

Abstract
The clinical benefit of monotherapy involving immune checkpoint inhibitors (ICIs) such as anti-programmed death-1 antibody (PD-1 Ab) is limited to small populations. We previously developed a telomerase-specific oncolytic adenovirus, Telomelysin (OBP-301), the safety of which was confirmed in a phase I clinical study. Here, we examined the potential of OBP-502, an OBP-301 variant, as an agent for inducing immunogenic cell death (ICD) and synergistically enhancing the efficacy of OBP-502 with PD-1 Ab using CT26 murine colon cancer and PAN02 murine pancreatic cancer cell lines. OBP-502 induced the release of ICD molecules such as adenosine triphosphate (ATP) and high-mobility group box protein 1 (HMGB1) from CT26 and PAN02 cells, leading to recruitment of CD8-positive lymphocytes and inhibition of Foxp3-positive lymphocyte infiltration into tumors. Combination therapy involving OBP-502 intratumoral administration and PD-1 Ab systemic administration significantly suppressed the growth of not only OBP-502-treated tumors but also tumors not treated with OBP-502 (so-called abscopal effect) in CT26 and PAN02 bilateral subcutaneous tumor models, in which active recruitment of CD8-positve lymphocytes was observed even in tumors not treated with OBP-502. This combined efficacy was similar to that observed in a CT26 rectal orthotopic tumor model involving liver metastases. In conclusion, telomerase-specific oncolytic adenoviruses are promising candidates for combined therapies with ICIs.
Keywords: immune checkpoint, programmed death-1, oncolytic adenovirus, combined immunotherapy, immunogenic cell death, tumor infiltrating lymphocytes, CD8, abscopal effect
Graphical Abstract
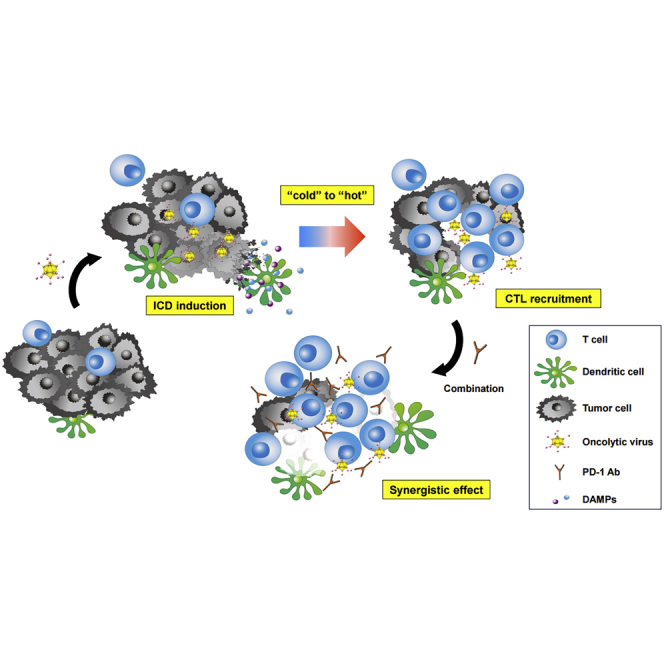
Kanaya and colleagues show that telomerase-specific oncolytic adenoviruses can facilitate cytotoxic T lymphocytes recruitment into tumor tissues through immunogenic cell death induction, leading to abscopal effects via activation of systemic antitumor immune responses, and combination therapy with anti-PD-1 antibody creates synergistic antitumor effects that even lead to tumor eradication.
Introduction
Cancer immunotherapy, a strategy that harnesses the immune system to combat tumors, has recently triggered a paradigm shift in the standard treatment of various cancers.1,2 In particular, immune checkpoint inhibitors (ICIs), such as blocking antibodies (Abs) to cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed death-1 (PD-1), and PD-1 ligand-1 (PD-L1), have dramatically improved clinical outcomes for patients with malignant tumors, such as melanoma, non-small cell lung cancer, Hodgkin’s lymphoma, and gastric cancer.3, 4, 5, 6 However, the clinical benefit of ICI monotherapy is limited to small populations exhibiting high expression of PD-L1, high numbers of tumor-infiltrating lymphocytes (TILs), and a high mutation burden.5,7,8 Therefore, a novel strategy involving combination therapy with various therapeutic drugs that enhance tumor immunogenicity is needed to further improve clinical outcomes.9
Immunogenic cell death (ICD) is a mechanism of cell death that leads to the induction of an effective antitumor immune response via the activation of dendritic cells (DCs) and T lymphocytes, unlike normal types of cell death such as apoptosis and necrosis. ICD is characterized by secretion of damage-associated molecular patterns (DAMPs), such as high-mobility group box protein 1 (HMGB1) and adenosine triphosphate (ATP).10,11 Agents that induce ICD have been recognized as ideal partners for combination therapies with ICIs that could clinically benefit a larger population of cancer patients. Oncolytic viruses are considered such an agent, along with various chemotherapeutics such as oxaliplatin and cyclophosphamide, in addition to radiation.12, 13, 14, 15, 16
We previously established a telomerase-specific oncolytic adenovirus (OBP-301, Telomelysin) in which the human telomerase reverse transcriptase (hTERT) promoter element drives the expression of the viral E1A and E1B genes.17 This gene modification enables OBP-301 to replicate selectively in tumor cells and induce tumor-specific oncolytic cell death. Moreover, following induction of oncolytic cell death, progeny viruses are capable of spreading to surrounding tumor cells to cause continued oncolytic cell death. A US Food and Drug Administration (FDA)-approved phase I clinical study confirmed the safety and biological activity of intratumoral administration of OBP-301 in patients with advanced solid tumors in the United States.18 Based on its safety profile and promising preclinical data regarding combination therapy with ionizing radiation, a phase I/II clinical trial is currently ongoing in Japan to evaluate the safety and efficacy of the combination for treating esophageal cancer (UMIN000010158).19
In the present study, we first examined the potential of our telomerase-specific oncolytic adenovirus as an ICD-inducing drug by assessing the secretion of ICD markers such as HMGB1 and ATP in vitro and recruitment of CD8-positive TILs on gastrointestinal tumors in vivo. We then examined the efficacy of combination therapy involving our oncolytic adenovirus and anti-PD-1 Ab using in vivo subcutaneous and orthotopic mouse models, focusing on abscopal effects mediated by the oncolytic adenoviruses via activation of the host immune system. Our findings demonstrate the promise of our telomerase-specific oncolytic adenovirus therapy, particularly in combination with an ICI. Moreover, these findings should facilitate the development of a novel oncolytic virus therapeutic strategy incorporating our adenovirus that will be a more attractive option for cancer treatment.
