supported by MEYS (LM2015062 Czech‐BioImaging) for their support with confocal and live imaging.
EMBO Reports (2020) 21: e48512
References
- 1. Riccomagno MM, Kolodkin AL (2015) Sculpting neural circuits by axon and dendrite pruning. Annu Rev Cell Dev Biol 31: 779–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnston MV (2004) Clinical disorders of brain plasticity. Brain Dev 26: 73–80 [DOI] [PubMed] [Google Scholar]
- 3. Vanderhaeghen P, Cheng HJ (2010) Guidance molecules in axon pruning and cell death. Cold Spring Harb Perspect Biol 2: a001859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Battum EY, Brignani S, Pasterkamp RJ (2015) Axon guidance proteins in neurological disorders. Lancet Neurol 14: 532–546 [DOI] [PubMed] [Google Scholar]
- 5. Yu F, Schuldiner O (2014) Axon and dendrite pruning in Drosophila . Curr Opin Neurobiol 27: 192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bagri A, Cheng HJ, Yaron A, Pleasure SJ, Tessier‐Lavigne M (2003) Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell 113: 285–299 [DOI] [PubMed] [Google Scholar]
- 7. Low LK, Liu XB, Faulkner RL, Coble J, Cheng HJ (2008) Plexin signaling selectively regulates the stereotyped pruning of corticospinal axons from visual cortex. Proc Natl Acad Sci USA 105: 8136–8141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM (1995) Collapsin‐induced growth cone collapse mediated by an intracellular protein related to UNC‐33. Nature 376: 509–514 [DOI] [PubMed] [Google Scholar]
- 9. Byk T, Dobransky T, Cifuentes‐Diaz C, Sobel A (1996) Identification and molecular characterization of Unc‐33‐like phosphoprotein (Ulip), a putative mammalian homolog of the axonal guidance‐associated unc‐33 gene product. J Neurosci 16: 688–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Minturn JE, Fryer HJ, Geschwind DH, Hockfield S (1995) TOAD‐64, a gene expressed early in neuronal differentiation in the rat, is related to unc‐33, a C. elegans gene involved in axon outgrowth. J Neurosci 15: 6757–6766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H et al (2002) CRMP‐2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 4: 583–591 [DOI] [PubMed] [Google Scholar]
- 12. Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K et al (2005) Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes Cells 10: 165–179 [DOI] [PubMed] [Google Scholar]
- 13. Yuasa‐Kawada J, Suzuki R, Kano F, Ohkawara T, Murata M, Noda M (2003) Axonal morphogenesis controlled by antagonistic roles of two CRMP subtypes in microtubule organization. Eur J Neurosci 17: 2329–2343 [DOI] [PubMed] [Google Scholar]
- 14. Balastik M, Zhou XZ, Alberich‐Jorda M, Weissova R, Ziak J, Pazyra‐Murphy MF, Cosker KE, Machonova O, Kozmikova I, Chen CH et al (2015) Prolyl isomerase Pin1 regulates axon guidance by stabilizing CRMP2A selectively in distal axons. Cell Rep 13: 812–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K (2005) GSK‐3beta regulates phosphorylation of CRMP‐2 and neuronal polarity. Cell 120: 137–149 [DOI] [PubMed] [Google Scholar]
- 16. Arimura N, Menager C, Kawano Y, Yoshimura T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M et al (2005) Phosphorylation by Rho kinase regulates CRMP‐2 activity in growth cones. Mol Cell Biol 25: 9973–9984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang H, Kang E, Wang Y, Yang C, Yu H, Wang Q, Chen Z, Zhang C, Christian KM, Song H et al (2016) Brain‐specific Crmp2 deletion leads to neuronal development deficits and behavioural impairments in mice. Nat Commun 7: 11773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Makihara H, Nakai S, Ohkubo W, Yamashita N, Nakamura F, Kiyonari H, Shioi G, Jitsuki‐Takahashi A, Nakamura H, Tanaka F et al (2016) CRMP1 and CRMP2 have synergistic but distinct roles in dendritic development. Genes Cells 21: 994–1005 [DOI] [PubMed] [Google Scholar]
