

Abstract
Dysregulation of lipid homeostasis is intimately associated with defects in insulin secretion, a key feature of type 2 diabetes. Here, we explore the role of the putative lipid transporter ABCA12 in regulating insulin secretion from β‐cells. Mice with β‐cell‐specific deletion of Abca12 display impaired glucose‐stimulated insulin secretion and eventual islet inflammation and β‐cell death. ABCA12's action in the pancreas is independent of changes in the abundance of two other cholesterol transporters, ABCA1 and ABCG1, or of changes in cellular cholesterol or ceramide content. Instead, loss of ABCA12 results in defects in the genesis and fusion of insulin secretory granules and increases in the abundance of lipid rafts at the cell membrane. These changes are associated with dysregulation of the small GTPase CDC42 and with decreased actin polymerisation. Our findings establish a new, pleiotropic role for ABCA12 in regulating pancreatic lipid homeostasis and insulin secretion.
Keywords: ABCA12, cholesterol homeostasis, insulin secretion, lipid rafts, type 2 diabetes
Subject Categories: Membrane & Intracellular Transport, Metabolism
The lipid transport protein ABCA12 plays pleiotropic roles in regulating glucose stimulated insulin secretion from beta cells, regulating the morphology and fusion of insulin granules, lipid raft abundance and the actin cytoskeleton.
Introduction
Insulin resistance and impaired β‐cell insulin secretion are two key features of type 2 diabetes (T2D). Insulin secretion usually worsens as disease progresses, and in later stages, T2D is often accompanied by inflammation and gradual loss of β‐cell mass due to apoptosis. Disturbances of systemic lipoprotein metabolism and cellular cholesterol transport are among key pathogenic mechanisms which drive impaired insulin sensitivity and secretion 1, 2. Accumulation of cholesterol in β‐cells due to inactivation of the cholesterol efflux regulators ABCG1 and/or ABCA1 causes disturbances in glucose metabolism and impaired glucose‐stimulated insulin secretion (GSIS) 3, 4. Several connections between cholesterol metabolism and impaired insulin secretion have been suggested, including high cellular levels of free cholesterol disturbing upstream glucose metabolism, diminishing β‐cell function and leading to β‐cell apoptosis (for review see ref. 1). While polymorphisms in ABCA1 are associated with increased risk of T2D 5, they also lead to low levels of high‐density lipoprotein, a recognised anti‐diabetic factor 6. The finding that patients with Tangier disease lacking ABCA1 have enhanced, rather than reduced, β‐cell insulin secretory capacity 7 further suggests that factors other than ABCA1 and ABCG1 may link cholesterol homeostasis and insulin secretion.
We recently described impaired cholesterol metabolism caused by deficiency of ABCA12 8, a lipid transporter best known as the gene mutated in harlequin ichthyosis (HI), a predominantly fatal skin disorder 9. Loss of ABCA12 correlates with defective loading of glucosylceramides into cutaneous lamellar bodies, the secretory organelles which fuse with the membrane of keratinocytes and help establish the organs’ waterproof barrier 9. We also showed that embryonic fibroblasts from Abca12 −/− mice were unable to activate cholesterol efflux in response to agonists of the nuclear receptor LXR, a key regulator of cholesterol homeostasis 10. We subsequently established that ABCA12 deficiency in macrophages enhanced interaction between LXRβ and ABCA1, resulting in ABCA1 degradation, reduction in cholesterol efflux and accumulation of cellular cholesterol 8.
Because ABCA12 acts upstream of cellular cholesterol regulators, this raises the possibility that it may also control insulin secretion in β‐cells—either independently or through ABCA1 or ABCG1. In addition, the capacity for ABCA12 deficiency to trigger inflammatory reactions and immune cell recruitment in other tissues 11 may contribute to similar pathogenic mechanisms operant in T2D. To test these hypotheses, we generated mice lacking Abca12 in β‐cells and identified a previously unrecognised role for the gene in regulating insulin secretion and pancreatic inflammation. Importantly, disease development does not appear to be dependent on the actions of ABCA1 or ABCG1. Instead, Abca12 deletion provoked a selective redistribution of cellular cholesterol, altering membrane lipid rafts and the associated F‐actin cytoskeleton and changing the normal morphogenesis of insulin‐containing secretory granules. These findings identify a unique and previously unrecognised role for ABCA12 in regulating homeostasis of β‐cells.
Results
ABCA12 expression and gene deletion in the mouse pancreas
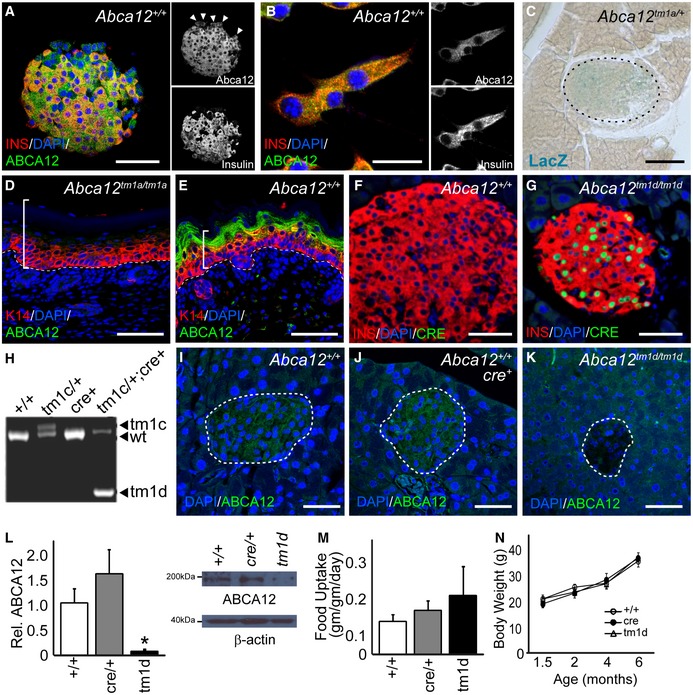
To examine the expression of ABCA12 in the pancreas, we performed immunofluorescent staining of ABCA12 in islets isolated from wild‐type mice (Fig 1A), the MIN6 mouse β‐cell line (Fig 1B) and in sections of human pancreas (Appendix Fig S1A). The expression of the protein was noted in most cells within the pancreas, and in all cases, co‐staining with insulin was observed, indicating that ABCA12 is present in murine pancreatic β‐cells. To investigate the role of ABCA12 in β‐cell function, we then generated an Abca12 LacZ gene trap reporter mouse using ES cells from EUCOMM (Abca12 tm1a(EUCOMM)Hmgu; hereafter Abca12 tm1a). The genome of these mice contains a splice acceptor‐LacZ cassette inserted in Abca12 intron 3, upstream of a floxed exon 4 (Appendix Fig S1B). This allele acts as both a reporter and a gene trap, halting protein translation. LacZ staining of frozen pancreas sections from mice carrying a single copy of this allele identified robust expression throughout the islets (with low levels of expression elsewhere in the organ; Fig 1C) confirming the β‐cell expression identified by immunofluorescence in wild‐type mice and cells.
Figure 1. Expression profiling of ABCA12 and generation of mice lacking ABCA12 in β‐cells.
-
AImmunofluorescent staining of ABCA12 (green) and insulin (red) in mouse islets showing extensive co‐localisation in β‐cells as well as expression in other islet populations (arrowheads, scale bar = 25 μm).
-
BCo‐expression of Abca12 (green) and insulin (red) in MIN6 cells (scale bar = 10 μm).
-
CLacZ staining (blue) to detect gene expression of the knocked‐in LacZ cassette from the Abca12 locus in islets (islet outlined, scale bar = 25 μm).
-
D, EABCA12 immunostaining and the development of hyperkeratotic skin disease in E18.5 Abca12 tm1a/tm1a mouse embryos (brackets indicate thickened epidermis, and dashed line indicates epidermal basement membrane, scale bar = 50 μm).
-
F, GImmunohistochemical detection of cre expression (green) in pancreatic islets from conditional mouse lines showing co‐expression with insulin (red) (scale bar = 50 μm).
-
HDetection of recombination of the Abca12 locus by PCR, indicating different alleles (tm1c, wild type and tm1d).
-
I–KDetection of ABCA12 expression (green) and deletion in pancreas sections from mice of indicated genotypes (islets outlined, scale bar = 25 μm).
-
LQuantitation of ABCA12 expression by densitometry of Western blots from purified pancreatic islets (values relative to wild type, n = 3–8 animals per genotype, mean ± SEM, *P = < 0.05, Student's t‐test), showing representative blot with actin loading control (right).
-
M, N(M) Average daily food intake of conditionally modified mice and (N) animal body weight (n = 4–10 animals/genotype, mean ± SEM).
To validate Abca12 tm1a as a loss of function allele, we generated homozygous gene trap embryos at embryonic day 18.5 and detailed hyperkeratotic cutaneous phenotypes (Fig 1D and E) which are consistent with loss of ABCA12 protein function 10. Because Abca12 tm1a/tm1a and Abca12 −/− mice die perinatally as a consequence of dehydration 10, we crossed Abca12 tm1a mice with a CAG‐flpe deleter to remove the Frt flanked LacZ reporter cassette and generate a conditional allele (Abca12 tm1c). β‐cell‐specific deletion of Abca12 was then achieved by crossing with mice expressing cre under a Rat insulin promoter 1 (Rip1‐cre; see Appendix Fig S1B). Cohorts of mice were produced which were homozygous for the β‐cell‐specific deletion of Abca12 (Abca12 tm1d/tm1d ;Rip1‐cre—hereafter Abca12 tm1d) as well cre only and wild‐type control mice. Strong nuclear Cre immunoreactivity was evident in islets from Abca12 tm1d mice compared to wild‐type controls (Fig 1F and G). PCR amplification across the Abca12 locus from Abca12 tm1d/+ ;Rip1‐cre islets and Abca12 tm1c/tm1c controls revealed efficient cre‐mediated recombination in the former strain (Fig 1H). Consistent with these results, ABCA12 expression was also found to be markedly reduced in Abca12 tm1d islets by immunofluorescence (Fig 1I–K) and Western blotting (Fig 1L). Hypothalamic ABCA12 expression and serum cortisol levels were unchanged in these mice (Appendix Fig S1C and D), indicating that hypothalamic function is not altered. Consistent with these findings, Abca12 tm1d mice housed in metabolic cages had no significant difference in food intake (Fig 1M) or increase in body weight over time (Fig 1N) when compared to control animals.
ABCA12 deficiency in β‐cells impairs insulin secretion
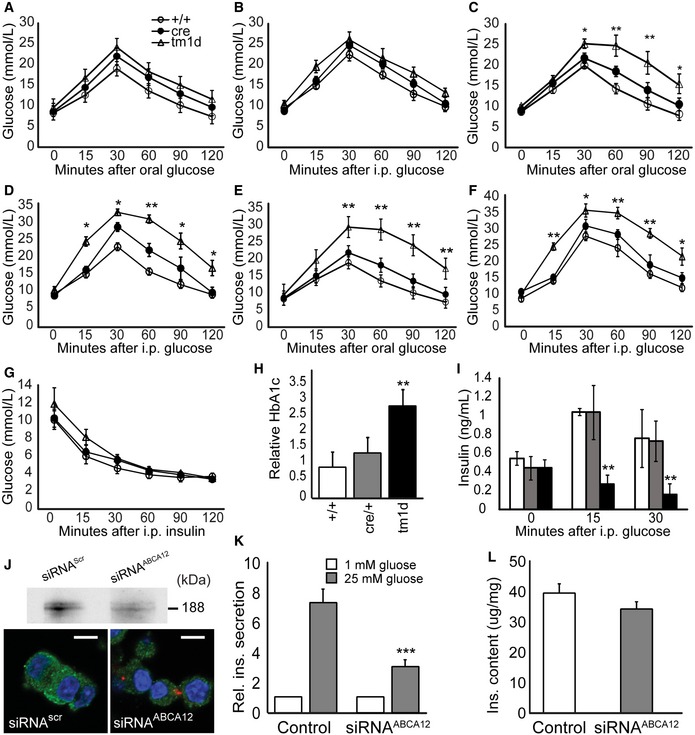
To investigate a role for Abca12 in regulating β‐cell function, we measured glucose tolerance through oral and intraperitoneal tests. In both assays, Abca12 tm1d mice had normal glucose tolerance at 6 weeks of age (Fig 2A and B) but exhibited significant glucose intolerance at 8 weeks of age (Fig 2C and D) which worsened at 16 weeks (Fig 2E and F). Insulin sensitivity was unaltered in Abca12 tm1d mice compared to controls during this period (Fig 2G). After 16 weeks, Abca12 tm1d mice also showed a threefold increase in glycated haemoglobin (Hb1Ac) levels (Fig 2H) which is consistent with increased circulating glucose levels. Plasma insulin levels during the glucose tolerance test were significantly lower in Abca12 tm1d mice 15 and 30 min after glucose challenge (Fig 2I). To investigate the possibility that these effects were a consequence of reduced β‐cell mass, we quantified evenly spaced pancreas sections from 16‐week Abca12 tm1d mice and controls. At this time point, there was no difference in β‐cell mass between genotypes (Appendix Fig S2A–C).
