

Abstract
p62/SQSTM1 is a multivalent protein that has the ability to cause liquid–liquid phase separation and serves as a receptor protein that participates in cargo isolation during selective autophagy. This protein is also involved in the non‐canonical activation of the Keap1‐Nrf2 system, a major oxidative stress response pathway. Here, we show a role of neighbor of BRCA1 gene 1 (NBR1), an autophagy receptor structurally similar to p62/SQSTM1, in p62‐liquid droplet formation and Keap1‐Nrf2 pathway activation. Overexpression of NBR1 blocks selective degradation of p62/SQSTM1 through autophagy and promotes the accumulation and phosphorylation of p62/SQSTM1 in liquid‐like bodies, which is required for the activation of Nrf2. NBR1 is induced in response to oxidative stress, which triggers p62‐mediated Nrf2 activation. Conversely, loss of Nbr1 suppresses not only the formation of p62/SQSTM1‐liquid droplets, but also of p62‐dependent Nrf2 activation during oxidative stress. Taken together, our results show that NBR1 mediates p62/SQSTM1‐liquid droplet formation to activate the Keap1‐Nrf2 pathway.
Keywords: autophagy, liquid droplet, NBR1, Nrf2, p62/SQSTM1
Subject Categories: Autophagy & Cell Death; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
SQSTM1/p62 can induce liquid‐liquid phase separation and serves as a cargo receptor during selective autophagy. NBR1 and p62 collaborate during the activation of the Keap1‐Nrf2 oxidative stress response pathway, promoting the formation of p62‐liquid droplets.

Introduction
When the concentration of molecules reaches a threshold, or when modifications increase the valency, liquid–liquid phase separation occurs, and similar components gather and concentrate to promote biochemical reactions or to sequester unwanted molecules. Such structures are called liquid droplets as they have liquid‐like properties, and the inside is a reversible structure in which fluidity and biochemical reactions are kept 1. There are many droplets of this kind in the cell such as stress granules, P‐granules, and nucleoli, and they control (the) integrated stress response mechanism(s), that is, the robustness that protects cells from harmful disturbances like environmental stresses 2. Liquid droplets are dynamic spherical structures that undergo fusion and fission events and are comparable in size to organelles. For this reason, they are often referred to as membraneless organelles 3. While the autophagy pathway mediates the turnover of some of these structures, like stress granules and P‐granules 4, 5, 6, suppression of autophagy is accompanied by the accumulation of structures in which liquid droplets are gelated and/or aggregated 7.
Macroautophagy (hereafter referred to as autophagy) is a cytoprotective mechanism that provides eukaryotic cells with building blocks for anabolic functions 7, 8. In brief, after the appearance of a stressful stimulus (metabolic stress, organelle damage, and invasive microbes), a double membrane arises to isolate the cytosolic material in a compartment called the autophagosome. The autophagosome then fuses with a lysosome, thereby promoting the degradation of the sequestered materials 9. While starvation‐induced autophagy is thought to randomly degrade cytosolic components, under certain circumstances, autophagosomes selectively surround and degrade specific cargoes. The latter is called selective autophagy and contributes to cellular homeostasis by degrading soluble proteins, liquid droplets, protein aggregates, excess or degenerated organelles, and invasive microbes 10. The molecular mechanism of autophagosome membrane formation in conventional and selective autophagy is considered to be common, but during selective autophagy, the “label of each cargo” or the presence of a “receptor protein” ensures selectivity. “Labeling of each cargo” means ubiquitination of cargo or the interaction of a receptor protein with cargo. “Receptor protein” refers to a group of proteins that bind to the cargo and to the autophagosomal proteins, i.e., the microtubule‐associated protein 1A/1B‐light chain 3 (LC3) or members of the GABA type A receptor‐associated protein (GABARAP) family. Receptor proteins are divided into ubiquitin‐binding receptor proteins that recognize the ubiquitin chain of the cargo, and cargo‐localized receptor proteins that interact directly with the cargo. Both types of receptor proteins have an LC3‐interacting motif (LIR) or a GABARAP‐interacting motif (GIM) and bind directly to the LC3, the GABARAP family, or both 11.
p62/SQSTM1 (hereafter referred to as p62) is a stress‐inducible protein able to change among binding partners and its cellular localization that forms liquid droplet structures in a context‐dependent manner. The protein was mainly defined as an ubiquitin‐binding receptor protein for selective autophagy 12. Besides this role, its ability to interact with multiple binding partners allows p62 to act as the main regulator and activator of nuclear factor erythroid 2‐related factor 2 (Nrf2), the mechanistic target of rapamycin complex 1 (mTORC1) and nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) signaling pathways, which links p62 to the oxidative defense system, nutrient‐sensing, and inflammation, respectively 13, 14. Recent reports revealed that once p62 binds to ubiquitin chains, it acquires liquid‐like properties 6. Such phase‐separated droplets allow the exchange of their components, including ubiquitin and LC3, with the surrounding environment. Consequently, the droplets can also function as nodes from which signaling cascades can be activated in the context of selective autophagy.
Here, we show a role of neighbor of BRCA1 gene 1 (NBR1), which serves as a ubiquitin‐binding autophagy receptor and is a binding partner of p62, in p62‐liquid droplet formation and p62‐mediated Nrf2 activation. Increasing levels of NBR1 stabilize p62 and promote the formation of liquid droplets resistant to autophagic degradation, leading to a robust induction of Nrf2 targets, including p62 itself. Loss of NBR1 attenuates p62‐mediated activation of Nrf2 in response to oxidative stress. Our results indicate that the collaboration between p62 and NBR1 stretches beyond their roles as selective autophagy receptors, and that NBR1 is a new player to orchestrate the antioxidant cell response.
Results
NBR1 increases the formation of p62‐liquid droplets
Autophagy receptors p62 and NBR1 have been shown to act cooperatively in different forms of selective autophagy 15, 16, 17, 18. This cooperation is also observed between p62 and other cargo receptors 19, 20. However, it is currently unknown if autophagy receptors have a direct effect on the relative abundance, cellular localization, and signaling roles of each other. As p62 and NBR1 share a common domain distribution and directly interact with each other, we wondered if NBR1 influences the autophagic and non‐autophagic functions of p62. With that aim, we exogenously expressed NBR1 in mouse primary hepatocytes using the adenovirus system. The overexpression of NBR1 increased not only total p62 protein levels, but also its Serine 349 and Serine 403 phosphorylated forms (Fig 1A), which are involved in Nrf2 activation 21 and ubiquitin‐binding followed by phase separation 6, respectively. Gene expression of p62, which is mainly regulated by the transcription factors Nrf2, NF‐κB, and the microphthalmia/transcription factor E family (MiT/TFE), was induced upon the overexpression of NBR1 (Fig 1B). Double‐immunofluorescent analysis with anti‐p62 and anti‐NBR1 antibodies was employed as a complementary approach to confirm the interplay between NBR1 and p62. As previously described 22, both proteins extensively co‐localized in the same punctate structures at endogenous levels, and the expression of NBR1 increased the size and number of such structures (Fig 1C). According to two recent reports 6, 23, the p62‐positive structures possess liquid‐like properties that are essential for its role as an autophagy receptor. To check whether the structures formed by NBR1 overexpression are also liquid‐like structures, we measured the exchange of components between the structures and the surrounding environment by a fluorescence recovery after photobleaching (FRAP) assay. To do this, we overexpressed C‐terminal GFP‐tagged NBR1, NBR1‐GFP in wild‐type primary mouse hepatocytes. Similar to non‐tagged NBR1, NBR1‐GFP completely co‐localized with p62‐positive structures (Fig EV1A). The signal intensity of the structures positive for NBR1‐GFP was recovered at 8–12 min after photobleaching (Fig 1D and Movie EV1), indicating that p62‐ and NBR1‐positive structures have liquid‐like properties. As a control and reference for NBR1 mobility, we expressed mCherry‐p62 in p62‐knockout mouse embryonic fibroblasts (MEFs) and performed the FRAP assay. Similar to the case of NBR1‐GFP (Fig 1D), we verified the recovery of signal intensity of the structures positive for mCherry‐p62 after photobleaching, and it was significantly enhanced by overexpression of GFP‐tagged NBR1 (Fig EV1B). Remarkably, such enhancement was also observed by the overexpression of an NBR1 mutant unable to interact with p62 (NBR1 D50R; Fig EV1B). Collectively, our data show that NBR1 induces phase separation of p62, promoting the formation of p62 bodies.
Figure 1. NBR1 promotes an increase in p62 levels, phosphorylation, and body formation.
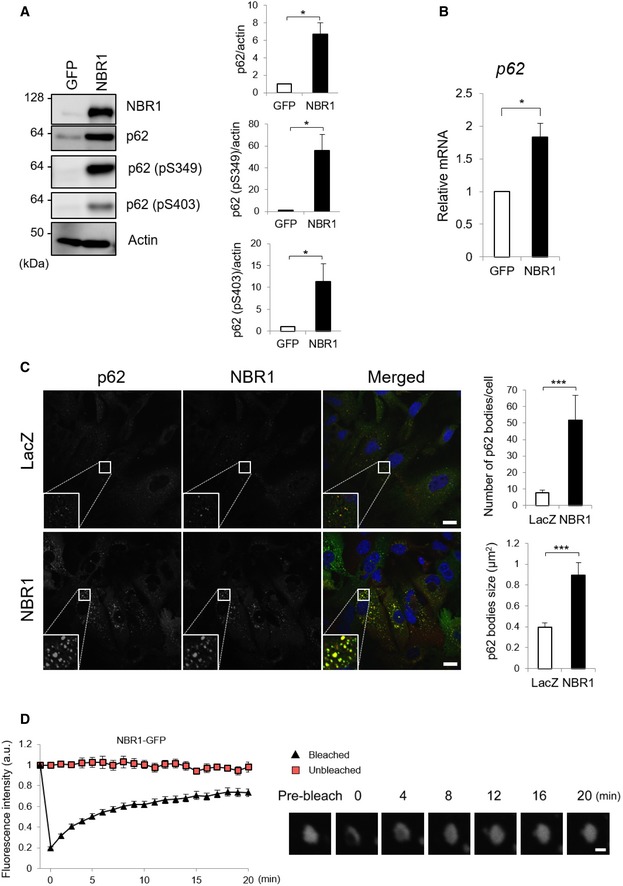
- Immunoblot analysis. Primary hepatocytes prepared from wild‐type mice were infected with adenovirus expressing GFP or NBR1 under the CAG promoter for 48 h. Cell lysates were prepared and subjected to immunoblot analysis with the indicated antibodies. Data shown are representative of three separate experiments. Bar graphs indicate the quantitative densitometric analysis of the indicated proteins relative to actin. Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
- Real‐time PCR. Total RNAs were prepared from primary hepatocytes described in (A). Values were normalized against the amount of mRNA in the hepatocytes expressing GFP. The experiments were performed three times. Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
- Immunofluorescence microscopy. Wild‐type hepatocytes were infected with LacZ or NBR1 adenovirus for 48 h and then immunostained with anti‐p62 and anti‐NBR1 antibodies. Each inset is a magnified image. Bars: 20 μm. The number and size of p62 bodies were measured in more than 100 cells. Data are shown as means ± SE. ***P < 0.001 as determined by Welch's t‐test.