- 19. Nakata K, Ujike H, Sakai A, Takaki M, Imamura T, Tanaka Y, Kuroda S (2003) The human dihydropyrimidinase‐related protein 2 gene on chromosome 8p21 is associated with paranoid‐type schizophrenia. Biol Psychiatry 53: 571–576 [DOI] [PubMed] [Google Scholar]
- 20. Liu Y, Pham X, Zhang L, Chen PL, Burzynski G, McGaughey DM, He S, McGrath JA, Wolyniec P, Fallin MD et al (2014) Functional variants in DPYSL2 sequence increase risk of schizophrenia and suggest a link to mTOR signaling. G3 (Bethesda) 5: 61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clark D, Dedova I, Cordwell S, Matsumoto I (2006) A proteome analysis of the anterior cingulate cortex gray matter in schizophrenia. Mol Psychiatry 11: 459–470, 423 [DOI] [PubMed] [Google Scholar]
- 22. Quach TT, Honnorat J, Kolattukudy PE, Khanna R, Duchemin AM (2015) CRMPs: critical molecules for neurite morphogenesis and neuropsychiatric diseases. Mol Psychiatry 20: 1037–1045 [DOI] [PubMed] [Google Scholar]
- 23. Braunschweig D, Krakowiak P, Duncanson P, Boyce R, Hansen RL, Ashwood P, Hertz‐Picciotto I, Pessah IN, Van de Water J (2013) Autism‐specific maternal autoantibodies recognize critical proteins in developing brain. Transl Psychiatry 3: e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S et al (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515: 209–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lord C, Elsabbagh M, Baird G, Veenstra‐Vanderweele J (2018) Autism spectrum disorder. Lancet 392: 508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bourgeron T (2015) From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 16: 551–563 [DOI] [PubMed] [Google Scholar]
- 27. Kitsukawa T, Shimizu M, Sanbo M, Hirata T, Taniguchi M, Bekku Y, Yagi T, Fujisawa H (1997) Neuropilin‐semaphorin III/D‐mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron 19: 995–1005 [DOI] [PubMed] [Google Scholar]
- 28. Taniguchi M, Yuasa S, Fujisawa H, Naruse I, Saga S, Mishina M, Yagi T (1997) Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron 19: 519–530 [DOI] [PubMed] [Google Scholar]
- 29. Chen H, Bagri A, Zupicich JA, Zou Y, Stoeckli E, Pleasure SJ, Lowenstein DH, Skarnes WC, Chedotal A, Tessier‐Lavigne M (2000) Neuropilin‐2 regulates the development of selective cranial and sensory nerves and hippocampal mossy fiber projections. Neuron 25: 43–56 [DOI] [PubMed] [Google Scholar]
- 30. Cheng HJ, Bagri A, Yaron A, Stein E, Pleasure SJ, Tessier‐Lavigne M (2001) Plexin‐A3 mediates semaphorin signaling and regulates the development of hippocampal axonal projections. Neuron 32: 249–263 [DOI] [PubMed] [Google Scholar]
- 31. Maimon R, Ionescu A, Bonnie A, Sweetat S, Wald‐Altman S, Inbar S, Gradus T, Trotti D, Weil M, Behar O et al (2018) miR126‐5p downregulation facilitates axon degeneration and NMJ disruption via a non‐cell‐autonomous mechanism in ALS. J Neurosci 38: 5478–5494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brown M, Jacobs T, Eickholt B, Ferrari G, Teo M, Monfries C, Qi RZ, Leung T, Lim L, Hall C (2004) Alpha2‐chimaerin, cyclin‐dependent Kinase 5/p35, and its target collapsin response mediator protein‐2 are essential components in semaphorin 3A‐induced growth‐cone collapse. J Neurosci 24: 8994–9004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou J, Wen Y, She L, Sui YN, Liu L, Richards LJ, Poo MM (2013) Axon position within the corpus callosum determines contralateral cortical projection. Proc