Figure 2. Deletion of Abca12 in β‐cells leads to glucose insensitivity.
-
A–FOral and intraperitoneal glucose tolerance testing in Abca12 tm1d mice at 6 (A, B), 8 (C, D) and 12 weeks (E, F) (mean ± SEM, n = 4–9 mice per group, *P = < 0.05, **P < 0.01 Student's t‐test).
-
GInsulin tolerance in 12‐week‐old mice (mean ± SEM, n = 4–9 mice per group).
-
HWhole blood Hb1Ac levels in 16‐week‐old Abca12 tm1d mice (mean ± SEM, n = 4–9 mice per group, **P < 0.01 Student's t‐test).
-
IAcute phase insulin secretion after intraperitoneal injection of glucose (mean ± SEM, n = 4–9 animals per group, white/grey/black = Abca12 +/+, cre/+ and Abca12 tm1d , respectively, mice at 8 weeks of age, **P < 0.01 Student's t‐test).
-
JSilencing of ABCA12 in MIN6 cells as assessed by Western blot of ABCA12 immuno‐precipitate (top panel) or confocal microscopy in cells transfected with Dy547‐siRNAABCA12 (lower panel, scale bar = 10 μm).
-
KGlucose‐stimulated insulin secretion (GSIS) from MIN6 cells after silencing of ABCA12 (mean ± SEM, n = 4 replicates from assays in quadruplicate, ***P < 0.0001, Student's t‐test).
-
LCellular insulin content in MIN6 cells after silencing of ABCA12 (mean ± SEM, n = 4 replicates from assays in quadruplicate).
To confirm the β‐cell‐specific impact of ABCA12 deficiency implied by our in vivo findings and to establish a cellular model to further investigate Abca12 function in β‐cells, we silenced Abca12 in MIN6 cells by transfection with siRNAABCA12. This reduced ABCA12 abundance by 70–80% compared to cells transfected with siRNAscr (Fig 2J, top panel). A direct relationship between siRNA uptake and ABCA12 depletion was confirmed using Dy547‐labelled Abca12 and control scrambled oligos: cells transfected with Dy547‐labelled siRNAABCA12 (with red dots) have lower abundance of ABCA12 compared with un‐transfected cells in the same viewing field or compared with cells transfected with siRNAscr (Fig 2J, lower panel). While ABCA12 deficiency had no effect on the basal level of insulin secretion at low concentrations of glucose, GSIS was severely impaired by ABCA12 deficiency (Fig 2K). Cell insulin content was unaffected by ABCA12 deficiency (Fig 2L). Analysis of previous reports of gene expression in mouse models and patients with T1D and T2D found that while ABCA12 was expressed in the pancreas, there was no correlation between the levels of gene expression and disease state 12, 13, 14.
Loss of Abca12 alters lipid homeostasis in pancreatic islets
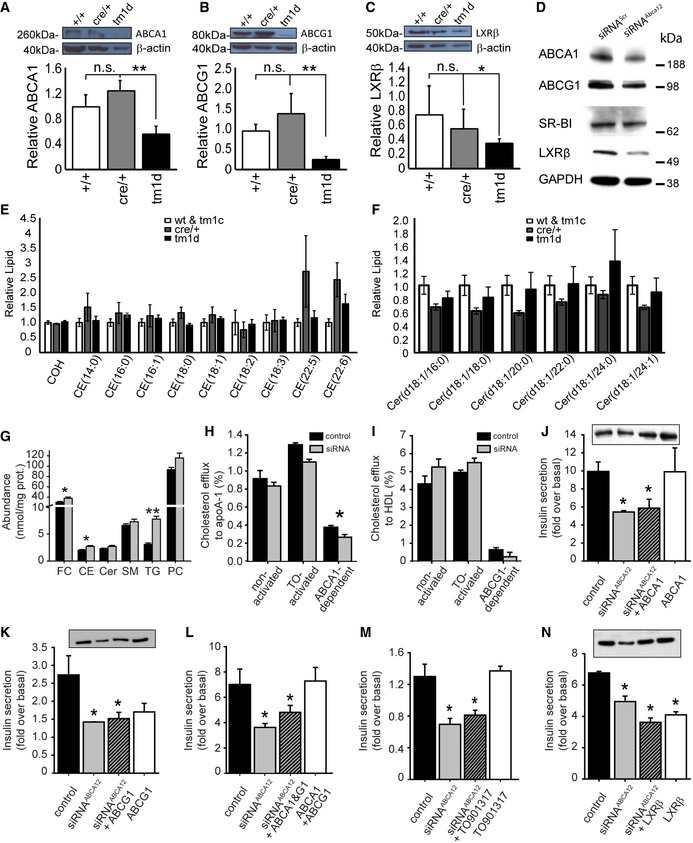
We have previously shown that ABCA12 can regulate the stability of ABCA1, ABCG1 and LXRβ in macrophages and fibroblasts 8, 10. To assess whether the defective GSIS we observed might be the result of changes in the abundance of these proteins, we performed Western blotting on extracts from isolated islets. Consistent with our previous findings 8, the levels of all three were reduced (but not ablated) in Abca12 tm1d islets (Fig 3A–C) and similar observations were made following silencing of ABCA12 in MIN6 cells (with no effect on the abundance of another important cholesterol transporter, SR‐B1) (Fig 3D). We then profiled the expression of key genes involved in the regulation of cholesterol metabolism in MIN6 cells (Abca1, Abca12, Abcg1, LdlR, Lxrb, Lxra, Scarb1, Hmgcr and Msr1) but found no statistically significant effect of ABCA12 deficiency (Appendix Fig S3A). Similar profiling of the expression of microRNA's involved in cholesterol metabolism found no effect on miR33b, miR145, miR144, miR126a, miR26a and miR10b and only very modest reductions in the levels of miR27a, miR33a, miR106b and miR758 (Appendix Fig S3B). We therefore propose that, as in other tissues, ABCA12's regulation of ABCA1 and ABCG1 levels in β‐cells is post‐translational.
Figure 3. Loss or depletion of ABCA12 results in defects in GSIS independently of changes in cholesterol levels and ABCA1, ABCG1 and LXRβ.
-
A–C(A) ABCA1, (B) ABCG1 and (C) LXRβ expression in islets of 8‐week‐old mice assessed by Western blotting (mean ± SEM, n = 3–6 mice per genotype, relative to β‐actin controls, *P < 0.05, **P = < 0.01, Student's t‐test).
-
DWestern blot of cell lysates of MIN6 cells after silencing of Abca12, probed with antibodies against ABCA1, ABCG1, SR‐B1 and LXRβ.
-
ETotal cholesterol (COH) or cholesteryl ester (CE) levels in isolated pancreatic islets of mice at 8 weeks of age with deletion of Abca12 in β‐cells (mean ± SEM n = 9, 5 and 5 mice per genotype, respectively).
-
FCeramides levels in isolated pancreatic islets of mice at 8 weeks of age with deletion of Abca12 in β‐cells (mean ± SEM n = 10, 4 and 5 mice per genotype, respectively).
-
GLipid levels in MIN6 cells in which ABCA12 had been depleted by siRNA treatment (mean ± SEM, n = 5 (biological replicates assayed in triplicate), FC = free cholesterol, CE = cholesterol ester, Cer = ceramide, SM = sphingomyelin, TG = triacylglycerol, PC = phosphatidylcholine, *P < 0.05, **P = < 0.01, Student's t‐test).
-
HCholesterol efflux to lipid‐free apoA‐I (30 μg/ml, 2 h) from MIN6 cells transfected with siRNAscr or siRNAABCA12 and activated or not with TO901317 (4 μM) (mean ± SEM, n = 4, (biological replicates assayed in triplicate), *P = < 0.05, Student's t‐test).
-
ICholesterol efflux to HDL (30 μg/ml, 2 h) from MIN6 cells transfected with siRNAscr or siRNAABCA12 and activated or not with TO901317 (4 μM); (mean ± SEM, n = 4 (biological replicates assayed in triplicate)).
-
JThe effect of overexpression of ABCA1 on GSIS from MIN6 cells with ABCA12 deficiency (mean ± SEM, n = 4 (biological replicates assayed in triplicate), *P = < 0.05, Student's t‐test), inset shows a Western blot of cell lysates probed with anti‐ABCA1 antibody in which the order of bands corresponds to the order of bars).
-
KThe effect of overexpression of ABCG1 on GSIS from MIN6 cells with ABCA12 deficiency (mean ± SEM, n = 4 (biological replicates assayed in triplicate), *P = < 0.05, Student's t‐test), inset shows a Western blot of cell lysate probed with anti‐ABCG1 antibodies in which the order of bands corresponds to the order of bars).
-
LThe effect of overexpression of ABCA1 and ABCG1 on GSIS from MIN6 cells with ABCA12 deficiency (mean ± SEM, n = 4 (biological replicates assayed in triplicate), *P = < 0.05, Student's t‐test).
-
MThe effect of activation of cells with TO901317 (4 μM) on GSIS from MIN6 cells with ABCA12 deficiency (mean ± SEM, n = 4 (biological replicates assayed in triplicate), *P = < 0.05, Student's t‐test).
-
NThe effect of overexpression of LXRβ on GSIS from MIN6 cells with ABCA12 deficiency (mean ± SEM, n = 4 (biological replicates), *P = < 0.05, Student's t‐test), inset shows a Western blot of cell lysates probed with anti‐LXRβ antibody in which the order of bands corresponds to the order of bars).
Combined deficiency of ABCA1 and ABCG1 has been shown to considerably reduce GSIS through increasing cellular cholesterol content 3, 4. To examine whether loss of ABCA12 in our mouse model impacted on cholesterol levels, we performed mass spectroscopy to quantify > 400 different lipid species in purified islets from mice at different ages. Despite the reduced levels of ABCA1 and ABCG1 proteins, at 8 weeks of age (when defects in glucose tolerance were noted), we observed no increase in cellular total, free or esterified cholesterol content in islets isolated from Abca12 tm1d mice compared to controls (Fig 3E). Given evidence in the skin that ABCA12 regulates ceramide levels 10, 15, 16, we also quantified these species in islets but found no differences between ABCA12‐deficient and control animals (Fig 3F). Similarly, ABCA12 deficiency in MIN6 cells was only accompanied by an increase in triglycerides and borderline changes in free cholesterol and cholesteryl esters (Fig 3G), nor did it result in dramatic changes in the levels of total ceramide (Fig 3G) or ceramide species (Appendix Fig S3C). Moreover, while inhibition of ceramide synthesis by Fumonisin B1 reduced cellular ceramide content (Appendix Fig S3D), this had no impact on the effect of ABCA12 deficiency on insulin secretion (Appendix Fig S3E). These findings suggest that, at least under basal conditions, ABCA12 does not regulate absolute levels of either cholesterol or ceramides in β‐cells.