Results
Cytotoxic Activity of OBP-502 against Murine Gastrointestinal Cancer Cell Lines
OBP-502, a variant of OBP-301, was employed in this study. The difference between OBP-301 and OBP-502 is that OBP-502 has the gene cassette expressing the RGD peptide in the E3 region (Figure S1A). The RGD peptide facilitates OBP-502 infection of murine tumor cells via interaction with the integrin αvβ5 expressed on tumor cells, because OBP-301 infection of murine cells such as CT26 and PAN02, which do not express the Coxsackie virus and adenovirus receptor (CAR) (Figure S2), is inefficient.
OBP-502 killed CT26 and PAN02 cells in a dose-dependent and time-dependent manner (Figure 1A; Figure S1B), and this cytotoxicity of OBP-502 was higher than OBP-301, especially at high doses, whereas no significant difference was observed between OBP301 and OBP-502 on human cancer cells such as TE4, GCIY, MIA PaCa-2, and HCT116 (Figure S1C). This cytotoxicity was associated with induction of apoptosis and autophagy following effective infection of murine tumor cells, which was shown by induction of E1A, upregulation of cleaved poly(ADP ribose) polymerase (PARP), and downregulation of p62 on western blot analysis (Figure 1B). The cytotoxic mechanism of OBP-502 was considered similar to that of OBP-301, an original oncolytic adenovirus.20 Interestingly, treatment with high doses of OBP-502 upregulated PD-L1 expression by CT26 and PAN02 cells (Figure 1B), particularly on the cell surface (Figure 1C).
Figure 1.

Cytotoxic Activity of OBP-502 against Murine Gastrointestinal Cancer Cell Lines
(A) Viability of CT26 and PAN02 cells was assessed using an XTT assay 3 days after OBP-502 treatment at the indicated doses (MOI). The percentage of viable cells relative to non-treated cells (0 MOI) was plotted. Error bars indicate 95% confidence intervals. (B) Whole-cell lysates of CT26 and PAN02 cells collected 3 days after OBP-502 treatment (0, 10, 100, 500, and 1,000 MOI) were subjected to western blot analysis of E1A, PARP, p62, PD-L1, and β-Actin expression. Cleaved PARP (C-PARP) and p62 indicate induction of apoptosis and autophagy, respectively. (C) CT26 and PAN02 cells treated with OBP-502 (1,000 MOI) were subjected to flow cytometry for analysis of PD-L1 expression on the cell surface 3 days after treatment.
Active Release of Immunogenic Molecules and Chemokines by OBP-502 In Vitro
OBP-502 significantly increased the release of ATP and HMGB1, which are known to be immunogenic molecules, by CT26 and PAN02 cells in vitro 24 h after treatment (Figures 2A and 2B) but did not affect the expression of calreticulin (CRT), another immunogenic molecule, in either cell line (Figure S3A). Intracellular HMGB1 levels also increased with OBP-502 treatment (Figures S3B and S3C). OBP-502 decreased β-catenin expression in CT26 and PAN02 cells (Figure S3D), which is reportedly associated with immune exclusion in the tumor microenvironment mediated by the Wnt/β-catenin pathway.21 Multi-cytokine and chemokine assays showed that at 24 h after treatment, OBP-502 markedly induced the release of several chemokines by CT26 cells, such as CCL5/RANTES and CXCL10/IP-10, which are known to play an important role in recruiting TILs22,23 (Figure 2C). Focusing on CCL5/RANTES based on multi-cytokine and chemokine assay results, OBP-502 significantly increased CCL5/RANTES release by CT26 and PAN02 cells in a dose-dependent manner (Figure 2D). These results suggested that OBP-502 has the potential to induce ICD via the active release of immunogenic molecules and chemokines, leading to activation of the host immune response against tumors.
Figure 2.

Active Release of Immunogenic Molecules and Chemokines by OBP-502 In Vitro
(A) Extracellular ATP secreted from CT26 and PAN02 cells was measured using a luminescence assay 24 h after OBP-502 treatment (0, 500, and 1,000 MOI). *p < 0.001. (B) Extracellular HMGB1 secreted from CT26 and PAN02 cells was measured using an ELISA 24 h after OBP-502 treatment (0, 500, and 1,000 MOI). *p < 0.001, **p < 0.005. (C) Cytokines and chemokines secreted from CT26 cells were measured using multi-cytokine and chemokine assays 24 h after OBP-502 treatment (0 and 1,000 MOI), and ratio of 1,000 MOI to 0 MOI was plotted for each cytokine or chemokine. (D) CCL5/RANTES secreted from CT26 cells and PAN02 cells was measured using an ELISA 24 h after OBP-502 treatment (0, 500, and 1,000 MOI). *p < 0.001.
Recruitment of CD8-Positive Lymphocytes to Tumors and Development of Acquired Antitumor Immunity Mediated by OBP-502
Immunohistochemical staining of CD8, CD11c, CD4, and Foxp3 on CT26 and PAN02 subcutaneous tumors indicated that OBP-502 treatment significantly increased the number of CD8-positive cells in CT26 and PAN02 tumors compared with PBS treatment, whereas OBP-502 treatment significantly increased the number of CD11c-positive cells in PAN02 tumors, but not CT26 tumors (Figures 3A–3C). OBP-502 treatment resulted in a significant decrease in the number of Foxp3-positive cells in CT26 tumors, but not PAN02 tumors, whereas no significant change was observed in the number of CD4-positive cells in either tumor type. Recruitment of CD8-positive cells and rejection of Foxp3-positive cells began to be observed 3 days after a single treatment with OBP-502, whereas no change in CD4-positive cells was observed (Figure S4). In cytotoxic T lymphocyte (CTL) assay, CD8-positive lymphocytes (effector) collected from the spleen of mice treated with OBP-502 showed significantly strong cytotoxicity against Colon26-GFP cells (target) at an effector/target ratio of 20:1 compared with PBS (Figures 3D and 3E).
Figure 3.

