

Abstract
Overexpressing Tau counteracts apoptosis and increases dephosphorylated β‐catenin levels, but the underlying mechanisms are elusive. Here, we show that Tau can directly and robustly acetylate β‐catenin at K49 in a concentration‐, time‐, and pH‐dependent manner. β‐catenin K49 acetylation inhibits its phosphorylation and its ubiquitination‐associated proteolysis, thus increasing β‐catenin protein levels. K49 acetylation further promotes nuclear translocation and the transcriptional activity of β‐catenin, and increases the expression of survival‐promoting genes (bcl2 and survivin), counteracting apoptosis. Mutation of Tau's acetyltransferase domain or co‐expressing non‐acetylatable β‐catenin‐K49R prevents increased β‐catenin signaling and abolishes the anti‐apoptotic function of Tau. Our data reveal that Tau preserves β‐catenin by acetylating K49, and upregulated β‐catenin/survival signaling in turn mediates the anti‐apoptotic effect of Tau.
Keywords: acetylation, acetyltransferase, anti‐apoptosis, Tau, β‐catenin
Subject Categories: Autophagy & Cell Death; Neuroscience; Post-translational Modifications, Proteolysis & Proteomics
Tau acetylates β‐catenin at K49, which stabilizes β‐catenin by inhibiting its phosphorylation and ubiquitination‐associated proteolysis. β‐catenin mediates the anti‐apoptotic effects of Tau by increasing the expression of survival‐promoting genes.
Introduction
Intracellular Tau accumulation forming neurofibrillary tangles is positively correlated with neurodegeneration and memory deterioration in Alzheimer's disease (AD) and other tauopathies 1, 2, 3, 4. The neurodegeneration observed in the AD brain takes over 20 years after the formation of tangles 5, although these neurons have been extensively exposed to an aging‐dependent increase in pro‐apoptotic environment. Then, why do these neurons (i.e., with accumulation of Tau) not go to acute apoptosis (occur in hours) but instead take a chronic degeneration?
As Tau is the major protein component of the tangles in the degenerated neurons, the role of Tau has been extensively studied. Previous reports show that overexpressing human wild‐type full‐length Tau (Tau40) renders the cells more resistant to apoptosis induced by various pro‐apoptotic factors both in vitro and in vivo 6, 7, 8, 9, 10, 11, 12. Meanwhile, the intracellular Tau accumulation also induces calcium overload, endoplasmic reticulum stress, mitochondrial dysfunction, and synaptic impairments 13, 14, 15, 16, 17, 18. Based on these observations, it has been proposed that Tau hyperphosphorylation and accumulation may play a dual role in leading the neurons to escape the acute apoptosis and simultaneously triggering a chronic degeneration 19, 20; importantly, the Tau‐induced apoptosis escape may serve as a precondition of AD neurodegeneration 21. Therefore, understanding the molecular mechanisms underlying apoptosis escape is important for designing strategies to prevent the apoptosis‐escaped neurons from degeneration. Accompanying the Tau‐induced apoptosis escape, the total β‐catenin level was remarkably increased with a reduced phosphorylation level of β‐catenin 6. It is currently not understood how Tau accumulation affects β‐catenin phosphorylation and preserves β‐catenin, and whether and how the increased β‐catenin mediates the anti‐apoptotic effects of Tau.
β‐catenin was originally discovered as a cell adhesion factor, mainly located in the cytoplasm 22. Subsequent studies reveal that β‐catenin participates in several cellular signaling pathways, such as Wnt signaling to regulate cell differentiation and proliferation 23, 24, 25. Gene mutation or upregulation of β‐catenin has been identified in cancers 26. Studies also show that elevation and the nuclear translocation of β‐catenin can prime cellular survival pathways 27, 28, 29. The N‐terminal (1–49 amino acids) of β‐catenin is the key region for its stability and the degradation by ubiquitin–proteasome system 30, and phosphorylation of β‐catenin at the N‐terminal Ser33, Ser37, Thr41, and Ser45 promotes its ubiquitination and degradation 30, 31, 32. Adjacent to the phosphorylation sites in the N‐terminal of β‐catenin, there are two lysine residuals (K49 and K19) which can be ubiquitinated and/or acetylated 30, 33, 34, 35, 36. Furthermore, overexpressing Tau decreases phosphorylation level with upregulation of β‐catenin 6, and Tau protein per se has the activity of acetyltransferase 37. It is well accepted that ubiquitination is the prerequisite for the proteasome‐associated degradation of proteins, and the ubiquitination and acetylation occur at the same amino acid residual in the proteins. Based on these observations, we speculate that Tau may directly acetylate β‐catenin, by which it inhibits the ubiquitination and/or phosphorylation of β‐catenin. Consequently, β‐catenin is preserved to prime the survival signaling and to mediate the anti‐apoptotic effect of Tau.
To test this idea, we demonstrate in the present study that Tau can directly and robustly acetylate β‐catenin both in vitro and in vivo. Acetylation of β‐catenin at K49 by Tau inhibits its phosphorylation, ubiquitination, and proteolysis and thus promotes its nuclease translocation leading to upregulation of β‐catenin/Wnt/survival signaling. The upregulation of β‐catenin/Wnt/survival signaling in turn mediates the anti‐apoptotic function of Tau.
Results
Tau can acetylate β‐catenin at K49 in vitro and in vivo
We reported previously that overexpressing Tau could upregulate β‐catenin 6, but the mechanism has been elusive. To clarify this, we first measured the effect of Tau on the transcription of β‐catenin. Overexpressing Tau did not significantly change the mRNA level of β‐catenin (Fig EV1A and B), although a significantly increased protein level of β‐catenin was shown in neuroblastoma (N2a) cells, the cultured primary hippocampal neurons, and human Tau transgenic mice (Fig EV1C–I). These data suggest that Tau‐induced β‐catenin elevation involves post‐translational modifications but not the transcription of β‐catenin.
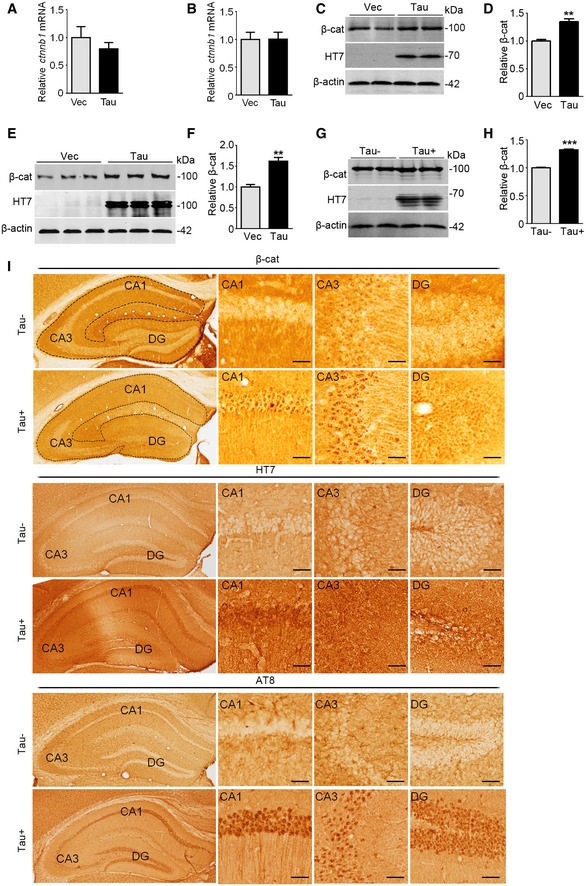
Figure EV1. Overexpressing Tau increases β‐catenin protein level without affecting the mRNA level of β‐catenin.
-
A, BOverexpressing Tau did not affect mRNA level of β‐catenin. HEK293 (A) and N2a (B) cells were transfected respectively with Tau40 (Tau) or the empty vector (Vec) for 48 h. β‐catenin mRNA level (ctnnb1) was detected by RT–PCR (n = 6–9 biological replicates each group).
-
C–HOverexpressing Tau increased β‐cat protein level in N2a cells (transfected with Tau40 or Vec for 48 h; C, D), cultured hippocampal neurons (cultured with 7 div and then transfected with AAV‐eGFP‐vector or AAV‐eGFP‐Tau for 5 div; E, F), and in the hippocampi of 10‐month‐old Tau transgenic mice (Tau+) (G, H) measured by Western blotting (n = at least three biological replicates each group).
-
IThe protein level of β‐cat protein (upper panels) was significantly increased in hippocampal cornu ammonis 1(CA1), cornu ammonis 3 (CA3), and dentate gyrus (DG) subsets of 10‐month‐old Tau transgenic mice (Tau+) compared with Tau knockout mice (Tau−), with significantly increased total Tau (HT7, middle panels) and the phosphorylated Tau (AT8, lower panels). Scale bar, 50 μm.
The N‐terminal (1–49 amino acids) of β‐catenin is the key region for its stability and the degradation by ubiquitin–proteasome system 30, and there are two lysine (K49 and K19) and four serine/therein (Ser33, Ser37, Thr41, and Ser45) residuals in this region of β‐catenin. A recent study shows that Tau has acetyltransferase activity and can promote its self‐acetylation 37. We speculate that Tau may acetylate β‐catenin and thus affects its phosphorylation and the ubiquitination‐related proteolysis. To test this, we first measured the acetylation of β‐catenin in Tau‐overexpressing systems. By immunoprecipitation, we observed that overexpressing Tau significantly increased acetylation level of β‐catenin compared with the empty vector control (Fig 1A and B). To identify the site acetylated by Tau, we used site‐specific mutagenesis to replace K49 and K19 with acetylation‐resistant arginine (K19R and K49R) in β‐catenin. We found that co‐expressing K49R or K49R/K19R double mutant almost abolished Tau‐induced acetylation whereas mutation at K19R only negligibly changed the acetylation level (Fig 1C and D), suggesting that Tau may predominantly acetylate β‐catenin at K49. Indeed, the following studies using K49‐specific antibody showed a significantly increased acetylation of β‐catenin at K49 in human Tau‐expressing N2a cells (Fig EV2A and B), in cultured hippocampal neurons (7 div) infected with AAV‐eGFP‐Tau for 5 div (Fig EV2C and D), in AAV‐eGFP‐Tau‐infected mouse hippocampus (Fig EV2E), and in human Tau transgenic mice (Fig 1E and F), while expressing the acetyltransferase activity domain‐deleted Tau‐K18(−) 37 abolished β‐catenin acetylation (Fig 1G and H). Furthermore, co‐expression of Tau and β‐catenin further increased the acetylation level of β‐catenin at K49 compared with expressing β‐catenin alone (Fig EV2F and G). The remarkably increased levels of total and/or K49‐acetylated β‐catenin were also detected in the hippocampal extracts of AD patients (Fig 1I and J), in which a positive correlation was detected between the increased phosphorylated Tau at pS199 or AT8 epitopes and the increased K49‐acetylated β‐catenin (Fig 1K and L). Furthermore, expressing pseudo‐phosphorylated Tau (TauS199E) further increased the K49 acetylation of β‐catenin (Fig 1M and N). These in vitro and in vivo data together strongly indicate that Tau may preserve β‐catenin by enhancing its acetylation at K49 and phosphorylation of Tau enhances its acetyltransferase activity toward K49‐β‐catenin.