Defective GSIS downstream of ABCA12 is not mediated by reduced ABCA1, ABCG1 or ABCLXRβ
Although the emergent defects in GSIS in Abca12 tm1d mice were not associated with an overt disruption in cholesterol homeostasis, we nonetheless used our cell model to examine whether ABCA1 and/or ABCG1 might otherwise contribute to this phenotype. As expected, reductions in the abundance of ABCA1 and ABCG1 in MIN6 cells due to ABCA12 deficiency resulted in modest decreases in cholesterol efflux to lipid‐free apolipoprotein A‐I (apoA‐I) (Fig 3H) but not to isolated high‐density lipoprotein (HDL) (Fig 3I). Indeed, comparison of efflux from naive cells or those exposed to the LXR agonist TO‐901317 (an activator of the ABCA1/G1 transporter expression) showed that reduction in ABCA12 levels reduced ABCA1‐dependent cholesterol efflux by only 25% and that ABCG1‐dependent efflux was not statistically significantly affected (Fig 3H and I). To confirm what we expected was a minimal contribution of ABCA1 and ABCG1 to defective GSIS, we then attempted to rescue this phenotype by their heterologous overexpression in MIN6 cells. While transfection with ABCA1 reversed the reduction in protein levels caused by concomitant silencing of ABCA12, they did not rescue defective GSIS (Fig 3J). Similar experiments employing overexpression of ABCG1 (or ABCG1 and ABCA1 together) also failed to restore normal GSIS (Fig 3K and L). The latter findings are consistent with a failure of the LXR agonist TO‐901317 to rescue secretion (Fig 3M). We also investigated whether reduced abundance of LXRβ might contribute to disease progression. LXRβ is the predominant LXR isotype in β‐cells 17 where it regulates genes involved in glucose metabolism (e.g. Glut4, Srebf‐1c) and plays a pivotal role in insulin secretion 18. However, while heterologous transfection restored the LXRβ abundance of ABCA12‐deficient cells, it failed to correct the impairment of GSIS (Fig 3N). It was noted that transfections with ABCG1 or LXRβ alone also reduced GSIS. The reason for this effect is unclear, but may be related to the effect of ABCG1 overexpression on secretory granules (see below). These experiments indicate that partial reductions in the abundance LXRβ, ABCA1 or ABCG1 are unlikely to be the mechanism underlying defective insulin secretion caused by loss of ABCA12.
Loss of Abca12 in β‐cells leads to transcriptional changes in pathways associated with secretion and the actin cytoskeleton
To flag potential regulators of the emergent phenotypes in our cell and mouse models, we analysed gene expression in isolated islets by RNA sequencing. At 5 weeks of age, Abca12 tm1d and cre control groups did not show genotype‐dependent clustering (Appendix Fig S4A) and only 3 genes (Nupr1, Fbxw15 and Hist1h2bn) were differentially expressed. However, by 8 weeks profound differences were evident between the transcriptomes of Abca12 tm1d and cre control islets and the genotypes clustered differentially (Fig 4A). Comparison of the two groups revealed 136 differentially expressed genes matching our filtering criteria (Fig 4B; see Materials and Methods), with 63 genes up‐regulated and 73 genes down‐regulated in Abca12 tm1d islets relative to control cre mice (Dataset EV1). KEGG pathway analysis of the DE gene list highlighted pancreatic secretion as the most altered pathway, along with other pancreas‐related functions including protein digestion and absorption and fat digestion and adsorption (Appendix Fig S4B). We also performed GO term cell component analysis and observed several recurring themes when cross‐comparing with KEGG classifications. In addition to secretory vesicle‐related terms (endocytic vesicle lumen, endoplasmic reticulum lumen, lysosomal lumen and Golgi lumen), a large number of actin‐related terms were also observed (actin‐based cell projection, cytoskeleton, actin cytoskeleton and focal adhesion; Appendix Fig S4C and D). Such vesicle trafficking and cytoskeletal remodelling pathways were further unified by overlapping KEGG terms such as Ras signalling pathway, Rap1 signalling pathway, PI3K‐Akt signalling pathway and calcium signalling pathway. Based on the clues provided by the gene expression analysis, we further investigated the effect of ABCA12 deficiency on vesicle trafficking and actin cytoskeleton.
Figure 4. Changes in gene expression and insulin granule fusion in Abca12tm1d mice.
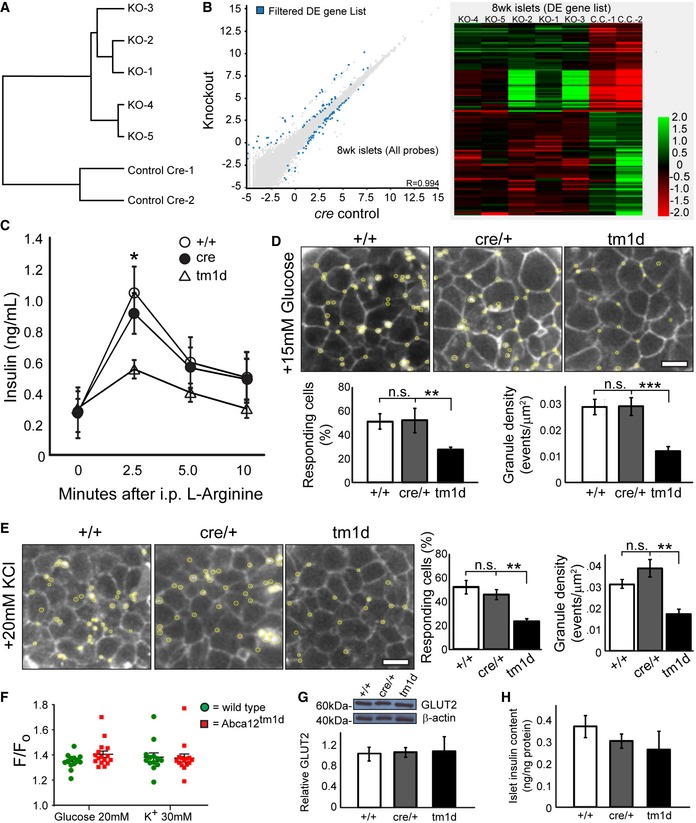
-
AClustering of gene expression by genotype in RNAseq of islets isolated of cre control and Abca12 tm1d mice at 8 weeks of age.
-
BScatter plot and heat map of differential gene expression between cre control and Abca12 tm1d mice at 8 weeks of age. Legend indicates log2 fold change values.
-
CCirculating glucose levels after administration of intraperitoneal l‐arginine in mice at 8 weeks of age (mean ± SEM, n = 3 mice per genotype, *P = < 0.05 of cre relative to tm1d, Student's t‐test).
-
DRepresentative images of insulin granule fusion events (yellow circles) in mice of indicated genotypes in response to administration of 15 mM glucose (scale bar = 10 μm). Quantification of the proportion of responding cells and the density of granule fusion events are presented in graphs in the lower panel (mean ± SEM, 8–10 islets/mouse, > 4 mice/genotype, **P = < 0.01, ***P = < 0.001 of cre relative to tm1d, Student's t‐test).
-
ERepresentative images of insulin granule fusion events (yellow circles) in mice of indicated genotypes in response to administration of 20 mM KCl (scale bar = 10 μm). Quantification of the proportion of responding cells and the density of granule fusion events are presented in graphs in the right panel (mean ± SEM, 8–10 islets/mouse, > 4 mice/genotype, **P = < 0.01 of cre relative to tm1d, Student's t‐test).
-
FAssessment of calcium flux measured by Fluo4 in response to either glucose or potassium in isolated islets from 11‐week‐old animals. Fluorescence responses were corrected for basal fill (F/F 0). (mean ± SEM, each data point represents the average of 10 cells/coverslip for 13–16 coverslips for each of 4 wild‐type and 5 Abca12 tm1d mice).
-
GThe expression of GLUT2 in isolated mouse islets at 8 weeks of age. The upper panel depicts a representative Western blot and the lower graph the quantitation of protein levels from three mice of each genotype, normalised to β‐actin (mean ± SEM).
-
HInsulin content of islets of Abca12 tm1d mice (mean ± SEM, n = 3 mice of each genotype, normalised to total protein content).
Abca12 regulates insulin granule secretion, morphology and function
To examine whether the changes in insulin trafficking and secretion indicated by RNA sequencing may contribute to the development of disease, we administered 0.3 mg/kg of the non‐glucose secretagogue l‐arginine to Abca12 tm1d mice. This resulted in significantly lower level of insulin secretion in response to stimulus compared to controls (Fig 4C), suggesting that loss of ABCA12 results in a secretory defect. To specifically profile insulin release from β‐cells, we then used two‐photon live cell imaging 19 to measure insulin granule fusion from cultured islets isolated from 8‐week‐old Abca12 tm1d mice and controls. Exposure of cells to 15 mM glucose revealed significant reductions in both the number of cells which responded and the density of fusion events (Fig 4D). To determine whether this was a consequence of defective glucose sensing or a more generalised defect in the capacity of insulin granules to fuse with the cell membrane, we profiled fusion events in response to 30 mM KCl and found similar decreases in both the number of responding cells and the number of vesicle fusion events (Fig 4E). These findings suggest that the impairment in insulin secretion in Abca12 deficient islets is not associated with defective nutrient sensing and secondary signal generation by the β‐cell and instead derives from cellular defects which impair insulin granule fusion. To confirm this was the case, we profiled calcium flux using Fluo‐4 in response to either glucose or potassium stimulation in isolated β‐cells from Abca12 tm1d and control mice at 11 weeks of age, but found no difference in F/Fo ratios for either treatment (Fig 4F). Consistent with this notion, we found that level of the GLUT2 glucose transporter (which participates in sensing extracellular glucose levels) was unchanged in Abca12 tm1d islets compared to controls (Fig 4G). Moreover, insulin content in islets lacking ABCA12 was also unchanged (Fig 4H). These findings identify an essential role for ABCA12 in regulating the normal release of insulin in response to glucose challenge and further indicate that loss of the protein results in a secretory defect which is not dependent on the capacity of the cell to sense glucose levels.
Abca12 regulates secretory granule morphology and insulin release in a cholesterol‐dependent manner
To examine whether the loss of ABCA12 resulted in changes in insulin granules, we examined their morphology using transmission electron microscopy. Quantification of 3,000 granules per genotype revealed that granule dysmorphology was a feature of in Abca12 tm1d β‐cells (Fig 5A–C), manifesting as an increase in mean insulin granule area (Fig 5D) and when plotted as a frequency distribution (Fig 5E). This phenotype is similar to observations of β‐cells of mice lacking Abcg1 20. In these animals, it is thought that intracellular redistribution of cholesterol from secretory granules to plasma membrane leads to an increase in insulin granule size and impaired insulin secretion. To examine the possibility that ABCA12 plays similar role and to establish whether such defects may underpin the changes in granule fusion observed in cultured Abca12 tm1d islets, 6‐week‐old Abca12 tm1d and control mice were fed a high cholesterol diet (HCD) (0.5% cholesterol, 6% fat by weight) for 4 weeks and analysis of granules was repeated. Dietary supplementation led to an increase in islet cholesterol levels when measured by mass spectroscopy (Fig 5F) and a partial restoration of mean granule area (Fig 5G) and size distribution (Fig 5H). These changes were accompanied by improvements in granule morphology (Fig 5I). Response to glucose challenge was improved in Abca12 tm1d fed a high cholesterol diet compared to those mice on normal chow, whereas a high cholesterol diet in cre control animals led to a worsening of GSIS, consistent with previous reports 21 (Fig 5J). As expected, insulin secretion in cultured Abca12 tm1d islets from mice fed a high cholesterol diet was improved relative to control animals (Fig 5K).
Figure 5. Loss of β‐cell ABCA12 leads to cholesterol‐modulated changes in insulin granule morphology.