Recruitment of CD8-Positive Lymphocytes to Tumors and Development of Acquired Antitumor Immunity Mediated by OBP-502
(A) Study protocol. In brief, CT26 or PAN02 subcutaneous tumors, intratumorally treated with OBP-502 (1 × 109 PFUs) or PBS three times each week, were harvested at 7 days after initiation of treatment for immunohistochemical staining. (B) Representative figures of immunohistochemical staining for CD8, CD11c, CD4, and Foxp3 in CT26 tumor tissues. Scale bar, 100 μm. (C) Median number of TILs expressing CD8, CD11c, CD4, and Foxp3 was statistically assessed from five selected fields. *p < 0.05, **p < 0.005, ***p < 0.001. (D) Representative figures of FACS for CTL assay in which cytotoxicity of CD8-positive lymphocytes (effector) harvested from the spleen of mice treated with OBP-502 (1 × 109 PFUs) or PBS on Colon26-GFP cells (target) was analyzed at different ratios of effector/target of 0:1, 10:1, and 20:1. Cells in the area surrounded by the black border are dead Colon26-GFP cells. (E) Percentage of dead cells in CTL assay was statistically assessed between PBS and OBP-502 (n = 3). *p < 0.05. (F) Protocol for the vaccination study. In brief, CT26 or PAN02 cells treated with OBP-502 (1,000 MOI) for 3 days were administered subcutaneously into the flank of BALB/c or C57/BL6 mice on days −7, −4, and −1 for vaccination, and CT26 or PAN02 cells (1 × 105 cells) were inoculated subcutaneously on day 0. (G) Tumor volume was monitored until day 28 and compared between PBS and vaccinated mice. *p < 0.05, **p < 0.005. (H) Representative figures of immunohistochemical staining for CD8-positive TILs in tumor tissues harvested 35 days after PAN02 inoculation in mice vaccinated with OBO-502-treated PAN02 cells or PBS. Scale bar, 100 μm. (I) Median number of TILs expressing CD8 was statistically assessed from five selected fields. *p < 0.05. E:T, effector/target.
In the vaccination study, compared with PBS-treated mice, tumor growth was significantly suppressed after inoculation of CT26 or PAN02 cells in mice vaccinated with OBP-502-treated cells (Figures 3F and 3G), and vaccination with OBP-502-treated cells actually increased the number of CD8-positive TILs in PAN02 tumors (Figures 3H and 3I). These results suggested that OBP-502 strongly activates antitumor immunity by recruiting CD8-positive lymphocytes to tumor tissues, and this effect was established as acquired antitumor immunity in the treated mice.
Antitumor Effects of Combination Therapy in a Subcutaneous Tumor Model
The antitumor effects of combination therapy involving OBP-502 and PD-1 Ab were evaluated using CT26 and PAN02 subcutaneous tumor models. PD-1 Ab was administered systemically 3 days after OBP-502 intratumoral injection, and this combination treatment was repeated three times each week (Figure 4A). The combination therapy significantly suppressed the growth of CT26 tumors compared with PBS and PD-1 Ab monotherapy, and surprisingly, 4 of 12 mice (33%) given the combination therapy became tumor free, whereas monotherapy with either OBP-502 or PD-1 Ab eradicated the tumors in 1 and 0 of the 12 mice, respectively (Figure 4B). When the tumor-free mice cured by combination therapy were re-challenged with CT26 inoculation, two of the four mice (50%) remained tumor free (Figure 4C). The combination therapy also significantly suppressed the growth of PAN02 tumors, which are reportedly ICI resistant,24 and one of seven mice (14%) became tumor free, but PD-1 Ab alone had no effect on PAN02 tumors (Figure 4D). Immunohistochemical staining showed that combination therapy led to recruitment of more CD8-positive TILs compared with controls at 28 days after initiation of treatment (Figure 4E). Although PD-1 Ab treatment or combination therapy with the Ab and another therapeutic agent are reportedly associated with severe adverse effects in several major organs, such as the lungs, pancreas, and thyroid gland,25 no signs of severe toxicity were observed in any major organs harvested 28 days after combination therapy against CT26 subcutaneous tumors (Figure S5). These results demonstrated the possibility that OBP-502 and PD-1 Ab combination therapy could become an attractive treatment option for gastrointestinal tumors because of its profound beneficial effects and favorable safety profile.
Figure 4.

Antitumor Effects of Combination Therapy in a Subcutaneous Tumor Model
(A) Study protocol. In brief, CT26 or PAN02 tumors were treated with OBP-502 (1 × 109 PFUs) intratumorally and/or PD-1 Ab (1st: 20 mg/kg, 2nd and 3rd: 10 mg/kg) intraperitoneally three times each week. (B) Volume of CT26 tumors was monitored until day 28 (n = 12). Statistical analysis was performed on day 21. Right table shows the number of mice in which tumors were completely eradicated in each treatment group. *p < 0.01, **p < 0.0005. (C) Four tumor-free mice cured by combination therapy of OBP-502 and PD-1 Ab in the experiment shown in (B) were re-challenged with CT26 inoculation (5 × 105 cells). Four naive mice were inoculated with CT26 cells (5 × 105 cells) and used as controls. Tumor volume was monitored until day 28. Right table shows the number of mice in which tumors did not develop in each group. **p < 0.005. (D) Volume of PAN02 tumors was monitored until day 28 (n = 7). Right table shows the number of mice in which tumors were completely eradicated in each treatment group. *p < 0.01. (E) Representative figures for each treatment group of immunohistochemical staining for CD8-positive TILs in tumor tissues harvested 28 days after initiation of treatment. Scale bar, 100 μm.
Abscopal Effects of Combination Therapy in a Bilateral Subcutaneous Tumor Model
The abscopal effect is an interesting phenomenon in which tumor shrinkage at metastatic sites can be achieved following application of local therapy, such as radiotherapy, and the immune system is considered to play a key role in this process. When we first assessed whether OBP-502 monotherapy induced an abscopal effect in a CT26 bilateral subcutaneous tumor model (Figure 5A), we found that OBP-502 intratumoral injection exhibited significant therapeutic effects not only at the treated site, but also at untreated sites, when compared with PBS (Figure 5B), and these effects involved efficient recruitment of CD8-positive cells (Figure S6A). However, the abscopal effect was diminished when the same experiment was performed using T cell-deficient BALB/c nude mice (Figure 5C; Figure S6B).
Figure 5.