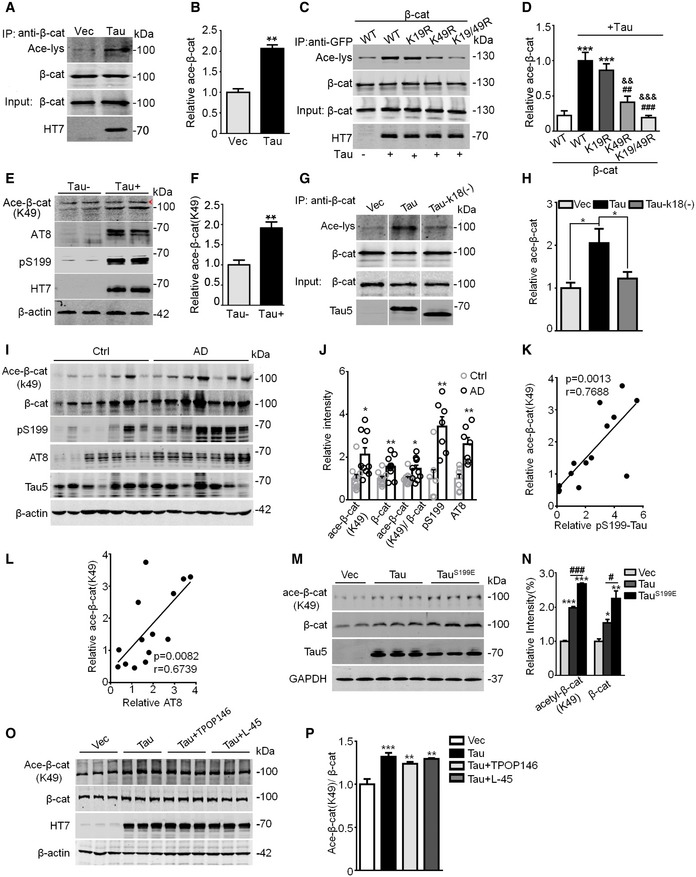
Figure 1. Tau acetylates β‐catenin at K49 in cells, mice, and human brains, and phosphorylation further increases its acetyltransferase activity.
-
A, BTau increased β‐catenin acetylation (Ace‐β‐cat) measured by immunoprecipitation using anti‐β‐cat, or Western blotting using anti‐β‐cat, anti‐acetylated lysine (Ace‐lys), and HT7 (probes specifically human total Tau). HEK293 cells transiently transfected with wild‐type Tau40 (Tau) or the empty vector (Vec) (n = 3 from three independent experiments).
-
C, Dβ‐cat mutation at K49 (K49R) but not K19 (K19R) abolished Tau‐induced acetylation. HEK293 cells were co‐transfected with eGFP‐β‐cat (WT, or K19R, or K49R, or K19R/K49R) and Tau or the empty vector for 48 h, and then immunoprecipitated using anti‐GFP and Western blotting using anti‐β‐cat, anti‐Ace‐lys, and HT7 (n = 3 from three independent experiments).
-
E, FThe increased β‐cat (K49) acetylation was detected in the hippocampal extracts of human Tau transgenic mice (Tau+) compared with the endogenous Tau knockout mice (Tau−), measured by Western blotting using acetylation site‐specific antibody (Ace‐β‐cat‐K49); arrowhead in panel (E) denoted non‐specific band identified by molecular weight. The remarkably increased levels of phosphorylated Tau at Ser199 (pS199) and AT8 epitopes were detected in Tau+ mice (at least three mice per group were used).
-
G, HK18 deletion of Tau (Tau‐k18(−)) attenuated Tau‐induced β‐cat acetylation. HEK293 cells were transfected with empty Vec, or Tau40 or Tau‐k18(−) for 48 h, and the ace‐β‐cat was measured by immunoprecipitation using anti‐β‐cat and Western blotting using anti‐ace‐lys, anti‐β‐cat, and Tau5 (detecting total Tau; n = 3 from three independent experiments).
-
I–LThe increased total and Ace‐β‐cat (K49) levels positively correlated with increased phosphorylated Tau pS199 and AT8 as detected by Western blotting in the hippocampal extracts of AD patients compared with age‐matched controls (n = 10 each group, detailed information for human brain samples was shown in Appendix Table S1). Pearson's analysis was used for correlation analysis (K and L).
-
M, NExpressing pseudo‐phosphorylated Tau (TauS199E) further increased the K49 acetylation of β‐catenin. N2a cells were transiently transfected with Vec, Tau40, or TauS199E for 48 h, levels of K49‐acetylated β‐cat, total β‐cat, and total Tau were detected by Western blotting (n = 3 biological replicates each group).
-
O, PInhibiting acetyltransferases CBP/P300 by TPOP146 (134 nM) or PCAF by L‐45 (126 nM) for 24 h did not significantly decrease the K49‐acetylated β‐cat level in Tau‐overexpressing N2a cells (n = 3 biological replicates each group).
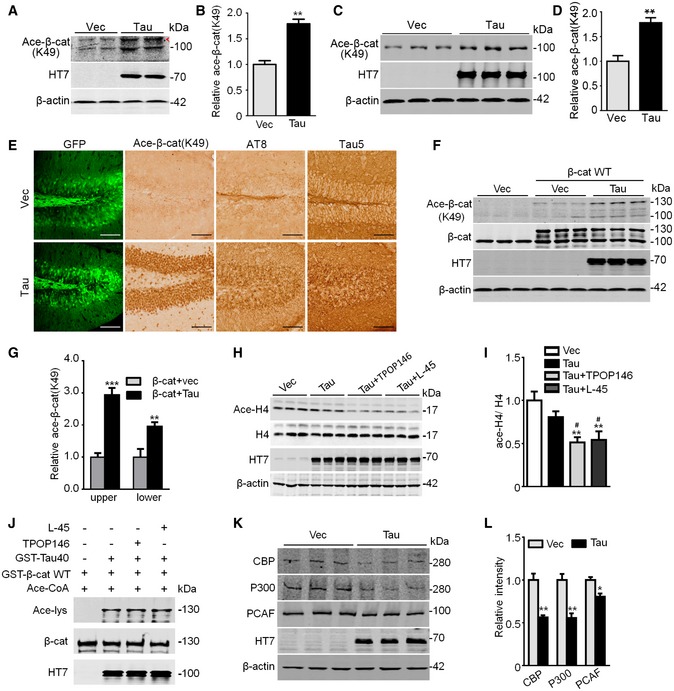
Figure EV2. Tau has a predominant role in acetylating β‐catenin at K49.
-
A–DN2a cells were transfected with Tau40 for 48 h (A, B), or the culture hippocampal neurons (7 div) were transfected with AAV‐eGFP‐Tau or AAV‐eGFP‐vector for 5 div (C, D). An increased Ace‐β‐cat (K49) was detected in the cell extracts by using anti‐K49‐Ace‐β‐cat antibody. The arrowhead in panel (A) denoted non‐specific bands identified by molecular weight (n = at least three biological replicates each group).
-
EThe increased Ace‐β‐cat (K49) in C57 mice after overexpressing AAV‐eGFP‐Tau40 for 1 month tested by immunohistochemical staining (n = 3, scale bars, 100 μm).
-
F, GCo‐expressing Tau40 and β‐cat further increased β‐cat K49 acetylation measured by Western blotting using Ace‐β‐cat (K49) antibody in HEK293 cells (n = at least three biological replicates each group); the “upper” denotes exogenous β‐catenin, and the “lower” denotes endogenous β‐cat.
-
H, IInhibiting acetyltransferases CBP/P300 significantly decreased acetylated H4 (Ace‐H4) level in N2a cells transfected with Tau40 or Vec for 48 h, and then treated with TPOP146 (134 nM) or L‐45 (126 nM) for 24 h (n = 3 biological replicates each group). The samples used for this figure were the same as those used for Fig 1O, and the blots of HT7 and β‐actin were shared.
-
JApplication of the inhibitor TPOP146 (134 nM) or L‐45 (126 nM) for 1 h did not change β‐cat acetylation by Tau measured by Western blotting using anti‐Ace‐lys in test tube (n = 3 from three independent experiments).
-
K, LOverexpressing Tau decreased the protein levels of acetyltransferases CBP, P300, and PCAF in N2a cells transfected with Tau40 or Vec for 48 h (n = 3 biological replicates each group).
Several acetyltransferases, including CREB‐binding protein (CBP), P300, and p300/CBP‐associated factor (PCAF), may also acetylate β‐catenin 33, 34, 35, 36. To verify whether Tau plays a dominant role in acetylating K49, we used inhibitors (TPOP146 and L‐45) of CBP/P300 38 and PCAF 39. Reduction of the acetylated H4 (Ace‐H4) confirmed the suppression of CBP, P300, and PCAF activities by TPOP146 or L‐45 (Fig EV2H and I). However, inhibiting CBP/P300 or PCAF only slightly decreased the acetylation level of K49‐β‐catenin by Tau (Fig 1O and P), which suggested a predominant role of Tau in acetylating β‐catenin at K49. We also demonstrated that incubating purified Tau with TPOP146 (134 nM) or L‐45 (126 nM) for 1 h did not affect its activity in acetylating β‐catenin (Fig EV2J). Interestingly, we also observed that overexpressing Tau decreased protein levels of CBP, P300, and PCAF (Fig EV2K and L), which further reduced the contribution of these acetyltransferases in β‐catenin acetylation at least in this Tau‐overexpressing system.