Natl Acad Sci USA 110: E2714–E2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu KY, He M, Hou QQ, Sheng AL, Yuan L, Liu F, Liu WW, Li G, Jiang XY, Luo ZG (2014) Semaphorin 3A activates the guanosine triphosphatase Rab5 to promote growth cone collapse and organize callosal axon projections. Sci Signal 7: ra81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang CL, Zhang L, Zhou Y, Zhou J, Yang XJ, Duan SM, Xiong ZQ, Ding YQ (2007) Activity‐dependent development of callosal projections in the somatosensory cortex. J Neurosci 27: 11334–11342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14: 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garey L (2010) When cortical development goes wrong: schizophrenia as a neurodevelopmental disease of microcircuits. J Anat 217: 324–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu XB, Low LK, Jones EG, Cheng HJ (2005) Stereotyped axon pruning via plexin signaling is associated with synaptic complex elimination in the hippocampus. J Neurosci 25: 9124–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Linke R, Pabst T, Frotscher M (1995) Development of the hippocamposeptal projection in the rat. J Comp Neurol 351: 602–616 [DOI] [PubMed] [Google Scholar]
- 40. Sahay A, Molliver ME, Ginty DD, Kolodkin AL (2003) Semaphorin 3F is critical for development of limbic system circuitry and is required in neurons for selective CNS axon guidance events. J Neurosci 23: 6671–6680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Basarsky TA, Parpura V, Haydon PG (1994) Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J Neurosci 14: 6402–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tran TS, Rubio ME, Clem RL, Johnson D, Case L, Tessier‐Lavigne M, Huganir RL, Ginty DD, Kolodkin AL (2009) Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature 462: 1065–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamashita N, Morita A, Uchida Y, Nakamura F, Usui H, Ohshima T, Taniguchi M, Honnorat J, Thomasset N, Takei K et al (2007) Regulation of spine development by semaphorin3A through cyclin‐dependent kinase 5 phosphorylation of collapsin response mediator protein 1. J Neurosci 27: 12546–12554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Petanjek Z, Judas M, Simic G, Rasin MR, Uylings HB, Rakic P, Kostovic I (2011) Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci USA 108: 13281–13286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bian WJ, Miao WY, He SJ, Qiu Z, Yu X (2015) Coordinated spine pruning and maturation mediated by inter‐spine competition for cadherin/catenin complexes. Cell 162: 808–822 [DOI] [PubMed] [Google Scholar]
- 46. Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TR, Hirsch SR (1998) Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry 65: 446–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ey E, Torquet N, Le Sourd AM, Leblond CS, Boeckers TM, Faure P, Bourgeron T (2013) The Autism ProSAP1/Shank2 mouse model displays quantitative and structural abnormalities in ultrasonic vocalisations. Behav Brain Res 256: 677–689 [DOI] [PubMed] [Google Scholar]
- 48. Hung AY, Futai K, Sala C, Valtschanoff JG, Ryu J, Woodworth MA, Kidd FL, Sung CC, Miyakawa T, Bear MF et al (2008) Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J Neurosci 28: 1697–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, Janssen AL, Udvardi PT, Shiban E, Spilker C et al (2012) Autistic‐like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486: 256–260 [DOI] [PubMed] [Google Scholar]
