

Abstract
Esophageal squamous cell carcinoma (ESCC) is a predominant cancer type in developing countries such as China, where ESCC accounts for approximately 90% of esophageal malignancies. Lacking effective and targeted therapy contributes to the poor 5-year survival rate. Recent studies showed that about 30% of ESCC cases have high levels of SOX2. Herein, we aim to target this transcription factor with aptamer. We established a peptide aptamer library and then performed an unbiased screening to identify several peptide aptamers including P42 that can bind and inhibit SOX2 downstream target genes. We further found that P42 overexpression or incubation with a synthetic peptide 42 inhibited the proliferation, migration, and invasion of ESCC cells. Moreover, peptide 42 treatment inhibited the growth and metastasis of ESCC xenografts in mouse and zebrafish. Further analysis revealed that P42 overexpression led to alternations in the levels of proteins that are important for the proliferation and migration of ESCC cells. Taken together, our study identified the peptide 42 as a key inhibitor of SOX2 function, reducing the proliferation and migration of ESCC cells in vitro and in vivo, and thereby offering a potential therapy against ESCC.
Keywords: esophageal squamous cell carcinoma, SOX2, peptide aptamers, peptide, drug screen
Graphical Abstract
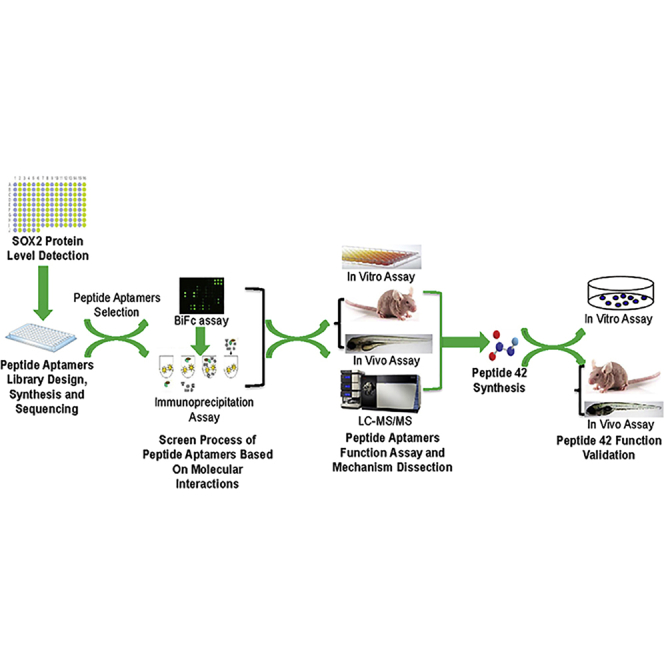
Liu and colleagues found that SOX2 protein is aberrantly expressed in ESCC clinical samples and cells, specific peptide aptamer and peptide against SOX2 are successfully obtained after library screen and synthesis, and their inhibitive roles in malignant processes are validated in vitro and in vivo, thereby providing a potential therapy against ESCC.
Introduction
Esophageal squamous cell carcinoma (ESCC) is one of the leading causes of cancer-related deaths worldwide and is the predominant histological subtype of esophageal cancer in China that leads to 90% of deaths among all esophageal cancer cases.1 Lack of early detection biomarkers and efficient treatments are considered to be the major contributors to this dismal outcome. Previous studies suggest that environmental (e.g., smoking, drinking, diet) and genetic factors promote malignancy development.1, 2, 3, 4, 5, 6 Recent progress in the study of genetic alternations in ESCC has brought new mechanistic insights.7 Exome sequencing results suggest that mutation of several genes involved in cell-cycle regulation (e.g., CDKN2A), apoptosis (e.g., TP53), and histone modification (e.g., KMT2D) is relatively common in ESCC. Genetic mutations can also lead to dysregulation of signaling pathways like Hippo and Notch.8 For example, the receptor NOTCH1 is a relatively commonly mutated gene, especially in Chinese ESCC patients.9 Interestingly, the transcription factor SOX2 is also commonly mutated in ESCCs with ∼25% samples showing genetic amplification.10,11 We previously used genetic mouse models to identify a novel mechanism by which SOX2 cooperates with inflammatory signals to transform basal stem cells in the esophagus, leading to ESCC initiation and progression.10
SOX2 belongs to the sex determining region Y (SRY)-like box (SOX) gene family. Previous studies have shown that SOX2 is important for the development and stem cell maintenance in the developing and adult esophagus.10,12 Consistently, SOX2 was found to repress respiratory fate and promote esophageal specification in part by suppressing the Wnt signaling pathway as evidenced by 3D organoid culture of human pluripotent stem cell-derived foregut progenitor cells.13,14 Notably, high levels of SOX2 protein have also been found in multiple malignancies, including lung cancer and breast cancer, and the levels are closely associated with clinical outcome.10,11,15, 16, 17 Further studies suggested that high levels of SOX2 promote multiple steps during cancer development, including initiation,18 maintenance,19,20 invasion, and metastasis.15,21 Therefore, targeting SOX2 protein can be explored for a potential therapeutic option for ESCC treatment.
Here, we aim to target SOX2 protein by using aptamer. We performed unbiased screening through bimolecular fluorescence complementation (BiFc) and identified several SOX2 binding aptamers. We then used in vitro and in vivo assays to test the efficacy of these candidates with a focus on an aptamer (aptamer P42). Our study further showed that P42 aptamer inhibited ESCC proliferation and migration, resulting in reduced tumor growth and metastasis in both mouse and zebrafish models. In addition, these tumor inhibitory effects are associated with changes in subsets of proteins as revealed by proteomics analysis.
Results
High Levels of SOX2 Are Closely Correlated with Poor Prognosis of ESCC
We previously showed that a significant portion of ESCCs express high levels of SOX2 in a relatively small number of human patients.10 In this study, we assessed the correlation with a larger collection of ESCCs. We examined SOX2 expression in an ESCC tissue microarray containing 75 cases and matched adjacent normal tissues (Figures 1A and 1B). We found that the levels of SOX2 were significantly higher in ESCCs than the adjacent tissues (n = 75; p < 0.001; Figure 1C). The majority of SOX2high ESCCs were from patients with diagnosis of cancer stage N and histopathological grade II (Table S1), suggesting high levels of SOX2 protein are correlated with aggressive cancer stage. In addition, we also detected low levels of SOX2 protein in HEEC and high levels in the immortalized human esophageal epithelial progenitor cell line EPC2 cells and ESCC lines (KYSE450, TE-1) (Figures 1D and 1E).
