

Abstract
Membranes are formed from a bilayer containing diverse lipid species with which membrane proteins interact. Integral, membrane proteins are embedded in this bilayer, where they interact with lipids from their surroundings, whilst peripheral membrane proteins bind to lipids at the surface of membranes. Lipid interactions can influence the function of membrane proteins, either directly or allosterically. Both experimental (structural) and computational approaches can reveal lipid binding sites on membrane proteins. It is, therefore, important to understand the free energies of these interactions. This affords a more complete view of the engagement of a particular protein with the biological membrane surrounding it. Here, we describe many computational approaches currently in use for this purpose, including recent advances using both free energy and unbiased simulation methods. In particular, we focus on interactions of integral membrane proteins with cholesterol, and with anionic lipids such as phosphatidylinositol 4,5-bis-phosphate and cardiolipin. Peripheral membrane proteins are exemplified via interactions of PH domains with phosphoinositide-containing membranes. We summarise the current state of the field and provide an outlook on likely future directions of investigation.
Keywords: free energy, lipid, membrane protein, molecular dynamics, simulation
Introduction
Biological membranes are formed from a bilayer containing different species of lipid, into which integral transmembrane proteins are inserted, whilst peripheral membrane proteins are bound to the surface. These membrane proteins play key roles in cell function via control of cellular transport, metabolism, and signalling. Many membrane proteins are of biomedical significance, and are therefore drug targets.
Protein–lipid interactions: structures and simulations
Membrane proteins interact with multiple lipid species. Some interactions play a structural role, e.g. stably anchoring and/or promoting oligomerization an integral protein within a bilayer [1,2], or targeting a peripheral protein to the surface of a specific cellular membrane. Other interactions influence the function of the protein, either allosterically [3–6] or via a direct functional role [7,8]. The nature of these lipid interactions is, therefore, the subject of both experimental and computational analyses [6].
Structural studies, using X-ray crystallography and/or cryo-electron microscopy, often require solubilisation of membrane proteins in detergents, which removes the majority of the bound lipids. Some protein–lipid interactions survive this process, allowing identification of lipids in the resultant density maps (e.g. [9–13]; Figure 1; Table 1). The use of nanodiscs, a portion of lipid bilayer encircled by either protein [14] or amphipathic polymer (SMALP) [15], may aid this purely structural approach (e.g. [16]). However, a combination of modest resolution and/or partial occupancy of the lipid site and the structural similarities between related lipid molecules can make unambiguous assignment of the identity of the bound lipid molecule non-trivial. In addition, it is uncertain to what extent interactions seen in a purified complex reflect those present in the native membrane environment.
Figure 1. Identification of protein–lipid interactions.

(A) Left: view of bovine AAC (PDB 1OKC), with the protein shown as a grey cartoon and densities for CDL shown in yellow surface. Middle: A bound CDL molecule from the crystal structure is shown in yellow, orange, and red spheres. Right: close-up of one of the CDL molecules: the CDL and contacting residues are shown as sticks, with aromatic, hydrophobic, and small amino acid sidechains coloured green, acidic sidechains red, basic sidechains blue, and polar uncharged sidechains in purple. (B) As (A), but for human β2AR (PDB 3D4S), showing the binding mode of two cholesterol molecules. (C) As (A) but for G. gallus Kir2.2 (PDB 3SPI) in complex with di-C8-PIP2 molecules.
Table 1. Where reported, errors are included for the free energy (FE) estimate. Note that the method of how the error is calculated varies between different studies.
| Lipid | Protein | PDBs | FE (kJ mol−1) | Ref. |
|---|---|---|---|---|
| Chol. | Smoothened | 5L7D | −10 | [67] |
| β2 AR inactive | 3D4S | −14.3, −14.6 | [65] | |
| β2 AR active | 3SN6 | −9.8, −13.3 | [65] | |
| A2AR inactive | 4EIY | −9.4, −9.6, −11.7 | [65] | |
| A2AR active | 3QAK | −14.4, −10.6, −9.9, −9.3 | [65] | |
| β2 AR | 3D4S | −53 ± 0.8 | [66] | |
| µ-opioid | 5C1M | −12 ± 3.8 | [66] | |
| 5-HT2B | 4NC3 | −18 ± 2.1 | [66] | |
| A2AR | 5IU4 | −5 ± 1 to −9 ± 3 | [51] | |
| A2AR-adenosine | 2YDO | −3, −5 | [64] | |
| PgP | Model | −26 ± 3, −6 ± 1, −4 ± 1 | [68] | |
| Kir2.1 | Model | −3.3 ± 6, −7.5 ± 8, −10.5 ± 6 | [69] | |
| PC2 | 6T9N | −12 ± 3 | [32] | |
| CDL | AAC | 1OKC | −20 ± 5 to −25 ± 5 | [29] |
| AAC | 1OKC | −22 | [47] | |
| AAC | 1OKC | −5 ± 2 to −14 ± 3 | [51] | |
| LeuT | 2A65 | −6 ± 3 to −9 ± 2 | [51] | |
| Cytochrome c oxidase | 1OCC/2OCC | −10 to −35 | [24,78] | |
| PIP2 | Kir2.2 (truncated) | 3SPI | −42 ± 5 | [47] |
| Kir2.2 (full) | 3SPI | −45 ± 6 to −48 ± 2 | [51] | |
| PC2 | 6T9N | −37 ± 3 | [32] | |
| A2AR inactive | 3EML | −10 to −20 | [44] | |
| A2AR active | 5G53 | −10 to −50 | [44] | |
| A2AR active + mini Gs | 5G53 | −20 to −80 | [44] | |
| EGFR transmembrane | 2M20 | −42 ± 5 | [75] |
Note that the method of how the error is calculated varies between different studies.