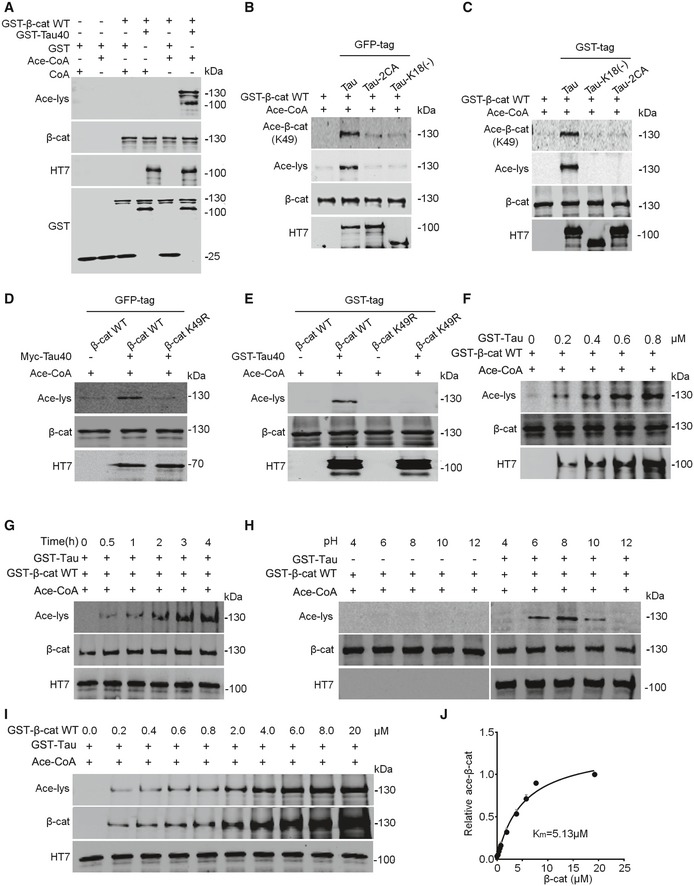
Tau directly acetylates β‐catenin in concentration‐, time‐, pH‐, and the acetyltransferase activity domain‐dependent manner
To confirm the direct effect of Tau on β‐catenin acetylation at K49 and characterize its enzymatic kinetics, we purified wild‐type Tau and the acetyltransferase activity domain‐deleted or domain‐mutated Tau, i.e., Tau‐k18(−) and Tau‐2CA (Fig EV3A) 37, the wild‐type β‐catenin and β‐catenin K49R proteins from eukaryocyte (HEK293) and prokaryocyte (Escherichia coli), respectively (Fig EV3B). Then, we incubated in test tube the purified wild‐type Tau with wild‐type β‐catenin and confirmed that Tau could directly acetylate β‐catenin (Fig 2A). However, incubating Tau‐k18(−) or Tau2CA with wild‐type β‐catenin (Fig 2B and C) and incubating wild‐type Tau with β‐catenin K49R abolished the acetylation of β‐catenin by Tau (Fig 2D and E). These data confirm that Tau can directly acetylate β‐catenin at K49 with activity domain‐dependent manner. Further kinetic studies showed that Tau could acetylate β‐catenin K49 in concentration‐, time‐, and pH‐dependent manner (Fig 2F–H), and the Km value for Tau and β‐catenin interaction was 5.13 μM (Fig 2I and J). The association of Tau with β‐catenin was also detected in HEK293 cells with stable or transient expression of Tau (Fig EV3C and D) and in mouse hippocampus (Fig EV3E and F) measured by immunoprecipitation and co‐localization by immunofluorescence staining in cultured primary hippocampal neurons (Fig EV3G). These data confirm that Tau can directly acetylate β‐catenin with concentration, pH, and time and its activity domain dependence.
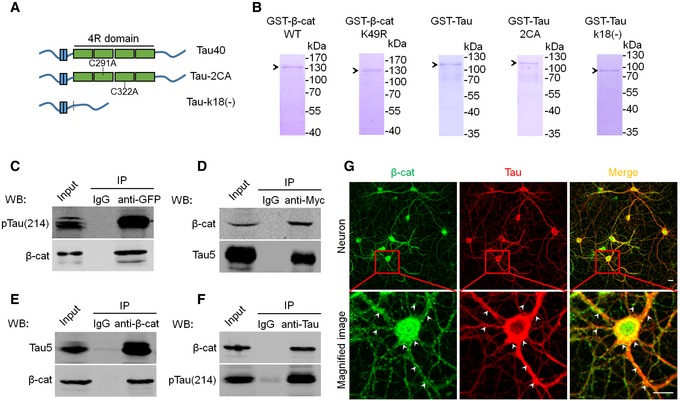
Figure EV3. Affinity‐purified Tau and β‐catenin proteins and the association of Tau with β‐catenin in vitro and in vivo .
-
AThe schematic shows full‐length Tau (Tau40), and the acetyltransferase activity domain double‐mutated Tau (cystine‐291/322 to alanine, Tau‐2CA) or the activity domain‐deleted Tau (Tau‐k18(−)).
-
BThe representative Coomassie blue staining pattern of the affinity‐purified β‐cat WT, β‐cat K49R, Tau40, Tau‐2CA, and Tau‐k18(−) from Escherichia coli using Ni‐NTA resin.
-
C, DAssociation of Tau with β‐cat in vitro. HEK293 cells with stable expression of Tau40 were transfected with eGFP‐β‐cat (C); or naïve HEK293 cells were transiently transfected with myc‐Tau40 (D). After 48 h, the cell extracts were immunoprecipitated using anti‐GFP or anti‐myc followed by Western blotting using Tau5, pS214, and β‐cat antibodies, respectively.
-
E, FAssociation of Tau with β‐cat in vivo. The mouse hippocampal extracts (3‐month‐old) were immunoprecipitated using anti‐β‐cat (E) or Tau5 (F) followed by Western blotting using Tau5 or pS214 and β‐cat antibodies, respectively.
-
GCo‐localization of Tau and β‐cat in primary hippocampal neurons infected with AAV‐Tau detected by co‐immunofluorescence labeling (scale bars, 10 μm); white arrowheads indicate co‐localization of Tau and β‐cat.
Source data are available online for this figure.
Figure 2. Tau directly acetylates β‐catenin in concentration‐, pH‐, time‐, and the acetyltransferase activity domain‐dependent manner.
-
ATau directly acetylates β‐cat in test tube. Both Tau40 and β‐cat were purified from Escherichia coli using Ni‐NTA resin (n = 3 from three independent experiments).
-
B, CMutation of Tau40 at acetyltransferase activity region abolishes its acetylation activity on β‐cat. eGFP‐Tau40, eGFP‐Tau‐2CA, and eGFP‐Tau‐k18(−) were purified from HEK293 cells (using anti‐GFP beads), and GST‐β‐cat was from E. coli (B), or both Tau40 and β‐cat were from E. coli (C) (n = 3 from three independent experiments).
-
D, EMutation of β‐cat K49 abolishes its acetylation by Tau. Both myc‐Tau40 and eGFP‐β‐cat were purified from HEK293 cells (using anti‐GFP or anti‐Myc beads) (D), or both from E. coli (E) (n = 3 from three independent experiments).
-
F–JKinetics of β‐cat acetylation by Tau. Tau acetylates β‐cat in concentration‐ (F), time‐ (G), pH‐ (H), and its acetyltransferase activity domain‐dependent manner. The affinity (Km) of Tau to β‐cat was measured by the initial rate of Tau as a function of the β‐cat concentrations (I, J). Michaelis–Menten parameters were determined using GraphPad Prism 6 (n = 3 from three independent experiments).
Source data are available online for this figure.
β‐catenin K49 acetylation inhibits its ubiquitylation‐associated proteolysis and decreases its phosphorylation
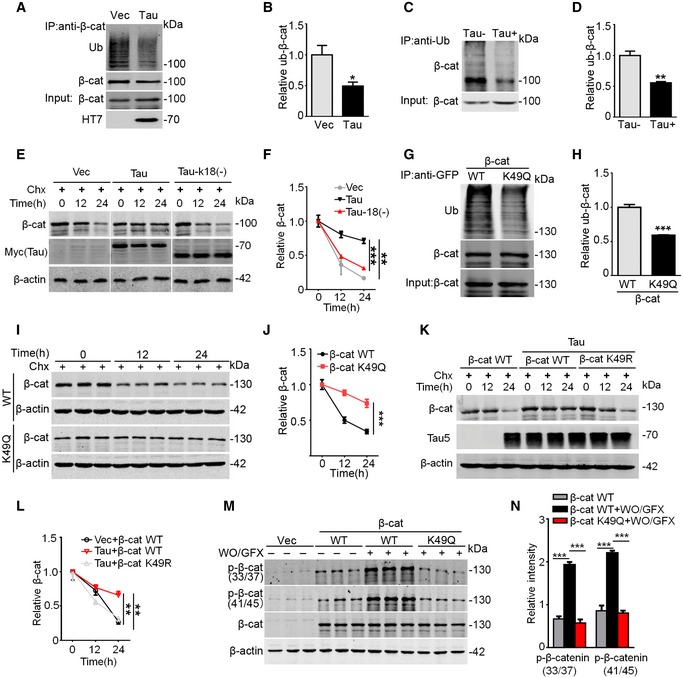
To further explore how acetylation leads to the accumulation of β‐catenin, we first measured the ubiquitination and proteolysis of β‐catenin. The results showed that overexpressing Tau decreased the ubiquitination level of β‐catenin in HEK293 cells (Fig 3A and B) and in human Tau transgenic mice (Fig 3C and D). Simultaneously, the proteolysis of β‐catenin was inhibited, while expressing Tau‐k18(−) restored the proteolysis of β‐catenin (Fig 3E and F). Furthermore, expression of the pseudo‐acetylated K49‐β‐catenin (i.e., K49Q) also decreased the ubiquitination with reduced degradation rate of β‐catenin (Fig 3G–J), whereas expressing K49R restored the degradation level of β‐catenin (Fig 3K and L). As acetylation and ubiquitination occur at the same lysine, we speculate that acetylation of β‐catenin may competitively block its ubiquitination and thus inhibits its degradation.