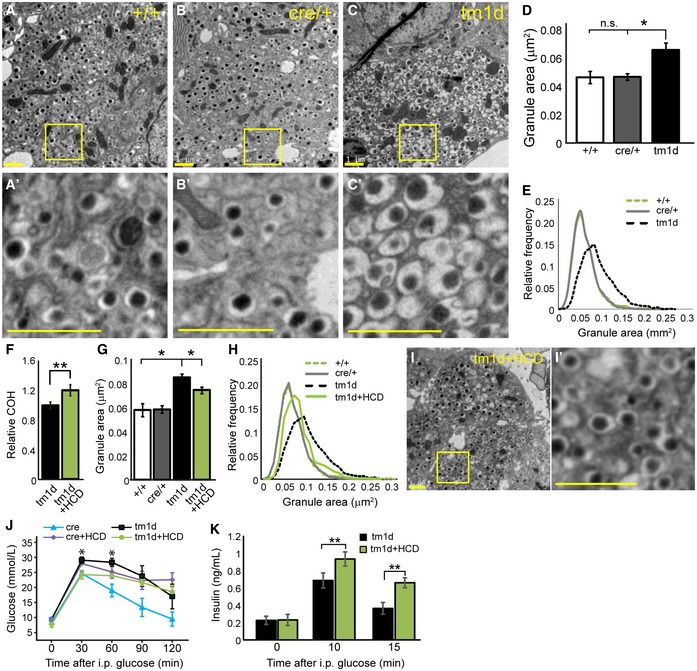
-
A–CTransmission electron microscopy of β‐cells from Abca12 tm1d and control mice at 8 weeks of age. Panels (A′–C′) depict higher resolution images from the boxed regions in (A–C) (scale bar = 1 μm).
-
DQuantitation of the area of 3,000 granules from each genotype (mean ± SEM, n = 4, 3 and 3 animals, respectively, *P = < 0.05 of cre relative to tm1d, Student's t‐test).
-
ERelative frequency distribution of granule areas from (D).
-
FAssessment of total cholesterol (COH) by mass spectroscopy in mice fed normal chow or a high cholesterol diet (HCD) (mean ± SEM, n = 5 and 4 animals, respectively, **P = < 0.01, Student's t‐test).
-
G, HQuantification of change in mean granule area in Abca12 tm1d on chow diet and Abca12 tm1d on high cholesterol diet islets assessed by transmission electron microscopy (n = 3,000 granules/genotype, mean ± SEM, n = 4, 3, 3 and 4 animals, respectively, *P = < 0.05, Student's t‐test) as an average and as a relative frequency distribution.
-
ICorrection in abnormal granule shape following high cholesterol feeding (bar = 1 μm), with I’ showing magnification of the boxed region indicated.
-
JGlucose tolerance test results following intraperitoneal glucose challenge in Abca12 tm1d and cre control mice fed normal chow or HCD (mean ± SEM, n = 3 mice per genotype, *P = < 0.05, for tm1d versus tm1d+HCD, Student's t‐test).
-
KSerum insulin in Abca12 tm1d ± HCD mice after i.p glucose challenge (mean ± SEM, n = 3 mice/genotype, **P = < 0.01, Student's t‐test).
Loss of Abca12 alters lipid raft formation, preventing normal glucose‐stimulated insulin secretion
In addition to secretory granules, another common structural element intersecting cholesterol metabolism and insulin secretion and glucose uptake are plasma membrane lipid rafts. Cholesterol enrichment of the plasma membrane reduces glucose transport into (and insulin secretion from) β‐cells and the rafts host many proteins involved in insulin secretion 22. To examine whether raft levels were altered by deletion of Abca12, we utilised confocal microscopy and FACS sorting. Both methods demonstrated that β‐cells isolated from Abca12 tm1d mice have an increased abundance of lipid rafts, consistent with a redistribution of cholesterol to the plasma membrane (Fig 6A and B). To study the impact of altered raft abundance on GSIS, we utilised our MIN6 model. As in our mouse studies, siRNA‐mediated knockdown of Abca12 increased the abundance of lipid rafts by 1.5‐fold compared with cells transfected with Fluorescein‐labelled siRNAScr or un‐transfected cells as shown by confocal microscopy (Fig 6C and D) and when assessed by flow cytometry (Fig 6E). To test the influence of ABCA12‐mediated regulation of lipid raft formation on GSIS, we treated MIN6 cells with MβCD (10 mM) for short period of time (5–10 min). Under these conditions, MβCD disrupts lipid rafts without affecting total cellular cholesterol content 23. Analysis of cells treated in this manner revealed that ABCA12‐mediated deficiency in GSIS was reversed by MβCD (Fig 6F).
Figure 6. Loss of Abca12 results in altered lipid raft levels and changes in F‐actin and CDC42 activity.
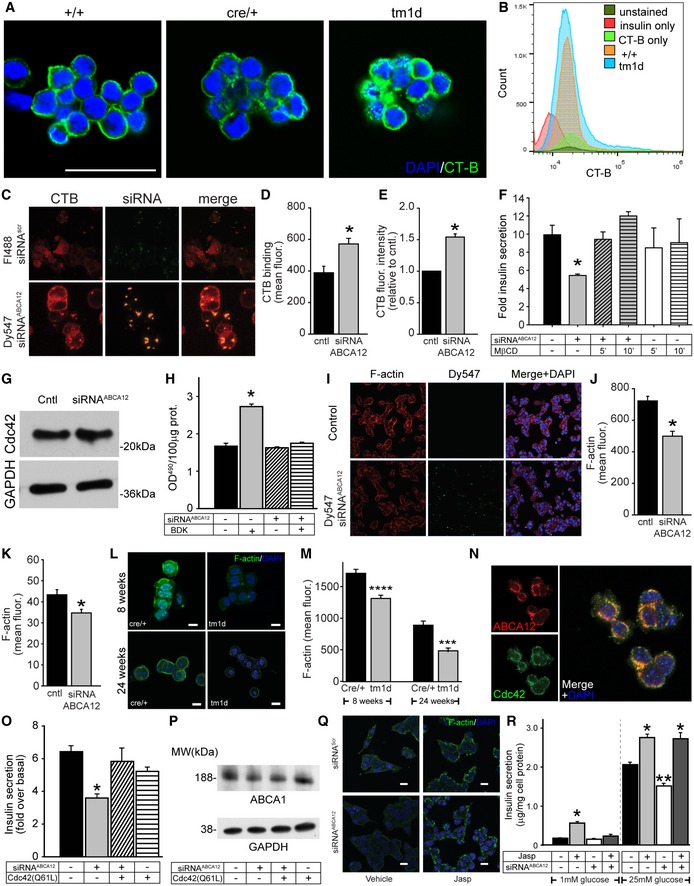
-
ADetection of lipid rafts in 8‐week‐old islets with recombinant cholera toxin subunit B (CT‐B) conjugated to Alexa Fluor 488 (green). Islets are co‐stained with DAPI (blue) (scale bar = 50 μm.).
-
BFACS profiling of dispersed islets from 12‐week‐old mice stained with insulin and CT‐B.
-
CConfocal images of rafts (using CT‐B‐Alexa Fluor 647) in MIN6 cells transfected with Fluorescein‐488‐labelled siRNAScr or Dy547‐labelled siRNAABCA12 (scale bar = 10 μm).
-
DQuantitation by confocal microscopy of the effect of ABCA12 deficiency on CT‐B binding to MIN6 cells (mean ± SEM, *P = < 0.05, Student's t‐test, n = 4 biological replicates, 50–100 cells for each).
-
EQuantitation by flow cytometry of the effect of ABCA12 deficiency on CT‐B binding to MIN6 cells (mean ± SEM, n = 3 biological replicates, *P = < 0.05, Student's t‐test).
-
FThe effect 10 mM MβCD on GSIS in MIN6 cells depleted or not for ABCA12 (incubation for 5 or 10 min as indicated, mean ± SEM, n = 4 (biological replicates), *P = < 0.05, Student's t‐test).
-
GWestern blot of the abundance of CDC42 in ABCA12‐deficient MIN6 cells.
-
HThe effect of ABCA12 deficiency on activation of CDC42 by bradykinin in MIN6 cells (BDK, 100 ng/ml, 4 min; *P = < 0.05, Student's t‐test, significance versus ABCA12‐deficient BDK‐activated cells are indicated, n = 4 biological replicates, mean ± SEM).
-
I, J(I) Confocal images of F‐actin in ABCA12‐deficient MIN6 cells (scale bar = 20 μm) and (J) quantification of abundance showing the effect of ABCA12 deficiency (mean ± SEM, *P = < 0.05, Student's t‐test, n = 4 biological replicates, 50–100 cells for each).
-
KF‐actin levels assessed by FACS sorting of labelled MIN6 cells transfected with siRNAABCA12 (mean ± SEM, n = 3 biological replicates, *P = < 0.05, Student's t‐test).
-
L, M(L) Representative image of F‐actin using LifeAct staining of purified islets isolated from Abca12 tm1d versus control mice (scale bar = 10 μm) and (M) quantitation of fluorescent signals from these samples (n = 3 biological replicates, mean ± SEM, ***P = < 0.001, ****P = < 0.0001 Student's t‐test).
-
NCo‐localisation between ABCA12 (red) and CDC42 (green) in MIN6 cells (scale bar = 10 μm).
-
OEffects of overexpression of a constitutively active form of CDC42 on GSIS from MIN6 cells with ABCA12 deficiency (mean ± SEM, n = 4 biological replicates, *P = < 0.05, Student's t‐test).
-
PThe effect of overexpression of constitutively active CDC42 on ABCA1 abundance in MIN6 cells with ABCA12 deficiency. Treatments are defined in the table below.
-
QEffects of Jasplakinolide on F‐actin levels (green) in siRNA‐treated cells (scale bar = 10 μm).
-
REffects of Abca12 knockdown and Jasplakinolide treatment on MIN6 cell in low and high glucose (mean ± SEM, n = 4 biological replicates, *P = < 0.05, **P = < 0.01, significance relative to untreated cells, Student's t‐test).
While it is possible that the increased abundance of rafts results from reduced abundance of ABCA1 and inhibited cholesterol efflux, failure of ABCA1 overexpression to reverse GSIS impairment caused by ABCA12 deficiency argues against this mechanism. Importantly when comparing the 8 KEGG pathways that were enriched in our DE gene list, we noticed that RAC was a member of 5/8 pathways, RHO a member of 4/8 pathways and CDC42 a member of 3/8 pathways, suggesting RAC/RHO/CDC42 pathways were well positioned to contribute to a possible actin‐related vesicle trafficking defect in Abca12 tm1d mice. While ABCA12 deficiency had no effect on CDC42 abundance (Fig 6G), its activation with bradykinin (100 ng/ml, 4 min) was impaired in ABCA12‐deficient cells (Fig 6H). One downstream effect of CDC42 activation is rearrangement of the actin cytoskeleton due to polymerisation 24 which, when reduced, causes increased formation of lipid rafts 25. To distinguish cells transfected with siRNAABCA12, we again used Dy547‐labelled siRNAABCA12 and found that in ABCA12‐deficient cells (green dots Fig 6I), the abundance of filamentous actin (F‐actin) was reduced when assayed by confocal microscopy (Fig 6J) or flow cytometry (Fig 6K). A considerable reduction in F‐actin abundance was also observed in β‐cells isolated from Abca12 tm1d mice at 8 weeks of age and also much later during disease progression at 24 weeks of age (Fig 6L and M). As an indication that ABCA12 may be a direct activator of CDC42, we found that there was considerable co‐localisation between the two proteins in MIN6 cells (Fig 6N). In an attempt to overcome the impairment of CDC42 activation and to provide further evidence for changes in the actin cytoskeleton playing a role in defective insulin secretion, we transfected cells with CDC42(Q61L), a constituently active protein variant 26. Unlike heterologous transfection with ABCA1, ABCG1 or LXRβ, this fully restored impairment of insulin secretion caused by ABCA12 deficiency (Fig 6O), without altering ABCA1 abundance (Fig 6P). Furthermore, restoration of F‐actin abundance using Jasplakinolide to stabilise it 27 also resulted in full restoration of impairment of insulin secretion on the background of ABCA12 deficiency (Fig 6Q and R). Thus, it appears that ABCA12 deficiency causes impede activation of CDC42 resulting in reduced actin polymerisation and increased abundance of lipid rafts. This suggests that the observed defects in GSIS derive from a new role for ABCA12 in controlling the cellular distribution of cholesterol, regulating the morphology and fusion of secretory granules and abundance of lipid rafts.