Computational approaches [17], especially molecular dynamics (MD) simulations [18], provide a powerful tool for analysis of protein–lipid interactions. Recent improvements in lipid force fields have greatly improved research in this area, however, these lipid force fields still have limitations which must be taken into consideration when analysing data (for recent discussions, see [19–21]). Parallel advances in MD algorithms and computer hardware now permit analysis of interactions that occur on relatively long (microsecond) timescales. For example, in certain coarse-grained (CG) biomolecular force fields, such as Martini [22], groups of atoms are modelled as a single bead or particle, allowing faster sampling of protein–lipid configurations compared with atomistic simulations. CG simulations have been successfully applied to predicting the location of a lipid binding site to a given membrane protein for several different systems (e.g. [23–26]; Table 1). These predictions have been validated by comparison with experimental structural and/or biophysical data for many cases, including e.g. phosphatidylinositol (4,5) bis-phosphate (PIP2) bound to Kir channels [27,28]; cardiolipin (CDL) bound to the ADP/ATP carrier [29,30], and to the LeuT transporter [1]; and PIP2 bound to Class A GPCRS [31] and to TRP channels [32]. MD simulations can also provide information on local enrichment of various lipid species in the bilayer around a given membrane protein [33].
Computational estimation of lipid-binding energetics
MD simulations have been demonstrated to predict binding sites, allowing structural aspects of protein–lipid interactions to be analysed (Figure 1 and Table 1). However, it is also important to understand the affinities of these interactions. The crowded nature of a cell membrane environment [34–36] means that many different species will jostle for position around a membrane protein, with many lipids potentially able to bind. Knowledge of the interaction free energies of different lipid species for a specific site on a protein will help to indicate how likely such an interaction is to occur within a native cell membrane, rather than e.g. in a detergent-solubilised experimental environment.
Importantly, lipids are not only potential ligands for a membrane protein, they also act as the surrounding environment. Thus, all lipid binding sites on the transmembrane surface of a protein are likely to be occupied by one or another lipid species. Therefore, it is important to be able to estimate the free energy of binding for a specific lipid to a given site compared with that of the other major lipid species in the surrounding bilayer.
Computational approaches to study the free energies of protein–lipid interactions can be broadly divided into two categories: those using long, unbiased MD simulations to estimate the probability (and hence free energy) of interactions directly (Figure 2A), and those which use biased (i.e. more targeted) MD simulations to compute the free energy of interaction of a specific lipid molecule with a protein.
Figure 2. Equilibrium and free energy calculations for protein–lipid interactions.
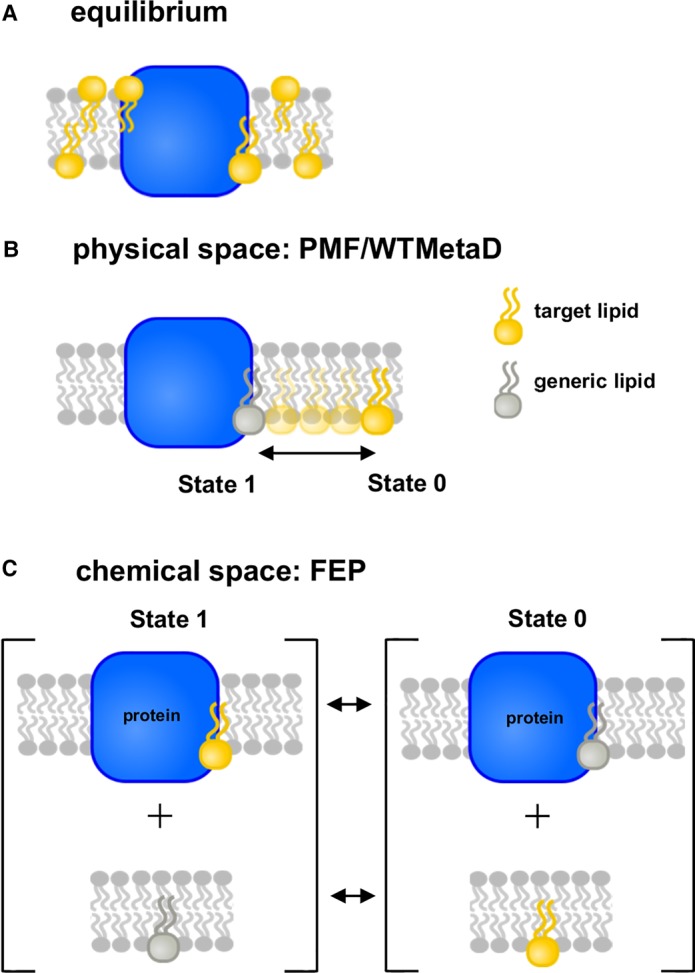
(A) Equilibrium simulations allow multiple protein–lipid events to occur in an unbiased fashion over long MD simulations. (B) Construction of a reaction co-ordinate in physical space requires sampling of the protein–lipid complex (State 1) and of the lipid free in membrane (State 0) as well as many intermediate positions of the lipid between the binding site and surrounding bilayer bulk. Note that in State 0, the lipid binding site on the protein will be occupied by a generic lipid. (C) Sampling States 1 and 0 in chemical space. This is done over two sets of simulations: first, whilst bound to the protein, the target lipid (yellow) is alchemically transformed into a background (generic e.g. PC; grey) lipid (upper panels). At the same time, a background lipid free in the bilayer is also transformed in a target lipid molecule. Combining the upper and lower calculations yields the free energy for binding of the target lipid relative to that of a background lipid.