Figure 3. β‐catenin K49 acetylation inhibits its ubiquitylation‐associated proteolysis and decreases its phosphorylation.
-
A, BExpressing Tau inhibited β‐cat ubiquitination in HEK293 cells, measured by immunoprecipitation using antibody against β‐cat and Western blotting using anti‐ubiquitination (anti‐Ub) and anti‐β‐cat, respectively (n = 3 from three independent experiments).
-
C, DOverexpressing Tau inhibited β‐cat ubiquitination in hippocampi of Tau transgenic mice, measured by immunoprecipitation using anti‐Ub and Western blotting using anti‐β‐cat, respectively (n = 3 from three independent experiments).
-
E, FExpressing Tau‐k18(−) attenuated Tau‐induced β‐cat elevation. HEK293 cells were transfected with Vec, or Tau‐40, or Tau‐k18(−) for 24 h, and then were treated with Chx for 12 h or 24 h, followed by Western blotting. β‐cat protein level was normalized to β‐actin (n = 4 biological replicates each group).
-
G, HK49 acetylation of β‐cat inhibited its ubiquitination. HEK293 cells were co‐transfected with HA‐ubiquitin and GFP‐β‐cat or pseudo‐acetylated K49‐β‐catenin (K49Q), and then, β‐cat was immunoprecipitated by anti‐GFP and blotted by anti‐Ub and anti‐β‐cat (n = 3 from three independent experiments).
-
I, JK49 acetylation of β‐cat inhibited its degradation. HEK293 cells were transfected with β‐cat WT or K49Q for 24 h and then treated with cycloheximide (Chx, 100 μg/ml) for 12 h or 24 h (n = 4 biological replicates each group).
-
K, Lβ‐cat mutation at K49 (K49R) restored the degradation of β‐catenin. HEK293 cells were transfected with β‐cat WT or co‐transfected with β‐cat WT and Tau40, or co‐transfected with β‐cat K49R and Tau40 for 24 h, and then treated with cycloheximide (Chx, 100 μg/ml) for 12 h or 24 h (n = 4 biological replicates each group).
-
M, NK49 acetylation of β‐cat inhibited its phosphorylation at N‐terminal domain. HEK293 cells were transfected with GFP‐vec, or eGFP‐β‐cat (WT) or K49Q for 48 h, and then treated with wortmannin (WO, 1 μM) plus GF109203X (GFX, 1 μM) to activate GSK‐3β for 1 h followed by Western blotting (n = 3 biological replicates each group).
As phosphorylation of β‐catenin at the N‐terminal Ser33, Ser37, Thr41, and Ser45 by casein kinase 1 (CK1) or glycogen synthase kinase‐3β (GSK‐3β) promotes its ubiquitination and degradation 30, 31, 32, we studied whether K49 acetylation affects phosphorylation of β‐catenin at Ser33/Ser37/Thr41/Ser45. The results showed that activation of GSK‐3β significantly increased β‐catenin phosphorylation at Ser33/37/45 and Thr41, whereas expressing K49Q abolished the phosphorylation of β‐catenin (Fig 3M and N). These data indicate that K49 acetylation also attenuates the phosphorylation of β‐catenin, which provides direct evidence in supporting our previous observation 6.
β‐catenin K49 acetylation mediates the anti‐apoptotic effect of Tau
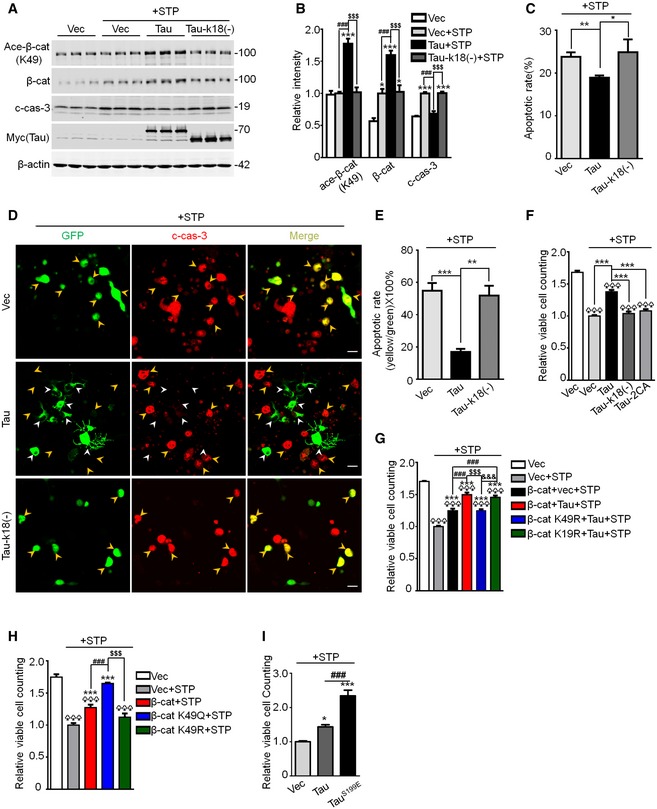
We reported previously that overexpressing Tau renders the cells more resistant to the apoptosis 6. Here, we found that overexpressing wild‐type Tau attenuated STP‐induced cleavage of caspase‐3 with upregulated β‐catenin K49 acetylation, while expressing Tau‐k18(−) abolished the effects of Tau on the caspase activation (Fig 4A and B). The anti‐apoptotic function of wild‐type Tau was also confirmed by the decreased apoptotic rate measured by flow cytometry (Fig 4C) and co‐immunofluorescence staining of Tau and the cleaved caspase‐3 (Fig 4D and E) as well as an increased cell viability measured by CCK8 assay (Fig 4F), while expressing acetyltransferase‐inactive Tau (Tau‐k18(−) or Tau‐2CA) abolished the anti‐apoptotic functions of Tau (Fig 4C–F). Furthermore, expressing non‐acetylation‐modifiable K49R but not K19R abolished the anti‐apoptotic effect of Tau (Fig 4G); expressing acetylation mimic K49Q further enhanced the anti‐apoptotic effect of β‐catenin, while expressing K49R abolished the anti‐apoptotic function of β‐catenin (Fig 4H); and overexpressing Tau or Tau‐k18(−) in N2a cells or primary cultured hippocampal neurons without STP treatment did not affect cell apoptosis (Fig EV4A and B). We also observed that overexpressing Ser199‐phospho‐mimic Tau (TauS199E) further augmented cell viability compared with the wild‐type Tau (Fig 4I). These data reveal that β‐catenin acetylation at K49 by Tau plays an essential role in mediating the anti‐apoptotic effect of Tau, and phosphorylation at Ser199 facilitates the anti‐apoptotic effect.
Figure 4. β‐catenin K49 acetylation mediates the anti‐apoptotic effect of Tau.
-
A, BExpressing Tau‐k18(−) abolished its effect on β‐cat K49 acetylation and c‐cas‐3 (cleaved caspase‐3). N2a cells were transfected with Vec, or Tau40, or Tau‐k18(−) for 48 h, and then treated with 0.5 μM staurosporine (STP, an apoptotic inducer) for 4 h, followed by Western blotting (n = 3 biological replicates each group).
-
C–FExpressing Tau‐k18(−) or Tau‐2CA abolished its anti‐apoptotic function. N2a cells, transfected with eGFP‐Vec, or eGFP‐Tau40, or eGFP‐Tau‐k18(−), or Tau‐2CA for 48 h, were then treated with 0.5 μM STP for 4 h followed by co‐staining with c‐cas‐3 (red) (D, E); N2a cells, transfected with myc‐Vec, or myc‐Tau40, or myc‐Tau‐k18(−), or myc‐Tau‐2CA for 48 h, were then treated with 0.5 μM STP for 4 h followed by flow cytometry using Annexin V‐FITC (C) or CCK8 assay (F). The apoptotic rate (E) was presented as the ratio of yellow (c‐casp‐3 and GFP co‐stained) to green (the total GFP‐expressing cells) staining. In panel (D) (middle row), the Tau expression (white arrowheads) was largely not co‐localized with c‐casp‐3 (yellow arrowheads) in the same visual field (n = 4–8 biological replicates each group; scale bar, 10 μm).
-
GNon‐acetylation‐modifiable mutation of β‐cat at K49 (K49R), not K19 (K19R), abolished the anti‐apoptotic effect of Tau. N2a cells were co‐transfected with Tau40 and β‐cat WT, or K49R, or K19R for 48 h, and then treated with 0.5 μM STP for 4 h. The cell viability was measured by CCK8 assay (n = 8 biological replicates each group).
-
HExpressing β‐cat K49Q most significantly increased cell viability. N2a cells were transfected with β‐cat WT or K49R or K49Q or the empty Vec for 48 h and then treated with 0.5 μM STP for 4 h. The cell viability was measured by CCK8 assay (n = 8 biological replicates each group).
-
IOverexpressing Ser199‐phosphorylation mimic Tau (TauS199E) further augmented cell viability. N2a cells were transfected with empty vec, Tau40, or TauS199E for 48 h and then treated with 0.5 μM STP for 4 h. The cell viability was measured by CCK8 assay (n = 8 biological replicates each group).
Figure EV4. Overexpressing Tau without apoptotic stimulation does not affect cell apoptosis.
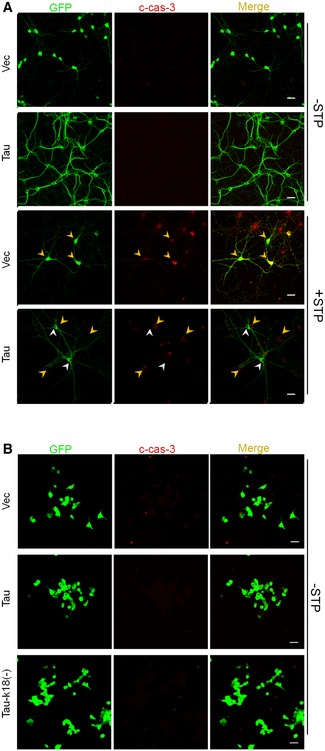
-
AThe hippocampal neurons (7 div) were transfected with AAV‐eGFP‐Tau for 5 div and then treated with 0.5 μM STP or the solvent for 4 h. Elevated cleaved caspase‐3 (c‐casp‐3, red) was shown in STP‐treated cells compared with the solvent‐treated controls; and the c‐casp‐3 (yellow arrowheads) was largely not co‐stained with Tau (white arrowheads) in the same visual field (n = 3 from three independent experiments; scale bar, 10 μm).