Figure 1.

The Levels of SOX2 Are Increased in ESCC Samples and Cell Lines
(A) Increased levels of SOX2 protein in ESCC samples (C) versus controls (CA) in tissue microarray. (B) A representative of ESCC/control pair. (C) The levels of SOX2 protein in ESCC cancer tissue are significantly higher than adjacent normal tissues (n = 75; p < 0.001). (D) SOX2 is enriched in ESCC cell lines (KYSE450, TE-1) and immortalized esophageal progenitor cell line (EPC2). Note SOX2 levels are relatively low in an esophageal epithelial cell line (HECC). Scale bars: 100 μm. (E) The levels of SOX2 protein were quantitated with western blot analysis. *p < 0.05, **p < 0.01, ***p < 0.001 versus control. Data are means ± SD.
Identification of Candidate Aptamers that Bind SOX2 through Screening a Peptide Aptamer Library
Aptamers have been shown to efficiently target proteins that are crucial for tumorigenesis.22, 23, 24 We asked whether we can target SOX2 with a similar strategy. We established an aptamer library based on BiFc,25 which allowed us to identify aptamers that bind SOX2 (Figures 2A and S1A). pBiFc-VC155-TrxA-MCS (multiple cloning site)-TrxA vector was generated and confirmed by restriction enzyme digestion and sequencing (Figure S1B). Subsequently, a library containing random nucleotide sequences coding for 33 peptide aptamers was synthesized and amplified (Figure S1C). The amplified sequences were then inserted into the construct pBiFc-VC155-TrxA-MCS-TrxA to generate the peptide aptamer library pBiFc-VC155-TrxA-peptide-TrxA. This library was subsequently used for peptide aptamer expression and BiFc screening after validation with bacterial liquid PCR and sequencing (Figure S1D).
Figure 2.

Establishment of a Peptide Aptamer Library and Screening for Specific Peptides Aptamers that Target SOX2 Protein
(A) Schematic of strategy generating peptide aptamer library. The vector expressing peptide aptamers was inserted, pBiFc-VC155-TrxA-MCS-TrxA, to establish the aptamer library, pBiFc-VC155-TrxA-peptide-TrxA, and SOX2-VENUS1-172 fusion protein is expressed with pBiFc-VN173-SOX2. If SOX2 binds potential aptamer, GFP will be produced based on BiFc and visualized with fluorescent microscope. (B) BiFc-based peptide aptamers P15, P18, P32, and P42 specifically bind SOX2 protein and generate green fluorescent signals in HEK293T cells. Scale bars: 50 μm. (C) Peptide aptamers P15, P18, and P42, but not P32, bind SOX2 protein as validated by co-immunoprecipitation. (D) The amino acid and nucleic acid sequences of peptide aptamers that specifically bind SOX2 protein.
Following co-transfection of the pBiFc-VC155-TrxA-peptide-TrxA aptamer library with pBiFc-VN173-SOX2, we found that the peptide aptamers P15, P18, P32, and P42 interact with SOX2 protein in the nucleus of HEK293T cells (Figure 2B). We then built FLAG- and Myc-tagged SOX2 constructs to further examine the interaction between SOX2 and the candidate aptamers (Figure S1E). Immunoprecipitation was performed and demonstrated that tagged SOX2 proteins interact with the peptide aptamers P15, P18, and P42, but not P32 (Figures 2C and 2D).
P42 Aptamer Suppresses the Proliferation, Migration, and Invasion of ESCC Cell Lines
To analyze whether the peptides aptamers screened from the library play inhibitory roles in ESCC cell lines, we established lentiviral vectors that allow overexpression of the peptide aptamers P15, P18, and P42 (Figure S2; Table S2). Two ESCC cell lines (KYSE450 and TE-1) and two normal esophageal cell lines (HEEC and EPC2) were infected with lentivirus carrying the overexpression constructs, and stable cell lines were established (Figure S3). We also generated stable cell lines harboring pCDH-CMV-Oligo-IRES-GFP-Puro as controls.
We next tested whether ectopic expression of the three peptide aptamers influences colony formation. Interestingly, ectopic expression of the peptide aptamers did not significantly affect colony formation efficiency (n = 3; p > 0.05; Figure 3A). Nevertheless, the number of colonies with a diameter greater than 0.5 mm was significantly reduced for KYSE450 cells that overexpress aptamers P15 and P42, but not P18, when compared with control (n = 3; p < 0.05 for P15 aptamer, p < 0.001 for P42 aptamer, and data not shown; Figure 3A). Consistently, overexpression of aptamers P15 and P42, but not P18, reduced the proliferation of KYSE450 cells as assessed by Cell Counting Kit-8 (CCK8) assay (n = 3; p < 0.05; Figure 3B). In addition, overexpression of P42 aptamers also led to significantly reduced proliferation of TE-1 cells at day 7 (n = 3; p < 0.001; Figure 3B).
Figure 3.

P42 Aptamer Inhibits the Proliferation of ESCC Cells In Vitro
(A) Ectopic expression of aptamers P42, P15, and P18 does not affect the colony numbers formed by KYSE450. However, the numbers of colonies with size larger than 0.5 mm are significantly reduced by aptamers P42 and P15, but not P18 (p < 0.05 for P15 aptamer and p < 0.001 for P42 aptamer; n = 3). Although it is statistically not significant, ectopic expression of P42 aptamer reduces the growth of colonies (size larger than 0.5 mm) formed by TE-1 cells (p > 0.05; n = 3). (B) Ectopic expression of aptamer P42 leads to reduced proliferation of KYSE450 and TE-1 cells as assessed by CCK8 assay (n = 3 per group; p < 0.05 for aptamers P15 and P42 in KYSE450 cells, p < 0.001 for aptamer P42 in TE-1 cells).
Next, we asked whether overexpression of these peptide aptamers affects cancer cell migration and invasion. Significantly, overexpression of P42 aptamer inhibited the migration of KYSE450 (n = 3; p < 0.05; Figure 4A). In keeping with this finding, P42 aptamer overexpression inhibits cell migration in wound-healing assay (79.9% [control] versus 56.11%; n = 3; p < 0.05; Figure 4A). Notably, overexpression of P18 increased the cell migration (79.9% [control] versus 97.23%; n = 3; p < 0.05; Figure 4A). In addition, P42 aptamer overexpression also led to reduced migration of TE-1 cells, and the healing index was reduced from 84.5% (control) to 56.55% (n = 3; p < 0.001; Figure 4B). We further tested whether overexpression of P42 aptamer inhibited the invasion of KYSE450 cells using transwell assay. The numbers of KYSE450 cells passing through the transwell membrane were significantly reduced upon ectopic expression of P42 aptamer (n = 3; p < 0.01; Figure 4C). Similarly, ectopic expression of P42 aptamer reduced the invasion of TE-1 (n = 3; p < 0.001; Figure 4D). Together these findings suggest that among the three aptamers, P42 aptamer shows consistent inhibitory effects on the proliferation and migration of ESCC cell lines upon overexpression.