- FRAP assay. Wild‐type hepatocytes cultured in glass‐bottom plates were infected with GFP‐NBR1 adenovirus for 48 h. The signal recovery after photobleaching was measured, quantified from at least three independent biological replicates, and is represented as means ± SE. Bar: 1 μm.
Source data are available online for this figure.
Figure EV1. Mobility analysis of structures positive for p62 and/or NBR1.

-
AWild‐type hepatocytes were infected with GFP or NBR1‐GFP adenovirus for 48 h and then immunostained with anti‐p62 antibody. Each inset is a magnified image. Bars: 20 μm.
-
B–DFRAP assay. p62‐knockout MEFs were co‐transfected mCherry‐p62 together with GFP, GFP‐NBR1, or GFP‐NBR1 D50R. Forty‐eight hours after the transfection, the signal recovery after photobleaching was measured and quantified (B). GFP‐NBR1 was transfected into wild‐type or p62‐knockout MEFs. Forty‐eight hours after the transfection, the signal recovery after photobleaching was measured and quantified (C). GFP‐p62 was transfected into wild‐type or Nbr1‐knockout MEFs. Forty‐eight hours after the transfection, the signal recovery after photobleaching was measured and quantified (D). Three independent biological replicates were performed on each experiment. Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
NBR1 regulates the Nrf2‐Keap1 pathway in a p62‐dependent manner
Since p62 is involved in the regulation of multiple signaling pathways including mTORC1, p38 MAP kinase, and Nrf2 13, we examined if p62‐phase separation due to overexpression of NBR1 affects such pathways. Expression of NBR1 in primary mouse hepatocytes did not have any effect on the conversion of LC3‐I to LC3‐II that is commonly used as a marker for autophagy fitness 24 (Fig EV2A). The phosphorylation of S6K, a substrate of mTORC1, was not affected by the expression of NBR1 (Fig EV2A). We did not observe any changes in phosphorylation of p38 indicating activation of p38 by NBR1 expression (Fig EV2A). Meanwhile, increased levels of NAD(P)H dehydrogenase, quinone 1 (Nqo1), an Nrf2 target were detected in the hepatocytes expressing NBR1 (Fig EV2A), suggesting that NBR1 could be involved in the activation of this signaling pathway.
Figure EV2. NBR1 influences Nrf2 activation but not other p62‐related pathways.
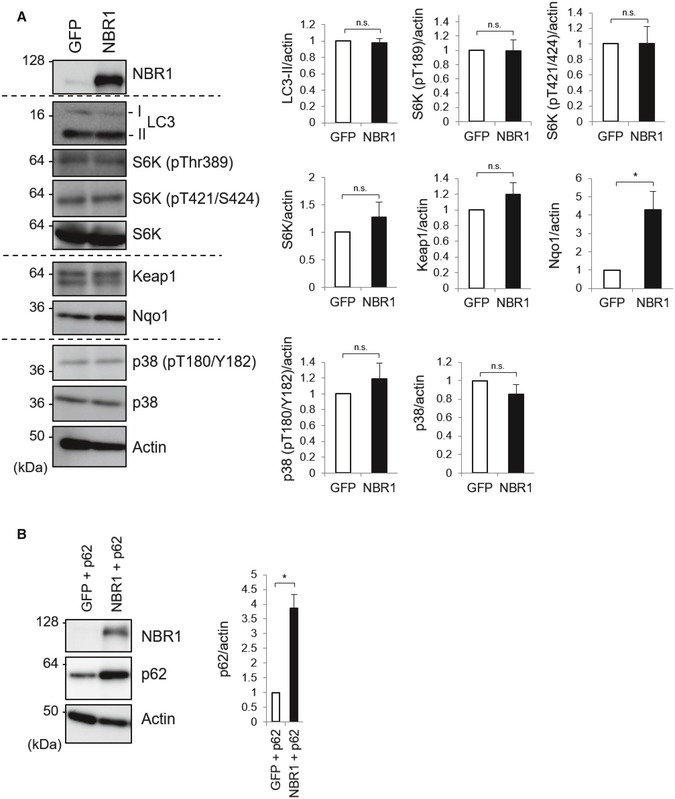
- The primary hepatocytes prepared from wild‐type mice and infected with adenovirus GFP or NBR1 for 48 h employed in Fig 1A were subjected to immunoblot analysis with the indicated antibodies. Data shown are representative of three separate experiments. Bar graphs indicate the quantitative densitometric analysis of the indicated proteins relative to actin. Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
- Primary hepatocytes prepared from p62 f/f; Alb‐Cre mice were infected with adenovirus expressing GFP or NBR1 in combination with p62 for 48 h. Cell lysates were prepared and subjected to immunoblot analysis with the indicated antibodies. Data shown are representative of three separate experiments. Bar graphs indicate the quantitative densitometric analysis of p62 relative to actin. Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
Source data are available online for this figure.
To confirm the role of NBR1 in the activation of Nrf2, we checked if NBR1 expression affected the nuclear translocation of Nrf2, which is a key step in the activation of the pathway 25. When mouse primary hepatocytes were infected with adenovirus against GFP‐tagged Nrf2 together with NBR1 but not GFP, total levels of GFP‐Nrf2 markedly increased (Fig 2A). As shown in Fig 2B, when NBR1 was expressed in primary mouse hepatocytes isolated from p62 f/f mice, the nuclear level of Nrf2 significantly increased compared with that in hepatocytes expressing GFP. In agreement with this biochemical analysis, translocation of GFP‐Nrf2 into the nucleus was significantly enhanced in mouse primary hepatocytes expressing NBR1 compared to those expressing LacZ (Fig 2C). Expectedly, gene expression of Nrf2 targets such as Nqo1, Glutamate‐cysteine ligase catalytic subunit (Gclc), and UDP‐glucose dehydrogenase (Ugdh) was markedly induced by the overexpression of NBR1, but not in the case of overexpression of GFP (Fig 2D). We verified up‐regulation of Nqo1, Gclc, and Ugdh proteins in the hepatocytes expressing NBR1 (Fig 2B), implying the functional activation of Nrf2.
Figure 2. NBR1 increases protein and mRNA levels of Nrf2 targets through p62.
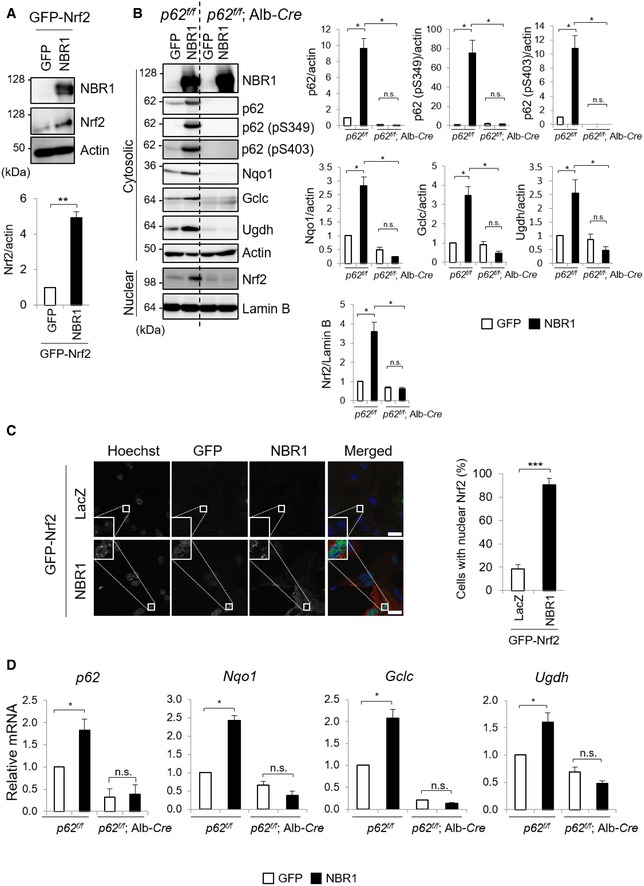
- Immunoblot analysis. Primary hepatocytes prepared from wild‐type mice were infected with adenovirus expressing GFP‐Nrf2 and GFP or NBR1 for 48 h. Total cell lysates were prepared and subjected to immunoblot analysis with the indicated antibodies. Data shown are representative of three separate experiments. Bar graphs indicate the quantitative densitometric analysis of the indicated proteins relative to actin. Data are shown as means ± SE. **P < 0.01 as determined by Welch's t‐test.
- Immunoblot analysis. Primary hepatocytes prepared from p62 f/f and p62 f/f; Alb‐Cre mice were infected with adenovirus against GFP or NBR1 for 48 h. Cytosolic and nuclear fractions were prepared and subjected to immunoblot analysis with the indicated antibodies. Data shown are representative of three separate experiments. Bar graphs indicate the quantitative densitometric analysis of the indicated proteins relative to actin (cytosolic proteins) or Lamin B (nuclear proteins). Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
- Immunofluorescence microscopy. Primary hepatocytes from wild‐type mice were infected with adenovirus expressing GFP‐Nrf2 and LacZ or NBR1 for 48 h. More than 100 cells were assayed for the nuclear signal of Nrf2. Data are shown as means ± SE. ***P < 0.001 as determined by Welch's t‐test. Bars: 10 μm.