Abscopal Effects of Combination Therapy in a Bilateral Subcutaneous Tumor Model
(A) Study protocol. In brief, in the bilateral subcutaneous tumor model, one side was treated with OBP-502 intratumorally (1 × 109 PFUs) three times each week, and the other side was left untreated with OBP-502. PD-1 Ab was administered intraperitoneally (1st: 20 mg/kg, 2nd and 3rd: 10 mg/kg) three times each week. (B) Volume of CT26 tumors treated with PBS or OBP-502 was monitored separately at the OBP-502-treated site and untreated site until day 24 (n = 7). *p < 0.001, **p < 0.05. (C) The same experiment shown in (B) was performed using BALB/c nude mice, and tumor volume was monitored until day 21 (n = 7). (D) Volume of CT26 tumors treated with PBS, monotherapy with OBP-502 or PD-1 Ab, or combination of both was monitored separately at the OBP-502-treated site and untreated site until day 24 (n = 6). *p < 0.005, **p < 0.05. (E) Representative figures for each treatment group of immunohistochemical staining for CD8-positive TILs in OBP-502-treated tumor tissues and untreated tumor tissues harvested 28 days after initiation of treatment. Scale bar, 100 μm. (F) Median number of CD8-positive TILs in OBP-502-treated tumor tissues and untreated tumor tissues was statistically assessed from five selected fields. *p < 0.005, **p < 0.001, ***p < 0.05. (G) Four mice rendered tumor free (OBP-502-treated site) by combination therapy with OBP-502 and PD-1 Ab in the experiment shown in (D) were re-challenged with CT26 inoculation (5 × 105 cells). Four naive mice were inoculated with CT26 cells (5 × 105 cells) and used as controls. Tumor volume was monitored until day 28. *p < 0.005.
When abscopal effects associated with combination therapy with PD-1 Ab were assessed using the same CT26 bilateral subcutaneous tumor model, combination therapy exhibited significant therapeutic effects at both the treated and the untreated site compared with control therapies (Figure 5D). Surprisingly, this combination therapy eradicated tumors at the treated site in 67% of mice (4/6) and at the untreated site in 33% of mice (2/6), whereas monotherapy with either agent eradicated no tumors at the treated or untreated sites in any of the mice. Combination therapy with OBP-502 strongly recruited CD8-positive TILs to both the treated and untreated sites compared with control therapies (Figures 5E and 5F), and combination therapy significantly decreased the number of Foxp3-positive TILs at the treated site, but not at the untreated site (Figure S7). Furthermore, when mice rendered tumor free (n = 4) by combination therapy against CT26 tumors were re-challenged with CT26 inoculation, all four mice remained tumor free (Figure 5G). These results suggested that OBP-502 intratumoral injection has the potential to produce abscopal effects via the activation of systemic antitumor immunity, and that these effects are enhanced by combination therapy with PD-1 Ab.
Antitumor Effects of Combination Therapy in a CT26 Orthotopic Rectal Tumor Model with Liver Metastases
Finally, in order to evaluate the therapeutic effects of combination therapy in a model more closely related to clinical practice than a bilateral subcutaneous tumor model, we developed a CT26 orthotopic rectal tumor model with liver metastases and treated the mice using the same protocol (Figure 6A). Kaplan-Meier analysis showed that combination therapy significantly prolonged survival (Figure 6B), and in vivo imaging system (IVIS) imaging demonstrated that the combination therapy actually suppressed the growth of liver metastases, as well as rectal tumors (Figures 6C–6E). The effectiveness of combination therapy against liver metastases was confirmed by macroscopic findings (Figure 6F) and hematoxylin and eosin staining (Figure 6G).
Figure 6.

Antitumor Effects of Combination Therapy in a CT26 Orthotopic Rectal Tumor Model with Liver Metastases
(A) Study protocol. In brief, in the CT26-Luc orthotopic rectal tumor model with liver metastases, CT26-Luc rectal tumors were treated with OBP-502 (1 × 109 PFUs) intratumorally and PD-1 Ab (1st: 20 mg/kg, 2nd and 3rd: 10 mg/kg) intraperitoneally three times each week. (B) Kaplan-Meier survival analysis (n = 8). *p < 0.05. (C) Representative IVIS images of mice 28 days after initiation of treatment with PBS, monotherapy with OBP-502 or PD-1 Ab, or combination of both. (D and E) Luminescence intensity of rectal tumors (D) and liver metastases (E) was measured using the IVIS imaging system on day 21 (n = 8). *p < 0.05, **p < 0.001. (F and G) Macroscopic findings (F) and H&E staining (G) of liver metastases harvested 14 days after initiation of treatment with PBS, monotherapy with OBP-502 or PD-1 Ab, or combination of both (twice, on days 0 and 7). Scale bar, 20 μm.
Discussion
Combined immunotherapy has become a hot topic in the field of cancer immunotherapy as clinicians have realized that the clinical benefits of ICI monotherapy are limited to a small proportion of patients. Worldwide, over 500 clinical trials evaluating combination therapy involving anti-PD-1/PD-L1 Ab with other therapeutic agents are currently ongoing, and the most common combination involves anti-CTLA-4 agents, followed by chemotherapy and radiotherapy.26 The combination of nivolumab and ipilimumab has been proved effective for patients with advanced melanoma or renal cell carcinoma in phase III clinical trials,27,28 and the combination of pembrolizumab and chemotherapy has also shown significant survival prolongation in patients with advanced non-small cell lung cancer.29 Another intriguing strategy is combination with oncolytic viruses, for which several clinical trials are currently ongoing.30 Talimogene laherparepvec (i.e., T-VEC), the first oncolytic herpes simplex virus, exhibited therapeutic benefits against advanced melanoma as a monotherapy in a phase III clinical trial,31 and recently demonstrated promising efficacy and acceptable safety in combination with pembrolizumab for patients with advanced melanoma in a phase Ib clinical trial.32 Coxsackie virus A21 (CVA21), an unmodified common cold RNA virus, also seems to have produced durable responses with minimal toxicity in combination with ipilimumab for patients with advanced melanoma in a phase Ib clinical trial.33
TIL-rich tumors respond to ICIs.34 From this viewpoint, preferred combination partners with ICIs include agents that are capable of inducing ICD. Along with some chemotherapeutics such as oxaliplatin and cyclophosphamide, oncolytic viruses are potent ICD-inducing agents.14,15 We previously reported that OBP-301, a telomerase-specific oncolytic adenovirus, releases a danger signal (i.e., uric acid) that triggers an immune response against human cancer cells.35 In addition, we showed that OBP-502, a variant of OBP-301, significantly increased the release of ATP and HMGB1 via the induction of autophagic and apoptotic cell death of murine colon cancer and pancreatic cancer cells in this study, indicating that OBP-502 is also a potent ICD inducer. Treatment with OBP-502, however, did not increase the expression of CRT, which is recognized as an ICD marker, along with ATP and HMGB1, although the reasons for this remain unclear. Various chemokines are also associated with the recruitment of immune cells.22,23 An oncolytic poxvirus and vaccinia virus expressing CXCL11 reportedly generated a systemic immune response and sensitized murine tumors to ICIs.36,37 We demonstrated in this study that OBP-502 increases the release of various chemokines, such as CCL5/RANTES and CXCL10/IP-10, both of which play an important role in recruiting CD8-positive TILs,38 and this is consistent with a previous report indicating that adenoviruses originally induce chemokines such as CCL5/RANTES and CXCL10/IP-10 following infection of cells.39 Through the release of these ICD molecules and chemokines, OBP-502 facilitates the recruitment of CD8-positive lymphocytes and DCs into CT26 and PAN02 tumor tissues, and this immune activation by OBP-502 is long lasting as acquired immunity. This effect is particularly important with respect to tumors such as PAN02, so-called cold tumors that normally have few TILs; therefore, we can say that OBP-502 has the potential to turn immunologically “cold” tumors into “hot” tumors.