-
BOverexpressing Tau40 or Tau‐k18(−) for 48 h without STP treatment did not significantly affect c‐casp‐3 in N2a cells measured by immunofluorescence staining (n = 3 from three independent experiments; scale bar, 10 μm).
β‐catenin K49 acetylation by Tau promotes its nuclear translocation and increases the expression of survival signaling
To further explore the mechanisms underlying β‐catenin acetylation by Tau in anti‐apoptotic, we measured the cellular distribution of β‐catenin. Co‐expression of Tau and β‐catenin increased nuclear translocation of β‐catenin, while co‐expressing Tau with K49R abolished the nuclear staining of β‐catenin (Fig 5A). Expressing K49Q increased the nuclear translocation of β‐catenin (Fig 5B–D) with an increased luciferase activity (Fig 5E). Furthermore, overexpressing wild‐type Tau increased mRNA levels of survival genes (bcl2 and survivin) and dkk1 (downstream of Wnt signaling), while expressing acetyltransferase‐inactive Tau (Tau‐k18(−) or Tau‐2CA) abolished elevation of survival genes with reduced luciferase activity in cells and the AD transgenic mice (Figs 5F and G, and EV5). Expressing phosphor‐mimic TauS199E further increased the transcription activity of Wnt/TCF/LEF1 (Fig 5H). The increased mRNA levels of dkk1, bcl2, and survivin were also detected in the AD brains compared with the age‐matched control (Fig 5I), and positive correlations between dkk1, or bcl2 or survivin and K49‐acetylated β‐catenin were shown by Pearson's analysis (Fig 5J–L). These data suggest that K49 acetylation increases the transcription activity of β‐catenin by promoting its nuclear translocation, which may underlie the anti‐apoptotic function of β‐catenin.
Figure 5. β‐catenin K49 acetylation promotes its nuclear translocation and increases the expression of survival signaling.
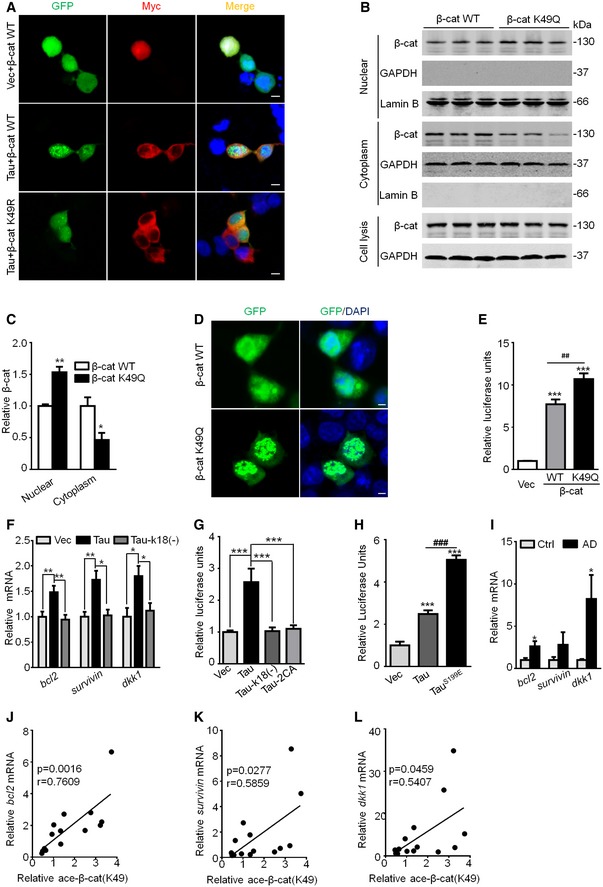
-
ACo‐expressing myc‐Tau and eGFP‐β‐catenin increased nuclear translocation of β‐catenin, while co‐expressing myc‐Tau and eGFP‐β‐cat K49R abolished the nuclear translocation of β‐catenin in HEK293 cells. The nuclei were stained using DAPI (n = 3 from three independent experiments, scale bar, 5 μm).
-
B–DExpressing β‐cat K49Q increased its nuclear translocation. HEK293 cells were transfected with eGFP‐β‐cat WT or K49Q for 48 h, and then, β‐cat protein level in different fractions was measured by Western blotting (B, C) and immunofluorescent labeling using a laser scanning confocal microscope (D). The nuclei (blue) was stained by DAPI (n = 3 from three independent experiments, scale bar, 2 μm).
-
EExpressing β‐cat K49Q further increased its transcription activity measured by dual TOPFlash/FOPFlash reporter luciferase assay after transient transfection of β‐cat WT, or K49Q, or Vec in HEK293 cells for 48 h, and the luciferase activity was normalized by β‐cat WT or K49Q protein level (n = 7 biological replicates each group).
-
FExpressing Tau‐k18(−) abolished Tau‐induced upregulation of mRNA levels of survival genes (bcl2 and survivin) and dkk1 (downstream of Wnt signaling) in N2a cells measured by Q‐PCR (n = 6–9 biological replicates each group).
-
GExpressing Tau‐k18(−) or Tau‐2CA abolished Tau‐induced Wnt/β‐catenin transcriptional activation measured by dual reporter luciferase activity assay in HEK293 cells (n = 7 biological replicates each group).
-
HExpressing phosphor‐mimic TauS199E (TauS199E) further increased the transcription activity of Wnt/TCF/LEF1 by Tau in HEK293 cells (n = 7 biological replicates each group).
-
I–LThe increased mRNA levels of bcl2, survivin, and dkk1 in the AD brains compared with the age‐/sex‐matched controls detected by Q‐PCR (n = 7 each group), and the positive correlations with the increased K49‐acetylated β‐catenin (see Fig 1I and J) analyzed by Pearson's analysis.
Figure EV5. Overexpressing Tau increases expression of survival genes.
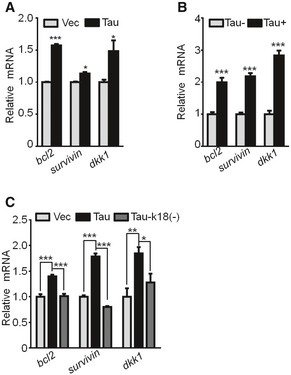
-
AThe hippocampal neurons (7 div) transfected with lenti‐GFP‐Tau or the empty vector for 5 div and the increased mRNA levels of bcl2 and survivin (survival molecular markers) and dkk1 (Wnt/β‐cat downstream marker) were detected by Q‐PCR (n = 6 biological replicates each group).
-
BThe increased mRNA levels of bcl2, survivin, and dkk1 in the hippocampi of 10‐month‐old Tau transgenic mice (Tau+) compared with Tau knockout mice (Tau−) detected by Q‐PCR (n = 4 biological replicates each group).
-
CExpressing Tau‐k18(−) abolishes Tau‐induced upregulation of bcl2, survivin, and dkk1 mRNA levels in HEK293 cells measured by Q‐PCR (n = 6–9 biological replicates each group).
Discussion
We reported previously that overexpressing Tau rendered cells more resistant to apoptosis with preservation of the dephosphorylated β‐catenin, but it was not known how β‐catenin was preserved, how β‐catenin was dephosphorylated, and how the preserved β‐catenin could mediate the anti‐apoptotic function of Tau. Here, we have provided novel evidence demonstrating that Tau can directly and robustly acetylate β‐catenin in concentration‐, time‐, pH‐, and the acetyltransferase activity domain‐dependent manner; acetylation of β‐catenin at K49 inhibits its phosphorylation at Ser33‐37‐45/Thr41 with significantly suppressed ubiquitination and proteolysis, leading to preservation of β‐catenin; the K49 acetylation promotes the nuclear translocation and the transcription activity of β‐catenin which mediates the anti‐apoptotic effect of Tau by increasing the expression of survival genes (Fig 6). These data reveal a new mechanism underlying Tau‐induced cell anti‐apoptosis.
Figure 6. The schematic diagram showing the mechanisms of Tau‐induced anti‐apoptosis.
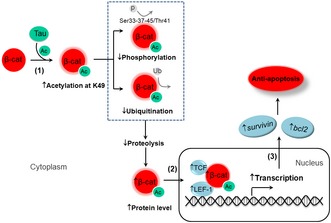
(1) Tau acetylates β‐catenin at K49, which inhibits K49 ubiquitination, Ser45/Thr41/Ser37/Ser33 phosphorylation and proteolysis of β‐catenin, leading to β‐catenin upregulation. (2 and 3) The increased β‐catenin is translocated into the nuclei (2) where it promotes transcription activity and increases expression of bcl2 and survivin leading to cellular anti‐apoptosis (3).
Elevation of β‐catenin can be caused by increased transcription or inhibited proteolysis. We observed that overexpressing Tau did not affect mRNA but induced degradation deficit of β‐catenin. β‐catenin is mainly degraded by ubiquitin/proteasome system 30, and ubiquitination and acetylation can competitively modify lysine 40, 41, 42, 43. Therefore, we tested whether these two post‐translational modifications oppositely influence the proteolysis nature of β‐catenin. By in vivo, in cultured cell lines/neurons and in test‐tube studies, we validated that Tau can acetylate β‐catenin and thus decrease its ubiquitination, leading to a reduced proteolysis and an increased protein level of β‐catenin. β‐catenin can be acetylated at K19, K49, and K354 33, 34, 35, 36, in which the N‐terminal K19 and K49 are putative lysine residuals for both acetylation and ubiquitination. We show that Tau preferentially acetylates K49 with negligible effect on K19, and pseudo‐acetylation of β‐catenin at K49 increases its self‐stabilization. However, ubiquitination level of K49 seems relatively low 30, 44, suggesting that the contribution of the inhibited ubiquitination by K49 acetylation to the impaired β‐catenin proteolysis may be limited. Therefore, other mechanisms, such as phosphorylation, may be involved in Tau‐induced self‐stabilization of β‐catenin. The alkaline pH dependence of Tau as an acetyltransferase, the binding properties of Tau to β‐catenin, and the effects of Tau aggregation on its biding to β‐catenin deserve further investigation.