Both approaches aim to compute the free energy difference between two states of a system: State 1 (Figure 2B) corresponds to a specific ‘target’ lipid bound to the protein, and State 0 to the target lipid free in the surrounding membrane. In State 0, a different (bulk or background) lipid species from the surrounding bilayer environment is likely to interact with the protein. Therefore, the quantity of interest is the ΔΔG of binding, where:
This is analogous to a binding site on a (water soluble) protein which can be occupied by either a ligand in the holo state or water molecules in the apo state.
Equilibrium simulation studies
Conceptually, the simplest approach to estimating free energies of protein–lipid binding is via long, unbiased simulations. These have the advantage that they are relatively straightforward to perform and analyse [37–40]. However, depending on the underlying free energy landscape they may require extended simulation times to adequately sample possible configurations of interest.
In principle, equilibrium simulations provide estimates of the relative probabilities of the lipid being bound (Pbound) or free (Pfree) (where Pbound = 1 − Pfree), calculated from the observed fraction of time a simulation system spends in the bound (State 1) and unbound (State 0) states. These can then be used to calculate the ΔΔG of the protein–lipid interaction:
It should be noted that for these probabilities to be properly sampled, multiple binding, and dissociation events must be observed, and the simulation time for this is difficult to be estimate in advance as it will depend on the specific protein–lipid interaction being modelled, and plausibly testing the limits of current MD simulations. If a sufficient number of binding and dissociation events occur, it may be possible to estimate kon and koff either directly from the simulation trajectories, possibly aided by e.g. Markov state models [41] or MetaDynamics [42]. Indeed, the latter approach has recently been applied to the calculation koff for ligand dissociation from a (water soluble) protein [43].
Equilibrium simulations also have the advantage that mixed lipid bilayers can readily be evaluated. In these cases, ΔΔG refers to the free energy of interaction of a target lipid in relation to the other lipid species in the system. This is, in turn, closer to a complex cell membrane than a free energy simulation based on simple single species lipid bilayer environment. In addition, multiple binding sites can be analysed simultaneously (Figure 2A).
Potential of mean force calculations
There are many more ‘targeted’ approaches to estimating free energies of protein–lipid interactions, of which potential of mean force (PMF) calculations are the most widely used (e.g. [24,29,44]). These can provide a cross-section through a more complex free energy landscape, showing the free energy of a lipid/protein system as a function of a typically one-dimensional reaction co-ordinate, e.g. the distance between the headgroup of a specific lipid molecule and its binding site on a membrane protein (i.e. the distance between States 1 and 0 in Figure 2B). Many methods are available (e.g. umbrella sampling [45] or replica exchange umbrella sampling [46,47]) to estimate the free energy as a function of the reaction co-ordinate.
To speed up convergence, it is typical to perform simulations with one copy of the target lipid, and the rest of the membrane modelled with a single generic lipid e.g. phosphatidylcholine (PC), rather than a complex lipid mixture. Therefore, comparisons of ΔGs for the target lipid with a generic lipid (often close to 0 kJ mol−1) allows estimation of ΔΔG between the two lipid species.
Free energy perturbation and absolute binding free energy calculations
More recently, studies have attempted to directly calculate ΔΔG using either free energy perturbation (FEP) [48] or absolute binding free energy (ABFE) [49] calculations. For these, the target lipid molecule is ‘alchemically’ transformed in silico into a generic lipid (see [50]), or fully removed from the system. If this is done to a target lipid whilst in complex with a protein and again to the same lipid species free in the bilayer, the difference between these values yields the free energy associated with the lipid binding to the protein. This permits modelling of the same states as by using PMF, but through a co-ordinate in chemical space (Figure 2C), rather than physical distance. Recent work suggests that this gives equivalent values to PMF analyses, while using less computational resource [51]. However, both approaches provide information on a single lipid site only, which needs to be identified prior to running calculations.