The mechanism underlying the abscopal effect remains unknown, even decades after this phenomenon was first reported in the 1970s as a rare unpredictable biological behavior involving simultaneous regression of distant untreated metastases after local radiotherapy.40 Systemic antitumor immune activation after local radiotherapy was recently recognized as a primary contributor to the abscopal effect,41 and combination therapies with ICIs such as ipilimumab reportedly increase the number of patients who can benefit from abscopal effects.42,43 Oncolytic virotherapy, mostly involving intratumoral injection, also reportedly induces abscopal effects.44 In this study, we demonstrated that OBP-502 has the potential to induce abscopal effects in a bilateral subcutaneous tumor model of CT26 and PAN02 cells. Furthermore, combination therapy employing OBP-502 and PD-1 Ab markedly suppressed tumor growth by recruiting CD8-positive lymphocytes, even into tumors not treated with OBP-502, and, surprisingly, eradicated some of these untreated CT26 or PAN02 tumors, whereas either monotherapy had little effect.
PD-L1 is generally considered a biomarker of anti-PD-1 therapy,45 and it is reportedly upregulated by INF-γ through the Janus kinase-signal transducer and activator of transcription pathway.46 Because some chemotherapies and radiotherapy reportedly upregulate PD-L1 expression on the surface of cancer cells,47,48 we found that OBP-502 upregulated PD-L1 expression both within and on the surface of CT26 and PAN02 cells. Although we did not investigate further the detailed mechanism in this study, stimulation of interferon-γ (IFN-γ) release by oncolytic adenoviruses, as we previously reported,35 is presumably associated with this PD-L1 upregulation, and PD-L1 upregulation by OBP-502 may be the mechanism underlying the synergistic effect of combination therapy with PD-1 Ab. We showed that OBP-502 reduced β-catenin in CT26 and PAN02 cells in vitro, and suppressed recruitment of Foxp3-positive cells in CT26 tumor tissues in vivo, which is another interesting aspect of OBP-502, but detailed mechanisms for these remain unclear.
Although the present study has demonstrated the promising antitumor activity of OBP-502 via immune activation, there were several discrepancies in this study. For example, OBP-502 showed decent cytotoxic effects on CT26 and PAN02 cells at a high dose in vitro (Figure 1A), but OBP-502 did not show significant antitumor effects on CT26 tumors in similar immune-deficient conditions in vivo using immune-deficient mice, although the tendency of an antitumor effect was observed for OBP-502 (Figure 5C). Another discrepancy is that combination therapy of OBP-502 and PD-1 Ab did not increase recruitment of CD8-positive TILs at treated sites in the CT26 bilateral subcutaneous tumor model, whereas combination therapy significantly increased recruitment of CD8-positive TILs at untreated sites (Figure 5F).
In the present study, we demonstrated that our telomerase-specific oncolytic adenovirus can facilitate CTL recruitment into tumor tissues through ICD induction after intratumoral administration, leading to abscopal effects via activation of systemic antitumor immune responses. Combination therapy with PD-1 Ab created synergistic antitumor effects that even led to tumor eradication. These findings support the contention that novel therapeutic strategies employing oncolytic virotherapy are ideal partners for ICIs, and that this combination can yield improved clinical benefits for patients with advanced tumors with distant metastases. We recently launched a multicenter, open-label phase I clinical study to evaluate the efficacy and safety of OBP-301 in combination with pembrolizumab in patients with advanced solid tumors (EPOC1505) (ClinicalTrials.org: NCT03172819), for which data from the present study will serve as proof of concept. We expect that oncolytic virotherapy will be the next great breakthrough in cancer therapy, following immunotherapy, and that combination therapy with ICIs will be the most promising therapeutic strategy.
Materials and Methods
Cell Lines and Cell Cultures
The murine colon cancer cell line CT26 derived from BALB/c mice was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), and the murine pancreatic cancer cell line PAN02 derived from C57BL/6 mice was purchased from the National Cancer Institute (Frederick, MD, USA). These cancer cell lines were cultured in RPMI supplemented with 10% fetal calf serum and 1% penicillin-streptomycin (100 U/mL). Neither cell line was cultured for more than 3 months following resuscitation. Cell authentication was not performed by the authors.
Oncolytic Adenovirus and ICI
OBP-502 (Figure S1), a variant of OBP-301, was employed in this study. OBP-502 has a mutant fiber containing the RGD peptide to facilitate infection of murine tumor cells via interaction with the integrin αvβ5 expressed on tumor cells, because OBP-301 infection of murine cells such as CT26 and PAN02, which do not express the CAR (Figure S2), is inefficient. Multiplicity of infection (MOI) and plaque-forming units (PFUs) were used as virus units in vitro and in vivo, respectively. Anti-PD-1 Ab (clone 4H2) (PD-1 Ab) was obtained from Ono Pharmaceutical.