Abca12 deficiency causes inflammation and reduction in β‐cell mass in older mice
To further profile the progression of disease caused by loss of ABCA12, we aged cohorts of mice to reveal a number of late‐onset phenotypes, which differ from those previously reported upon deletion of Abca1 or Abcg1. Islets from 16‐week‐old Abca12 tm1d mice still had unchanged levels of cholesterol and cholesterol esters (Fig 7A), but were found to contain increased levels of total triglycerides (Fig 7B) affecting all the species examined (Fig 7C). Ageing of mice to 24 weeks further revealed progressive increases in the levels of stearic acid, GM3 lipid species and total GM3 lipids compared to controls (Appendix Fig S5A–C). Both triglycerides and stearic acid are known to play pro‐inflammatory roles in other models of T2D and are known to cause β‐cell death 28, 29. To examine whether deletion of ABCA12 resulted in islet inflammation, we profiled the expression of IL‐1β 30, 31, identifying a significant upregulation (Fig 7D and E). Consistent with the known link between inflammation and β‐cell depletion, at 24 weeks of age we observed a significant reduction in β‐cell mass (Fig 7F), increases in apoptosis (Fig 7G), glucagon‐positive cells (Fig 7H) and macrophage infiltration (Fig 7I). Similar to what was observed in 8‐week‐old mice, loss of ABCA12 was associated with sharp reduction in the abundance of F‐actin (Fig 6L and M). These features were associated with an elevation in fasting blood glucose in aged Abca12 tm1d mice compared to controls (Fig 7J). Taken together, these results indicate that long‐term ABCA12 deficiency in islets is accompanied by inflammation and cell death which are coincident with the progressive accumulation of pro‐inflammatory lipid species.
Figure 7. Ageing of Abca12tm1d mice leads to the development of lipid imbalance and progressive inflammatory disease.
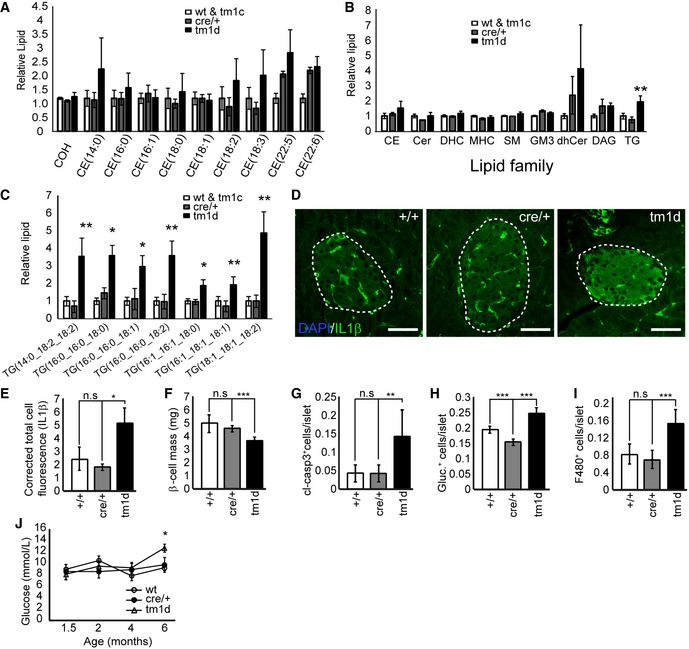
-
AMass spectroscopy analysis of levels of cholesterol and cholesteryl esters in purified islets from Abca12 tm1d mice versus control animals at 16 weeks of age (mean ± SEM, n = 10, 4 and 5 mice per genotype, respectively; COH = cholesterol, CE = cholesteryl ester).
-
BMass spectroscopy analysis of lipids families in purified islets from Abca12 tm1d mice versus control animals at 16 weeks of age (mean ± SEM, n = 10, 4 and 5 mice per genotype, respectively, **P = < 0.01 Student's t‐test); CE = cholesteryl ester, Cer = ceramide, DHC (Hex2Cer) = dihexosylceramide, MHC (HexCer) = monohexosylceramide, SM = sphingomyelin, GM3 = GM3 ganglioside, dhCer = dihydroceramide, DG = diacylglycerol, TG = triacylglycerol).
-
CMass spectroscopy analysis of triglyceride species in purified islets from Abca12 tm1d mice versus control animals at 16 weeks of age (mean ± SEM, n = 10, 4 and 5 mice per genotype, respectively, *P = < 0.05, **P = < 0.01, Student's t‐test).
-
DImmunohistochemical staining of levels IL‐1β expression in sections of pancreas from Abca12 tm1d mice versus control animals at 24 weeks of age (bar = 25 μm, islets outlined).
-
EQuantification of islet‐specific IL1β staining from sections of Abca12tm1d, Cre and wild‐type mice (mean ± SEM, n = 3, 3 and 4 mice of each genotype, respectively; *P = < 0.05, Student's t‐test).
-
F–JAnalysis of Abca12 tm1d pancreata from mice at 24 weeks of age compared to control mice assessing β‐cell mass (F), cleaved caspase 3 (G), glucagon (H) and F480+ macrophage populations (I) (mean ± SEM, n = 3–9 mice per genotype, **P = < 0.01, ***P = < 0.001, Student's t‐test). (J) Fasting blood glucose levels coincident with the emergence of inflammatory phenotypes (mean ± SEM, n = 3, 4 and 5 mice per genotype, respectively, *P = < 0.05, Student's t‐test).
Discussion
The role of ABCA12 in regulating lipid metabolism in epidermal keratinocytes is well established 9, but little is known about the function of this widely expressed protein in other cell types. We found that ABCA12 is expressed abundantly in β‐cells and have investigated its influence on insulin secretion. Through a combination of in vivo and in vitro studies, we show that deletion or depletion of ABCA12 in β‐cells results in defective glucose‐stimulated insulin secretion. In mice, conditional deletion of Abca12 precipitates the progressive development of glucose intolerance which is first evident at 8 weeks and develops further as animals age. Both in vivo and in vitro studies indicate that this is a β‐cell‐specific effect associated with a failure in the normal maturation and fusion of insulin‐containing secretory granules and altered regulation of lipid rafts and the actin cytoskeleton.
We have previously reported that loss of ABCA12 reduces the levels of ABCA1 in macrophages and that this has considerable impacts on cholesterol levels 8. For this reason (and because glucose intolerance is a feature of mice lacking pancreatic ABCA1 3, 4), we initially assumed that accumulation of cellular cholesterol was likely to drive the observed defects in GSIS. However, ABCA12 deficiency had no appreciable impact on total levels of cellular cholesterol in islets of conditionally modified mice and only modest effects on cholesterol levels or efflux in MIN6 cell models. Importantly, we showed that restoring the abundance of ABCA1 failed to rescue the impairment of insulin secretion in vitro. Similarly, correction in levels of ABCG1 and LXRβ also failed to restore normal GSIS. It is likely that the modest reduction in ABCA1/G1 abundance due to ABCA12 deficiency (in contrast to ablation in ABCA1/G1 knockout animals) is insufficient to cause accumulation of cholesterol in quantities necessary to impair insulin secretion.
On this basis, we investigated the possibility that ABCA12 might play a different role in regulating cellular responses to glucose. Transcriptome analysis flagged considerable impacts on both secretion and the arrangement of the actin cytoskeleton as possible mechanisms for the action of the protein. Moreover, we found that ABCA12 is required for the normal genesis of insulin granules, which appear larger in conditionally modified β‐cells. A potential role for ABCA12 in regulating the lipid composition and maturation of these cellular organelles parallels a known function of the protein in the skin, where it regulates the loading of the lamellar granules central to the transport of lipids from the Golgi to the intercellular space between terminally differentiating keratinocytes 9. A role for ABCA12 in regulating granule lipid content is further supported by the partial reversal in secretory defects (and granule morphology) found upon feeding mice with cholesterol‐rich diet. Although the improvement in insulin secretion by cholesterol‐rich diet may seem counterintuitive, it is important to recognise that the impact of this diet on insulin secretion in control mice was negative, suggesting that maintaining an appropriate balance in cholesterol levels and distribution is critical to normal insulin secretion. Most of the studies on the effect of dietary lipids on insulin secretion have been undertaken using high‐fat (rather than high cholesterol) dietary regimens 32, 33. The failure of heterologous ABCA1 to rescue these changes argues strongly that this is a direct effect of ABCA12, and while similar cholesterol driven rescue of GSIS has been reported in β‐cells lacking ABCG1 20, the failure of restoration of ABCG1 levels to correct GSIS indicates that this is likely to be a parallel mechanism specific to ABCA12.
The changes in insulin granule morphology associated with ABCA12 loss of function are consistent with depletion in cholesterol in these organelles. In this respect, while we found that loss of ABCA12 has a limited impact on total cholesterol levels, it may result in the intracellular redistribution of this lipid to plasma membrane lipid rafts—an effect noted both in vivo and in vitro. The observation that the transient normalisation in lipid rafts was able to rescue defects in secretion argues strongly that in addition to the observed differences in insulin granule maturation, their fusion and secretion may be impacted by alterations in lipid rafts at the plasma membrane. The function of these lipid domains in regulating secretory granule fusion is intimately linked with the regulation of the actin cytoskeleton, disruption of which was flagged in our transcriptomic analysis of islets lacking ABCA12. It was previously reported that activation of the Rho‐GTPase CDC42 leads to actin polymerisation with formation of filamentous actin (F‐actin) 34 and that this cytoskeletal restructuring reduces the abundance of lipid rafts 25, 35. We propose that as well as regulating the maturation of insulin secretory granules, ABCA12 deficiency also impacts cytoskeletal rearrangements leading to increased abundance of lipid rafts and impairment of efficient granule fusion and insulin release. This is supported by the reduction in F‐actin levels we observe, both in vivo and in vitro, in association with altered raft abundance. Moreover, depletion of ABCA12 renders CDC42 refractory to the activation mediated by the actions of bradykinin. The importance of these changes to insulin secretion is further illustrated by the rescue in GSIS mediated by the transfection of a constitutively active variant of the CDC42. Reports that ABCA1 can mediate CDC42 activation 36, 37 set a precedent for the ABC protein family to act in this manner. However, whether these effects are mediated by a direct interaction of ABCA12 and CDC42 (as has been proposed for ABCA1 37) or by downstream consequences of changes in lipid distribution remains to be determined.
The unique actions of ABCA12 in regulating insulin secretion and pancreatic function are further reflected in the emergence of inflammatory and apoptotic phenotypes in aged Abca12 tm1d mice. Notably, such effects have not been reported in mice lacking Abcg1 or Abca1 3, 7. These phenotypes develop alongside the progressive accumulation of several potentially toxic and pro‐inflammatory lipid species including triglycerides, stearic acid and GM3 gangliosides in islets. Previous studies have established that triglycerides up‐regulate IL‐1β‐induced nitric oxide (NO) production, leading to β‐cell apoptosis. Furthermore, treatment with leptin or troglitazone (which lower TG content) enhanced β‐cell survival 38. Both observations are consistent with the phenotypes observed in our mice. Accumulation of stearic acid and other long‐chain saturated fatty acids has also been shown to be pro‐apoptotic factors in β‐cells 39, and there is a large body of evidence demonstrating increased pancreatic saturated fatty acid content generates lipotoxic conditions that promote apoptosis in both INS‐1 cells and human islets 40, 41. While the influence of GM3 accumulation in the development of T2D is unclear, altered pancreatic islet ganglioside composition has been documented in NOD mice, where they act as putative autoantigens that mediate β‐cell destruction by the immune system 42. When viewed in light of our recent findings that loss of cutaneous Abca12 can result an in utero sterile inflammatory response 11, our results suggest that gradual accumulation of dysregulated lipid species can contribute to an inflammation‐driven worsening of disease phenotypes in ageing Abca12 tm1d mice.