Figure 4.

P42 Aptamer Inhibits the Migration and Invasion of ESCC Cells In Vitro
(A and B) Ectopic expression of P42 aptamer inhibits the migration of (A) KYSE450 and (B) TE-1 cells as revealed by wound-healing assay (n = 3 per group; p < 0.05 for KYSE450 and p < 0.001 for TE-1 cells). Note P18 overexpression promotes the migration of KYSE450 cells. Scale bars: 500 μm. (C and D) P42 aptamer overexpression suppresses the invasion of (C) KYSE450 and (D) TE-1 cells in transwell assay (n = 3 per group; p < 0.01 for KYSE450 and p < 0.001 for TE-1 cells). Scale bars: 100 μm. *p < 0.05, **p < 0.01, ***p < 0.001 versus control. Data are means ± SD.
P42 Aptamer Treatment Reduced the Growth and Metastasis of ESCC Xenografts in Mouse and Zebrafish Models
We next asked whether ectopic expression of P42 peptide aptamers reduces tumor growth with using a xenograft model. KYSE450 cells expressing P42 aptamers were inoculated into immunocompromised nude mice. Ectopic expression of P42 aptamers reduced the tumor weight by 86% (0.37 ± 0.1 g versus 0.05 ± 0.045 g; n = 3, p < 0.05; Figures 5A–5C). To test whether ectopic expression of P42 aptamer can also block metastasis of ESCC cells in vivo, we injected the perivitelline space of zebrafish embryos with KYSE450 cells that overexpress P42 aptamer. Ectopic expression of P42 aptamer resulted in a significant decrease in the numbers of metastatic foci (14 ± 4 versus 5 ± 1; n = 5, p < 0.05; Figures 5D and 5E). Together these results suggest that ectopic P42 aptamer reduced ESCC metastasis in vivo.
Figure 5.

Ectopic Expression of P42 Aptamer Suppresses the Growth of Xenografts in Mice and Metastasis of KYSE450 Cells in Zebrafish
(A and B) Representative tumors formed in mice following injection with KYSE450 cells that stably express P15, P18, or P42 aptamers. (C) Ectopic expression of P42 aptamer significantly reduces the weight of xenografts initiated by KYSE450 cells more than other peptide aptamers (n = 3 per group; p < 0.05). (D and E) P42 aptamer overexpression significantly inhibits the metastasis of KYSE450 cells more than control peptide aptamer along the body of zebrafish. Metastatic foci were examined with fluorescent microscopy (n = 5 per group; p < 0.05). *p < 0.05, **p < 0.01, ***p < 0.001 versus control. Data are means ± SD.
Protein Profile of KYSE450 Cells following Ectopic Expression of P42 Aptamer
To better understand the underlying mechanism of aptamer-induced tumor inhibitory effects, we performed proteomics to globally survey changes in the levels of protein upon P42 aptamer overexpression in KYSE450 cells. A total of 133 proteins were upregulated and 99 proteins were downregulated in response to ectopic expression of P42 aptamer. Further analysis revealed that the levels of 34 and 24 proteins were increased and decreased by more than 1.5-fold, respectively (Figures 6A and 6B). Notably, SQRDL and MAU2 are the most dramatically upregulated and downregulated proteins among these changed proteins, respectively. Moreover, Gene Ontology (GO) analysis indicated significant changes in multiple biological processes, including cellular, single-organism, biological regulation, metabolic process, and response to stimulus. Notably, a significant number of proteins have been previously associated with binding, catalytic activity, and molecular function regulation (Table S3). In addition, the majority of upregulated proteins (e.g., IFI35) were previously located and shown to be enriched in the cytoplasm,26 whereas the majority of downregulated proteins (e.g., ZNF330) were located in the nucleus27 (Figure 6C). Cellular component-based clustering analysis revealed that downregulated proteins include several major components of cell and plasma membrane (e.g., SLC38A1), cell surface (e.g., LY6K), cell periphery (e.g., SLC7A11), extracellular matrix component (e.g., FREM1), and nucleosome (e.g., H1FX). By contrast, ectopic P42 aptamer induced upregulation of mast cell granule and secretory granule, extracellular exosome, and some vesicles including extracellular and secretory vesicles.
Figure 6.

P42 Aptamer Overexpression Induces Changes in the Levels of Proteins that Have Been Implicated in Cancer Progression
(A) Proteins that either increase or decrease by >1.5-fold upon ectopic expression of P42 aptamer in KYSE450 cells. (B) Top 15 proteins that are either upregulated or downregulated upon P42 aptamer ectopic expression. (C) Subcellular localization of the upregulated and downregulated proteins upon P42 aptamer ectopic expression.
P42 aptamer overexpression seemed to lead to changes in the levels of proteins essential for tumor growth. For example, downregulated proteins include those involved in multiple steps of tumorigenesis, e.g., cell migration (SLC7A11), cell growth (CPNE1), and cell proliferation (CCND1). In addition, the protein levels of cell junction proteins (CAV1) and growth factors (DOCK10) are also downregulated. By contrast, upregulated proteins include those involved in cell differentiation (DICER1), adhesion (CEACAM5), and activation (IGBP1). Kyoto Encyclopedia of Genes and Genomes (KEGG)-based clustering analysis demonstrated that ectopic P42 aptamer induced downregulation of proteins pertinent to the metabolism of arachidonic acid, tryptophan, and glutamate. The proteomic dataset also suggests that the glycolysis and gluconeogenesis pathways were suppressed following aptamer overexpression. Together these findings are consistent with the reduced growth of cancer cells upon aptamer treatment.
Synthetic Peptide 42 Also Shows Tumor Inhibitory Effects in ESCC Cells Both In Vitro and In Vivo
We next synthesized a peptide 42 (sP42) and control peptide, both including a cell-penetrating peptide (YGRKKRRQRRR) and a fluorescent molecule TAMRA. The main structure is similar to P42 aptamer identified in the peptide aptamer library. The two synthetic peptides (sP42 and control) also contain a constrained scaffold structure in addition to a disulfide bond between the two cysteines (Table S4).