Cell Viability Assay
CT26 and PAN02 cells were seeded in 96-well plates (1 × 103 cells/well) (n = 5) and treated with OBP-502 at the indicated MOI. Cell viability was determined 3 days after treatment using a Cell Proliferation Kit II (XTT) (Roche Diagnostics) according to the manufacturer’s protocol.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue samples cut at 4 μm were deparaffinized in xylene and rehydrated using a graded ethanol series. After blocking endogenous peroxidases by incubation with 3% H2O2 for 10 min, the samples were boiled in citrate buffer or EDTA buffer for 14 min in a microwave oven for antigen retrieval. The samples were incubated with primary Abs for 1 h at room temperature or overnight at 4°C and then with peroxidase-linked secondary Ab for 30 min at room temperature. Samples were stained with 3,3-diaminobenzidine for signal generation, counterstained with Mayer’s hematoxylin, and then dehydrated and mounted onto coverslips. Abs to CD8 (eBioscience, San Diego, CA, USA), CD11c (Abcam, Cambridge, MA, USA), CD4 (eBioscience), and Foxp3 (eBioscience) were used. The number of cells expressing CD8, CD11c, CD4, and Foxp3, which indicate CTLs, DCs, helper T lymphocytes, and regulatory T lymphocytes, respectively, was determined from five randomly selected fields.
Flow Cytometry
Cells were incubated with Ab to PD-L1 (BioLegend, San Diego, CA, USA) for 30 min on ice and analyzed using a fluorescence-activated cell sorting (FACS) Array (BD Biosciences, San Jose, CA, USA).
ATP and HMGB1 Assays
CT26 and PAN02 cells were treated with OBP-502 (0, 500, and 1,000 MOI) for 24 h (n = 5), after which levels of extracellular ATP and HMGB1 in the supernatants were measured using an ENLITEN ATP assay (Promega, Madison, WI, USA) and HMGB1 ELISA Kit II (Shino-Test, Kanagawa, Japan), respectively, according to the manufacturers’ protocols.
Multi-cytokine and Chemokine Assays
CT26 and PAN02 cells were treated with OBP-502 (0, 500, and 1,000 MOI) for 24 h (n = 5), after which various cytokines and chemokines in the supernatants were measured using a mouse cytokine array (R&D Systems, Minneapolis, MN, USA) and CCL5/RANTES ELISA kit (R&D Systems), respectively, according to the manufacturers’ protocols.
Western Blot Analysis
Proteins extracted from whole-cell lysates were electrophoresed on 10%–15% SDS-polyacrylamide gels and transferred onto Hybond-polyvinylidene difluoride transfer membranes (GE Healthcare UK, UK). The membranes were incubated with primary Abs against adenovirus type 5 E1A (BD Biosciences, Franklin Lakes, NJ, USA), poly(ADP ribose) polymerase (PARP) (Cell Signaling Technology), p62 (SQSTM1) (Medical & Biological Laboratories [MBL], Nagoya, Japan), PD-L1 (Abcam, Cambridge, MA, USA), and β-Actin (Sigma-Aldrich, Saint Louis, MO, USA), followed by peroxidase-linked secondary Ab. The Amersham ECL chemiluminescence system (GE Healthcare UK) was used to detect the peroxidase activity of the bound Ab. Equal loading of samples was confirmed using β-Actin.
In Vivo Experiments
CT26 cells (1 × 106 cells) were subcutaneously injected into the flanks of 6-week-old female BALB/c mice and BALB/c nude mice, and PAN02 cells (1 × 106 cells) were subcutaneously injected into the flanks of 6-week-old female C57BL/6 mice. Treatment was initiated when tumors reached a diameter of approximately 5 mm. The perpendicular diameter of each tumor was then measured twice per week, and tumor volume was calculated using the following formula: tumor volume (mm3) = a × b2 × 0.5, where a represents the longest diameter, b represents the shortest diameter, and 0.5 is a constant used to calculate the volume of an ellipsoid.
To evaluate the effect of OBP-502 on TILs, we treated CT26 and PAN02 subcutaneous tumors intratumorally with OBP-502 (1 × 109 PFUs) or PBS three times per week. The mice were sacrificed 7 days after initiation of treatment to investigate the effect on TILs.
CT26 and PAN02 subcutaneous tumors were treated intratumorally with OBP-502 injection (1 × 109 PFUs) and intraperitoneal PD-1 Ab injection (1st: 20 mg/kg, 2nd/3rd: 10 mg/kg) three times each week,49 and tumor volume was monitored until day 28 to assess the therapeutic efficacy of OBP-502 and PD-1 Ab combination therapy. Tumor-free mice cured after combination therapy were subcutaneously re-challenged with inoculation of CT26 cells (5 × 105 cells).
CT26 and PAN02 bilateral subcutaneous tumor models were prepared in BALB/c mice or BALB/c nude mice (for CT26) and C57BL/6 mice (for PAN02) to investigate abscopal effects of OBP-502 alone or in combination therapy with PD-1 Ab. One side was treated with OBP-502 intratumorally three times each week, and the other side was left untreated. PD-1 Ab was administered intraperitoneally three times each week. Tumor volume was monitored until days 21–24.
To establish an orthotopic rectal tumor model with liver metastases, we inoculated CT26 cells stably expressing luciferase (CT26-Luc) into the submucosal layer of the rectum (1 × 106 cells) of BALB/c mice, followed by injection of 5 × 105 cells via the portal vein 2 days after rectum inoculation. Mice were treated with OBP-502 intratumorally and PD-1 Ab intraperitoneally three times each week starting 5 days after portal vein injection. Tumor growth was monitored using an IVIS imaging system (Xenogen), and survival was assessed by Kaplan-Meier analysis.
Mice were housed in a specific pathogen-free environment in the Department of Animal Resources of Okayama University. All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of Okayama University.
CTL Assay
The GFP-expressing murine colon cancer cell line Colon26-GFP (1 × 106 cells), kindly provided by Dr. Hoffmann of AntiCancer (San Diego, CA), was subcutaneously injected into the flanks of 6-week-old female BALB/c mice. Colon26-GFP subcutaneous tumors were treated intratumorally with PBS or OBP-502 injection (1 × 109 PFUs) three times a week, and the mice were sacrificed 7 days after initiation of treatment (n = 3). Splenocytes, collected by homogenization of the spleen, were treated by red blood cell (RBC) lysis buffer (BioLegend) and debris removal solution kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Then splenocytes were co-incubated with Colon26-GFP cells, which were irradiated with 100 Gy and incubated with INF-γ (100 U/mL) (Wako Pure Chemical Industries, Osaka, Japan) for 5 days for stimulation. After CD8-positive selection by CD8 (TIL) MicroBeads (Miltenyi Biotec) and LS Columns (Miltenyi Biotec), Colon26-GFP cells (target) were incubated with the CD8-positive cells (Effector) for 4 h at 37°C at different effector/target ratios (0:1, 10:1, and 20:1). After staining with Zombie NIR (BioLegend) to label dead cells, cells were analyzed with FACS Array (BD Biosciences).