- Total RNAs were prepared from primary hepatocytes described in (B). Values were normalized against the amount of mRNA in the p62 f/f hepatocytes expressing GFP. The experiments were performed at least three times. Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
Source data are available online for this figure.
p62 has the ability to activate Nrf2 under stress conditions through the competitive binding to Kelch‐like ECH‐associated protein 1 (Keap1), which is an adaptor protein of Cullin 3‐based ubiquitin ligase for Nrf2 26. To examine the effect of p62 in the NBR1‐mediated Nrf2 activation, we isolated hepatocytes from p62 f/f mice that expressed the Cre recombinase under the control of the albumin promoter (p62 f/f; Alb‐Cre), resulting in the loss of the protein in the hepatocytes (Fig 2B). Loss of p62 completely suppressed nuclear translocation of Nrf2 upon NBR1 expression (Fig 2B). As a result, induction of Nrf2‐target genes, which was observed in p62‐competent hepatocytes, was abolished at both mRNA and protein levels by the ablation of p62 (Fig 2B and D).
Immunofluorescent analysis with anti‐Keap1, anti‐NBR1, and anti‐p62 antibodies revealed their extensive co‐localization in the same droplets (Fig 3A). We verified that immunoprecipitants with anti‐NBR1 antibody contained both Keap1 and p62 (Fig 3B). Both the translocation of Keap1 onto the droplets and the interaction of NBR1 with Keap1 were dependent on p62 as they were lost after the expression of Cre recombinase in primary hepatocytes isolated from p62 f/f mice (Fig 3A and B). Surprisingly, NBR1 formed foci even in p62‐knockout hepatocytes (Fig 3A). The FRAP assay showed extremely low fluorescence recovery of the GFP‐NBR1 structures in p62‐deficient MEFs (Fig EV1C), suggesting that in the absence of p62, NBR1 was aggregated. In the next series of experiments, we sought to find out whether phosphorylation of p62 at Serine 349 and the phase separation ability of p62 are required for NBR1‐mediated Nrf2 activation. To do this, we utilized two p62 mutants: phosphorylation‐defective p62 S349A and phase separation‐defective p62 K7A/D69A. The overexpression of NBR1 in p62‐deficient primary hepatocytes resulted in increased levels of exogenous wild‐type p62 and p62 S349A mutant (Fig 3C). The amount of p62 K7A/D69A tended to be also up‐regulated by the overexpression of NBR1, but the level was much milder than in the case of wild‐type p62 and the p62 S349A mutants (Fig 3C). In addition to the phosphorylation‐defective p62 mutant, p62 K7A/D69A hardly showed phosphorylation at Ser349, indicating that oligomerization and/or phase separation is required for phosphorylation (Fig 3C). Accordingly, both mutants hampered the induction of Nrf2 targets triggered by NBR1 overexpression (Fig 3D), and the effect was especially drastic in the case of p62 K7A/D69A (Fig 3D). These results suggest that p62 phase separation and phosphorylation are both needed for the full activation of Nrf2 mediated by NBR1.
Figure 3. Phase separation and phosphorylation of p62 are needed for full activation of Nrf2 mediated by NBR1.
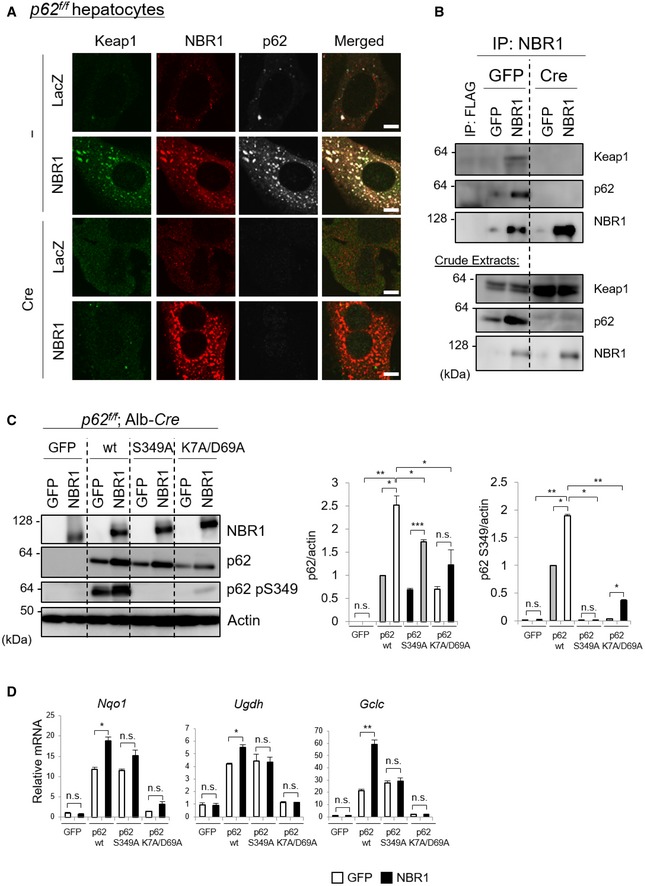
- Immunofluorescence microscopy. Primary hepatocytes prepared from p62 f/f mice were infected with or without adenovirus Cre recombinase for 48 h and then additionally infected with adenovirus LacZ or NBR1. Twenty‐four hours after the infection, the hepatocytes were immunostained with anti‐Keap1, anti‐p62, and anti‐NBR1 antibodies. Each inset is a magnified image. Bars: 10 μm.
- Immunoprecipitation analysis. Crude extracts were prepared from primary hepatocytes as described in (A) and then immunoprecipitated with anti‐FLAG (as a negative control) or anti‐NBR1 antibody. The resulting samples were analyzed by immunoblot analysis. Data shown are representative of three separate experiments.
- Primary hepatocytes prepared from p62 f/f; Alb‐Cre mice were co‐infected with adenovirus GFP or NBR1 in combination with wild‐type p62 or the mutants for 48 h. Cell lysates were prepared and subjected to immunoblot analysis with the indicated antibodies. Data shown are representative of three separate experiments. Bar graphs indicate the quantitative densitometric analysis of the indicated proteins relative to actin. Data are shown as means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001 as determined by Welch's t‐test.
- RT–qPCR analysis. Total RNAs were prepared from hepatocytes described in (C). Values were normalized against the amount of mRNA in the hepatocytes expressing GFP. The experiments were performed three times. Data are shown as means ± SE. *P < 0.05 and **P < 0.01 as determined by Welch's t‐test.
Source data are available online for this figure.
NBR1 stabilizes p62 by preventing its autophagic degradation
How does NBR1 achieve the accumulation of p62? Since p62 is one of Nrf2 targets, we speculated that NBR1‐mediated Nrf2 activation contributes to this. To validate this idea, we isolated primary mouse hepatocytes from Nrf2 f/f and Nrf2 f/f; Alb‐Cre mice (Fig 4A). Unexpectedly, the expression of NBR1 in hepatocytes lacking Nrf2 still had an ability to increase the levels of both total p62 and its phosphorylated forms albeit their levels were slightly but significantly lower than those in Nrf2‐competent hepatocytes (Fig 4A). Like gene expression of typical Nrf2‐target genes, the induction of p62 transcript upon overexpression of NBR1 was repressed by the loss of Nrf2 (Fig 4B). In Nrf2‐deficient hepatocytes, the accumulation of p62 remained about 60% of Nrf2‐competent hepatocytes (Fig 4A), suggesting that ~ 40% of the accumulation of p62 may be derived from gene expression. p62‐liquid droplets were observed even in Nrf2‐knockout hepatocytes, and their number and size increased upon expression of NBR1 (Fig 4C). While comparable in number, the size of NBR1‐induced p62 droplets in Nrf2‐ablated hepatocytes was smaller compared with those in control hepatocytes (Fig 4C), suggesting that lower levels of p62 protein affect the growing/maturation of p62‐liquid droplets. These results indicate that Nrf2 partially participates to the accumulation of p62 under NBR1‐expressing conditions through induction of the p62 transcript. Actually, we confirmed that NBR1 is able to up‐regulate even the protein level of exogenously expressed p62 (Fig EV2B).
Figure 4. NBR1 enhances the protein levels of p62 partially independent on Nrf2.
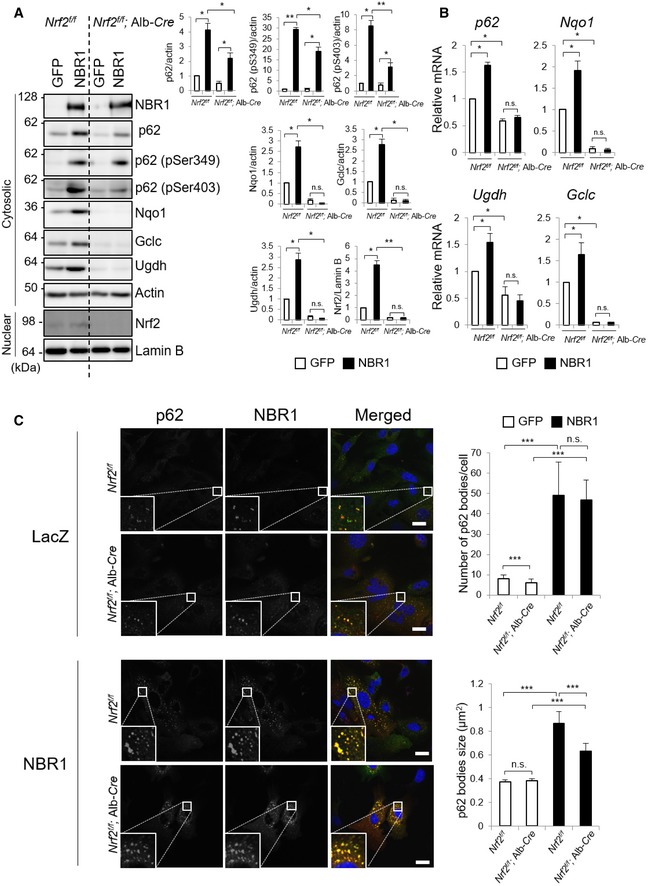
- Immunoblot analysis. Primary hepatocytes prepared from Nrf2 f/f and Nrf2 f/f; Alb‐Cre mice were infected with adenovirus expressing GFP or NBR1 for 48 h. Cytosolic and nuclear fractions were prepared and subjected to immunoblot analysis with the indicated antibodies. Data shown are representative of three separate experiments. Bar graphs indicate the quantitative densitometric analysis of the indicated proteins relative to actin (cytosolic proteins) or Lamin B (nuclear proteins). Data are shown as means ± SE. *P < 0.05 and **P < 0.01 as determined by Welch's t‐test.