β‐catenin degradation by ubiquitin–proteasome system is also regulated by phosphorylation, and the phosphorylation of β‐catenin by casein kinase 1 (CK1) and GSK‐3β accelerates its degradation 30, 31, 32. CK1 can phosphorylate β‐catenin at Ser45 which is structurally adjoining K49, and the phosphorylation at Ser45 is the prerequisite for the further phosphorylation of β‐catenin by GSK‐3β 32, 45, 46. Our previous study showed that overexpressing human Tau decreased the phosphorylation level of β‐catenin 6, but we did not know which sites and how the phosphorylation was suppressed. Here, we further verified that Tau could inhibit phosphorylation of β‐catenin by increasing its acetylation at K49, and we also identified the phosphorylation sites of β‐catenin at Ser33/Ser37/Thr41/Ser45. We speculate that the increased acetylation of β‐catenin at K49 can inhibit its phosphorylation at Ser45 and consequently inhibit the phosphorylation at the adjoining Thr41, Ser37, and Ser33. These results perfectly explained the puzzle observed in our previous study, in which a significantly activated GSK‐3β with a reduced β‐catenin phosphorylation was shown 6, although it is known that β‐catenin is an active substrate of GSK‐3β 32, 45, 46. This novel evidence also supports the previously proposed substrate competition of GSK‐3β in phosphorylating Tau and β‐catenin 6, 19.
As an important component of Wnt signaling 23, upregulation of β‐catenin enhances the Wnt and thus plays a crucial role in development and carcinogenesis 26, 33, 34. There are several target genes of Wnt/β‐catenin signaling that can inhibit cell apoptosis 47, 48, 49. Here, we show that Tau increased mRNA levels of survival‐promoting factors (bcl2 and survivin), and the increase was abolished by mutation of Tau at acetyltransferase activity region. We also demonstrated that acetylation of β‐catenin at K49 was required for the role of β‐catenin in mediating the anti‐apoptotic effect of Tau. Furthermore, Tau could interact with β‐catenin both in vitro and in vivo, and deletion of the acetyltransferase activity at the microtubule‐binding domain abolished Tau‐induced acetylation of β‐catenin with abolishment of the downstream upregulation of the transcription activity. A previous study showed that overexpressing full‐length Tau1–441 and Tau1–230 fragment blocked apoptosis with Akt activation, while expressing Tau1–44 fragment induced cell death with involvement of NMDA receptors and ROS in cerebellar granule cells 50. It was also reported that PHP‐Tau, constructed by changing the codons for S198, S199, S202, T231, S235, S396, S404, S409, S413, and S422 to glutamate, induced cell death, while the survival rate in the cells with expression of wild‐type Tau was higher than the control group 51. Therefore, multiple mechanisms must be involved in the anti‐apoptotic function of Tau proteins. Furthermore, like other intrinsically disordered proteins, Tau must be able to interact directly or indirectly with many proteins including several members of signaling cascades. Therefore, it would be definitely important to relate the Tau interactome with its anti‐apoptotic activity to obtain a coherent view of this function.
The role of apoptosis in the cell loss of the AD brains is not a clear‐cut issue. It was reported that apoptosis is not the major form of cell loss in the AD brains 52. It was also observed that both pro‐apoptotic factors (caspase‐3, Bax, caspase‐8, and caspase‐9) and anti‐apoptotic factors (Bcl‐2 and Bcl‐x) were increased in the AD brains 53, 54, 55, 56. Furthermore, the number of apoptosis‐like nuclear morphology was lower in the AD brains than the age‐matched controls 57. As regards the role of Tau in cell apoptosis, some studies showed that overexpressing full‐length wild‐type Tau blocked apoptosis50, 51 that was in agreement with our previous findings 6. Some other studies showed Tau was required for Aβ‐ or stress‐induced brain pathologies in AD mouse models 58, 59, 60, 61, which suggests a pathological role of Tau. The discrepancy regarding the protective or detrimental role of Tau may depend on different experimental conditions. Though overexpressing Tau rendered the cells more resistant to the chemically induced acute apoptosis, the increasing intracellular Tau could simultaneously impair multiple cellular functions, such as mitochondrial dynamic 14, 15, synaptic transmission 62, and autophagy 63. Therefore, we have proposed that Tau overexpression/accumulation may play a dual role in leading the neurons to escape an acute apoptosis and simultaneously triggering a chronic neurodegeneration 19, 21.
Taken together, we found that Tau can directly acetylate β‐catenin at K49 and thus inhibit its proteolysis by repressing K49 ubiquitination and Ser45/Thr41/Ser37/Ser33 phosphorylation. The increased nuclear translocation/activation of β‐catenin mediates the anti‐apoptotic effect of Tau by promoting expression of survival signals, such as bcl2 and survivin.
Materials and Methods
Plasmids, chemicals, transgenic mice, and human brain tissue
The human β‐catenin‐eGFP plasmid was a gift of Dr. Alpha Yap (Addgene plasmid #71367; a G245A mutation was found during sequencing, and it was repaired before application). The plasmid pEGFP‐Tau‐2N4R (Tau40), encoding human Tau, was a generous gift of Dr. Fei Liu (Jiangsu Key Laboratory of Neuroregeneration, Nantong, China). The plasmids of Tau and Tau‐k18(−) were generated by PCR and cloned in a myc‐tagged pcDNA3.0 vector/EGFP N1 vector in ECORI and BamHI restriction sites. Mutation of β‐catenin at lysine‐19 and lysine‐49 into arginine (K19R, K49R) or glutamine (K19Q, K49Q) and Tau at cysteine‐291 and cysteine‐322 into alanine (Tau‐2CA) was carried out using the Mut Express II Fast Mutagenesis Kit by following the manufacturer's instructions (Vazyme Biotech, Nanjing, China). Cycloheximide (Chx) was from Sigma‐Aldrich (St. Louis, MO, USA). TPOP146 and L‐45 dihydrochloride (L‐45) were from MedChemExpress (Shanghai, China), and all other reagents were from Tocris (Bristol, UK). Bicinchoninic Acid Protein Detection Kit was from Pierce. Reagents for cell culture were from Gibco (Grand Island, NY, USA). The human transgenic mice (STOCK Mapttm1(EGFP)Klt Tg(MAPT)8cPdav/J, stock number 004808) and Tau knockout mice (STOCK Mapttm1(EGFP)Klt/J, stock number 004779) were from Jackson laboratory. All animal experiments were performed according to the “Policies on the Use of Animals and Humans in Neuroscience Research,” revised, and approved by the Society for Neuroscience in 1995, and the animal study was approved by the Academic Review Board of Tongji Medical College, Huazhong University of Science and Technology. Post‐mortem human brain samples were provided by Dr. Chao Ma of Human Brain Bank, Chinese Academy of Medical Sciences, and Peking Union Medical College, Beijing, China, and Dr. Keqiang Ye of the Emory Alzheimer's Disease Research Center, United States (Appendix Table S1). AD was diagnosed according to the criteria of the Consortium to Establish a Registry for AD and the National Institute on Aging. Diagnoses were confirmed by the presence of amyloid plaques and neurofibrillary tangles in formalin‐fixed tissue. The study was approved by the Biospecimen Committee, and informed consent was obtained from the subjects.
Apoptosis induction
Before administration of apoptotic inducers, the cells were cultured in serum‐free medium for 12 h. To induce apoptosis, cells were treated with 0.5 μM staurosporine (STP) for 4 h in serum‐free medium. Apoptosis was assayed using an Annexin V–PI staining kit by following the manufacturer's procedure (BD Pharmingen, San Diego, CA), and the apoptotic rate was measured by flow cytometry using standardized program of the instrument (FACSCalibur, BD Biosciences, San Jose, CA). Cell viability was also measured by CCK8 assay by following the manufacturer's instructions (Dojindo, Shanghai, China).
Luciferase assay
The luciferase assay was performed as described previously64. Cells were transfected with 0.3 μg of TOPFlash or FOPFlash (Upstate, USA) along with 10 ng of pRL‐SV40 Renilla luciferase reporter vector (Promega, USA) by using Neofect™ DNA transfection reagent kit (Neofect Biotech, Beijing, China). Cells co‐transfected with Tau40, or Tau‐k18(−), or Tau‐2CA, or wild‐type β‐catenin (β‐catenin WT), or β‐catenin K49Q, or β‐catenin K49R and TOPFlash plus pRL‐SV40 or FOPFlash reporter plus pRL‐SV40 were cultured for 48 h, and the cell lysate was used to measure luciferase reporter gene expression using the Dual‐Luciferase Reporter Assay System (Promega, USA). The luciferase activity of each sample was normalized against Renilla.
Cell culture, transfection, and drug treatment
HEK293 cells were cultured in DMEM supplemented with 10% (vol/vol) fetal bovine serum (FBS). N2a cell were cultured in 45% (vol/vol) DMEM and 45% (vol/vol) Opti‐MEM with 10% (vol/vol) FBS. All the cultured cells were grown at 37°C in a humid atmosphere containing 5% (vol/vol) CO2. Transfection of plasmids in cells was carried out with Neofect™ DNA transfection reagent kit (Neofect Biotech, Beijing, China) by following the manufacturer's instructions. TPOP146 (134 nM) and L‐45 dihydrochloride (126 nM), dissolved in DMSO (0.1%), were used to inhibit respectively the activity of CBP/P300 and PCAF.
Primary hippocampal neuron culture
The primary hippocampal neuron culture was performed as described previously 13. The hippocampi were dissected from embryonic rats at days 17–18, gently minced in Hank's buffered saline solution, and then suspended in 0.25% (vol/vol) trypsin solution at 37°C for 15 min. The neurons were plated in culture dishes coated with 100 μg/ml poly‐d‐lysine and cultured for 7 days in vitro (div) in neurobasal medium supplemented with 2% (vol/vol) B‐27 and 1× GlutaMAX for immunofluorescence.