Statistical Analysis
Statistical analysis was performed using JMP software (SAS Institute, Cary, NC, USA). Student’s t test was used to assess the significance of differences in most continuous variables, except for quantitative immunohistochemical analyses of CD8, CD4, Foxp3, and CD11c and analyses of tumor growth in the orthotopic tumor model, for which the Wilcoxon signed-rank test was used. The log rank test was used for Kaplan-Meier survival analyses. The p values <0.05 were considered significant.
Author Contributions
N.K. and S. Kuroda designed research studies. N.K., Y.K., K.K., T.T., M.H., T.M., T.K., and K.A. conducted experiments. S. Kuroda, S. Kikuchi, M.N., S. Kagawa, H.T., and T.F. interpreted the results. H.M. and Y.U. provided reagents. N.K. and S. Kuroda wrote the manuscript. All authors reviewed the manuscript.
Conflicts of Interest
Y.U. is President and CEO of Oncolys BioPharma, Inc., the manufacturer of OBP-301 (Telomelysin). H.T. and T.F. are consultants of Oncolys BioPharma, Inc. The other authors have no potential conflicts of interest to disclose.
Acknowledgments
The authors would like to thank Tomoko Sueishi and Tae Yamanishi for their excellent technical support. This work was supported by AMED (grant no. JP17lk0201023 to T.F.) and JSPS KAKENHI (grant no. JP16K19893 to S. Kuroda).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.01.003.
Supplemental Information
References
- 1.Sharma P., Allison J.P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen D.S., Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Weber J.S., D’Angelo S.P., Minor D., Hodi F.S., Gutzmer R., Neyns B., Hoeller C., Khushalani N.I., Miller W.H., Jr., Lao C.D. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 4.Horn L., Spigel D.R., Vokes E.E., Holgado E., Ready N., Steins M., Poddubskaya E., Borghaei H., Felip E., Paz-Ares L. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057) J. Clin. Oncol. 2017;35:3924–3933. doi: 10.1200/JCO.2017.74.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahmer J., Reckamp K.L., Baas P., Crinò L., Eberhardt W.E., Poddubskaya E., Antonia S., Pluzanski A., Vokes E.E., Holgado E. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang Y.K., Boku N., Satoh T., Ryu M.H., Chao Y., Kato K., Chung H.C., Chen J.S., Muro K., Kang W.K. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:2461–2471. doi: 10.1016/S0140-6736(17)31827-5. [DOI] [PubMed] [Google Scholar]
- 7.Haanen J.B.A.G. Converting Cold into Hot Tumors by Combining Immunotherapies. Cell. 2017;170:1055–1056. doi: 10.1016/j.cell.2017.08.031. [DOI] [PubMed] [Google Scholar]
- 8.Le D.T., Uram J.N., Wang H., Bartlett B.R., Kemberling H., Eyring A.D., Skora A.D., Luber B.S., Azad N.S., Laheru D. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma P., Allison J.P. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 10.Kroemer G., Galluzzi L., Kepp O., Zitvogel L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 11.Kepp O., Senovilla L., Vitale I., Vacchelli E., Adjemian S., Agostinis P., Apetoh L., Aranda F., Barnaba V., Bloy N. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology. 2014;3:e955691. doi: 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartlett D.L., Liu Z., Sathaiah M., Ravindranathan R., Guo Z., He Y., Guo Z.S. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer. 2013;12:103. doi: 10.1186/1476-4598-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zamarin D., Holmgaard R.B., Subudhi S.K., Park J.S., Mansour M., Palese P., Merghoub T., Wolchok J.D., Allison J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014;6:226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiocca E.A., Rabkin S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014;2:295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfirschke C., Engblom C., Rickelt S., Cortez-Retamozo V., Garris C., Pucci F., Yamazaki T., Poirier-Colame V., Newton A., Redouane Y. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity. 2016;44:343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golden E.B., Apetoh L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015;25:11–17. doi: 10.1016/j.semradonc.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Kawashima T., Kagawa S., Kobayashi N., Shirakiya Y., Umeoka T., Teraishi F., Taki M., Kyo S., Tanaka N., Fujiwara T. Telomerase-specific replication-selective virotherapy for human cancer. Clin. Cancer Res. 2004;10:285–292. doi: 10.1158/1078-0432.ccr-1075-3. [DOI] [PubMed] [Google Scholar]
- 18.Nemunaitis J., Tong A.W., Nemunaitis M., Senzer N., Phadke A.P., Bedell C., Adams N., Zhang Y.A., Maples P.B., Chen S. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol. Ther. 2010;18:429–434. doi: 10.1038/mt.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuroda S., Fujiwara T., Shirakawa Y., Yamasaki Y., Yano S., Uno F., Tazawa H., Hashimoto Y., Watanabe Y., Noma K. Telomerase-dependent oncolytic adenovirus sensitizes human cancer cells to ionizing radiation via inhibition of DNA repair machinery. Cancer Res. 2010;70:9339–9348. doi: 10.1158/0008-5472.CAN-10-2333. [DOI] [PubMed] [Google Scholar]
- 20.Tazawa H., Yano S., Yoshida R., Yamasaki Y., Sasaki T., Hashimoto Y., Kuroda S., Ouchi M., Onishi T., Uno F. Genetically engineered oncolytic adenovirus induces autophagic cell death through an E2F1-microRNA-7-epidermal growth factor receptor axis. Int. J. Cancer. 2012;131:2939–2950. doi: 10.1002/ijc.27589. [DOI] [PubMed] [Google Scholar]
- 21.Pai S.G., Carneiro B.A., Mota J.M., Costa R., Leite C.A., Barroso-Sousa R., Kaplan J.B., Chae Y.K., Giles F.J. Wnt/beta-catenin pathway: modulating anticancer immune response. J. Hematol. Oncol. 2017;10:101. doi: 10.1186/s13045-017-0471-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Comerford I., McColl S.R. Mini-review series: focus on chemokines. Immunol. Cell Biol. 2011;89:183–184. doi: 10.1038/icb.2010.164. [DOI] [PubMed] [Google Scholar]