Metadynamics
MetaDynamics allows more extensive sampling of complex free energy landscapes. For example, in well-tempered metadynamics (WTMetaD) [52], the sampling of a free energy landscape is enhanced in a history-dependent manner [53] to ensure that the simulation system spends less time in energy minima, thus significantly speeding up the overall process. If applied to a patch of the membrane around a protein of interest, with a collective variable (CV) corresponding to the distance between the lipid and protein in the bilayer plane, this can be used to compute the binding poses and free energies for multiple lipid binding sites around a protein. In test calculations, this approach revealed all four PIP2 binding sites on Kir2.2 and all three CDL sites on AAC (see below), and yielded free energies of interaction in good agreement with both PMF and FEP/ABFE [51], albeit at a greater computational cost.
Application to specific lipids
Computational studies of protein–lipid interactions have largely focussed on two main classes of lipid: cholesterol, which as a relatively hydrophobic and rigid lipid species is amenable to free energy calculations, and more complex anionic lipids e.g. CDL and PIP2, which interact with a range of membrane proteins in bacterial and mitochondrial inner membranes [54], and in eukaryotic cell membranes [55,56], respectively.
Binding of cholesterol to GPCRs and other membrane proteins
Cholesterol is present in high (>30%) concentrations in eukaryotic plasma membranes [57]. Cholesterol interacts with many different proteins, including G-protein coupled receptors (GPCRs) and ion channels [6]. GPCRs constitute one of the largest classes of membrane proteins, and their key roles in many biological processes mean that they make up approximately one-third of all drug targets [58]. Many GPCRs have been shown to bind [59,60] and have been suggested to be functionally modulated by cholesterol [61–63]. These include the intensively studied adenosine 2A (A2AR) and β2-adrenergic (β-2AR) receptors.
The free energies of cholesterol–GPCR interactions have been addressed in many simulation studies (Figure 3). Cholesterol interactions with A2AR have been estimated by both WTMetaD and ABFE calculations using the Martini CG force field [51], yielding values from ∼ −5 to −10 kJ mol−1. Cholesterol–GPCR interactions have also been modelled using unbiased equilibrium simulations. Sub-microsecond duration atomistic simulations of A2AR in a 30% cholesterol and 70% PC membrane [64] yielded free energies of interaction between −3 and −5 kJ mol−1 for different sites, in good agreement with the values from the Martini CG estimates above. Extended (50 µs) CG simulations in membranes containing 15% cholesterol [65] yielded interaction free energies of −9 to −12 kJ mol−1 for the agonist-free state of the A2AR. Thus, for class A GPCRs a consensus emerges (despite using different estimation methods and force fields) of free energies of interaction in the range −5 to −12 kJ mol−1.
Figure 3. Computational estimates of protein–lipid interaction free energies.

(A) Summary of estimates of free energies for integral protein interactions of cholesterol (green), CDL (purple), and PIP2 (orange). The data for this graph were derived from the range of simulation studies (both coarse-grained and atomistic) discussed in the main text. Data are plotted as box and whiskers, showing the 5–95 percentile range. Data plotted in Prism 7 (GraphPad). (B) Cholesterol, CDL, and PIP2, indicating the likely charge state of the anionic lipids.
Other free energy calculation approaches have also been used to estimate the strength of cholesterol interactions. Salari et al. [66] used ABFE with the atomistic CHARMM36 force field to obtain values of cholesterol binding to β-2AR that is much stronger than in other studies (∼ −53 kJ mol−1). Interestingly, they obtained free energies for other different GPCRs, µ-opioid (∼ −12 kJ mol−1) and 5-HT2B (∼ −18 kJ mol−1) more in line with the energies obtained from other calculations.
Comparisons with proteins other than Class A GPCRs also suggest that cholesterol binds with modest (i.e. ∼ −10 kJ mol−1) free energies to surface-exposed sites on TM domains. CG calculations yielded free energies of −10 kJ mol−1 for cholesterol interactions with the TM domain of the Class F GPCR Smoothened [67]. Comparable values (−4 to −6 kJ mol−1) were obtained by CG simulations for two different cholesterol binding sites on the ABC transporter P-glycoprotein transporter [68]. Interestingly, this study also revealed a third, more deeply buried, site with a stronger interaction of −25 kJ mol−1. Similarly, atomistic simulations employing molecular mechanics/Poisson–Boltzmann surface area (MM-PBSA) calculations suggest binding energies of −3, −7.5, and −10.5 kJ mol−1 of cholesterol to three distinct sites on Kir2.1 [69]. Recent CG simulations based on cholesterol bound to the ciliary TRP channel PC2 [32] yielded an interaction free energy of −12 kJ mol−1. Thus, these systems also reveal relatively modest binding free energies for cholesterol. This is also in agreement with a recent docking study of cholesterol to a range of different GPCRs which concluded that cholesterol does not tend to occupy a single, well-defined conformational state but when bound to a TM surface site can adopt a range of binding poses [70], consistent with a modest free energy of interaction (Figure 3).