- Real‐time PCR. Total RNAs were prepared from primary hepatocytes described in (A). Values were normalized against the amount of mRNA in the Nrf2 f/f hepatocytes expressing GFP. The experiments were performed at least three times. Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
- Immunofluorescence microscopy. Hepatocytes isolated from Nrf2 f/f and Nrf2 f/f; Alb‐Cre mice were infected with LacZ or NBR1 adenovirus for 48 h and then immunostained with anti‐p62 and anti‐NBR1 antibodies. Each inset is a magnified image. Bars: 20 μm. The number and size of p62 bodies were measured in more than 100 cells. Data are shown as means ± SE. ***P < 0.001 as determined by Welch's t‐test.
Source data are available online for this figure.
Although p62 is degraded through alternative pathways under certain situations 27, 28, this protein is mainly and selectively degraded by autophagy 29. Thus, a possible explanation for the increase in the protein levels of p62 observed in Fig 4A is that NBR1 prevents autophagic degradation of p62. To examine this hypothesis, we conducted an autophagy‐flux assay. The treatment of control GFP‐expressing hepatocytes with bafilomycin A1 (BafA1), an inhibitor of autophagosome–lysosome fusion, caused increased levels of LC3‐II and p62 (Fig 5A). As in control hepatocytes, we observed prominent accumulation of LC3‐II in wild‐type NBR1‐ or NBR1 D50R‐expressing hepatocytes by the treatment of BafA1 (Fig 5A). In striking contrast to the control hepatocytes, the treatment of BafA1 did not further augment increased levels of p62 due to the overexpression of NBR1 (Fig 5A), suggesting the blockage of p62 degradation. The inhibition of autophagic degradation of p62 by NBR1 overexpression was not merely caused by an increase in total level of p62 protein, as exogenously expressed p62 was sensitive to the treatment of BafA1 (Fig 5A). As a complementary approach to confirm the blockage of p62 degradation through autophagy, we employed a tandem fluorescent reporter assay 30. In this assay, we expressed mCherry‐GFP‐p62 in the presence or absence of NBR1 or the NBR D50R mutant in p62‐deficient MEFs. If the mCherry‐GFP‐p62 is transported into the lysosome, only the red signal is detected due to high sensitivity of GFP to the low pH. As shown in Fig 5B, about 40% of the total dots displayed only red signals in control MEFs. The overexpression of NBR1 dramatically decreased the number of red‐only dots, and most p62 puncta possessed both colors (Fig 5B). This was observed even in the case of NBR1 D50R (Fig 5B). These results suggest that NBR1 inhibits selective autophagy of p62 rather than general autophagy.
Figure 5. NBR1 prevents the autophagic degradation of p62.
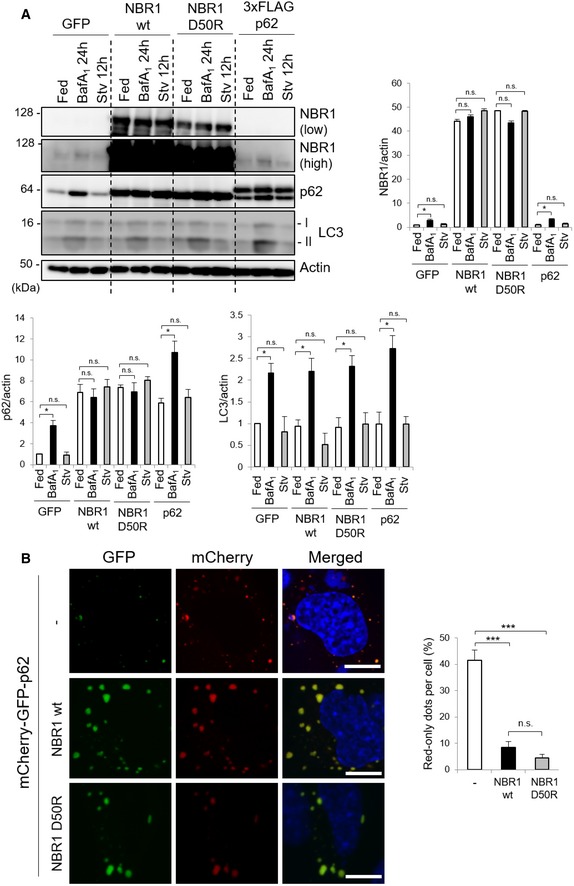
- Immunoblot analysis. Primary hepatocytes prepared from wild‐type mice were infected with the indicated adenovirus for 48 h and subsequently treated with 100 nM bafilomycin A1 (BafA1) for 24 h or cultured in amino acid‐deprived medium (Stv) for 12 h. Cell lysates were prepared and subjected to immunoblot analysis with the indicated antibodies. Data shown are representative of three separate experiments. Bar graphs show the quantitative densitometric analysis of the indicated proteins relative to actin. Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
- Fluorescence microscopy. p62‐deficient MEFs were co‐transfected with mCherry‐GFP‐p62 in combination with wild‐type NBR1 or NBR1 D50R mutant. Forty‐eight hours after the transfection, fluorescence images were observed. Bars: 10 μm. The number of puncta positive for only red signal in each MEFs was counted in more than 100 cells. Bar graphs are represented as means ± SE. ***P < 0.001 as determined by Welch's t‐test.
Source data are available online for this figure.
NBR1 is a stress‐inducible protein indispensable for full activation of the p62‐Keap1‐Nrf2 system
We have shown a role of NBR1 in p62‐mediated activation of Nrf2. While gene expression of p62 is induced in response to different forms of stress 31, 32, 33, the expression pattern of NBR1 largely remains unclear. To investigate the expression of endogenous NBR1 upon the exposure to oxidative stress, we selected the treatment with sodium arsenite (As [III]) as an oxidative stimulus, since it is known to activate Nrf2 and induce the expression of p62 34. The amount of NBR1 protein dramatically increased when primary mouse hepatocytes were exposed to As (III) (Fig 6A). Unlike in a previous report suggesting that Nrf2 modulates the expression of NBR1 in aging mice brains 35, the increase in NBR1 upon the exposure of As [III] was observed even in the absence of p62 or Nrf2 (Fig 6A). Real‐time PCR analysis revealed that Nrf2 is not responsible for NBR1 expression in hepatocytes in response to As (III) (Fig 6B). This suggested that, despite its similarities, the induction of NBR1 during oxidative stress relies on (a) different transcription factor(s). In agreement with NBR1 overexpression, As (III)‐mediated induction of NBR1 increased the size and number of p62‐liquid droplets (Fig 6C).
Figure 6. NBR1 is induced by oxidative stress.
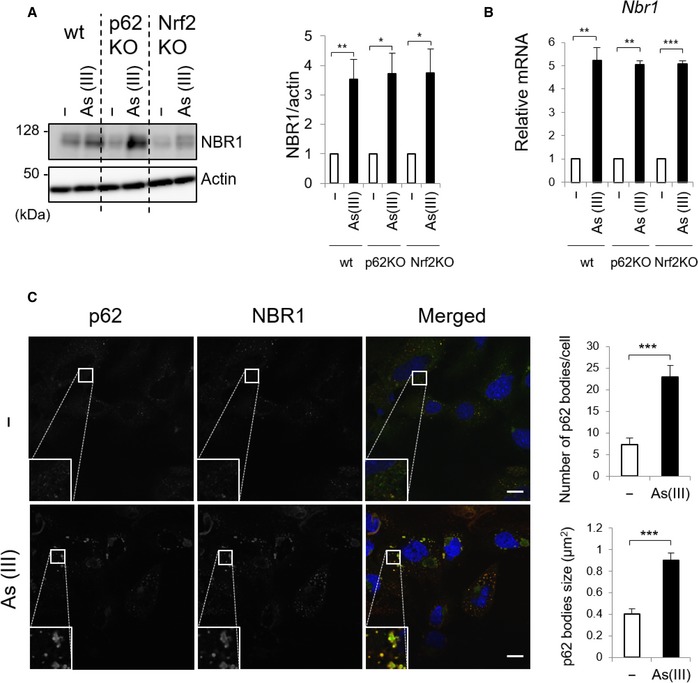
- Immunoblot analysis. Primary hepatocytes were isolated from wild‐type (wt), p62 f/f; Alb‐Cre (p62KO) and Nrf2 f/f; Alb‐Cre (Nrf2KO) mice. The hepatocytes were challenged with 10 μM sodium arsenite [As (III)] for 12 h. Cell lysates were prepared and subjected to immunoblot analysis with anti‐NBR1 and anti‐actin antibodies. Data shown are representative of three separate experiments. Bar graphs indicate the quantitative densitometric analysis of the indicated proteins relative to actin. Data are shown as means ± SE. *P < 0.05 and **P < 0.01 as determined by Welch's t‐test.
- Real‐time PCR. Total RNAs were prepared from primary hepatocytes described in (A). Values were normalized against the amount of mRNA in each non‐treated hepatocytes. The experiments were performed three times. Data are shown as means ± SE. **P < 0.01 and ***P < 0.001 as determined by Welch's t‐test.
- Immunofluorescence microscopy. Primary hepatocytes isolated from wild‐type mice were challenged with 10 μM sodium arsenite [As (III)] for 12 h and then immunostained with anti‐p62 and anti‐NBR1 antibodies. Each inset is a magnified image. Bars: 20 μm. The number and size of p62 bodies were measured in more than 100 cells. Data are shown as means ± SE. ***P < 0.001 as determined by Welch's t‐test.
Source data are available online for this figure.