- 23.Zlotnik A., Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 24.Luheshi N.M., Coates-Ulrichsen J., Harper J., Mullins S., Sulikowski M.G., Martin P., Brown L., Lewis A., Davies G., Morrow M., Wilkinson R.W. Transformation of the tumour microenvironment by a CD40 agonist antibody correlates with improved responses to PD-L1 blockade in a mouse orthotopic pancreatic tumour model. Oncotarget. 2016;7:18508–18520. doi: 10.18632/oncotarget.7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naidoo J., Page D.B., Li B.T., Connell L.C., Schindler K., Lacouture M.E., Postow M.A., Wolchok J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015;26:2375–2391. doi: 10.1093/annonc/mdv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang J., Shalabi A., Hubbard-Lucey V.M. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 2018;29:84–91. doi: 10.1093/annonc/mdx755. [DOI] [PubMed] [Google Scholar]
- 27.Wolchok J.D., Chiarion-Sileni V., Gonzalez R., Rutkowski P., Grob J.J., Cowey C.L., Lao C.D., Wagstaff J., Schadendorf D., Ferrucci P.F. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017;377:1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Motzer R.J., Tannir N.M., McDermott D.F., Arén Frontera O., Melichar B., Choueiri T.K., Plimack E.R., Barthélémy P., Porta C., George S., CheckMate 214 Investigators Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018;378:1277–1290. doi: 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gandhi L., Rodríguez-Abreu D., Gadgeel S., Esteban E., Felip E., De Angelis F., Domine M., Clingan P., Hochmair M.J., Powell S.F., KEYNOTE-189 Investigators Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 30.Marshall H.T., Djamgoz M.B.A. Immuno-Oncology: Emerging Targets and Combination Therapies. Front. Oncol. 2018;8:315. doi: 10.3389/fonc.2018.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 32.Ribas A., Dummer R., Puzanov I., VanderWalde A., Andtbacka R.H.I., Michielin O., Olszanski A.J., Malvehy J., Cebon J., Fernandez E. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017;170:1109–1119.e10. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coxsackievirus A21 Synergizes with Checkpoint Inhibitors. Cancer Discov. 2017;7:OF9. doi: 10.1158/2159-8290.CD-NB2017-051. [DOI] [PubMed] [Google Scholar]
- 34.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J.M., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Endo Y., Sakai R., Ouchi M., Onimatsu H., Hioki M., Kagawa S., Uno F., Watanabe Y., Urata Y., Tanaka N., Fujiwara T. Virus-mediated oncolysis induces danger signal and stimulates cytotoxic T-lymphocyte activity via proteasome activator upregulation. Oncogene. 2008;27:2375–2381. doi: 10.1038/sj.onc.1210884. [DOI] [PubMed] [Google Scholar]
- 36.Francis L., Guo Z.S., Liu Z., Ravindranathan R., Urban J.A., Sathaiah M., Magge D., Kalinski P., Bartlett D.L. Modulation of chemokines in the tumor microenvironment enhances oncolytic virotherapy for colorectal cancer. Oncotarget. 2016;7:22174–22185. doi: 10.18632/oncotarget.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jonas B.A. Combination of an oncolytic virus with PD-L1 blockade keeps cancer in check. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aan2781. eaan2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harlin H., Meng Y., Peterson A.C., Zha Y., Tretiakova M., Slingluff C., McKee M., Gajewski T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartman Z.C., Black E.P., Amalfitano A. Adenoviral infection induces a multi-faceted innate cellular immune response that is mediated by the toll-like receptor pathway in A549 cells. Virology. 2007;358:357–372. doi: 10.1016/j.virol.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 40.Ehlers G., Fridman M. Abscopal effect of radiation in papillary adenocarcinoma. Br. J. Radiol. 1973;46:220–222. doi: 10.1259/0007-1285-46-543-220. [DOI] [PubMed] [Google Scholar]
- 41.Rodríguez-Ruiz M.E., Vanpouille-Box C., Melero I., Formenti S.C., Demaria S. Immunological Mechanisms Responsible for Radiation-Induced Abscopal Effect. Trends Immunol. 2018;39:644–655. doi: 10.1016/j.it.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Postow M.A., Callahan M.K., Barker C.A., Yamada Y., Yuan J., Kitano S., Mu Z., Rasalan T., Adamow M., Ritter E. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012;366:925–931. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hiniker S.M., Chen D.S., Knox S.J. Abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012;366:2035. doi: 10.1056/NEJMc1203984. author reply 2035–2036. [DOI] [PubMed] [Google Scholar]
- 44.Ricca J.M., Oseledchyk A., Walther T., Liu C., Mangarin L., Merghoub T., Wolchok J.D., Zamarin D. Pre-existing Immunity to Oncolytic Virus Potentiates Its Immunotherapeutic Efficacy. Mol. Ther. 2018;26:1008–1019. doi: 10.1016/j.ymthe.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Topalian S.L., Taube J.M., Anders R.A., Pardoll D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mimura K., Teh J.L., Okayama H., Shiraishi K., Kua L.F., Koh V., Smoot D.T., Ashktorab H., Oike T., Suzuki Y. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109:43–53. doi: 10.1111/cas.13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Der Kraak L., Goel G., Ramanan K., Kaltenmeier C., Zhang L., Normolle D.P., Freeman G.J., Tang D., Nason K.S., Davison J.M. 5-Fluorouracil upregulates cell surface B7-H1 (PD-L1) expression in gastrointestinal cancers. J. Immunother. Cancer. 2016;4:65. doi: 10.1186/s40425-016-0163-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dovedi S.J., Cheadle E.J., Popple A.L., Poon E., Morrow M., Stewart R., Yusko E.C., Sanders C.M., Vignali M., Emerson R.O. Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by Both Resident and Infiltrating Polyclonal T-cell Populations when Combined with PD-1 Blockade. Clin. Cancer Res. 2017;23:5514–5526. doi: 10.1158/1078-0432.CCR-16-1673. [DOI] [PubMed] [Google Scholar]
- 49.Terawaki S., Chikuma S., Shibayama S., Hayashi T., Yoshida T., Okazaki T., Honjo T. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J. Immunol. 2011;186:2772–2779. doi: 10.4049/jimmunol.1003208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.