- 50. Binder MS, Lugo JN (2017) NS‐Pten knockout mice show sex‐ and age‐specific differences in ultrasonic vocalizations. Brain Behav 7: e00857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reynolds CD, Nolan SO, Jefferson T, Lugo JN (2016) Sex‐specific and genotype‐specific differences in vocalization development in FMR1 knockout mice. NeuroReport 27: 1331–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nakamura H, Yamashita N, Kimura A, Kimura Y, Hirano H, Makihara H, Kawamoto Y, Jitsuki‐Takahashi A, Yonezaki K, Takase K et al (2016) Comprehensive behavioral study and proteomic analyses of CRMP2‐deficient mice. Genes Cells 21: 1059–1079 [DOI] [PubMed] [Google Scholar]
- 53. Kjelstrup KG, Tuvnes FA, Steffenach HA, Murison R, Moser EI, Moser MB (2002) Reduced fear expression after lesions of the ventral hippocampus. Proc Natl Acad Sci USA 99: 10825–10830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Luo L, O'Leary DD (2005) Axon retraction and degeneration in development and disease. Annu Rev Neurosci 28: 127–156 [DOI] [PubMed] [Google Scholar]
- 55. Riccomagno MM, Hurtado A, Wang H, Macopson JG, Griner EM, Betz A, Brose N, Kazanietz MG, Kolodkin AL (2012) The RacGAP beta2‐Chimaerin selectively mediates axonal pruning in the hippocampus. Cell 149: 1594–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tapia JC, Wylie JD, Kasthuri N, Hayworth KJ, Schalek R, Berger DR, Guatimosim C, Seung HS, Lichtman JW (2012) Pervasive synaptic branch removal in the mammalian neuromuscular system at birth. Neuron 74: 816–829 [DOI] [PubMed] [Google Scholar]
- 57. Brill MS, Kleele T, Ruschkies L, Wang M, Marahori NA, Reuter MS, Hausrat TJ, Weigand E, Fisher M, Ahles A et al (2016) Branch‐specific microtubule destabilization mediates axon branch loss during neuromuscular synapse elimination. Neuron 92: 845–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xu NJ, Henkemeyer M (2009) Ephrin‐B3 reverse signaling through Grb4 and cytoskeletal regulators mediates axon pruning. Nat Neurosci 12: 268–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cocchi E, Drago A, Serretti A (2016) Hippocampal pruning as a new theory of schizophrenia etiopathogenesis. Mol Neurobiol 53: 2065–2081 [DOI] [PubMed] [Google Scholar]
- 60. Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22: 389–442 [DOI] [PubMed] [Google Scholar]
- 61. Venkova K, Christov A, Kamaluddin Z, Kobalka P, Siddiqui S, Hensley K (2014) Semaphorin 3A signaling through neuropilin‐1 is an early trigger for distal axonopathy in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 73: 702–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. De Winter F, Vo T, Stam FJ, Wisman LA, Bar PR, Niclou SP, van Muiswinkel FL, Verhaagen J (2006) The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol Cell Neurosci 32: 102–117 [DOI] [PubMed] [Google Scholar]
- 63. Demyanenko GP, Mohan V, Zhang X, Brennaman LH, Dharbal KE, Tran TS, Manis PB, Maness PF (2014) Neural cell adhesion molecule NrCAM regulates Semaphorin 3F‐induced dendritic spine remodeling. J Neurosci 34: 11274–11287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schwarting GA, Kostek C, Ahmad N, Dibble C, Pays L, Puschel AW (2000) Semaphorin 3A is required for guidance of olfactory axons in mice. J Neurosci 20: 7691–7697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Friedel RH, Plump A, Lu X, Spilker K, Jolicoeur C, Wong K, Venkatesh TR, Yaron A, Hynes M, Chen B et al (2005) Gene targeting using a promoterless gene trap vector (“targeted trapping”) is an efficient method to mutate a large fraction of genes. Proc Natl Acad Sci USA 102: 13188–13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pozas E, Pascual M, Nguyen Ba‐Charvet KT, Guijarro P, Sotelo C, Chedotal A, Del Rio JA, Soriano E (2001) Age‐dependent effects of secreted Semaphorins 3A, 3F, and 3E on developing hippocampal axons: in vitro effects and phenotype of Semaphorin 3A (‐/‐) mice. Mol Cell Neurosci 18: 26–43 [DOI] [PubMed] [Google Scholar]