Similar to P42 aptamer, treatment with the sP42 did not significantly affect the colony-forming efficiency in KYSE450 cells (n = 3; p > 0.05; Figure 7A). However, the number of colonies with a diameter of greater than 0.5 mm was reduced (n = 3; p < 0.001; Figure 7A). Similar results were obtained when TE-1 cells were cultured with sP42 (data not shown). Further analysis revealed that sP42 treatment significantly reduced the proliferation of KYSE450 and TE-1 cells at day 5 (n = 3; p < 0.01 for KYSE450 cells and p < 0.001 for TE-1 cells; Figure 7B). Notably, the inhibitory effect is comparable with SOX2 knockdown in KYSE450 and TE-1 cells (Figure 7B). Moreover, sP42 treatment also inhibited the migration of KYSE450 and TE-1 cells, and the healing index was decreased from 84.4% to 20.9% and from 93.9% to 54.6%, respectively, as revealed by the wound-healing assay (n = 3; p < 0.001 for KYSE450 cells and p < 0.01 for TE-1 cells; Figure 7C). Similar to P42 aptamer, sP42 also inhibited the invasion of KYSE450 and TE-1 cells as assessed with transwell assay. The number of KYSE450 and TE-1 cells that passed through the transwell membrane was dramatically reduced after incubating with sP42 (n = 3; p < 0.001; Figure 7D). Together these results suggest that the synthetic peptide 42 exhibits inhibitory effects on the proliferation, migration, and invasion of cultured ESCC cells, similar to what have been observed with using P42 aptamer.
Figure 7.

Synthetic Peptide 42 (sP42) Inhibits the Proliferation, Migration, and Invasion of ESCC Cells In Vitro
(A) sP42 application does not lead to reduction in the total number of KYSE450 colonies (p > 0.05), but the numbers of colonies larger than 0.5 mm are significantly reduced (n = 3 per group; p < 0.001). (B) Addition of sP42 to culture medium significantly reduces the proliferation of KYSE450 and TE-1 cells (n = 3 per group; p < 0.05 for KYSE450 cells and p < 0.001 for TE-1 cells). Note that the efficacy is comparable with SOX2 knockdown in KYSE450 and TE-1 cells (n = 3 per group; p < 0.05 for KYSE450 cells). (C) sP42 application leads to reduced migration of KYSE450 and TE-1 cells (n = 3 per group; p < 0.001 for KYSE450 cells and p < 0.01 for TE-1 cells). Scale bars: 500 μm. (D) sP42 application reduces the invasion of KYSE450 and TE-1 cells (n = 3 per group; p < 0.001 for KYSE450 cells and TE-1 cells). Scale bars: 100 μm. *p < 0.05, **p < 0.01, ***p < 0.001 versus control. Data are means ± SD.
We next assessed whether sP42 also reduced the growth of ESCC xenografts. We found that sP42 treatment led to the reduction of tumor weight from 0.58 ± 0.02 to 0.15 ± 0.04 g (n = 4; p < 0.01; Figures 8A–8C). Moreover, sP42 treatment promoted tumor cell differentiation, resulting in increased keratin pearls and intercellular bridges (Figure 8D). sP42 treatment also inhibited the metastasis of KYSE450 cells in the zebrafish xenograft model (n = 5; p < 0.01; Figure 8E). Consistent with sP42 inhibitory effects on cell proliferation and invasion, the protein levels of cell-cycle proteins (cyclin-dependent kinase 4 [CDK4], CCND1) and the metallopeptidase protein MMP8 were decreased upon treatment with sP42. Of note is that the levels of SOX2 protein were not dramatically changed, suggesting that binding of sP42 and SOX2 did not affect SOX2 protein stability (Figure 8F).
Figure 8.

sP42 Treatment Reduces the Growth of Xenografts Initiated by KYSE450 Cells and Inhibits the Metastasis of ESCC Cells in Zebrafish
(A and B) sP42 treatment dramatically reduces the growth of xenografts than control peptide. sP42 was injected into xenografts every 2 days for 10 times. (C) sP42 treatment reduces the growth of KYSE450 cell-initiated xenograft more than control peptide (n = 4 per group; p < 0.01). (D) sP42 application promotes terminal differentiation of cancer cells with the increased presence of keratin pearls and intercellular bridges. Scale bars: 100 μm. (E) sP42 treatment significantly inhibits the metastasis of KYSE450 cells along the axis of zebrafish (n = 5 per group; p < 0.01). (F) sP42 treatment leads to reduced protein levels of CCND1, CDK4, and MMP8, but not SOX2 protein. *p < 0.05, **p < 0.01, ***p < 0.001 versus control. Data are means ± SD.
Discussion
SOX2 is involved in multiple steps of cancer development, including initiation, proliferation, and metastasis. Increased levels of SOX2 are also correlated with therapeutic resistance and stemness maintenance. Therefore, targeting SOX2 protein or its function offers an attractive option for blocking cancer formation at various stages. Here, we identified multiple aptamers that interact with SOX2 and block its function, leading to reduced growth of ESCC cells and xenografts. In addition, our synthetic aptamer sP42 exhibits similar tumor inhibitory effects in both mouse and zebrafish xenograft models. Further analysis revealed that aptamer treatments altered the levels of multiple proteins that are important for the growth, migration, and invasion of cancer cells.
Peptide aptamers have been found to effectively disrupt the function of target proteins, and they are used for inhibiting proteins such as CDK2,28 Nr13,29 and mutant p53.30 Inhibition of heat shock protein 27 with aptamer also leads to inhibition of tumor progression.31 Moreover, several peptides have also been tested as anti-cancer drugs, and they showed specific binding to their target proteins or protein complexes including Bax,32 the NOTCH transcription complex,33 and YAP.34 Importantly, several aptamer peptides including buserelin, AN-152, and dactinomycin are currently tested in clinics for treating multiple cancers.35 Herein we first demonstrated that SOX2 is amplified in ESCC biopsies, which is consistent with our previous findings.10 We then searched for aptamers that can bind SOX2, and we built an aptamer library based on the BiFc methodology that allows us to identify SOX2 binding aptamers. Initially, we found that peptide aptamers P15, P18, and P42 can interact with SOX2 protein in the nuclei of cells, and we validated the interaction with immunoprecipitation assay. We chose P42 for further characterization of its function in tumor inhibition.15 We found that ectopic expression of P42 aptamer and synthetic P42 aptamer led to inhibition of multiple malignancy processes, including proliferation, migration, and invasion.