This study establishes a new and unique role for ABCA12 in regulating insulin secretion. Deletion of the protein results in the development of progressive glucose intolerance and of inflammatory disease and apoptosis. These changes derive, at least in part, from alterations in insulin granule composition, maturation and fusion. We find that these changes are also accompanied by alterations in the distribution of cellular cholesterol from the insulin granules to plasma membrane lipid rafts. The ability to rescue defects in insulin release by altering cytoskeleton, secretory granules and lipid rafts highlights the pleiotropic role played by ABCA12 in regulating insulin secretion from β‐cells. Multiple lines of evidence indicate that these phenotypes are mediated directly by the actions of ABCA12 and not by other co‐regulated lipid transporters of the same protein family. As with many putative lipid transport proteins like ABCA1, the exact mechanism(s) by which ABCA12 regulates redistribution of cholesterol in β‐cells remains to be elucidated. One possibility is that ABCA12 is required specifically for fusion of the secretory granules with lipid rafts during exocytosis. In its absence, lipids may still move from granule to rafts releasing cholesterol‐depleted granules back into cytoplasm. Another possibility is that ABCA12 is involved in intracellular cholesterol trafficking and the absence of ABCA12 disrupts loading of granules with cholesterol, resulting in an imbalance favouring the accumulation of cholesterol‐rich lipid rafts. These two mechanisms are not mutually exclusive, and both may be shaped by ABCA12 directly or through modification of the actin cytoskeleton. Further biochemical studies aimed at establishing the capacity of ABCA12 to transport different lipid moieties in both the maturing insulin granules and the plasma membrane will therefore be required to better establish the mode of action of this new player with a central role in regulating insulin release in the pancreas.
Materials and Methods
Animals
We generated an Abca12 LacZ gene trap reporter mouse (Abca12 tm1a), using ES cells from EUCOMM. CAG‐flpe mice were obtained from the Australian Phenomics Network and used to generate floxed Abca12 mice. We generated β‐cell‐specific Abca12 knockout mice and controls by crossing floxed Abca12 mice with, B6.Cg‐Tg (Ins2‐cre)25 Mgn/J mice expressing Cre under the Rat Insulin Promoter 1 (Rip1Cre) (Jackson Laboratories). Mice received a standard laboratory chow or a high cholesterol diet (SF16‐017, a variant of SF00‐219 with 0.5% cholesterol by weight and 6% fat; Specialty Feeds, Glen Forrest, WA, Australia).
Physiological and metabolic experiments
We performed intraperitoneal and oral glucose tolerance tests on 5‐h fasted mice by injecting 3 mg/kg glucose or administering 50 μl 200 mg/ml glucose solution. Insulin tolerance tests were performed by injecting mice fasted for 5 h with 1 U/kg recombinant human insulin. To determine how ABCA12 regulated insulin secretion, we injected fasted mice with 3 mg/kg glucose or 0.3 mg/kg l‐arginine and measured serum insulin by ELISA (Crystal Chem). Mice were housed in an environmentally controlled Promethion Metabolic Caging System at the Monash Metabolic Phenotyping facility for 4 days to measure metabolic parameters. Insulin secretion was performed in vitro on hand‐picked islets isolated after intra‐ductal collagenase injection. Briefly, pancreata were perfused with sterile 0.35 mg/ml Collagenase P (Roche) in complete HBSS via bile duct cannulation. Perfused pancreata were harvested and digested in a 37°C water bath for 16 min, vigorously agitated for 1 min, washed with serum‐free RPMI and filtered through a sterile 50‐μm mesh. Tubes were spun down at 0.1 g for 2 min and the pellet resuspended in 10 ml Histopaque (Sigma) topped with 5 ml of RPMI at room temperature. Islets were separated by gradient centrifugation at 0.4 g for 15 min at 22°C, immediately removed from the interface and washed with 40 ml RPMI. Islets were spun down at 1,000 rpm for 2 min and recovered in CMRL (10% HI‐FBS, 2 mM glutamine, pen/strep) for 1 h at 37°C, 5% CO2, handpicked into fresh media with the aid of a dissecting microscope and cultured overnight in CMRL. Islets were plated at a density of 20 islets per well in Krebs‐Ringer bicarbonate buffer containing 3 mM glucose for 2 h, then incubated with buffer containing 30 mM glucose or 30 mM KCl. After 1 h, media was removed and islets were lysed in Pierce IP Lysis Buffer (Thermo Fisher Scientific) and insulin levels determined by ELISA (Crystal Chem). Insulin secretion was expressed as per cent of islet insulin content and normalised to basal levels to allow comparison between multiple experiments.
Cells
Murine pancreatic β‐cell line MIN6 was obtained from AddexBio Technologies (San Diego, CA 92117 USA). MIN6 cells were maintained in Dulbecco's modified Eagle's medium (with 25 mM glucose and 2 mM l‐glutamine) supplemented with 10% foetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 100 μg/ml sodium pyruvate and 50 μM 2‐mercaptoethanol. Early passage cells (between passage 17 and 23) were used in the experiments. MIN6 β‐cells were transfected with murine ABCA12‐specific siRNA or scrambled siRNA. Fluorophore Dy547‐conjugated ABCA12‐specific siRNA was obtained from Millennium Science using oligonucleotides against the following ABCA12 sequences: GCAGGAUCGUUCAGUACUAUU. Fluorescein‐labelled scramble siRNA was from Santa Cruz Biotechnology. Transfection was performed using Lipofectamine RNAi MAX (Invitrogen) following manufacturer's protocol, and transfected cells were processed 48 h post‐transfection. ABCA1, ABCG1 and LXRβ sequences were inserted into pCMV6‐AC‐GFP plasmid (OriGene). Constitutively active form pcDNA3‐EGFP‐Cdc42 (Q61L) was a gift from Dr Klaus Hahn (Addgene plasmid # 12600). Transfection with plasmids was conducted using Lipofectamine LTX (Invitrogen) following the instructions by the manufacturer. When indicated, Fumonisin B1 (Sigma) was added to the cells at indicated concentration after transfection.
Islet dispersion and lipid raft detection
Isolated islets were dispersed into single cells via incubation with 0.1 mg/ml bovine trypsin solution + 2 mM EDTA for 5 min at 37°C. Dispersed islets were then fixed by addition of pre‐warmed 4% PFA in PBS for 15 min at room temperature. Fixation was terminated by adding 1 ml of ice‐cold PBS, and fixed cells were pelleted at 0.11 g for 5 min. Cells were then resuspended in blocking buffer (0.2% w/v BSA + 0.1% Triton × 100) for 10 min, and lipid rafts were labelled using the Vybrant® Alexa Fluor® 488 or Alexa Fluor 647 Lipid Raft Labelling Kit (Thermo Fisher Scientific), according to the manufacturer's instructions. Cells were then stained for insulin and analysed by flow cytometry using a CytoFLEX (Beckman Coulter). Gates were determined via fluorescence minus 1 gating, and data were analysed using FlowJo (V10) software. Insulin granule fusion and β‐cell mass were quantified as previously described 43. Average insulin granule area was calculated by measuring cross‐sectional area of 3,000 granules for each genotype using ImageJ software. Proportions of cleaved caspase‐3, glucagon‐positive and F480‐positive cells/islet were calculated from at least 10 evenly spaced pancreas sections across at least three biological replicates per genotype.
Measurement of insulin secretion and content
MIN6 cells were seeded into 12‐well plate and incubated for insulin secretion assays, and insulin was measured by ELISA as described 4, 44. In brief, following pre‐incubation for 2 h in Krebs‐Ringer bicarbonate Hepes buffer with 1 mM glucose (KRBB; 110.8 mM NaCl, 4.87 mM KCl, 25.7 mM NaHCO3, 1.22 mM KH2PO4, 1.21 mM MgSO4, 2.5 mM CaCl2, 10 mM Hepes and 0.35% BSA, pH 7.4), the cells were incubated for 45 min in KRBB, containing low (1 mM) or stimulatory (25 mM) glucose concentrations. When indicated, methyl‐β‐cyclodextrin (1 mM) or Jasplakinolide (1 μM, 2 h) was added to KRBB. The supernatant fractions were obtained after the 45‐min static incubation. Cellular insulin was extracted in an acid–ethanol solution (75% ethanol/1.5% HCl, v/v) and sonicated for 20 s on ice. Insulin in supernatants and cell extracts was measured using an ELISA kit (Millipore) following the protocol described by the manufacturer. Secretion was normalised to protein and expressed as a fold increase over basal–stimulated secretion.
HDL and apolipoprotein A‐I
High‐density lipoprotein (HDL) was isolated by sequential centrifugation in the 1.083 < d < 1.21 g/l density range in KBr solutions. Purified human apoA‐I was a kind gift of CSL.
Cholesterol efflux
Cholesterol efflux was measured as described previously 8. Briefly, MIN6 cells were incubated in growth medium containing [3H] cholesterol (75 kBq/ml) for 48 h. Cells were then washed with PBS and incubated for 18 h in serum‐free medium in the presence or absence of LXR agonist TO901317 (final concentration 4 μM). ApoA‐I or isolated HDL was then added to the final concentration of 30 μg/ml, and cells were incubated for 2 h at 37°C. The efflux was calculated as a proportion of radioactivity moved by acceptors from cells to medium (minus passive efflux to medium). ABCA1‐ or ABCG1‐dependent efflux was defined as a difference in the efflux to apoA‐I or HDL from cells activated or non‐activated with TO901317.
Western blot analysis
Antibodies used were generated against ABCA1 (Abcam, ab7360), ABCG1 (Abcam, ab52617), SR‐B1 (Abcam, ab106572), LXRβ (Abcam ab28479), GLUT2 (Abcam, ab54460), GAPDH (Abcam, ab8245) and CDC42 (BD Transduction Laboratories, #610928). For ABCA12 analysis, cellular proteins were biotinylated with 2 mM EZ‐Link NHS‐LC‐Biotin (Thermo Fisher Scientific) at room temperature for 1 h. Cells were then lysed with RIPA lysis buffer, and 150 μg each of the lysates was incubated with 1.5 μg goat anti‐ABCA12 antibody (Santa Cruz Biotechnology). Complexes of ABCA12 protein and antibody were precipitated with Sepharose‐protein G beads. An amount of protein equivalent to 150 μg of original protein lysate was run on the NuPAGE Bis‐Tris gels followed by Western blot and detected with HRP‐conjugated streptavidin.
Real‐time PCR
PCR primers were obtained from TaqMan Gene Expression Assays with following gene assay IDs (Mm00442646_m1, Mm00550501_m1, Mm00613683_m1, Mm00437390_m1, Mm00443451_m1, Mm00437262_m1, Mm00450234_m1, Mm00446214_m1, Mm00440169_m1, Mm01282499_m1, Mm00441480_m1, Mm_00446229_m1, Mm01245502_m1, Mm00809442_s1, Mm00504086_m1 and Mm00456425_m1). Real‐time PCRs were performed on 7500 Fast System from ABI Applied Biosystems. The relative amount of mRNA was calculated using the comparative C T method. Gene expression was normalised to 18S rRNA.
microRNA assay
The expression of 10 microRNAs was investigated by RT–PCR using TaqMan microRNA assays (primers ID 001990, 002676, 002216, 000405, 000408, 002218, 002135, 002085, 000442, 002278; Life Technologies) and normalised to the expression level of the U6 small nuclear RNA (ID 001973).
Electron microscopy
Primary mouse islets were isolated, fixed in phosphate‐buffered 4% PFA, 2.5% glutaraldehyde and post‐fixed with 1% osmium tetroxide, dehydrated in acetone and embedded in EPON. Digital images were acquired using the Hitachi Field Emission Transmission Electron Microscope HF‐3300 at Monash Micro Imaging. For islet granule analysis, insulin granules were identified by the characteristic appearance of a zinc‐insulin condensed core with surrounding halo.