- 67. Schwarz Q, Waimey KE, Golding M, Takamatsu H, Kumanogoh A, Fujisawa H, Cheng HJ, Ruhrberg C (2008) Plexin A3 and plexin A4 convey semaphorin signals during facial nerve development. Dev Biol 324: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bretin S, Reibel S, Charrier E, Maus‐Moatti M, Auvergnon N, Thevenoux A, Glowinski J, Rogemond V, Premont J, Honnorat J et al (2005) Differential expression of CRMP1, CRMP2A, CRMP2B, and CRMP5 in axons or dendrites of distinct neurons in the mouse brain. J Comp Neurol 486: 1–17 [DOI] [PubMed] [Google Scholar]
- 69. Niisato E, Nagai J, Yamashita N, Abe T, Kiyonari H, Goshima Y, Ohshima T (2012) CRMP4 suppresses apical dendrite bifurcation of CA1 pyramidal neurons in the mouse hippocampus. Dev Neurobiol 72: 1447–1457 [DOI] [PubMed] [Google Scholar]
- 70. Yamane M, Yamashita N, Hida T, Kamiya Y, Nakamura F, Kolattukudy P, Goshima Y (2017) A functional coupling between CRMP1 and Nav1.7 for retrograde propagation of Semaphorin3A signaling. J Cell Sci 130: 1393–1403 [DOI] [PubMed] [Google Scholar]
- 71. Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, Pickering S, Simmons D, Rastan S, Walsh FS et al (2000) Neuropilin‐2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron 25: 29–41 [DOI] [PubMed] [Google Scholar]
- 72. Ip JP, Fu AK, Ip NY (2014) CRMP2: functional roles in neural development and therapeutic potential in neurological diseases. Neuroscientist 20: 589–598 [DOI] [PubMed] [Google Scholar]
- 73. Tobe BTD, Crain AM, Winquist AM, Calabrese B, Makihara H, Zhao WN, Lalonde J, Nakamura H, Konopaske G, Sidor M et al (2017) Probing the lithium‐response pathway in hiPSCs implicates the phosphoregulatory set‐point for a cytoskeletal modulator in bipolar pathogenesis. Proc Natl Acad Sci USA 114: E4462–E4471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dominguez‐Iturza N, Lo AC, Shah D, Armendariz M, Vannelli A, Mercaldo V, Trusel M, Li KW, Gastaldo D, Santos AR et al (2019) The autism‐ and schizophrenia‐associated protein CYFIP1 regulates bilateral brain connectivity and behaviour. Nat Commun 10: 3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Silva AI, Haddon JE, Ahmed Syed Y, Trent S, Lin TE, Patel Y, Carter J, Haan N, Honey RC, Humby T et al (2019) Cyfip1 haploinsufficient rats show white matter changes, myelin thinning, abnormal oligodendrocytes and behavioural inflexibility. Nat Commun 10: 3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu X, Malenfant P, Reesor C, Lee A, Hudson ML, Harvard C, Qiao Y, Persico AM, Cohen IL, Chudley AE et al (2011) 2p15‐p16.1 microdeletion syndrome: molecular characterization and association of the OTX1 and XPO1 genes with autism spectrum disorders. Eur J Hum Genet 19: 1264–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Weimann JM, Zhang YA, Levin ME, Devine WP, Brulet P, McConnell SK (1999) Cortical neurons require Otx1 for the refinement of exuberant axonal projections to subcortical targets. Neuron 24: 819–831 [DOI] [PubMed] [Google Scholar]