Our xenograft models further confirmed that aptamer P42 inhibits tumor growth and metastasis, presumably through inhibiting SOX2 function as its downstream targets, such as CDK4,15,36 cyclin D1,15,37 and MMP8, were reduced. Our further analysis with proteomics suggests that aptamer P42 treatments resulted in decreased levels of many proteins involved in cell proliferation, migration, and invasion. Notably, the levels of SQRDL and MAU2 were the most dramatically upregulated and downregulated proteins upon P42 aptamer treatment, respectively. SQRDL has been shown to regulate sulfur metabolism and is a key component of the hydrogen sulfide oxidation pathway in the human mitochondria.38 SQRDL is downregulated in colorectal cancer and non-small cell lung cancer (NSCLC),39 suggesting that SQRDL plays an inhibitory role in cancer progression. On the other hand, the chromatid cohesion factor homolog MAU2 forms a heterodimeric complex with Nipped-B-like protein (Nipbl) and is critical for loading the cohesin onto chromatin during G1/S phase in yeast and telophase in mammals.40 This process is important for maintaining proper chromatid cohesion, chromatin structure, and accurate chromosome segregation.41, 42, 43 MAU2 can also be directly recruited to DNA damage sites and involved in DNA repair,44 suggesting that MAU2 are correlated with cancer progression. Notably, ectopic expression of P42 aptamer does not affect the proliferation of the esophageal epithelium HEEC cells and human esophageal progenitor EPC2 cells (Figures S4A and S4B). Similarly, we did not observe abnormalities when P42 aptamer is overexpressed in EPC2 cells (Figure S4C). This is consistent with findings that the aptamers do not interfere with the protein levels of SOX2, and these findings also suggest that normal esophageal epithelium and ESCC cell have distinct proliferation mechanisms.
In summary, our findings supported that SOX2 protein can be an effective therapeutic target. Both the aptamer P42 and synthetic P42 display apparent inhibitory effects on the growth and metastasis of ESCC cells in vitro and in vivo. Our results further demonstrate that the aptamers do not affect the normal epithelial cells while maintaining high efficacy against ESCCs.
Materials and Methods
Chemicals
FLAG immunoprecipitation kit (Catalog [Cat] No. FLAGIPT1) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Monoclonal antibody anti-Myc was purchased from Beyotime Biotechnology (Cat No. AM926; China). Monoclonal antibody anti-FLAG was purchased from Abmart company (Cat No. M20008; China). Monoclonal antibody anti-SOX2 was obtained from Cell Signaling Technology (Cat No. 4900; USA). Polyclonal rabbit anti-SOX2 was purchased from SEVEN HILLS (Cat No. WRAB-1236; Cincinnati, OH, USA). Monoclonal antibody anti-Ki67 was purchased from Cell Signaling Technology (Cat No. 12202; USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Cat No. ab136817; Abcam, Cambridge, MA, USA) and HRP-conjugated goat anti-mouse IgG (Cat No. ZB-2305; ZSGB-Bio, China) were used as secondary antibodies for detecting proteins. Goat anti-Rabbit IgG (H+L) superclonal secondary antibody (Cat No. A27034) and goat anti-Rabbit IgG (H+L) superclonal secondary antibody (Cat No. A-11037) were purchased from Thermo Fisher Scientific. Puromycin was purchased from Santa Cruz Biotechnology (Cat No. sc-108071; Dallas, TX, USA). All other chemicals were obtained from Sigma-Aldrich.
Cell Lines, Cell Culture, and Mice
Human ESCC cell lines KYSE450 and TE-1 were purchased from ATCC (Manassas, VA, USA), and EPC2 cell line was human telomerase reverse transcriptase (hTERT) immortalized with functionally intact p53 and p16 and was maintained as previously described,45 HEEC, a normal esophageal epithelial cell line, was purchased from the cell bank of Chinese Academy of Sciences. KYSE450 and TE-1 cells were cultured in DMEM or RPMI-1640 medium containing 10% fetal bovine serum (FBS). Stable cell lines were established after lentiviral infection and puromycin selection as described in our previous study.10,15 Six-week-old nude mice were purchased from SLRC Laboratory Company (Shanghai, China). All of the protocols on human tissue examination and animal experiments were approved by the Medical Ethics Committee of 900 Hospital of the Joint Logistics Team.
Library Construction
A nucleotide sequence for TrxA-MCS-TrxA containing the enzyme recognition sites was designed and obtained after performing high-fidelity PCR, followed by inserting TrxA-MCS-TrxA DNA into pBiFc-VC155 for pBiFc-VC155-TrxA-MCS-TrxA construction, and an MCS was inserted into a protein scaffold to form a constrained structure. Thirty-three random nucleotides sequences were also designed and inserted into pBiFc-VC155-TrxA-MCS-TrxA to establish the library, which could drive the expression of peptide aptamers containing 11 random amino acids, after nucleotide synthesis, high-fidelity PCR, and ligation. The library construct pBiFc-VC155-TrxA-peptide-TrxA was identified by PCR and sequencing, and pBiFc-VN173-SOX2 that could drive the expression of target protein SOX2 was also successfully obtained; all sequences and primers designed for these constructs were listed in Table S5.
BiFc Assay
We used BiFc method to confirm the interaction between SOX2 and peptide aptamers. HEK293T cells are maintained in DMEM supplemented with 10% FBS and penicillin/streptomycin in 12-well plates, and in each combined group, 0.2 μg of each construct, which expresses fused protein to VN173 or VC155 that was listed in Table S5, was co-transfected into HEK293T cells by using Lipofectamine 2000 (Cat No. 11668-019; Invitrogen). For ratio analysis, 50 ng of pDsRed2-C1 that encodes red fluorescent protein was co-transfected as an internal control to measure the BiFC efficiency of fragments derived from the Venus. For BiFC assay, transfected cells were fixed with 4% paraformaldehyde and permeabilized in PBST (PBS with 0.5% Triton X-100), followed by incubation with PBS containing DAPI and acquiring images by using fluorescent microscope (Nikon).