To assess the physiological relevance of NBR1 in p62‐dependent activation of Nrf2, we generated a line of conditional knockout mice for Nbr1, in which Nbr1 is deleted in a Cre recombinase‐dependent manner (Fig EV3A and B). Mice homozygous for the Nbr1 flox allele (referred to as Nbr1 f/f mice) were bred with a line of transgenic (Tg) mice, EIIa‐Cre Tg that express the Cre recombinase under the control of the EIIa promoter in the early embryo. Heterozygous mice (referred to Nbr1 +/−) were obtained by crossbreeding of Nbr1 f/+; EIIa‐Cre with C57BL mice. We isolated MEFs from Nbr1 +/+, Nbr1 +/−, and Nbr1 −/− mice and confirmed that neither Nbr1 transcript nor NBR1 protein was detectable in Nbr1 −/− MEFs (Fig EV3C and D), indicating the effective recombination of the Nbr1 flox allele in conditional knockout mice. Next, we generated hepatocyte‐specific Nbr1‐knockout mice, Nbr1 f/f; Alb‐Cre by crossbreeding Nbr1 f/f with an Alb‐Cre Tg mice. Primary mouse hepatocytes were isolated from each wild‐type, p62 f/f; Alb‐Cre and Nbr1 f/f; Alb‐Cre mouse. Immunofluorescent analysis with anti‐LC3 antibody showed the appearance of LC3‐positive dots corresponding to autophagosomes, and the number increased under nutrient‐deprived conditions regardless of genotypes (Fig EV3E). We then examined the degradation of long‐lived proteins. In wild‐type hepatocytes, nutrient deprivation significantly enhanced the degradation, which was suppressed by the addition of lysosomal inhibitors, E64d and pepstatin A (EP; Fig EV3F). Degradation of long‐lived proteins was also observed in Nbr1‐deficient hepatocytes, and the level was comparable to those in wild‐type and p62‐deficient hepatocytes (Fig EV3F). These results indicate that loss of Nbr1 does not affect general autophagy.
Figure EV3. Generation of Nbr1‐conditional knockout mice.
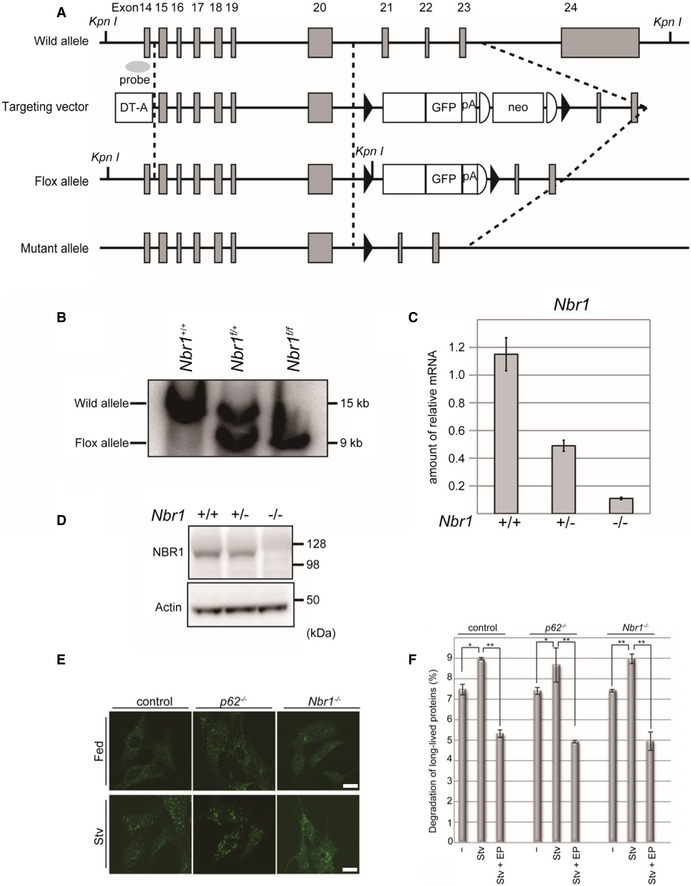
- Schematic representation of the targeting vector and the targeted allele of the Nbr1 gene. The coding exons numbered in accordance with the initiation site as exon 1 are depicted by gray boxes. Exon 21 was fused to a cDNA fragment encoded by exons 22, 23, and 24 (aa 2,523–2,964), and GFP and polyA signal sequence was added. Neo‐resistant gene cassette (neo) with FLP sequences (representing semi‐ellipsoid) was ligated behind the polyA signal sequence. The black triangles indicate the loxP sequence. The probe for Southern blot analysis is shown as a gray ellipse.
- Southern blot analysis of genomic DNA extracted from mice tails. Wild‐type and Flox alleles are indicated.
- Expression of the Nbr1 transcript in MEFs. Transcripts from the indicated genotypes were detected by real‐time PCR analysis. Data are shown as means ± SE calculated from three independent biological replicates.
- MEFs from the indicated genotypes were subjected to immunoblot analysis with the anti‐NBR1 and anti‐actin antibodies. Data shown are representative of three separate experiments.
- Immunofluorescence microscopy. Primary hepatocytes from the indicated genotypes were isolated, cultured in full media (Fed) or amino acid‐deprived (Stv) for 12 h, and then subjected to immunostaining with an anti‐LC3 antibody. Bars: 20 μm.
- Hepatocytes from wild‐type, p62‐, and Nbr1‐knockout mice were isolated and labeled with [14C] leucine for 24 h, and degradation of long‐lived protein in deprived (Stv) or non‐deprived (−) condition was measured. E64d and pepstatin (EP) were added as indicated. Data are shown as means ± SE. *P < 0.05, **P < 0.01 as determined by Welch's t‐test derived from three independent biological replicates.
Source data are available online for this figure.
Nbr1‐deficient mouse livers showed no obvious phenotypes (Fig EV4). We did not observe any changes in both the mRNA and protein levels of typical Nrf2‐target genes in the Nbr1‐knockout livers (Fig EV4A and B). Nbr1 f/f; Alb‐Cre mice showed a normal liver weight per unit of body weight ratio (Fig EV4C), and no increased serum hepatic enzymes such as AST, ALT, and ALP were observed by loss of Nbr1 (Fig EV4D). Hematoxylin and eosin stain indicated no tissue damage (Fig EV4E), suggesting that the loss of Nbr1 has no effect under basal conditions. Finally, we sought whether stress‐induced NBR1 expression contributes to p62‐mediated Nrf2 activation or not. With this aim, primary hepatocytes isolated from Nbr1 f/f mice were infected by adenoviral vector for Cre recombinase, or GFP as a negative control. NBR1 in the hepatocytes was completely deleted by the expression of Cre recombinase, but not GFP (Fig 7A). We observed the accumulation of total and phosphorylated p62 in both NBR1‐competent and incompetent hepatocytes when challenged with As (III), but the induction levels in Nbr1‐deficient hepatocytes were lower than in control hepatocytes (Fig 7A). More importantly, gene expression of Nrf2 targets upon exposure to As (III) was significantly suppressed by loss of Nbr1 (Fig 7B). Furthermore, deletion of Nbr1 resulted in a slight but significant reduction on the number and size of p62 liquid droplets during basal conditions, which was exacerbated when the hepatocytes were subjected to As (III) treatment (Fig 7C). Loss of Nbr1 in MEFs had no effect on recovery of signal intensity of structures positive for GFP‐p62 after photobleaching (Fig EV1D), suggesting that deletion of NBR1 does not affect the quality of the p62‐liquid droplets. Taken together, these results indicate that NBR1 is an oxidative stress‐inducible protein and needed for full activation of the p62‐Keap1‐Nrf2 pathway through the enhancement of p62‐droplet formation.
Figure EV4. Phenotypic analysis of the Nbr1 f/f; Alb‐Cre mice.

- Immunoblot analysis. Total homogenates were prepared from the livers of 6‐week‐old Nbr1 f/f (n = 3, biological replicates) and Nbr1 f/f; Alb‐Cre mice (n = 3, biological replicates). These were subjected to immunoblotting using the indicated antibodies. Bar graphs show the amounts of the indicated proteins measured by densitometry relative to actin. Data are shown as means ± SE determined by Welch's t‐test.
- Gene expression of Nrf2 targets in Nbr1‐deficient livers. Total RNA was prepared from the livers of 6‐week‐old Nbr1 f/f (n = 3) and Nbr1 f/f; Alb‐Cre mice (n = 3). Values were normalized against the amount of mRNA in the livers of Nbr1 f/f mice. The experiments were performed three times. Data are shown as means ± SE determined by Welch's t‐test.
- Liver weights (% per body weight) of mice described in (A). Data are shown as means ± SE determined by Welch's t‐test from three independent biological replicates.
- Liver function tests of the mice used in (A). The serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase (ALP) were measured. IU/l, international units/liter. Data are shown as means ± SE determined by Welch's t‐test from three independent biological replicates.
- Hematoxylin and eosin staining. Liver sections from 6‐week‐old Nbr1 f/f and Nbr1 f/f; Alb‐Cre mice were prepared and stained with H&E. Data are representative of three separate experiments. Bar: 50 μm.
Source data are available online for this figure.
Figure 7. Deletion of NBR1 reduces p62‐mediated Nrf2 activation under oxidative stress.
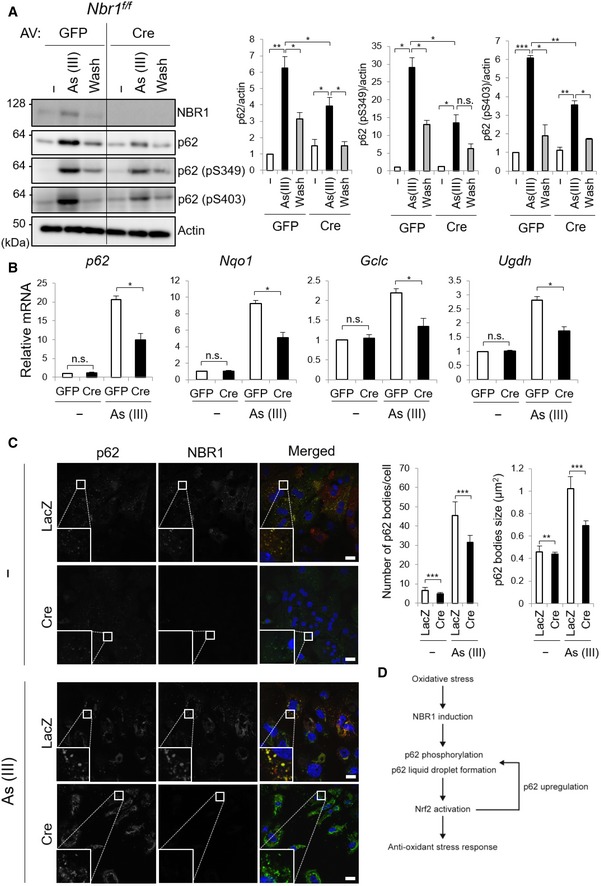
- Immunoblot analysis. Primary hepatocytes were isolated from Nbr1 f/f mice and then infected with adenovirus GFP or Cre for 48 h. Thereafter, the hepatocytes were divided into three groups: (i) cultured in regular medium (−), (ii) treated with 10 μM As (III) for 12 h [As (III)], and (iii) after treated with 10 μM As (III) for 12 h, cultured in regular medium for another 12 h (Wash). Cell lysates were prepared and subjected to immunoblot analysis with indicated antibodies. Data shown are representative of three separate experiments. Bar graphs indicate the quantitative densitometric analysis of the indicated proteins relative to actin. Data are shown as means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001 as determined by Welch's t‐test.