- 78. Li Z, Jagadapillai R, Gozal E, Barnes G (2019) Deletion of semaphorin 3F in interneurons is associated with decreased GABAergic neurons, autism‐like behavior, and increased oxidative stress cascades. Mol Neurobiol 56: 5520–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Degano AL, Pasterkamp RJ, Ronnett GV (2009) MeCP2 deficiency disrupts axonal guidance, fasciculation, and targeting by altering Semaphorin 3F function. Mol Cell Neurosci 42: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF (2011) Efficient design and assembly of custom TALEN and other TAL effector‐based constructs for DNA targeting. Nucleic Acids Res 39: e82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, Bogdanove AJ (2012) TAL Effector‐Nucleotide Targeter (TALE‐NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res 40: W117–W122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kasparek P, Krausova M, Haneckova R, Kriz V, Zbodakova O, Korinek V, Sedlacek R (2014) Efficient gene targeting of the Rosa26 locus in mouse zygotes using TALE nucleases. FEBS Lett 588: 3982–3988 [DOI] [PubMed] [Google Scholar]
- 83. Magiera MM, Bodakuntla S, Ziak J, Lacomme S, Marques Sousa P, Leboucher S, Hausrat TJ, Bosc C, Andrieux A, Kneussel M et al (2018) Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J 37: e100440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sherazee N, Alvarez VA (2013) DiOlistics: delivery of fluorescent dyes into cells. Methods Mol Biol 940: 391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Haddad‐Tovolli R, Szabo NE, Zhou X, Alvarez‐Bolado G (2013) Genetic manipulation of the mouse developing hypothalamus through in utero electroporation. J Vis Exp e50412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ionescu A, Zahavi EE, Gradus T, Ben‐Yaakov K, Perlson E (2016) Compartmental microfluidic system for studying muscle‐neuron communication and neuromuscular junction maintenance. Eur J Cell Biol 95: 69–88 [DOI] [PubMed] [Google Scholar]
- 87. Kleele T, Marinkovic P, Williams PR, Stern S, Weigand EE, Engerer P, Naumann R, Hartmann J, Karl RM, Bradke F et al (2014) An assay to image neuronal microtubule dynamics in mice. Nat Commun 5: 4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Brill MS, Marinkovic P, Misgeld T (2013) Sequential photo‐bleaching to delineate single Schwann cells at the neuromuscular junction. J Vis Exp e4460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Marinkovic P, Godinho L, Misgeld T (2015) Imaging acute neuromuscular explants from Thy1 mouse lines. Cold Spring Harb Protoc 2015: pdb prot087692 [DOI] [PubMed] [Google Scholar]
- 90. Kerschensteiner M, Reuter MS, Lichtman JW, Misgeld T (2008) Ex vivo imaging of motor axon dynamics in murine triangularis sterni explants. Nat Protoc 3: 1645–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bures J, Fenton AA, Kaminsky Y, Zinyuk L (1997) Place cells and place navigation. Proc Natl Acad Sci USA 94: 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Burghardt NS, Park EH, Hen R, Fenton AA (2012) Adult‐born hippocampal neurons promote cognitive flexibility in mice. Hippocampus 22: 1795–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Stuchlik A, Petrasek T, Prokopova I, Holubova K, Hatalova H, Vales K, Kubik S, Dockery C, Wesierska M (2013) Place avoidance tasks as tools in the behavioral neuroscience of learning and memory. Physiol Res 62(Suppl 1): S1–S19 [DOI] [PubMed] [Google Scholar]
- 95. D'Hooge R, De Deyn PP (2001) Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev 36: 60–90 [DOI] [PubMed] [Google Scholar]
- 96. Wesierska M, Dockery C, Fenton AA (2005) Beyond memory, navigation, and inhibition: behavioral evidence for hippocampus‐dependent cognitive coordination in the rat. J Neurosci 25: 2413–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bahník Š (2014) Carousel Maze Manager (Version 0.4.0) Software. Retrieved from https://github.com/bahniks/CM_Manager_0_4_0 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Review Process File