Immunoprecipitation and Western Blot
To examine the interactions between SOX2 protein and peptide aptamers, pCMV-Tag2B-SOX2, pcDNA3.1-myc-hisC-TrxA-Peptide-TrxA constructs were generated with high-fidelity PCR using primers listed in Table S6, and the sequence was confirmed by DNA sequencing (Figure S1E). A total of 5 μg pCMV-Tag2B-SOX2 and 5 μg pcDNA3.1-myc-hisC-TrxA-Peptide-TrxA were co-transfected into HEK293T cells with polyethylenimine (PEI) reagent, and HEK293T cells transfected with pCMV-Tag2B-SOX2 were used as controls. Forty-eight hours after transfection with the constructs, proteins were extracted and immunoprecipitation was performed with the FLAG Immunoprecipitation Kit according to the manufacturer’s manual. FLAG and myc antibodies were used for the identification of target protein.
Tissue Hematoxylin and Eosin Staining, Immunofluorescence Assay, Cell Proliferation, and Colony Formation Assays
Peptide Design and Synthesis
Peptides were synthesized by Dg peptides (Hangzhou, China), in which a fluorescent molecule TAMRA and a penetrating peptide sequence were synthesized at the N terminus of peptides, respectively (Table S4). The synthesized peptides were then resuspended in water and stored at −20°C.
Wound-Healing Assay
Cells expressing peptide aptamers or control vectors were seeded in six-well plates and grown to 90% confluence. The cell monolayers were then scrapped with a sterile pipette tip, the floating cells were removed, and the cultures were maintained in DMEM supplemented with 5% FBS. The wound area was recorded at 0, 48, and 72 h after scrapping. The healing index was calculated and analyzed by using the formula: (S0 − Sn)/S0 × 100%, which represents the ability of cellular migration. S0 and Sn represent the blank area at 0 and n hours after scraping, respectively.
Matrigel Invasion Assay
Invasive assays were performed in inserts of 24-well plates (Cat No. 3422; Corning, NY, USA). The insert membrane was coated with 12 μL of ice-cold Matrigel Basement Membrane Matrix (Cat No. 356234; BD Company, USA). Cells were seeded on the insert and cultured with 200 μL of medium containing 5% FBS. The lower chamber was filled with 500 μL of medium containing 20% FBS. The culture was maintained for 48 h. The cells were then subsequently fixed with methanol and stained with 1% crystal violet. Cells at the lower end of the insert membrane were counted under the microscope and photographed for analysis.
Xenograft and Metastasis Studies
A total of 1 × 107 KYSE450 cells were re-suspended in RPMI/Matrigel mixture, which were subcutaneously injected into the flanks of 6-week-old BALB/c nude mice and were maintained in specific pathogen-free environment. Five female nude mice per cohort (P42 ectopic expression and control) were randomly divided to receive cancer cells, and tumors were collected 7 weeks after cell inoculation. To further test the anti-cancer role of peptide 42 in nude mice, KYSE450 cells mixed with matrigel were subcutaneously injected into the flanks of 6-week-old male BALB/c nude mice, followed by injecting 0.04 μg/μL peptide 42 and control peptide into the tumors when they are visible. After 1 month, tumors were harvested, fixed in 4% PFA, and sectioned for hematoxylin and eosin (H&E) staining.
To evaluate the effect of P42 aptamer and peptide 42 on metastatic capability, fertilized eggs of zebrafish were developed for 24 h, and then the embryos were cultured with 1-phenyl-2-thiourea (PTU), an inhibitor of melanin production, in salt culture medium. These embryos were then harvested for injecting tumor cells after 48 h, and the perivitelline space of each embryo was injected with 200 KYSE450 cells by a pressure microinjector. For analyzing the effect caused by synthetic peptide 42, KYSE450 cells were cultured with 0.02 μg/μL peptide 42 or control peptide for 24 h. After injection with cancer cells, these embryos were cultured in salt culture medium containing PTU at 34°C and were anesthetized after 48 h. The cells were then photographed under a fluorescent microscope, and the metastatic foci were analyzed.
Proteomics and Bioinformatics Analyses
KYSE450 cells were sonicated three times on ice using a high-intensity ultrasonic processor (Scientz) in lysis buffer containing 8 M urea and 1% protease inhibitor cocktail. The remaining debris was removed after centrifugation at 12,000 × g at 4°C for 10 min. After that, the supernatant was collected and the protein concentration was determined by a BCA kit according to the manual instructions. The protein solution was reduced by 5 mM dithiothreitol for 30 min at 56°C and alkylated with 11 mM iodoacetamide for 15 min at room temperature in darkness. The protein samples were diluted by adding 100 mM triethylammonium bicarbonate buffer (TEAB) to a urea concentration of less than 2M, followed by the addition of trypsin at 1:50 trypsin-to-protein mass ratio for the first digestion overnight and then a 1:100 trypsin-to-protein mass ratio for a second 4-h digestion. After this, the peptides were desalted by Strata X C18 SPE column (Phenomenex) and vacuum-dried. Peptides in 0.5 M TEAB were reconstituted and processed according to the manufacturer’s protocol of TMT kit/iTRAQ kit.
The tryptic peptides were subsequently fractionated into fractions by high pH reverse-phase high-performance liquid chromatography (HPLC) using Agilent 300 Extend C18 column (5-μm particles, 4.6 mm inner diameter [ID], 250 mm length). In brief, the peptides were initially separated with a gradient of 8%–32% acetonitrile (pH 9.0) for over 60 min into 60 fractions, and then the peptides were combined into 18 fractions and dried by vacuum centrifugation and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The resulting data of MS/MS were processed by using the Maxquant search engine (v.1.5.2.8). Bioinformatics analyses were performed using annotation and functional enrichment, and the annotation methods include GO annotation, KEGG pathway annotation, and subcellular localization. The functional enrichment contains enrichment of GO analysis, enrichment of pathway analysis, and enrichment-based clustering.
Statistical Analysis
The data represent at least three independent experiments by using cells or extracts from a minimum of three separate isolations. Differences between the groups were compared by using analysis of variance for repeated measures. All statistical analyses were performed by using GraphPad PRISM v.5.0 software. Data were presented as means ± standard deviation (SD). Statistical significance between the two groups was calculated by unpaired Student’s t test, and p values <0.05 were considered to be significant.
Author Contributions
K.L. conceived the study, prepared the figures, and contributed to the writing of the manuscript. F.X., T.Z., R.Z., A.G., Y.C., and S.Z. conducted cell culture, western blot, and function experiments. K.L., J.Q., and X.L. analyzed the data and performed the statistical analysis. Z.X., J.L., X.H., L.S., W.H., J.W., H.L., W.E.-R., A.Z., and X.C. planned the project and assisted with experiments. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The research was supported by the following grants: National High Technology Research and Development Program of China (863 Program, no. 2014AA020541 to K.L.); the National Natural Science Foundation of China (nos. 81772994 and 81302068 to K.L.); the International Collaborative Project of Fujian Province (2017I0014 to K.L.); and the Program for the Top Young Innovative Talents of Fujian Province (2016RCLKC to K.L).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.01.012.