- Real‐time PCR. Total RNAs were prepared from primary hepatocytes described in (A). Values were normalized against the amount of mRNA in the Nbr1 f/f hepatocytes expressing GFP under normal culture conditions. The experiments were performed at least three times. Data are shown as means ± SE. *P < 0.05 as determined by Welch's t‐test.
- Immunofluorescence microscopy. Nbr1 f/f hepatocytes were infected with LacZ or Cre adenovirus for 48 h and, when indicated, challenged with 10 μM As (III) for 12 h. The cells were then immunostained with anti‐p62 and anti‐NBR1 antibodies. Each inset is a magnified image. Bars: 20 μm. The number and size of p62 bodies were measured in more than 100 cells. Data are shown as means ± SE. **P < 0.01 and ***P < 0.001 as determined by Welch's t‐test.
- Role of NBR1 in the p62‐liquid droplets and the p62‐mediated Nrf2 activation. Oxidative stress up‐regulates the level of NBR1, which enhances the phosphorylation and liquid droplet formation of p62. The resulting p62 droplets are resistant to autophagic degradation and serve as signaling nodes for the activation of Nrf2. Since p62 is one of the targets of Nrf2, increased level of p62 in cooperation with NBR1 promotes the formation of liquid droplets and contributes to the persistent activation of Nrf2 against oxidative stress.
Source data are available online for this figure.
Discussion
A decade has passed since the identification of NBR1 as a selective autophagy receptor 22, and yet, little attention has been paid to its functional characterization. Previous research revealed that NBR1 and p62 interact with each other through their PB1 domains 22 and act collaboratively in the autophagic clearance of protein aggregates 22, 36, peroxisomes 37, and invading pathogens 38. Herein, we show that NBR1 is induced in response to stress in order to promote phase separation and phosphorylation of p62 and that the resulting droplets are resistant to autophagic degradation and serve as signaling hubs for the p62‐mediated activation of Nrf2 (Fig 7D).
In agreement with previous in vitro observations 23, NBR1 facilitated the formation of p62‐liquid bodies in cells (Fig 1). How does NBR1 promote phase separation of p62? Since phase separation can happen when a concentration threshold of the components is reached, an easy explanation could be that the relative abundance of p62 and its binding partners surpass the concentration threshold, thus resulting in phase separation. As the overexpression of NBR1 increased the amounts of p62 via the activation of Nrf2 (Figs 2, 3, 4), it is plausible that the concentration of components (i.e., NBR1 and p62) is one factor influencing the formation of p62 bodies. Another possible mechanism is an increase in the valency of p62 by posttranslational modification. Posttranslational modifications including phosphorylation have been extensively identified as regulators of phase separation 39, 40, 41. In agreement with such criteria, the overexpression of NBR1 was accompanied by the robust phosphorylation of p62 at Serine 349 and Serine 403 (Fig 1). Serine 349 phosphorylation is required for the competitive interaction with Keap1 21, while the phosphorylation of Serine 403 enhances the binding affinity of p62 to ubiquitin chains 42, which is required for phase separation 6, 23. Thus, it is likely that the increase in the phosphorylation states of p62 produced by NBR1 positively influences the formation of p62 bodies.
Unexpectedly, like wild‐type NBR1, the p62‐binding defective D50R mutant had the ability to enhance the fluorescence recovery of mCherry‐p62 bodies (Fig EV1B), suggesting that NBR1 enhances liquid‐droplet formation of p62 bodies regardless of p62 binding. Although NBR1 formed foci in p62‐knockout hepatocytes (Fig 3A), the FRAP assay showed extremely low fluorescence recovery of the structures positive for GFP‐NBR1 in p62‐deficient MEFs (Fig EV1C), suggesting that in the absence of p62, NBR1 was aggregated. On the other hand, while loss of Nbr1 decreased p62 droplets in size and number under stress conditions (Fig 7C), it had no effect on the recovery of signal intensity of structures positive for GFP‐p62 after photobleaching (Fig EV1D), suggesting that deletion of NBR1 does not affect the quality of the p62‐liquid droplets. Based on these results, NBR1 may titrate components that prevent the formation of p62 droplets and then enhance the liquid‐like properties of p62 bodies.
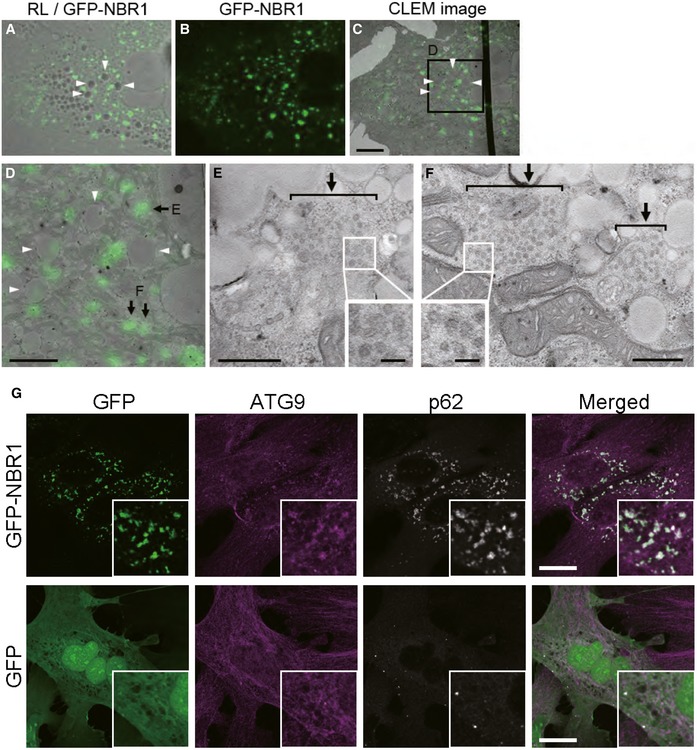
Remarkably, p62 bodies induced by NBR1 overexpression were resistant to autophagic degradation (Fig 5). Since the overexpression of NBR1 had no effect on the turnover of LC3‐II, conventional autophagy is thought to be intact. How do these structures escape autophagic degradation? The formation of the autophagosome along the interface of phase‐separated p62 requires sequential and antagonistic steps. First, a complex between p62 and FAK family kinase‐interacting protein of 200 kDa (FIP200) is formed to initiate selective autophagy of p62 43. This complex is dependent on phosphorylation of p62 at Serine 349, which is located in close proximity to the LIR. This complex is mutually exclusive to the one formed between p62 and LC3, so the binding of LC3 occurs after dissociation from FIP200. Which step(s) does NBR1 inhibit? Since the overexpression of NBR1 enhanced both phase separation of p62 and the phosphorylation of Serine 349 (Fig 1), it is plausible that p62 droplets are primed to recruit FIP200. Thus, NBR1 might prevent the translocation of FIP200 onto p62 bodies. Alternatively, the persistent phosphorylation of p62 at Serine 349 by the overexpression of NBR1 may inhibit the transition of FIP200 to LC3‐II. Actually, p62 is de‐phosphorylated at Serine 349 in response to the removal of As (III) 44, and the phospho‐defective mutant of p62 is resistant to autophagy 43, suggesting the involvement of de‐phosphorylation in LC3 binding. Another possibility is that NBR1 titrates ATG components that are essential for the degradation of p62 droplets. Actually, we observed excessive accumulation of ATG9‐positive vesicles onto p62 droplets when NBR1 was overexpressed (Fig EV5A–G). In this scenario, it may be explainable that NBR1 D50R, which does not bind to p62, also inhibits autophagic degradation of p62 droplets through segregation of ATG9 from p62 droplets. Further analysis is needed to clarify how NBR1 regulates the removal of p62‐liquid droplets.
Figure EV5. Assembly of ATG9‐vesicles onto p62 droplets by NBR1 overexpression.
-
A–FCorrelative light‐electron microscopy (CLEM). Wild‐type hepatocytes were infected with GFP‐NBR1 adenovirus for 48 h, and then, NBR1‐positive structures (arrows) were identified by CLEM. The reflected light image (A) was aligned with the EM image (C) using 4 lipid droplets (arrowheads). The boxed region in C is enlarged and shown in D. Note that numerous small vesicles (approximately 40–50 nm in diameter; insets in E and F) are present in the GFP‐NBR1‐positive structures (arrows in E and F). Bars: 10 μm (C), 5 μm (D), 500 nm (E and F), and 100 nm (insets).
-
GImmunofluorescence microscopy. Wild‐type hepatocytes were infected with GFP or GFP‐NBR1 adenovirus for 48 h and then immunostained with anti‐ATG9 antibody. Each inset is a magnified image. Bars: 20 μm. The overexpression of GFP‐NBR1, but not GFP, induced the translocation of Atg9 onto the GFP‐NBR1‐positive structures.