Experimental data against which to compare these calculations are relatively sparse. For example, it has been suggested for the β2AR that low affinity (∼ −10 kJ mol−1) and high affinity (∼ −50 kJ mol−1) cholesterol sites may exist, by NMR and by thermostability assays, respectively [71]. However, the relationship of these binding modes to the computational analyses described above (and to the 3–5 cholesterol binding sites on the β2AR indicated by structural studies) remains to be established.
Binding of anionic lipids to membrane proteins
Many membrane proteins interact specifically with anionic lipids, including e.g. CDL and PIP2. PIP2 is a signalling lipid found in the eukaryotic cell membrane and has been shown to with a wide range of receptors [6], ion channels [56], and transport proteins [72]. These interactions have been the subject of range of simulation studies [18].
Inward rectifying potassium channels (Kirs) provide a well-studied example of activation of an ion channel by PIP2 binding [11]. The energetics of these interactions has been explored using PMF calculations, giving free energies of −42 kJ mol−1 (for Kir2.2 truncated at the transmembrane region) [47] and of −45 kJ mol−1 (for full length Kir2.2) [51]. The free energy of interaction of PIP2 with the full length channel has also been estimated by FEP (−48 kJ mol−1) and by WTMetaD (−45 kJ mol−1) [51]. This can be compared with e.g. the free energy of interaction of PIP2 with the transmembrane domain of a TRP channel (PC2) of ∼ −35 kJ mol−1 [32]. As can be seen in Figure 1C, the PIP2 binding site is formed of a cluster of basic sidechains located close to the interface between the membrane and cytosolic regions. These values of the free energy of interaction may be compared with an experimental estimate of −33 kJ mol−1 derived from an EC50 for activation of Kir.2 1 by di-C8-PIP2 [73].
PIP2 interactions have also been investigated with many receptors including GPCRs [31]. For example, free energies for binding to the A2AR receptor range from −10 to −80 kJ mol−1 depending on the conformational state of the receptor [44]. This state dependence of binding to the receptor suggests PIP2 may be an allosteric regulator of A2AR, as has been suggested experimentally to be the case for other anionic lipids and GPCRs [74]. PIP2 binding free energy has also been estimated at −42 kJ mol−1 for a model of the TM domain of the EGFR (a receptor tyrosine kinase) [75]. Together, these data suggest that PIP2 binds tightly to membrane proteins at cationic sites at the inner leaflet/cytoplasmic interface systems. In support of this, lengthy simulations suggest that PIP2 remains bound for 10 s of microsecond (unpublished data), making a direct sampling of multiple binding/dissociation events computationally challenging.
CDL is an important anionic lipid present in bacterial and inner mitochondrial membranes. CDL binds to many different proteins including the ADP/ATP carrier protein, AAC (also known as the adenine nucleotide translocator, ANT) [9,76,77]. AAC transports ADP and ATP across the mitochondrial inner membrane, allowing regulation of mitochondrial adenine nucleotide concentration. AAC binds and is activated by CDL [9,77]. This interaction has been probed using PMFs, giving a values of the order of −20 kJ mol−1 depending on which of the three quasi-equivalent binding sites is being considered [29,47]. Comparable strengths of interaction of CDL with other proteins as estimated using PMF calculations include binding to cytochrome c oxidase (∼ −10 to −35 kJ mol−1) [24,78] and also to LeuT, a bacterial inner membrane transporter protein (∼ −6 kJ mol−1) [51].
Equilibrium simulations have been used to estimate mean residence times of bound CDL molecules for the mitochondrial F-ATPase [25] (residence times of ∼ 0.5 µs), for AAC (∼ 1–2 µs) [30], for cytochrome c oxidase (∼ 50–60 µs) [24] and for the bacterial SecYEG translocon (∼ 1–2 µs) [26]. In combination with e.g. analysis using Markov state models [41] such simulations could in principle be used to estimate on and off rates for lipids and hence binding affinities.
General trends can be identified from the summary of calculated free energies in Figure 3. As might be anticipated, there is an overall dependence of the strength of protein–lipid interactions on the net charge on the lipid headgroup. However, the range of values for a given lipid species is large (e.g. not all cholesterol sites are the same — see Table 1). Also, for some systems (e.g. PC2 [32]) PIP2 is predicted to bind more strongly than PIP3 (this is also seen experimentally for e.g. A2AR [31]). So although there is an overall dependence on net charge, this is not the sole determinant of the strength of the interaction.