In test‐tube acetylation assays
Acetyltransferase activity of Tau was confirmed by incubating in test tube the purified β‐catenin and purified human wild‐type full‐length Tau (Tau40) proteins from eukaryocyte (HEK293) and prokaryocyte (E. coli), respectively. For the prokaryotic purification, the cDNAs of Tau, Tau‐k18(−), Tau‐2CA, β‐catenin (WT), and β‐catenin (K49R) were cloned respectively in a GST‐His‐tagged PET‐41a(+) vector and amplified in Rosetta2(de3)pLysS competent cells, and then affinity‐purified by using Ni‐NTA resin. For the eukaryotic purification, the plasmid β‐catenin‐eGFP (WT), β‐catenin‐eGFP (K49R), and myc‐Tau, myc‐Tau‐k18(−), and myc‐Tau2CA were transfected respectively in HEK293 cell for 48 h and then affinity‐purified by using anti‐GFP or anti‐myc beads. For the in test‐tube acetylation assay, 50 nM each of the purified Tau proteins, 1.5 μM each of the purified β‐catenin, and 1 mM acetyl CoA (Sigma) were incubated at 37°C for 2 h with constant shaking. The reaction buffer contains 50 mM HEPES (pH 8.0), 10% glycerol, 1 mM dithiothreitol (DTT), and 10 mM sodium butyrate. The acetylation level of β‐catenin was analyzed by Western blotting using anti‐acetylated lysine antibody. The immunoreactive bands were quantified by using the Odyssey Infrared Imaging System (LI‐COR Biosciences, Lincoln, NE, USA). Michaelis–Menten parameters were determined using GraphPad Prism 6.
Quantitative RT–PCR
Total RNA was extracted from cultured cells, or mouse brains or the post‐mortem human brains using TRIzol reagent (Life Technologies, Carlsbad, CA). The cDNA was generated using a PrimeScript RT Reagent Kit (Takara Bio, Dalian, China), and quantitative PCR was performed using an SYBR Premix Ex Taq (Tli RNase H Plus) (Takara Bio, Dalian, China). The primers used in this paper are listed in Appendix Table S2. RT–PCR was performed in a StepOnePlus Real‐Time PCR Detection System (Life Technologies, NY, USA).
Western blotting
Western blotting was performed by following the method routinely employed in our laboratory 65. Briefly, the total cellular proteins were extracted using RIPA lysis buffer (Beyotime, Shanghai, China). The cytoplasmic and nuclear proteins were fractionated using the Nucl‐Cyto‐Mem Preparation Kit (Applygen Technologies Inc) by following the manufacturer's instructions. After electrophoresis (10% SDS/PAGE), the proteins were transferred onto the nitrocellulose membrane and blocked using 5% bovine serum albumin (BSA) for 1 h at room temperature. The membranes were then incubated with specific primary antibody (Appendix Table S3), and the immunoreactive bands were visualized by using the Odyssey Infrared Imaging System (LI‐COR Biosciences, Lincoln, NE, USA).
Immunohistochemistry
The mice were anesthetized and transcardially perfused with normal saline, followed by 4% paraformaldehyde (PFA). The dissected brains were post‐fixed for 48 h and then cryoprotected with 30% sucrose and then in optimum cutting temperature (OCT) compound for cryostat sectioning. The brains were sliced 30 μM coronally by using a freezing microtome (CM1900, Leica, Germany). The brain slices were soaked in PBS‐3%H2O2‐0.5%Triton for 30 min and then blocked by 3% BSA for another 30 min. The primary antibodies (Appendix Table S3) were applied in blocking solution and incubated at 4°C overnight, followed by a biotinylated secondary antibody for 1 h at 37°C, horseradish peroxidase‐labeled secondary antibody for 1 h at 37°C, and then DAB staining. The images were observed under a microscope (Nikon, 90i, Tokyo, Japan).
Immunofluorescence
The cultured neurons were fixed in 4% (vol/vol) paraformaldehyde for 15 min and permeabilized in phosphate buffer solution (PBS) containing 0.5% Triton X‐100. Non‐specific binding was blocked by incubating in PBS containing 0.1% Triton X‐100 and 5% (wt/vol) BSA for 1 h. The primary antibodies (Appendix Table S3) were then applied in blocking solution and incubated at 4°C overnight. The secondary antibodies conjugated to Alexa Fluor 488/568 (Thermo Fisher Scientific) were added to the coverslip for 1 h at room temperature, and then, Hoechst 33258 (Sigma, St. Louis, MO, USA) (1:1,000) was used for the nuclear staining for 5 min. The coverslips were washed and mounted onto slides. The images were observed with a laser confocal microscope (710; Zeiss, Germany).
Immunoprecipitation
The dissected hippocampi or HEK293 cell was homogenized on ice in RIPA buffer (Beyotime, Shanghai, China) at 4°C and then centrifuged at 13,000 × g for 15 min. A total of 200 μl supernatants containing about 200 μg total proteins were incubated with primary antibodies (Appendix Table S3) overnight followed by addition of protein G agarose (Millipore, Darmstadt, Germany) for 2 h (at 4°C on rotation). The agarose beads were washed three times and resuspended in 40 μl of SDS‐loading buffer, and then denatured at 95°C for 10 min. The immunoprecipitants were analyzed by Western blotting.
Statistics
Data were analyzed using commercial software (SPSS version 21.0 for Windows, SPSS Inc., Chicago, IL, USA). The two‐way ANOVA or one‐way ANOVA or a Student's t‐test was used to determine the different means among groups. The data were expressed as mean ± SEM, and the level of significance was set at P < 0.05.
Author contribution
J‐ZW designed research; EL, QZ, A‐JX, XL, ML, JY, SL, Z‐PX, LL, QW, DK, YY, and H‐LL performed experiments; EL, QZ, X‐CW, YY, G‐PL, and J‐ZW analyzed data; H‐LL and J‐ZW wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Drs. Chao Ma and Keqiang Ye for the human brain tissue, and Drs. Alpha Yap and Fei Liu for plasmids. This work was supported in parts by the Ministry of Science and Technology of China (2016YFC1305800); the Natural Science Foundation of China (91632305, 91632111, 31730035, 81721005, 91949205, and 31171028); Guangdong Provincial Key S&T Program (2018B030336001); and the China Human Brain Bank Consortium.
EMBO Reports (2020) 21: e48328
Contributor Information
Hong‐Lian Li, Email: lihonglian@mail.hust.edu.cn.
Jian‐Zhi Wang, Email: wangjz@mail.hust.edu.cn.
References
- 1. Iqbal K, Grundke‐Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, Wisniewski HM, Alafuzoff I, Winblad B (1986) Defective brain microtubule assembly in Alzheimer's disease. Lancet 2: 421–426 [DOI] [PubMed] [Google Scholar]
- 2. Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42: 631–639 [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82: 239–259 [DOI] [PubMed] [Google Scholar]
- 4. Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17: 5–21 [DOI] [PubMed] [Google Scholar]
- 5. Morsch R, Simon W, Coleman PD (1999) Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol 58: 188–197 [DOI] [PubMed] [Google Scholar]
- 6. Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q, Wang XC, Chen XQ, Yang Y, Zhang JY et al (2007) Phosphorylation of tau antagonizes apoptosis by stabilizing beta‐catenin, a mechanism involved in Alzheimer's neurodegeneration. Proc Natl Acad Sci USA 104: 3591–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang HH, Li HL, Liu R, Zhang Y, Liao K, Wang Q, Wang JZ, Liu SJ (2010) Tau overexpression inhibits cell apoptosis with the mechanisms involving multiple viability‐related factors. J Alzheimers Dis 21: 167–179 [DOI] [PubMed] [Google Scholar]
- 8. Wang ZF, Yin J, Zhang Y, Zhu LQ, Tian Q, Wang XC, Li HL, Wang JZ (2010) Overexpression of tau proteins antagonizes amyloid‐beta‐potentiated apoptosis through mitochondria‐caspase‐3 pathway in N2a cells. J Alzheimers Dis 20: 145–157 [DOI] [PubMed] [Google Scholar]
- 9. Liu XA, Song J, Jiang Q, Wang Q, Tian Q, Wang JZ (2012) Expression of the hyperphosphorylated tau attenuates ER stress‐induced apoptosis with upregulation of unfolded protein response. Apoptosis 17: 1039–1049 [DOI] [PubMed] [Google Scholar]
- 10. Yeh PA, Chang CJ, Tu PH, Wilson HI, Chien JY, Tang CY, Su MT (2010) Phosphorylation alters tau distribution and elongates life span in Drosophila. J Alzheimers Dis 21: 543–556 [DOI] [PubMed] [Google Scholar]
- 11. Rzechorzek NM, Connick P, Livesey MR, Borooah S, Patani R, Burr K, Story D, Wyllie DJA, Hardingham GE, Chandran S (2016) Hypothermic preconditioning reverses tau ontogenesis in human cortical neurons and is mimicked by protein phosphatase 2A inhibition. EBioMedicine 3: 141–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ittner A, Chua SW, Bertz J, Volkerling A, van der Hoven J, Gladbach A, Przybyla M, Bi M, van Hummel A, Stevens CH et al (2016) Site‐specific phosphorylation of tau inhibits amyloid‐beta toxicity in Alzheimer's mice. Science 354: 904–908 [DOI] [PubMed] [Google Scholar]
- 13. Yin Y, Gao D, Wang Y, Wang ZH, Wang X, Ye J, Wu D, Fang L, Pi G, Yang Y et al (2016) Tau accumulation induces synaptic impairment and memory deficit by calcineurin‐mediated inactivation of nuclear CaMKIV/CREB signaling. Proc Natl Acad Sci USA 113: E3773–E3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li XC, Hu Y, Wang ZH, Luo Y, Zhang Y, Liu XP, Feng Q, Wang Q, Ye K, Liu GP et al (2016) Human wild‐type full‐length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep 6: 24756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu Y, Li XC, Wang ZH, Luo Y, Zhang X, Liu XP, Feng Q, Wang Q, Yue Z, Chen Z et al (2016) Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 7: 17356–17368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu ZC, Chu J, Lin L, Song J, Ning LN, Luo HB, Yang SS, Shi Y, Wang Q, Qu N et al (2016) SIL1 rescued bip elevation‐related tau hyperphosphorylation in ER stress. Mol Neurobiol 53: 983–994 [DOI] [PubMed] [Google Scholar]