What is the physiological role of NBR1‐mediated p62 bodies? We speculate that NBR1 shifts p62 bodies from autophagic cargo clusters to signaling nodes. Specifically, these signaling nodes seem to be specialized on the activation of the Keap1‐Nrf2 antioxidant pathway, as other p62‐related signaling pathways were hardly affected by the overexpression of NBR1 (Fig EV2). The fact that the NBR1‐mediated activation of Nrf2 is lost in p62‐deficient cells (Fig 2) indicates that the role of NBR1 in this pathway is achieved by increasing p62 protein levels and phosphorylation states. Most importantly, NBR1 was induced during oxidative stress in an Nrf2‐independent manner (Fig 6), implying that a rate‐limiting step of NBR1‐mediated Nrf2 activation is an induction of the expression of NBR1. NBR1 is located on a region of chromosome 17q21.1 that is in close proximity to the BRCA1 tumor suppressor gene. In fact, in mice, Nbr1 and Brca1 are separated by just < 300 base pairs and share a common bidirectional promoter 45, 46. Besides an established role as tumor suppressor 47, BRCA1 also participates in the orchestration of the oxidative stress response in multiple manners 48, including the stabilization of Nrf2 49. Potential transcription factors regulating the BRCA1/NBR1 bidirectional promoter include specificity protein 1 (Sp1), CCAAT‐enhancer‐binding proteins (C/EBPs), and cAMP response element‐binding (CREB) 45, all of them involved in the antioxidant defense pathway 50, 51, 52. Although further research is required to clarify the mechanism, it could be envisioned that during an oxidative threat, stress‐inducible transcription factors would be recruited to the bidirectional promoter to secure the activation of the master antioxidant regulator Nrf2 by the action of both NBR1 and BRCA1.
Material and Methods
Cell culture
Isolation and immortalization of the MEFs employed in this study was described previously 21, 44. Cells were grown in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS), 5 U/ml penicillin, and 50 μg/ml streptomycin. Twenty‐four hours after seeding, cells were transfected using FuGene HD (Promega) following the manufacturer's instructions.
Mice
p6253 and Nrf254‐conditional knockout mice with C57BL/6 genetic background were previously described. Generation of Nbr1‐conditional knockout mice was performed as described 55. Mice were housed in specific pathogen‐free facilities, and the Ethics Review Committee for Animal Experimentation of Niigata University and Juntendo University approved the experimental protocol.
Isolation of mouse primary hepatocytes
Six‐month‐old mice were anesthetized with isoflurane, and the mouse livers were perfused by reverse flow through the vena cava using 20 ml of ex‐perfusate solution (pH 7.4, 140 mM NaCl, 5 mM KCl, 0.5 mM NaH2PO4, 10 mM HEPES, 4 mM NaHCO3, 50 mM glucose, and 0.5 mM GEDTA) and 30 ml of perfusate solution (pH 7.6, 70 mM NaCl, 6.7 mM KCl, 100 mM HEPES, 5 mM CaCl2, 35 units collagenase). After dissection, hepatocyte suspension was washed three times in HBSS (Gibco, Thermo Fisher Scientific) and the cells were grown in collagen‐coated plates with William's Medium E (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS.
Immunoblot analysis
Cells were lysed in ice‐cold TNE buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA) containing 1% Triton X‐100 and protease inhibitors. Nuclear and cytoplasmic fractions were prepared using the NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific). Samples were separated by SDS–PAGE and then transferred to polyvinylidene difluoride (PVDF) membranes. For immunoprecipitation analysis, cells were lysed by 200 μl of TNE, and the lysate was then centrifuged at 10,000 g for 10 min at 4°C to remove debris. Then, 800 μl of TNE and 1 μg of anti‐NBR1 antibody (4BR; Santa Cruz Biotechnology) were added to the lysate, and the mixture was mixed under constant rotation for 12 h at 4°C. The immunoprecipitates were washed five times with ice‐cold TNE. The complex was boiled for 10 min in LDS sample buffer in the presence of 2‐mercaptoethanol to elute proteins and centrifuged at 10,000 g for 5 min. Antibodies against NBR1 (4BR; Santa Cruz Biotechnology), p62 (GP62‐C; Progen Biotechnik GmbH), LC3B (#2775; Cell Signaling Technology), T421/S424‐phosphorylated S6K (#9204; Cell Signaling Technology), T389‐phosphorylated S6K (#9205; Cell Signaling Technology), S240/S244‐phosphorylated S6 (#2215; Cell Signaling Technology) T180/Y182‐phosphorylated p38 (#9211; Cell Signaling Technology), Actin (MAB1501R; Merck Millipore Headquarters), Lamin B (M‐20; Santa Cruz Biotechnology), Nqo1 (ab34173; Abcam), Ugdh (ab155005; Abcam), Gclc (ab41463; Abcam), Nrf2 (H‐300; Santa Cruz Biotechnology), and Keap1 (10503‐2‐AP; Proteintech Group) were purchased from the indicated suppliers and employed at 1:500 dilution. Anti‐S349‐phosphorylated p62 polyclonal antibody was raised in rabbits by using the peptide Cys + KEVDP(pS)TGELQSL as an antigen 21. Blots were then incubated with horseradish peroxidase‐conjugated secondary antibody (Goat Anti‐Mouse IgG [H + L], 115‐035‐166; Jackson ImmunoResearch; Goat Anti‐Rabbit IgG [H + L] 111‐035‐144; Goat Anti‐Guinea Pig IgG [H + L]) and visualized by chemiluminescence.
Quantitative real‐time PCR (qRT–PCR)
Using the Transcriptor First‐Strand cDNA Synthesis Kit (Roche Applied Science, Indianapolis, IN, USA), cDNA was synthesized from 1 μg of total RNA. Quantitative PCR was performed using the LightCycler® 480 Probes Master mix (Roche Applied Science) on a LightCycler® 480 (Roche Applied Science). Signals were normalized against Gusb (β‐glucuronidase). The sequences of the primers employed are provided in Table EV1.
Immunofluorescence microscopy
Cells grown on coverslips were fixed in 4% paraformaldehyde in PBS for 10 min, permeabilized with 0.1% Triton X‐100 in PBS for 5 min, blocked with 0.1% (w/v) gelatin (Sigma‐Aldrich) in PBS for 45 min, and then incubated overnight with primary antibodies diluted 1:200 in gelatin/PBS. After washing, cells were incubated with Goat anti‐Guinea pig IgG (H + L) Cross‐Adsorbed Secondary Antibody, Alexa Fluor 488 (A11073, Thermo Fisher Scientific), and Goat anti‐Mouse IgG (H + L) Highly Cross‐Adsorbed Secondary Antibody, Alexa Fluor 647 (A21236, Thermo Fisher Scientific) at a dilution ratio of 1:1000 for 60 min. Cells were imaged using a confocal laser‐scanning microscope (Olympus, FV1000) with a UPlanSApo ×60 NA 1.40 oil objective lens. The number and size of p62 bodies were analyzed with the ImageJ 1.51 software (Wayne Rasband, National Institutes of Health, USA).
To measure the FRAP, cells were grown in 35 mm glass base dishes (Iwaki, Japan). p62 bodies were bleached for 3 s using a laser intensity of 70% at 480 nm, and then, the recovery was recorded for the indicated time.
Protein degradation assay
The assay was performed essentially as described 55. In brief, hepatocytes isolated from the indicated genotypes were plated at 5 × 104 cells/well in collagen‐coated 24‐well plates and cultured in Williams’ E medium with 10% FBS for 24 h. Cells were incubated with this medium also containing 0.5 μCi/ml [14C]leucine for 24 h to label long‐lived proteins. After washing with Williams’ E containing 2 mM of unlabeled leucine, cells were incubated with the medium for 2 h to allow degradation of short‐lived proteins and minimize the incorporation of labeled leucine. Next, the cells were washed again with PBS and incubated at 37°C with Krebs‐Ringer bicarbonate medium and Williams’ E/10% FCS in the presence or absence of protease inhibitors (10 μg/ml E64d and pepstatin). After 4 h, aliquots of the medium were taken and a one‐tenth volume of 100% trichloroacetic acid was added to each aliquot. The mixtures were centrifuged at 12,000 g for 5 min, and the acid‐soluble radioactivity was determined using a liquid scintillation counter. At the end of the experiment, the cultures were washed twice with PBS, and 1 ml of cold trichloroacetic acid was added to fix the cell proteins. The fixed cell monolayers were washed with trichloroacetic acid and dissolved in 1 ml of 1 N NaOH at 37°C. Radioactivity in an aliquot of 1 N NaOH was determined by liquid scintillation counting. The percentage of protein degradation was calculated according to the procedures published 56.
Histological examination
Excised liver tissues were fixed by immersion in 0.1 M phosphate buffer (PB, pH 7.4) containing 4% paraformaldehyde and 4% sucrose. Each liver was carefully dissected and processed for paraffin embedding, and then, sections were prepared for hematoxylin and eosin staining. Images were acquired with a BX51 microscope (Olympus, Tokyo, Japan).
Correlative light and electron microscopy (CLEM)
The detailed correlative light and electron microscopy (CLEM) method was recently described 57. Briefly, wild‐type hepatocytes were cultured on coverslips coated with 150‐μm grids (Matsunami Glass Ind., Ltd.) and infected with GFP‐NBR1 adenovirus for 48 h. They were fixed with 2% PA–0.5% glutaraldehyde (GA) in 0.1 M PB (pH 7.4), and the fluorescence images were obtained using a confocal microscope (FV1000; Olympus). They were fixed again with 2% PA and 2% GA in 0.1 M PB (pH 7.4), processed for the reduced‐osmium method, and embedded in Epon812. Areas containing cells of interest were trimmed, cut as serial ultrathin sections, and analyzed with an electron microscope (EM; JEM1400EX; JEOL). Alignment of images of light microscopy and EM was performed using Photoshop (Adobe Systems Inc., San Jose, CA).
Author contributions
PS‐M and MK conceived the study. PS‐M, Y‐sS, SW, and SK performed the experiments. PS‐M and MK wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Terje Johansen (University of Tromsø) for his help with the fluorescent analysis of autophagy flux. We also thank Katsuyuki Kanno and Takayuki Yabe (Fukushima Medical University School of Medicine) for their help in the CLEM analysis. P.S.‐M. is supported by a Grant‐in‐Aid for JSPS Research Fellows (P18099). Y.‐S.S. is supported by a Grant‐in‐Aid for Scientific Research (C) (19K15043). S.K. is supported by a Grant‐in‐Aid for Early‐Career Scientists (18K15043). M.K. is supported by a Grant‐in‐Aid for Scientific Research on Innovative Areas (19H05706), a Grant‐in‐Aid for Scientific Research (B) (18H02611), the Japan Society for the Promotion of Science (an A3 foresight program), and the Takeda Science Foundation (to M.K.).