Confocal microscopy
MIN6 cells grown in μ‐slide 8‐well chamber slide (Ibidi) for 2 days were fixed with 4% paraformaldehyde in PBS for 10 min, permeabilised with 0.2% Triton X‐100 in PBS and blocked with 2% BSA in PBS for 1 h at room temperature. The abundance of lipid rafts was assessed using the Vybrant lipid raft labelling kit (Life Technologies) according to manufacturer's instructions. Briefly, cells were incubated with Alexa Fluor 488‐ or Alexa Fluor 647‐labelled CT‐B for 10 min on ice, washed, cross‐linked by incubation with anti‐CT‐B antibody for 15 min at 4°C and fixed with 4% paraformaldehyde for 15 min at room temperature. F‐actin was visualised either with Phalloidin‐iFluor 488 or with LifeAct‐TagGFP2 protein staining according to manufacturer's instructions (Abcam ab176753 or Ibidi, 60112, respectively). Briefly, cells were fixed with paraformaldehyde, washed with PBS, permeabilised with 0.1% Triton X‐100 for 3–5 min and then washed again with PBS. Cells were incubated with phalloidin‐conjugate or LifeAct‐TagGFP2 working solution for 50 min and washed to remove excess stain. When needed, cells were counterstained with Hoechst 33342 (Invitrogen, Melbourne, Australia). Primary islet beta cells were incubated with 2 μg/ml LifeAct‐TagGFP2 for 30 min after paraformaldehyde fixation and Triton X‐100 permeabilisation.
For co‐localisation of ABCA12 and CDC42, fixed and permeabilised MIN6 cells were incubated with primary mouse anti‐Cdc42 (BD Transduction, #610928) and rabbit anti‐ABCA12 antibodies (Abcam) overnight at 4°C. Cells were washed with PBS, CDC42 signal was enhanced by incubation with biotin‐conjugated secondary anti‐mouse IgG, then detected with Alexa Fluor 633‐conjugated streptavidin (Invitrogen), and ABCA12 signal was detected with Alexa Fluor‐conjugated anti‐rabbit IgG (Invitrogen). After being washed with PBS, cell nuclei were stained with DAPI (Vector Laboratories, Inc., Burlingame, CA). Slides were imaged at 60× water lens with a Nikon A1r microscope (Nikon Instruments Inc). Images were analysed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Freshly dissected pancreas tissue was fixed in 10% formalin for 20 h, dehydrated, cleared and embedded in paraffin. The paraffin‐embedded tissue was rehydrated by washing two times xylene at 10 min each, two times absolute ethanol at 5 min each and 95% ethanol at 5 min, then rinsed with tap water, immersed in Tris‐buffered saline. After slides were hydrated, antigens were unmasked in 10 mM sodium citrate, pH 6, with 0.05% Tween‐20, blocked with protein blocking agent (Thermo Fisher Scientific). Insulin was stained with monoclonal primary antibody (Abcam) and Alexa Fluor 568 anti‐mouse secondary antibody (Invitrogen). ABCA12 was stained with polyclonal primary antibody (Abcam) and Alexa Fluor 488 anti‐rabbit secondary antibody (Invitrogen). The slide was mounted with VECTASHIELD anti‐fade mounting medium with DAPI (Vector Laboratories), sealed with nail polish. The fluorescence was visualised under 20× lens using BX61 microscope (Olympus).
Two‐photon imaging of granule fusion
To investigate insulin secretory defects in Abca12 tm1d islets, we used 2‐photon live cell imaging, as previously described 45. Briefly, isolated islets were cultured for 2–3 days and prior to imaging were bathed in an Na‐rich solution of 3 mmol/l glucose for 30 min (37°C, 95/5% air/CO2). Islets were then transferred to 3 mmol/l glucose solution containing 800 μmol/l sulforhodamine B (SRB). Extracellular glucose and KCl concentrations were increased (15 mM and 20 mM, respectively), and imaging was performed at 34°C, 3 cell layers deep into the islet, with exocytic fusion events recorded as the entry of SRB into each fused granule. Islets were excited at 950 nm, and total number of fusion events occurring within the 2‐photon volume (~ 40 × 50 × 1 μm) was recorded over 20 min. Average number and density of fusion events were recorded from at least five different sections, across 8–10 islets, in at least four mice per genotype.
Measurement of calcium flux
Isolated islet cells on glass coverslips were studied at 24 and 48 h following isolation. Coverslips were transferred to physiological saline solution (PSS) medium containing NaCl 111 mM, KCl 5 mM, NaHCO3 25 mM, KH2PO4 1.2 mM, MgSO4 1.2 mM, CaCl2 2 mM, Hepes 10 mM, BSA 3%, glucose 1 mM and adjusted to pH 7.4 for 15 min at 37°C. They were then transferred to PSS containing Fluo‐4 AM 2 μM and pluronic 0.01% (both from molecular probes) at 23°C for 15 min, in the dark. The cells on coverslips were transferred to a Warner tissue bath mounted on an Olympus IX71 inverted confocal microscope and continuously superfused with PSS at 33°C for 15 min to allow cleavage of Fluo‐4‐AM by cytoplasmic esterases. The cells were illuminated by a Kr/Ar laser at 488 nm, and the light passed through a Yokogawa CSU22 Nipkow spinning disc (Yokogawa Australia Pty. Ltd., Macquarie Park New South Wales, Australia) to a high sensitivity electron‐multiplying Andor iXon CCD (Andor Inc., Belfast, Ireland). Fluorescence images were acquired every 900 ms.
Regions of interest (ROI) were identified, and, of the ~50 cells in view, 10 were chosen on the basis of displaying no fluctuations in cytoplasmic calcium in the 3 min before application of 20 mM glucose to the superfusing PSS. A 5‐min application of high glucose was followed by a return to normal PSS, followed by a 15‐s application of PSS containing 30 mM KCl (NaCl replacement) at 10 min. Pixel intensities were analysed using Andor iQ 1.9 controller software (Andor). Fluorescence responses were corrected for basal fill (F/F 0).
Flow cytometry
MIN6 cells grown in 12‐well plates were transfected with Fluorescein‐labelled scrambled siRNA or Dy547‐labelled ABCA12‐specific siRNA. Lipid rafts were stained with cholera toxin subunit B with Alexa Fluor 647 or Alexa Fluor 488 conjugate at 0.5 μg/ml. After fixation and permeabilisation, actin filaments were stained with a CytoPainter Phalloidin‐iFluor 647 reagent (Abcam) at 1:1,000 dilution and washed once with PBS. CT‐B binding was measured as mean fluorescence intensity (MFI). Treated cells were immediately analysed using the BD FACS Canto II flow cytometer (BD Bioscience), and data were analysed with DiVA software.
CDC42 activity assay
Activity of CDC42 was determined as described by Yao et al 46. Twenty‐four hours after transfection of cells with either siRNAABCA12 or siRNAScr, cells were washed twice with serum‐free DMEM and left in serum‐free media for 24 h for starvation to decrease the basal level of active CDC42. After starvation, cells were washed twice with warm serum‐free DMEM and simulated with bradykinin (100 ng/ml, 4 min). The level of active CDC42 was estimated using CDC42 G‐LISA Activation Assay Kit (Cytoskeleton Inc; cat # BK127) according to manufacturer's instructions. Briefly, activated cells were lysed with CDC42 lysis buffer, protein content estimated and samples were snap‐frozen. Lysates were added to CDC42‐GTP binding plate, incubated at 4°C for 15 min, washed and incubated with antigen‐presenting buffer for 2 min before adding anti‐CDC42 primary antibodies. After 30‐min incubation, plates were washed and secondary antibody‐HRP conjugate was added and incubated for 30 min. After washing the plate, HRP detection reagent was added, and after 15‐min incubation at 37°C, signal was measured at 490 nm using a microplate spectrophotometer.
Lipidomics
MIN6 cells were transfected with ABCA12‐specific siRNA or scrambled siRNA. Cells were washed with PBS, resuspended into 20 mM Tris/500 mM NaCl solution containing 0.1 mM butylated hydroxytoluene and sonicated. Ten microlitres (20 μg of protein) each of the supernatant were subjected to lipid extraction and analysis using mass spectrometry as described previously 8. The analysis was performed by liquid chromatography electrospray ionisation‐tandem mass spectrometry (LC ESI‐MS/MS) using an Agilent 1200 liquid chromatography system (Agilent Technologies) and Applied Biosystems API 4000 Q/TRAP mass spectrometer with a turbo‐ion spray source (350°C) and Analyst 1.5 and MultiQuant data systems (AB SCIEX). Lipid concentrations were calculated by relating the peak area of each species to the peak area of the corresponding internal standard (one per group). Data are presented as individual species or the sum total of each lipid family. Output was normalised to the abundant cellular membrane lipid, phosphatidylcholine. Isolated islets were sonicated in Tris‐NaCl, lipid extracted and analysed as described above. Collective data for MS‐based experiments are available in Dataset EV1, Tables 1–9.
RNA sequencing
Whole islets were isolated and total RNA purified using the RNAqueous Kit (Ambion). Samples were further DNAse treated with the TURBO DNA‐free kit (Ambion) as per the manufacturer's instructions. RNA integrity and concentration were determined by Bioanalyszer and Qubit. RNA sequencing was performed on 5‐week islet RNA samples using the services of the Australian Genome Research Facility (AGRF) using polyA library preparation and Illumina HiSeq 100 bp single‐end reads. PolyA‐based cDNA libraries were prepared from 8‐week islet RNA samples using NEB Next Ultra Directional RNA Library Prep Kit for Illumina RNA sequencing (E7420S) and sequenced using the services of GeneWiz on Illumina HiSeqX10 machines in 150 bp paired‐end format. RNA sequencing data were processed with Skewer adaptor trimmer and mapped with HiSat2 to Mus musculus GRCm38_v90 genome assembly. The resulting ordered Bam files were analysed in Seqmonk v1.43. Samples are grouped as KO (Abca12 tm1d mice) or control Cre (Rip‐Cre and Abca12tm1c/+ Rip1‐Cre mice). A minimum of 13 million reads was obtained per sample, and library duplication and QC metrics were assessed. Differentially expressed genes between groups were determined using the count‐based DeSeq2 method with a multiple testing‐corrected P‐value cut‐off of P < 0.05 and independent filtering applied. This list was further filtered following count/total sequences log2 transformation selecting for fold change ratios greater or equal to 1.5. Filtered DE gene lists were further analysed for significantly (adjusted P values < 0.05) enriched pathway changes using online tools, http://String-db.org, http://genome.jp/kegg/pathway.html and http://amp.pharm.mssm.edu/Enrichr/.
Statistics
All data are shown as mean ± standard error of means (SEM) unless stated otherwise. Statistical significance of the differences was assessed in SigmaStat or GraphPad Prism software packages by unpaired Student's t‐test or one‐way ANOVA when data followed normal distribution or Mann–Whitney U‐test on ranks. Significance is indicated a *P < 0.05, **P < 0.01, ***P < 0.0001, ****P < 0.00001. Cell‐based experiments were conducted in quadruplicates and repeated 2–5 times, mouse experiments utilised animal numbers as indicated. When normalisation of data was difficult and the experiments were performed in replicates, representative experiments out of 2–3 similar experiments are shown.
Study approval
All animal procedures complied with standards set under Australian guidelines for animal welfare and experiments were subject to Monash University animal welfare ethics review (Approval #MARP/2016/164).