Contributor Information
Kuancan Liu, Email: liukuancan@163.com.
Jianwen Que, Email: jq2240@cumc.columbia.edu.
Supplemental Information
References
- 1.Liang H., Fan J.H., Qiao Y.L. Epidemiology, etiology, and prevention of esophageal squamous cell carcinoma in China. Cancer Biol. Med. 2017;14:33–41. doi: 10.20892/j.issn.2095-3941.2016.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prabhu A., Obi K.O., Rubenstein J.H. The synergistic effects of alcohol and tobacco consumption on the risk of esophageal squamous cell carcinoma: a meta-analysis. Am. J. Gastroenterol. 2014;109:822–827. doi: 10.1038/ajg.2014.71. [DOI] [PubMed] [Google Scholar]
- 3.Liu X., Wang X., Lin S., Yuan J., Yu I.T. Dietary patterns and oesophageal squamous cell carcinoma: a systematic review and meta-analysis. Br. J. Cancer. 2014;110:2785–2795. doi: 10.1038/bjc.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang F., Syrjänen S., Wang L., Syrjänen K. Infectious agents in the etiology of esophageal cancer. Gastroenterology. 1992;103:1336–1348. doi: 10.1016/0016-5085(92)91526-a. [DOI] [PubMed] [Google Scholar]
- 5.Chen T., Cheng H., Chen X., Yuan Z., Yang X., Zhuang M., Lu M., Jin L., Ye W. Family history of esophageal cancer increases the risk of esophageal squamous cell carcinoma. Sci. Rep. 2015;5:16038. doi: 10.1038/srep16038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qu Y., Zhang S., Cui L., Wang K., Song C., Wang P., Zhang J., Dai L. Two novel polymorphisms in PLCE1 are associated with the susceptibility to esophageal squamous cell carcinoma in Chinese population. Dis. Esophagus. 2017;30:1–7. doi: 10.1111/dote.12463. [DOI] [PubMed] [Google Scholar]
- 7.Liu K., Zhao T., Wang J., Chen Y., Zhang R., Lan X., Que J. Etiology, cancer stem cells and potential diagnostic biomarkers for esophageal cancer. Cancer Lett. 2019;458:21–28. doi: 10.1016/j.canlet.2019.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y.B., Chen Z.L., Li J.G., Hu X.D., Shi X.J., Sun Z.M., Zhang F., Zhao Z.R., Li Z.T., Liu Z.Y. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 9.Song Y., Li L., Ou Y., Gao Z., Li E., Li X., Zhang W., Wang J., Xu L., Zhou Y. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–95. doi: 10.1038/nature13176. [DOI] [PubMed] [Google Scholar]
- 10.Liu K., Jiang M., Lu Y., Chen H., Sun J., Wu S., Ku W.Y., Nakagawa H., Kita Y., Natsugoe S. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell. 2013;12:304–315. doi: 10.1016/j.stem.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bass A.J., Watanabe H., Mermel C.H., Yu S., Perner S., Verhaak R.G., Kim S.Y., Wardwell L., Tamayo P., Gat-Viks I. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009;41:1238–1242. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Que J., Okubo T., Goldenring J.R., Nam K.T., Kurotani R., Morrisey E.E., Taranova O., Pevny L.H., Hogan B.L. Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development. 2007;134:2521–2531. doi: 10.1242/dev.003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trisno S.L., Philo K.E.D., McCracken K.W., Catá E.M., Ruiz-Torres S., Rankin S.A., Han L., Nasr T., Chaturvedi P., Rothenberg M.E. Esophageal organoids from human pluripotent stem cells delineate Sox2 functions during esophageal specification. Cell Stem Cell. 2018;23:501–515.e7. doi: 10.1016/j.stem.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y., Yang Y., Jiang M., Huang S.X., Zhang W., Al Alam D., Danopoulos S., Mori M., Chen Y.W., Balasubramanian R. 3D modeling of esophageal development using human PSC-derived basal progenitors reveals a critical role for Notch signaling. Cell Stem Cell. 2018;23:516–529.e5. doi: 10.1016/j.stem.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu K., Xie F., Gao A., Zhang R., Zhang L., Xiao Z., Hu Q., Huang W., Huang Q., Lin B. SOX2 regulates multiple malignant processes of breast cancer development through the SOX2/miR-181a-5p, miR-30e-5p/TUSC3 axis. Mol. Cancer. 2017;16:62. doi: 10.1186/s12943-017-0632-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCaughan F., Pole J.C., Bankier A.T., Konfortov B.A., Carroll B., Falzon M., Rabbitts T.H., George P.J., Dear P.H., Rabbitts P.H. Progressive 3q amplification consistently targets SOX2 in preinvasive squamous lung cancer. Am. J. Respir. Crit. Care Med. 2010;182:83–91. doi: 10.1164/rccm.201001-0005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudin C.M., Durinck S., Stawiski E.W., Poirier J.T., Modrusan Z., Shames D.S., Bergbower E.A., Guan Y., Shin J., Guillory J. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santini R., Pietrobono S., Pandolfi S., Montagnani V., D’Amico M., Penachioni J.Y., Vinci M.C., Borgognoni L., Stecca B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene. 2014;33:4697–4708. doi: 10.1038/onc.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leis O., Eguiara A., Lopez-Arribillaga E., Alberdi M.J., Hernandez-Garcia S., Elorriaga K., Pandiella A., Rezola R., Martin A.G. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354–1365. doi: 10.1038/onc.2011.338. [DOI] [PubMed] [Google Scholar]
- 20.Basu-Roy U., Seo E., Ramanathapuram L., Rapp T.B., Perry J.A., Orkin S.H., Mansukhani A., Basilico C. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene. 2012;31:2270–2282. doi: 10.1038/onc.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiang R., Liao D., Cheng T., Zhou H., Shi Q., Chuang T.S., Markowitz D., Reisfeld R.A., Luo Y. Downregulation of transcription factor SOX2 in cancer stem cells suppresses growth and metastasis of lung cancer. Br. J. Cancer. 2011;104:1410–1417. doi: 10.1038/bjc.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu K., Lin B., Lan X. Aptamers: a promising tool for cancer imaging, diagnosis, and therapy. J. Cell. Biochem. 2013;114:250–255. doi: 10.1002/jcb.24373. [DOI] [PubMed] [Google Scholar]