EMBO Reports (2020) 21: e48902
References
- 1. Dolgin E (2018) What lava lamps and vinaigrette can teach us about cell biology. Nature 555: 300–302 [DOI] [PubMed] [Google Scholar]
- 2. Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18: 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boeynaems S, Alberti S, Fawzi NL, Mittag T, Polymenidou M, Rousseau F, Schymkowitz J, Shorter J, Wolozin B, Van Den Bosch L et al (2018) Protein phase separation: a new phase in cell biology. Trends Cell Biol 28: 420–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buchan JR, Kolaitis RM, Taylor JP, Parker R (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153: 1461–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y, Yan L, Zhou Z, Yang P, Tian E, Zhang K, Zhao Y, Li Z, Song B, Han J et al (2009) SEPA‐1 mediates the specific recognition and degradation of P granule components by autophagy in C. elegans . Cell 136: 308–321 [DOI] [PubMed] [Google Scholar]
- 6. Sun D, Wu R, Zheng J, Li P, Yu L (2018) Polyubiquitin chain‐induced p62 phase separation drives autophagic cargo segregation. Cell Res 28: 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- 8. Kaur J, Debnath J (2015) Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16: 461–472 [DOI] [PubMed] [Google Scholar]
- 9. Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349–364 [DOI] [PubMed] [Google Scholar]
- 10. Gatica D, Lahiri V, Klionsky DJ (2018) Cargo recognition and degradation by selective autophagy. Nat Cell Biol 20: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mizushima N (2018) A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol 20: 521–527 [DOI] [PubMed] [Google Scholar]
- 12. Sanchez‐Martin P, Komatsu M (2018) p62/SQSTM1 ‐ steering the cell through health and disease. J Cell Sci 131: jcs222836 [DOI] [PubMed] [Google Scholar]
- 13. Sanchez‐Martin P, Saito T, Komatsu M (2019) p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J 286: 8–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moscat J, Karin M, Diaz‐Meco MT (2016) p62 in cancer: signaling adaptor beyond autophagy. Cell 167: 606–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamark T, Kirkin V, Dikic I, Johansen T (2009) NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8: 1986–1990 [DOI] [PubMed] [Google Scholar]
- 16. Kirkin V, Lamark T, Johansen T, Dikic I (2009) NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 5: 732–733 [DOI] [PubMed] [Google Scholar]
- 17. Aki T, Funakoshi T, Unuma K, Uemura K (2013) Impairment of autophagy: from hereditary disorder to drug intoxication. Toxicology 311: 205–215 [DOI] [PubMed] [Google Scholar]
- 18. Lahiri P, Schmidt V, Smole C, Kufferath I, Denk H, Strnad P, Rulicke T, Frohlich LF, Zatloukal K (2016) p62/sequestosome‐1 is indispensable for maturation and stabilization of mallory‐denk bodies. PLoS ONE 11: e0161083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang R, Varela M, Vallentgoed W, Forn‐Cuni G, van der Vaart M, Meijer AH (2019) The selective autophagy receptors Optineurin and p62 are both required for zebrafish host resistance to mycobacterial infection. PLoS Pathog 15: e1007329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cemma M, Kim PK, Brumell JH (2011) The ubiquitin‐binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria‐associated microdomains to target Salmonella to the autophagy pathway. Autophagy 7: 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T et al (2013) Phosphorylation of p62 activates the Keap1‐Nrf2 pathway during selective autophagy. Mol Cell 51: 618–631 [DOI] [PubMed] [Google Scholar]
- 22. Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P et al (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33: 505–516 [DOI] [PubMed] [Google Scholar]
- 23. Zaffagnini G, Savova A, Danieli A, Romanov J, Tremel S, Ebner M, Peterbauer T, Sztacho M, Trapannone R, Tarafder AK et al (2018) p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J 37: e98308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 12: 1–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suzuki T, Yamamoto M (2017) Stress‐sensing mechanisms and the physiological roles of the Keap1‐Nrf2 system during cellular stress. J Biol Chem 292: 16817–16824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12: 213–223 [DOI] [PubMed] [Google Scholar]
- 27. Mejlvang J, Olsvik H, Svenning S, Bruun JA, Abudu YP, Larsen KB, Brech A, Hansen TE, Brenne H, Hansen T et al (2018) Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol 217: 3640–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song P, Li S, Wu H, Gao R, Rao G, Wang D, Chen Z, Ma B, Wang H, Sui N et al (2016) Parkin promotes proteasomal degradation of p62: implication of selective vulnerability of neuronal cells in the pathogenesis of Parkinson's disease. Protein Cell 7: 114–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy‐deficient mice. Cell 131: 1149–1163 [DOI] [PubMed] [Google Scholar]
- 30. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145 [DOI] [PubMed] [Google Scholar]
- 31. Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H et al (2012) KrasG12D‐induced IKK2/beta/NF‐kappaB activation by IL‐1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 21: 105–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T (2010) p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element‐driven gene transcription. J Biol Chem 285: 22576–22591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shah P, Trinh E, Qiang L, Xie L, Hu WY, Prins GS, Pi J, He YY (2017) Arsenic induces p62 expression to form a positive feedback loop with Nrf2 in human epidermal keratinocytes: implications for preventing arsenic‐induced skin cancer. Molecules 22: E194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang M, Ji C, Pallo S, Rahman I, Johnson GVW (2018) Nrf2 mediates the expression of BAG3 and autophagy cargo adaptor proteins and tau clearance in an age‐dependent manner. Neurobiol Aging 63: 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Odagiri S, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K (2012) Autophagic adapter protein NBR1 is localized in Lewy bodies and glial cytoplasmic inclusions and is involved in aggregate formation in alpha‐synucleinopathy. Acta Neuropathol 124: 173–186 [DOI] [PubMed] [Google Scholar]
- 37. Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott‐Schwartz J et al (2013) NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci 126: 939–952 [DOI] [PubMed] [Google Scholar]
- 38. Shi J, Fung G, Piesik P, Zhang J, Luo H (2014) Dominant‐negative function of the C‐terminal fragments of NBR1 and SQSTM1 generated during enteroviral infection. Cell Death Differ 21: 1432–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aumiller WM Jr, Keating CD (2016) Phosphorylation‐mediated RNA/peptide complex coacervation as a model for intracellular liquid organelles. Nat Chem 8: 129–137 [DOI] [PubMed] [Google Scholar]
- 40. Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R, Pelkmans L (2013) Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 152: 791–805 [DOI] [PubMed] [Google Scholar]
- 41. Petri S, Grimmler M, Over S, Fischer U, Gruss OJ (2007) Dephosphorylation of survival motor neurons (SMN) by PPM1G/PP2C gamma governs Cajal body localization and stability of the SMN complex. J Cell Biol 179: 451–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 44: 279–289 [DOI] [PubMed] [Google Scholar]
- 43. Turco E, Witt M, Abert C, Bock‐Bierbaum T, Su MY, Trapannone R, Sztacho M, Danieli A, Shi X, Zaffagnini G et al (2019) FIP200 claw domain binding to p62 promotes autophagosome formation at ubiquitin condensates. Mol Cell 74: 330–346.e311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eino A, Kageyama S, Uemura T, Annoh H, Saito T, Narita I, Waguri S, Komatsu M (2015) Sqstm1‐GFP knock‐in mice reveal dynamic actions of Sqstm1 during autophagy and under stress conditions in living cells. J Cell Sci 128: 4453–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu CF, Chambers JA, Solomon E (1997) Complex regulation of the BRCA1 gene. J Biol Chem 272: 20994–20997 [DOI] [PubMed] [Google Scholar]
- 46. Whitehouse C, Chambers J, Catteau A, Solomon E (2004) Brca1 expression is regulated by a bidirectional promoter that is shared by the Nbr1 gene in mouse. Gene 326: 87–96 [DOI] [PubMed] [Google Scholar]
- 47. Silver DP, Livingston DM (2012) Mechanisms of BRCA1 tumor suppression. Cancer Discov 2: 679–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yi YW, Kang HJ, Bae I (2014) BRCA1 and oxidative stress. Cancers (Basel) 6: 771–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, Joshi PA, Wakeham A, Molyneux SD, Martin B et al (2013) BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med 210: 1529–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thon M, Al Abdallah Q, Hortschansky P, Scharf DH, Eisendle M, Haas H, Brakhage AA (2010) The CCAAT‐binding complex coordinates the oxidative stress response in eukaryotes. Nucleic Acids Res 38: 1098–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee B, Cao R, Choi YS, Cho HY, Rhee AD, Hah CK, Hoyt KR, Obrietan K (2009) The CREB/CRE transcriptional pathway: protection against oxidative stress‐mediated neuronal cell death. J Neurochem 108: 1251–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, Ratan RR (2003) Sp1 and Sp3 are oxidative stress‐inducible, antideath transcription factors in cortical neurons. J Neurosci 23: 3597–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Harada H, Warabi E, Matsuki T, Yanagawa T, Okada K, Uwayama J, Ikeda A, Nakaso K, Kirii K, Noguchi N et al (2013) Deficiency of p62/Sequestosome 1 causes hyperphagia due to leptin resistance in the brain. J Neurosci 33: 14767–14777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xue P, Hou Y, Chen Y, Yang B, Fu J, Zheng H, Yarborough K, Woods CG, Liu D, Yamamoto M et al (2013) Adipose deficiency of Nrf2 in ob/ob mice results in severe metabolic syndrome. Diabetes 62: 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y et al (2005) Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 169: 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gronostajski RM, Pardee AB (1984) Protein degradation in 3T3 cells and tumorigenic transformed 3T3 cells. J Cell Physiol 119: 127–132 [DOI] [PubMed] [Google Scholar]
- 57. Arai R, Waguri S (2019) Improved electron microscopy fixation methods for tracking autophagy‐associated membranes in cultured mammalian cells. Methods Mol Biol 1880: 211–221 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7