Membrane surface recognition: PH domains
Membrane signalling and trafficking in eukaryotic cells is regulated by many peripheral membrane protein domains which associate with membrane surfaces, in a lipid-dependent fashion [55]. Considerations of space preclude a more general survey (see e.g. [79–83] for this), so we will focus on one particular example, that of PH (pleckstrin homology) domains. PH domains are a large and well-characterized family present in many membrane recognition proteins, which can bind to phosphatidylinositol-phosphates (mainly PIP2 and/or PIP3) in membranes in a specific fashion [84]. Cooperativity in binding with other anionic lipid species may aid the recruitment of PH domains to particular cell membranes [85].
Simulation studies have provided a detailed picture of structural and dynamic aspects of the interaction of PH domains with PIP-containing lipid bilayers [86–92]. These studies demonstrate that simulations can predict the interactions of PH domains with lipid bilayers, reproducing the experimental structural aspects seen in interactions of these domains with soluble inositol phosphates which correspond to the headgroups of PIP lipid molecules.
More recently, CG simulations have been used to estimate the free energy of interactions of PH domains with PIP molecules in lipid bilayers. For example, CG simulation studies of the GRP1 PH domain with a PIP3-containing lipid bilayer (Figure 4, [93]), with the data suggesting that both canonical and alternative modes of interaction are possible. This was subsequently extended to 12 different PH domains [94] for which structural and energetic experimental data were available. Free energies of the interaction of PH domains with a single PIP molecule in a bilayer ranged from −5 to −40 kJ mol−1. Surprisingly, these calculated free energies were generally smaller than those estimated experimentally. Recent simulations [95] suggest that this apparent discrepancy reflects the interaction of a PH domain with multiple PIP molecules when binding to a PIP-containing bilayer. A comparison of the dependence of PH-membrane binding energies on the number of PIP molecules interacting with the PH domain suggests that local nanoscale clustering of PIP molecules can control the strength of this interaction. Thus, binding of a single PH domain to 3 or more PIP molecules yields interaction free energies consistent with experimental estimates of binding affinity of the GRP1 PH domain. Simulations of the PH domain of ACAP1BAR-PH protein [96] and of the PH domain of Bruton's tyrosine kinase [92] have also suggested more than a single PIP binding site.
Figure 4. Energetics of interaction of the GRP1 PH domain with a PIP3-containing lipid bilayer.
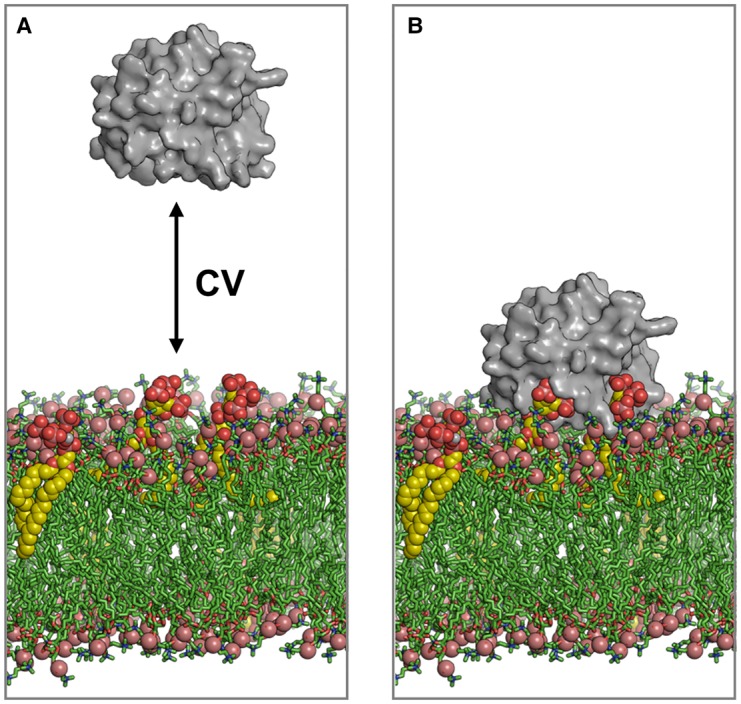
(A) Simulations of the interaction of the GRP1 PH domain (grey) with a lipid bilayer. The double-headed arrow indicates the reaction co-ordinate for estimation of the free energy landscape (PMF) as a function of the distance of the PH domain from the target lipid molecule(s). (B) The GRP1 PH domain is shown bound to a PIP3-containing bilayer. PIP3 molecules are shown as yellow spheres. Lipids are shown as green sticks. Simulation co-ordinates from [95].
These examples show how simulations can contribute to our quantitative understanding of membrane recognition by lipid-binding domains. It will be important to extend such studies to other common membrane recognition domains such as C2 domains [83]. Future challenges include extending free energy calculations to include the interactions of more complex multi-domain membrane binding proteins such as the PIP-phosphatase PTEN [97], the dimeric PIP kinase PIP5K1A [98], and the Arf/GEF complex [99].