Author contributions
Conceptualisation, GMU, PT, HET, IMS, DS and HCP; Methodology, GMU, YF, NM, PT, HET, PJM, IMS, DS and HCP; Formal Analysis, GMU, YF, DLC, PJM, IMS, DS and HCP; Investigation, GMU, YF, DLC, NM, LKJ, HL, MST, WJG, NAM, JMW, MD, SF, PJM, PPD, IMS and HCP; Resources, PT, HET, PJM, IMS and DS; Data Curation, GMU, YF and NM; Writing—Original Draft, GMU, DLC and IMS, DS; Writing—Review and Editing, GMU, IMS and DS; Visualisation, GMU, YF, IMS and DS; Supervision, IMS and DS; Project Administration, IMS and DS; Funding Acquisition, IMS and DS. The Smyth and Sviridov laboratories contributed equally to this study—the order of the joint first authorship is a reflection of the reversed joint last authorship.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Review Process File
Acknowledgements
The study was supported by the grants from the National Health and Medical Research Council of Australia (GNT1020281, GNT1036352, GNT1123554) and supported in part by the Victorian Government's OIS Program. The authors acknowledge the assistance of the Monash Microimaging Platform, Monash Animal Research Platform, Histology Platform and Monash FlowCore. IMS is supported by a Senior Research Fellowship of the National Health and Medical Research Council (APP1106516), and GMU acknowledges the support of an Australian Postgraduate Award.
EMBO Reports (2020) 21: e48692
Data availability
RNAseq datasets are available as a GEO resource with the identifier GSE140379 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140379). MS quantitation is provided as part of Dataset EV1 (Tables 1–9).
References
- 1. Brunham LR, Kruit JK, Verchere CB, Hayden MR (2008) Cholesterol in islet dysfunction and type 2 diabetes. J Clin Invest 118: 403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. von Eckardstein A, Sibler RA (2011) Possible contributions of lipoproteins and cholesterol to the pathogenesis of diabetes mellitus type 2. Curr Opin Lipidol 22: 26–32 [DOI] [PubMed] [Google Scholar]
- 3. Kruit JK, Wijesekara N, Westwell‐Roper C, Vanmierlo T, de Haan W, Bhattacharjee A, Tang R, Wellington CL, Lutjohann D, Johnson JD et al (2012) Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired β‐cell function. Diabetes 61: 659–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, Marsh BJ, Rodrigues B, Johnson JD, Parks JS et al (2007) beta‐cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med 13: 340–347 [DOI] [PubMed] [Google Scholar]
- 5. Kruit JK, Brunham LR, Verchere CB, Hayden MR (2010) HDL and LDL cholesterol significantly influence beta‐cell function in type 2 diabetes mellitus. Curr Opin Lipidol 21: 178–185 [DOI] [PubMed] [Google Scholar]
- 6. Drew BG, Rye KA, Duffy SJ, Barter P, Kingwell BA (2012) The emerging role of HDL in glucose metabolism. Nat Rev Endocrinol 8: 237–245 [DOI] [PubMed] [Google Scholar]
- 7. Rickels MR, Goeser ES, Fuller C, Lord C, Bowler AM, Doliba NM, Hegele RA, Cuchel M (2015) Loss‐of‐function mutations in ABCA1 and enhanced β‐Cell secretory capacity in young adults. Diabetes 64: 193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fu Y, Mukhamedova N, Ip S, D'Souza W, Henley Katya J, DiTommaso T, Kesani R, Ditiatkovski M, Jones L, Lane Rachael M et al (2013) ABCA12 regulates ABCA1‐dependent cholesterol efflux from macrophages and the development of atherosclerosis. Cell Metab 18: 225–238 [DOI] [PubMed] [Google Scholar]
- 9. Akiyama M, Sugiyama‐Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, Tsuji‐Abe Y, Tabata N, Matsuoka K, Sasaki R et al (2005) Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest 115: 1777–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smyth I, Hacking DF, Hilton AA, Mukhamedova N, Meikle PJ, Ellis S, Slattery K, Collinge JE, de Graaf CA, Bahlo M et al (2008) A mouse model of harlequin ichthyosis delineates a key role for abca12 in lipid homeostasis. PLoS Genet 4: e1000192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cottle DL, Ursino GMA, Ip SCI, Jones LK, Ditommaso T, Hacking DF, Mangan NE, Mellett NA, Henley KJ, Sviridov D et al (2015) Fetal inhibition of inflammation improves disease phenotypes in harlequin ichthyosis. Hum Mol Genet 24: 436–449 [DOI] [PubMed] [Google Scholar]
- 12. Marselli L, Thorne J, Dahiya S, Sgroi DC, Sharma A, Bonner‐Weir S, Marchetti P, Weir GC (2010) Gene expression profiles of Beta‐cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS One 5: e11499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calderon B, Carrero JA, Miller MJ, Unanue ER (2011) Entry of diabetogenic T cells into islets induces changes that lead to amplification of the cellular response. Proc Natl Acad Sci USA 108: 1567–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stewart TP, Kim HY, Saxton AM, Kim JH (2010) Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J x TALLYHO/JngJ) F2 mice. BMC Genom 11: 713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishibashi Y, Kohyama‐Koganeya A, Hirabayashi Y (2013) New insights on glucosylated lipids: metabolism and functions. Biochim Biophys Acta 1831: 1475–1485 [DOI] [PubMed] [Google Scholar]
- 16. Ip SCI, Cottle DL, Jones LK, Weir JM, Kelsell DP, O'Toole EA, Meikle PJ, Smyth IM (2017) A profile of lipid dysregulation in harlequin ichthyosis. Br J Dermatol 177: e217–e219 [DOI] [PubMed] [Google Scholar]
- 17. Calkin AC, Tontonoz P (2012) Transcriptional integration of metabolism by the nuclear sterol‐activated receptors LXR and FXR. Nat Rev Mol Cell Biol 13: 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gerin I, Dolinsky VW, Shackman JG, Kennedy RT, Chiang S‐H, Burant CF, Steffensen KR, Gustafsson J‐Å, MacDougald OA (2005) LXRβ is required for adipocyte growth, glucose homeostasis, and β cell function. J Biol Chem 280: 23024–23031 [DOI] [PubMed] [Google Scholar]
- 19. Low JT, Mitchell JM, Do OH, Bax J, Rawlings A, Zavortink M, Morgan G, Parton RG, Gaisano HY, Thorn P (2013) Glucose principally regulates insulin secretion in mouse islets by controlling the numbers of granule fusion events per cell. Diabetologia 56: 2629–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sturek JM, Castle JD, Trace AP, Page LC, Castle AM, Evans‐Molina C, Parks JS, Mirmira RG, Hedrick CC (2010) An intracellular role for ABCG1‐mediated cholesterol transport in the regulated secretory pathway of mouse pancreatic beta cells. J Clin Invest 120: 2575–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Haan W, Bhattacharjee A, Ruddle P, Kang MH, Hayden MR (2014) ABCA1 in adipocytes regulates adipose tissue lipid content, glucose tolerance, and insulin sensitivity. J Lipid Res 55: 516–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dirkx R Jr, Solimena M (2012) Cholesterol‐enriched membrane rafts and insulin secretion. J Diabetes Investig 3: 339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zidovetzki R, Levitan I (2007) Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta 1768: 1311–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chadda R, Howes MT, Plowman SJ, Hancock JF, Parton RG, Mayor S (2007) Cholesterol‐sensitive Cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway. Traffic 8: 702–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chichili GR, Rodgers W (2009) Cytoskeleton‐membrane interactions in membrane raft structure. Cell Mol Life Sci 66: 2319–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nalbant P, Hodgson L, Kraynov V, Toutchkine A, Hahn KM (2004) Activation of endogenous Cdc42 visualized in living cells. Science 305: 1615–1619 [DOI] [PubMed] [Google Scholar]
- 27. Posey SC, Bierer BE (1999) Actin stabilization by jasplakinolide enhances apoptosis induced by cytokine deprivation. J Biol Chem 274: 4259–4265 [DOI] [PubMed] [Google Scholar]
- 28. Zhou YP, Grill V (1995) Long term exposure to fatty acids and ketones inhibits B‐cell functions in human pancreatic islets of Langerhans. J Clin Endocrinol Metab 80: 1584–1590 [DOI] [PubMed] [Google Scholar]
- 29. Shimabukuro M, Zhou YT, Levi M, Unger RH (1998) Fatty acid‐induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA 95: 2498–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Neill CM, Lu C, Corbin KL, Sharma PR, Dula SB, Carter JD, Ramadan JW, Xin W, Lee JK, Nunemaker CS (2013) Circulating levels of IL‐1B+IL‐6 cause ER stress and dysfunction in islets from prediabetic male mice. Endocrinology 154: 3077–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dinarello CA, Donath MY, Mandrup‐Poulsen T (2010) Role of IL‐1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 17: 314–321 [DOI] [PubMed] [Google Scholar]
- 32. Eto K, Yamashita T, Matsui J, Terauchi Y, Noda M, Kadowaki T (2002) Genetic manipulations of fatty acid metabolism in beta‐cells are associated with dysregulated insulin secretion. Diabetes 51(Suppl 3): S414–S420 [DOI] [PubMed] [Google Scholar]
- 33. Imai Y, Dobrian AD, Morris MA, Taylor‐Fishwick DA, Nadler JL (2016) Lipids and immunoinflammatory pathways of beta cell destruction. Diabetologia 59: 673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701 [DOI] [PubMed] [Google Scholar]
- 35. Sezgin E, Levental I, Mayor S, Eggeling C (2017) The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol 18: 361–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nofer J‐R, Remaley AT, Feuerborn R, Wolinnska I, Engel T, von Eckardstein A, Assmann G (2006) Apolipoprotein A‐I activates Cdc42 signaling through the ABCA1 transporter. J Lipid Res 47: 794–803 [DOI] [PubMed] [Google Scholar]
- 37. Tsukamoto K, Hirano K, Tsujii K, Ikegami C, Zhongyan Z, Nishida Y, Ohama T, Matsuura F, Yamashita S, Matsuzawa Y (2001) ATP‐binding cassette transporter‐1 induces rearrangement of actin cytoskeletons possibly through Cdc42/N‐WASP. Biochem Biophys Res Commun 287: 757–765 [DOI] [PubMed] [Google Scholar]
- 38. Shimabukuro M, Koyama K, Lee Y, Unger RH (1997) Leptin‐ or troglitazone‐induced lipopenia protects islets from interleukin 1beta cytotoxicity. J Clin Invest 100: 1750–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eitel K, Staiger H, Brendel MD, Brandhorst D, Bretzel RG, Haring HU, Kellerer M (2002) Different role of saturated and unsaturated fatty acids in beta‐cell apoptosis. Biochem Biophys Res Commun 299: 853–856 [DOI] [PubMed] [Google Scholar]
- 40. Acosta‐Montano P, Garcia‐Gonzalez V (2018) Effects of dietary fatty acids in pancreatic beta cell metabolism, implications in homeostasis. Nutrients 10: E393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moffitt JH, Fielding BA, Evershed R, Berstan R, Currie JM, Clark A (2005) Adverse physicochemical properties of tripalmitin in beta cells lead to morphological changes and lipotoxicity in vitro . Diabetologia 48: 1819–1829 [DOI] [PubMed] [Google Scholar]
- 42. Misasi R, Dionisi S, Farilla L, Carabba B, Lenti L, Di Mario U, Dotta F (1997) Gangliosides and autoimmune diabetes. Diabetes Metab Rev 13: 163–179 [DOI] [PubMed] [Google Scholar]
- 43. Do OH, Gunton JE, Gaisano HY, Thorn P (2016) Changes in beta cell function occur in prediabetes and early disease in the Lepr db mouse model of diabetes. Diabetologia 59: 1222–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sharma G, Prossnitz ER (2011) Mechanisms of estradiol‐induced insulin secretion by the G protein‐coupled estrogen receptor GPR30/GPER in pancreatic beta‐cells. Endocrinology 152: 3030–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Do OH, Low JT, Thorn P (2015) Lepr(db) mouse model of type 2 diabetes: pancreatic islet isolation and live‐cell 2‐photon imaging of intact islets. J Vis Exp 99: e52632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yao H, Duan M, Yang L, Buch S (2013) Nonmuscle myosin light‐chain kinase mediates microglial migration induced by HIV Tat: involvement of beta1 integrins. FASEB J 27: 1532–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Review Process File
Data Availability Statement
RNAseq datasets are available as a GEO resource with the identifier GSE140379 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140379). MS quantitation is provided as part of Dataset EV1 (Tables 1–9).