- 17. Kimura T, Yamashita S, Fukuda T, Park JM, Murayama M, Mizoroki T, Yoshiike Y, Sahara N, Takashima A (2007) Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild‐type human tau. EMBO J 26: 5143–5152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild‐type mice. Mol Neurodegener 6: 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang JZ, Liu F (2008) Microtubule‐associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol 85: 148–175 [DOI] [PubMed] [Google Scholar]
- 20. Wang JZ, Wang ZH, Tian Q (2014) Tau hyperphosphorylation induces apoptotic escape and triggers neurodegeneration in Alzheimer's disease. Neurosci Bull 30: 359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang Y, Wang JZ (2018) Nature of Tau‐associated neurodegeneration and the molecular mechanisms. J Alzheimers Dis 62: 1305–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCrea PD, Turck CW, Gumbiner B (1991) A homolog of the armadillo protein in Drosophila (plakoglobin) associated with E‐cadherin. Science 254: 1359–1361 [DOI] [PubMed] [Google Scholar]
- 23. Peifer M, Rauskolb C, Williams M, Riggleman B, Wieschaus E (1991) The segment polarity gene armadillo interacts with the wingless signaling pathway in both embryonic and adult pattern formation. Development 111: 1029–1043 [DOI] [PubMed] [Google Scholar]
- 24. Noordermeer J, Klingensmith J, Perrimon N, Nusse R (1994) dishevelled and armadillo act in the wingless signalling pathway in Drosophila . Nature 367: 80–83 [DOI] [PubMed] [Google Scholar]
- 25. MacDonald BT, Tamai K, He X (2009) Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morin PJ (1999) beta‐catenin signaling and cancer. BioEssays 21: 1021–1030 [DOI] [PubMed] [Google Scholar]
- 27. Wang H, MacNaughton WK (2005) Overexpressed beta‐catenin blocks nitric oxide‐induced apoptosis in colonic cancer cells. Cancer Res 65: 8604–8607 [DOI] [PubMed] [Google Scholar]
- 28. Chen YZ (2004) APP induces neuronal apoptosis through APP‐BP1‐mediated downregulation of beta‐catenin. Apoptosis 9: 415–422 [DOI] [PubMed] [Google Scholar]
- 29. Toledo EM, Colombres M, Inestrosa NC (2008) Wnt signaling in neuroprotection and stem cell differentiation. Prog Neurobiol 86: 281–296 [DOI] [PubMed] [Google Scholar]
- 30. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) beta‐catenin is a target for the ubiquitin‐proteasome pathway. EMBO J 16: 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW (1997) Serine phosphorylation‐regulated ubiquitination and degradation of beta‐catenin. J Biol Chem 272: 24735–24738 [DOI] [PubMed] [Google Scholar]
- 32. Hagen T, Vidal‐Puig A (2002) Characterisation of the phosphorylation of beta‐catenin at the GSK‐3 priming site Ser45. Biochem Biophys Res Commun 294: 324–328 [DOI] [PubMed] [Google Scholar]
- 33. Levy L, Wei Y, Labalette C, Wu Y, Renard CA, Buendia MA, Neuveut C (2004) Acetylation of beta‐catenin by p300 regulates beta‐catenin‐Tcf4 interaction. Mol Cell Biol 24: 3404–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kemler R (2000) The p300/CBP acetyltransferases function as transcriptional coactivators of beta‐catenin in vertebrates. EMBO J 19: 1839–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wolf D, Rodova M, Miska EA, Calvet JP, Kouzarides T (2002) Acetylation of beta‐catenin by CREB‐binding protein (CBP). J Biol Chem 277: 25562–25567 [DOI] [PubMed] [Google Scholar]
- 36. Ge X, Jin Q, Zhang F, Yan T, Zhai Q (2009) PCAF acetylates {beta}‐catenin and improves its stability. Mol Biol Cell 20: 419–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cohen TJ, Friedmann D, Hwang AW, Marmorstein R, Lee VM (2013) The microtubule‐associated tau protein has intrinsic acetyltransferase activity. Nat Struct Mol Biol 20: 756–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Popp TA, Tallant C, Rogers C, Fedorov O, Brennan PE, Muller S, Knapp S, Bracher F (2016) Development of selective CBP/P300 benzoxazepine bromodomain inhibitors. J Med Chem 59: 8889–8912 [DOI] [PubMed] [Google Scholar]
- 39. Moustakim M, Clark PG, Trulli L, Fuentes de Arriba AL, Ehebauer MT, Chaikuad A, Murphy EJ, Mendez‐Johnson J, Daniels D, Hou CD et al (2017) Discovery of a PCAF bromodomain chemical probe. Angew Chem Int Ed Engl 56: 827–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Freiman RN, Tjian R (2003) Regulating the regulators: lysine modifications make their mark. Cell 112: 11–17 [DOI] [PubMed] [Google Scholar]
- 41. Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP (2002) MDM2‐HDAC1‐mediated deacetylation of p53 is required for its degradation. EMBO J 21: 6236–6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jin YH, Jeon EJ, Li QL, Lee YH, Choi JK, Kim WJ, Lee KY, Bae SC (2004) Transforming growth factor‐beta stimulates p300‐dependent RUNX3 acetylation, which inhibits ubiquitination‐mediated degradation. J Biol Chem 279: 29409–29417 [DOI] [PubMed] [Google Scholar]
- 43. Gronroos E, Hellman U, Heldin CH, Ericsson J (2002) Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell 10: 483–493 [DOI] [PubMed] [Google Scholar]
- 44. Winer IS, Bommer GT, Gonik N, Fearon ER (2006) Lysine residues Lys‐19 and Lys‐49 of beta‐catenin regulate its levels and function in T cell factor transcriptional activation and neoplastic transformation. J Biol Chem 281: 26181–26187 [DOI] [PubMed] [Google Scholar]
- 45. Frame S, Cohen P (2001) GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hagen T, Di Daniel E, Culbert AA, Reith AD (2002) Expression and characterization of GSK‐3 mutants and their effect on beta‐catenin phosphorylation in intact cells. J Biol Chem 277: 23330–23335 [DOI] [PubMed] [Google Scholar]
- 47. Inestrosa NC, Varela‐Nallar L (2014) Wnt signaling in the nervous system and in Alzheimer's disease. J Mol Cell Biol 6: 64–74 [DOI] [PubMed] [Google Scholar]
- 48. Kim PJ, Plescia J, Clevers H, Fearon ER, Altieri DC (2003) Survivin and molecular pathogenesis of colorectal cancer. Lancet 362: 205–209 [DOI] [PubMed] [Google Scholar]
- 49. Zhang T, Otevrel T, Gao Z, Gao Z, Ehrlich SM, Fields JZ, Boman BM (2001) Evidence that APC regulates survivin expression: a possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res 61: 8664–8667 [PubMed] [Google Scholar]
- 50. Amadoro G, Serafino AL, Barbato C, Ciotti MT, Sacco A, Calissano P, Canu N (2004) Role of N‐terminal tau domain integrity on the survival of cerebellar granule neurons. Cell Death Differ 11: 217–230 [DOI] [PubMed] [Google Scholar]
- 51. Fath T, Eidenmuller J, Brandt R (2002) Tau‐mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J Neurosci 22: 9733–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stadelmann C, Bruck W, Bancher C, Jellinger K, Lassmann H (1998) Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. J Neuropathol Exp Neurol 57: 456–464 [DOI] [PubMed] [Google Scholar]
- 53. Nagy ZS, Esiri MM (1997) Apoptosis‐related protein expression in the hippocampus in Alzheimer's disease. Neurobiol Aging 18: 565–571 [DOI] [PubMed] [Google Scholar]
- 54. Giannakopoulos P, Kovari E, Savioz A, de Bilbao F, Dubois‐Dauphin M, Hof PR, Bouras C (1999) Differential distribution of presenilin‐1, Bax, and Bcl‐X(L) in Alzheimer's disease and frontotemporal dementia. Acta Neuropathol 98: 141–149 [DOI] [PubMed] [Google Scholar]
- 55. Satou T, Cummings BJ, Cotman CW (1995) Immunoreactivity for Bcl‐2 protein within neurons in the Alzheimer's disease brain increases with disease severity. Brain Res 697: 35–43 [DOI] [PubMed] [Google Scholar]
- 56. Tortosa A, Lopez E, Ferrer I (1998) Bcl‐2 and Bax protein expression in Alzheimer's disease. Acta Neuropathol 95: 407–412 [DOI] [PubMed] [Google Scholar]
- 57. Woodhouse A, Dickson TC, West AK, McLean CA, Vickers JC (2006) No difference in expression of apoptosis‐related proteins and apoptotic morphology in control, pathologically aged and Alzheimer's disease cases. Neurobiol Dis 22: 323–333 [DOI] [PubMed] [Google Scholar]
- 58. Roberson ED, Scearce‐Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2007) Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science 316: 750–754 [DOI] [PubMed] [Google Scholar]
- 59. Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ et al (2011) Amyloid‐beta/Fyn‐induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci 31: 700–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA et al (2010) Dendritic function of tau mediates amyloid‐beta toxicity in Alzheimer's disease mouse models. Cell 142: 387–397 [DOI] [PubMed] [Google Scholar]
- 61. Lopes S, Vaz‐Silva J, Pinto V, Dalla C, Kokras N, Bedenk B, Mack N, Czisch M, Almeida OF, Sousa N et al (2016) Tau protein is essential for stress‐induced brain pathology. Proc Natl Acad Sci USA 113: E3755–E3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li XG, Hong XY, Wang YL, Zhang SJ, Zhang JF, Li XC, Liu YC, Sun DS, Feng Q, Ye JW et al (2019) Tau accumulation triggers STAT1‐dependent memory deficits by suppressing NMDA receptor expression. EMBO Rep 20: e47202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Feng Q, Luo Y, Zhang XN, Yang XF, Hong XY, Sun DS, Li XC, Hu Y, Li XG, Zhang JF et al (2019) MAPT/Tau accumulation represses autophagy flux by disrupting IST1‐regulated ESCRT‐III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy 10.1080/15548627.2019.1633862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang Q, Sun ZX, Allgayer H, Yang HS (2010) Downregulation of E‐cadherin is an essential event in activating beta‐catenin/Tcf‐dependent transcription and expression of its target genes in Pdcd4 knockdown cells. Oncogene 29: 128–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu E, Xie AJ, Zhou Q, Li M, Zhang S, Li S, Wang W, Wang X, Wang Q, Wang JZ (2017) GSK‐3beta deletion in dentate gyrus excitatory neuron impairs synaptic plasticity and memory. Sci Rep 7: 5781 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5