Outlook
It is evident that computational methods for studying the energetics of protein–lipid interactions are now established, and are poised to be extended to a wider range of membrane proteins and lipid species in order to address a range of key functional questions. In particular, as increasing computational power allows the description of systems at higher accuracies, such as including polarisability using mean-field corrections [100] or polarisable force fields [101], these analyses are likely to improve in scope and accuracy. It is also evident that we need both more and better experimental data against which to evaluate and compare predictions from simulations. Data are needed both from in vitro experiments (ideally in model membranes rather than in detergents) [102–104] and from in vivo studies, e.g. single-molecule experiments in live cell membranes [105]. A comparison of such data with simulations will provide a more quantitative understanding of specific protein–lipid interactions in a complex (both compositionally and laterally inhomogeneous) membrane environment. Ongoing improvements in simulation methods and computational power should allow us to extended CG simulations of free energies of interaction to atomistic (but converged!) descriptions. This will require us to account for both dynamic ionisation states of anionic lipids and the binding of divalent metal ions [106–108]. Together, this approach will lead to mechanistic insights into lipids as allosteric ligands, insights which will enable lipid sites to be more fully exploited as druggable sites on biomedically important membrane protein targets.
Perspectives
Interactions with specific lipids influence the structure and function of membrane proteins. Identification and characterisation of these interactions is important for our understanding of the biology of these systems, and in the development of novel therapeutics.
Assessment of the free energies of protein–lipid interactions is central to determining whether the binding is physiologically relevant, especially within the complex environment of the biological membrane. Evaluation of protein–lipid interactions is possible using MD simulations, either through free energy calculations or lengthy unbiased simulations.
As computer power increases and simulation algorithms improve, so too will the scope and accuracy of these analyses.
Acknowledgements
R.A.C., P.J.S., and M.S.P.S. are funded by Wellcome (208361/Z/17/Z). Research in P.J.S.’s laboratory is funded by the M.R.C. (MR/S009213/1) and BBSRC (BB/P01948X/1, BB/R002517/1, BB/S003339/1); research in M.S.P.S.’s group is funded by BBSRC, EPSRC, and Wellcome. This project was also supported by ARCHER via the UK High-End Computing Consortium for Biomolecular Simulation, HECBioSim (http://hecbiosim.ac.uk), supported by EPSRC [grant no. EP/R029407/1].
Abbreviations
- MD
molecular dynamics
- CG
coarse-grained
- PIP2
phosphatidylinositol (4,5) bis-phosphate
- CDL
cardiolipin
- PMF
potential of mean force
- PC
phosphatidylcholine
- FEP
free energy perturbation
- WTMetaD
well-tempered metadynamics
- CV
collective variable
- GPCRs
G-protein coupled receptors
- A2AR
adenosine 2A receptor
- β-2AR
β2-adrenergic receptor
- MM-PBSA
molecular mechanics/Poisson–Boltzmann surface area
Author contribution
All authors contributed to the writing and editing of the manuscript.
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Gupta K., Donlan J.A.C., Hopper J.T.S., Uzdavinys P., Landreh M., Struwe W.B. et al. (2017) The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 541, 421–424 10.1038/nature20820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parker J.L., Corey R.A., Stansfeld P.J. and Newstead S. (2019) Structural basis for substrate specificity and regulation of nucleotide sugar transporters in the lipid bilayer. Nat. Commun. 10, 4657 10.1038/s41467-019-12673-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wen P.C., Mahinthichaichan P., Trebesch N., Jiang T., Zhao Z.Y., Shinn E. et al. (2018) Microscopic view of lipids and their diverse biological functions. Curr. Opin. Struct. Biol. 51, 177–186 10.1016/j.sbi.2018.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corradi V., Sejdiu B.I., Mesa-Galloso H., Abdizadeh H., Noskov S.Y., Marrink S.J. et al. (2019) Emerging diversity in lipid–protein interactions. Chem. Rev. 119, 5775–5848 10.1021/acs.chemrev.8b00451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manna M., Nieminen T. and Vattulainen I. (2019) Understanding the role of lipids in signaling through atomistic and multiscale simulations of cell membranes. Ann. Rev. Biophys. 48, 421–439 10.1146/annurev-biophys-052118-115553 [DOI] [PubMed] [Google Scholar]
- 6.Duncan A.L., Song W. and Sansom M.S.P. (2020) Lipid-dependent regulation of ion channels and GPCRs: insights from structures and simulations. Ann. Rev. Pharmacol. Toxicol. 60 10.1146/annurev-pharmtox-010919-023411 [DOI] [PubMed] [Google Scholar]