- 23.Yoon S., Wu X., Armstrong B., Habib N., Rossi J.J. An RNA Aptamer Targeting the Receptor Tyrosine Kinase PDGFRα Induces Anti-tumor Effects through STAT3 and p53 in Glioblastoma. Mol. Ther. Nucleic Acids. 2019;14:131–141. doi: 10.1016/j.omtn.2018.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoon S., Huang K.W., Andrikakou P., Vasconcelos D., Swiderski P., Reebye V., Sodergren M., Habib N., Rossi J.J. Targeted Delivery of C/EBPα-saRNA by RNA Aptamers Shows Anti-tumor Effects in a Mouse Model of Advanced PDAC. Mol. Ther. Nucleic Acids. 2019;18:142–154. doi: 10.1016/j.omtn.2019.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu C.D., Chinenov Y., Kerppola T.K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 26.Shirai K., Shimada T., Yoshida H., Hayakari R., Matsumiya T., Tanji K., Murakami M., Tanaka H., Imaizumi T. Interferon (IFN)-induced protein 35 (IFI35) negatively regulates IFN-β-phosphorylated STAT1-RIG-I-CXCL10/CCL5 axis in U373MG astrocytoma cells treated with polyinosinic-polycytidylic acid. Brain Res. 2017;1658:60–67. doi: 10.1016/j.brainres.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 27.de Melo I.S., Jimenez-Nuñez M.D., Iglesias C., Campos-Caro A., Moreno-Sanchez D., Ruiz F.A., Bolívar J. NOA36 protein contains a highly conserved nucleolar localization signal capable of directing functional proteins to the nucleolus, in mammalian cells. PLoS ONE. 2013;8:e59065. doi: 10.1371/journal.pone.0059065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colas P., Cohen B., Jessen T., Grishina I., McCoy J., Brent R. Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature. 1996;380:548–550. doi: 10.1038/380548a0. [DOI] [PubMed] [Google Scholar]
- 29.Nouvion A.L., Thibaut J., Lohez O.D., Venet S., Colas P., Gillet G., Lalle P. Modulation of Nr-13 antideath activity by peptide aptamers. Oncogene. 2007;26:701–710. doi: 10.1038/sj.onc.1209832. [DOI] [PubMed] [Google Scholar]
- 30.Guida E., Bisso A., Fenollar-Ferrer C., Napoli M., Anselmi C., Girardini J.E., Carloni P., Del Sal G. Peptide aptamers targeting mutant p53 induce apoptosis in tumor cells. Cancer Res. 2008;68:6550–6558. doi: 10.1158/0008-5472.CAN-08-0137. [DOI] [PubMed] [Google Scholar]
- 31.Gibert B., Hadchity E., Czekalla A., Aloy M.T., Colas P., Rodriguez-Lafrasse C., Arrigo A.P., Diaz-Latoud C. Inhibition of heat shock protein 27 (HspB1) tumorigenic functions by peptide aptamers. Oncogene. 2011;30:3672–3681. doi: 10.1038/onc.2011.73. [DOI] [PubMed] [Google Scholar]
- 32.Guo B., Zhai D., Cabezas E., Welsh K., Nouraini S., Satterthwait A.C., Reed J.C. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- 33.Moellering R.E., Cornejo M., Davis T.N., Del Bianco C., Aster J.C., Blacklow S.C., Kung A.L., Gilliland D.G., Verdine G.L., Bradner J.E. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiao S., Wang H., Shi Z., Dong A., Zhang W., Song X., He F., Wang Y., Zhang Z., Wang W. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. 2014;25:166–180. doi: 10.1016/j.ccr.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Araste F., Abnous K., Hashemi M., Taghdisi S.M., Ramezani M., Alibolandi M. Peptide-based targeted therapeutics: Focus on cancer treatment. J. Control. Release. 2018;292:141–162. doi: 10.1016/j.jconrel.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Kwan K.Y., Shen J., Corey D.P. C-MYC transcriptionally amplifies SOX2 target genes to regulate self-renewal in multipotent otic progenitor cells. Stem Cell Reports. 2015;4:47–60. doi: 10.1016/j.stemcr.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y., Shi L., Zhang L., Li R., Liang J., Yu W., Sun L., Yang X., Wang Y., Zhang Y., Shang Y. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J. Biol. Chem. 2008;283:17969–17978. doi: 10.1074/jbc.M802917200. [DOI] [PubMed] [Google Scholar]
- 38.Libiad M., Yadav P.K., Vitvitsky V., Martinov M., Banerjee R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 2014;289:30901–30910. doi: 10.1074/jbc.M114.602664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang S., Jin J., Tian X., Wu L. hsa-miR-29c-3p regulates biological function of colorectal cancer by targeting SPARC. Oncotarget. 2017;8:104508–104524. doi: 10.18632/oncotarget.22356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watrin E., Schleiffer A., Tanaka K., Eisenhaber F., Nasmyth K., Peters J.M. Human Scc4 is required for cohesin binding to chromatin, sister-chromatid cohesion, and mitotic progression. Curr. Biol. 2006;16:863–874. doi: 10.1016/j.cub.2006.03.049. [DOI] [PubMed] [Google Scholar]
- 41.Rhodes J.M., McEwan M., Horsfield J.A. Gene regulation by cohesin in cancer: is the ring an unexpected party to proliferation? Mol. Cancer Res. 2011;9:1587–1607. doi: 10.1158/1541-7786.MCR-11-0382. [DOI] [PubMed] [Google Scholar]
- 42.Murayama Y., Uhlmann F. Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature. 2014;505:367–371. doi: 10.1038/nature12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill V.K., Kim J.S., Waldman T. Cohesin mutations in human cancer. Biochim. Biophys. Acta. 2016;1866:1–11. doi: 10.1016/j.bbcan.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bot C., Pfeiffer A., Giordano F., Manjeera D.E., Dantuma N.P., Ström L. Independent mechanisms recruit the cohesin loader protein NIPBL to sites of DNA damage. J. Cell Sci. 2017;130:1134–1146. doi: 10.1242/jcs.197236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okawa T., Michaylira C.Z., Kalabis J., Stairs D.B., Nakagawa H., Andl C.D., Johnstone C.N., Klein-Szanto A.J., El-Deiry W.S., Cukierman E. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev. 2007;21:2788–2803. doi: 10.1101/gad.1544507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.