- 7.Bushell K.S.R., Pike A.C.W., Falzone M.E., Rorsman N.J.G., Ta C.M., Corey R.A. et al. (2019) The structural basis of lipid scrambling and inactivation in the endoplasmic reticulum scramblase TMEM16K. Nat. Commun. 10, 3956 10.1038/s41467-019-11753-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caffalette C.A., Corey R.A., Sansom M.S.P., Stansfeld P.J. and Zimmer J. (2019) A lipid gating mechanism for the channel-forming O antigen ABC transporter. Nat. Commun. 10, 824 10.1038/s41467-019-08646-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pebay-Peyroula E., Dahout-Gonzalez C., Kahn R., Trezeguet V., Lauquin G.J. and Brandolin G. (2003) Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426, 39–44 10.1038/nature02056 [DOI] [PubMed] [Google Scholar]
- 10.Hunte C. and Richers S. (2008) Lipids and membrane protein structures. Curr. Opin. Struct. Biol. 18, 406–411 10.1016/j.sbi.2008.03.008 [DOI] [PubMed] [Google Scholar]
- 11.Hansen S.B., Tao X. and MacKinnon R. (2011) Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498 10.1038/nature10370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drachmann N.D., Olesen C., Moller J.V., Guo Z., Nissen P. and Bublitz M. (2014) Comparing crystal structures of Ca2+-ATPase in the presence of different lipids. FEBS J. 281, 4249–4262 10.1111/febs.12957 [DOI] [PubMed] [Google Scholar]
- 13.Norimatsu Y., Hasegawa K., Shimizu N. and Toyoshima C. (2017) Protein–phospholipid interplay revealed with crystals of a calcium pump. Nature 545, 193–198 10.1038/nature22357 [DOI] [PubMed] [Google Scholar]
- 14.Hagn F., Nasr M.L. and Wagner G. (2018) Assembly of phospholipid nanodiscs of controlled size for structural studies of membrane proteins by NMR. Nat. Protoc. 13, 79–98 10.1038/nprot.2017.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juarez J.F.B., Harper A.J., Judge P.J., Tonge S.R. and Watts A. (2019) From polymer chemistry to structural biology: the development of SMA and related amphipathic polymers for membrane protein extraction and solubilisation. Chem. Phys. Lipids 221, 167–175 10.1016/j.chemphyslip.2019.03.008 [DOI] [PubMed] [Google Scholar]
- 16.Gao Y., Cao E.H., Julius D. and Cheng Y.F. (2016) TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351 10.1038/nature17964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee A.G. (2018) A database of predicted binding sites for cholesterol on membrane proteins, deep in the membrane. Biophys. J. 115, 522–532 10.1016/j.bpj.2018.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedger G. and Sansom M.S.P. (2016) Lipid interaction sites on channels, transporters and receptors: recent insights from molecular dynamics simulations. Biochim. Biophys. Acta 1858, 2390–2400 10.1016/j.bbamem.2016.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyubartsev A.P. and Rabinovich A.L. (2016) Force field development for lipid membrane simulations. Biochim. Biophys. Acta 1858, 2483–2497 10.1016/j.bbamem.2015.12.033 [DOI] [PubMed] [Google Scholar]
- 20.Ollila O.H.S. and Pabst G. (2016) Atomistic resolution structure and dynamics of lipid bilayers in simulations and experiments. Biochim. Biophys. Acta 1858, 2512–2528 10.1016/j.bbamem.2016.01.019 [DOI] [PubMed] [Google Scholar]
- 21.Poger D., Caron B. and Mark A.E. (2016) Validating lipid force fields against experimental data: progress, challenges and perspectives. Biochim. Biophys. Acta 1858, 1556–1565 10.1016/j.bbamem.2016.01.029 [DOI] [PubMed] [Google Scholar]
- 22.Marrink S.J. and Tieleman D.P. (2013) Perspective on the Martini model. Chem. Soc. Rev. 42, 6801–6822 10.1039/c3cs60093a [DOI] [PubMed] [Google Scholar]
- 23.Arnarez C., Mazat J.-P., Elezgaray J., Marrink S.-J. and Periole X. (2013) Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J. Am. Chem. Soc. 135, 3112–3120 10.1021/ja310577u [DOI] [PubMed] [Google Scholar]
- 24.Arnarez C., Marrink S.J. and Periole X. (2013) Identification of cardiolipin binding sites on cytochrome c oxidase at the entrance of proton channels. Sci. Rep. 3, 1263 10.1038/srep01263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duncan A.L., Robinson A.J. and Walker J.E. (2016) Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Natl Acad. Sci. U.S.A. 113, 8687–8692 10.1073/pnas.1608396113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corey R.A., Pyle E., Allen W.J., Watkins D.W., Casiraghi M., Miroux B. et al. (2018) Specific cardiolipin-SecY interactions are required for proton-motive force stimulation of protein secretion. Proc. Natl Acad. Sci. U.S.A. 115, 7967–7972 10.1073/pnas.1721536115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stansfeld P.J., Hopkinson R., Ashcroft F.M. and Sansom M.S.P. (2009) PIP2-binding site in Kir channels: definition by multiscale biomolecular simulations. Biochem. 48, 10926–10933 10.1021/bi9013193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt M.R., Stansfeld P.J., Tucker S.J. and Sansom M.S.P. (2013) Simulation-based prediction of phosphatidylinositol 4,5-bisphosphate binding to an ion channel. Biochemistry 52, 279–281 10.1021/bi301350s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hedger G., Rouse S.L., Domański J., Chavent H., Koldsø H. and Sansom M.S.P. (2016) Lipid loving ANTs: molecular simulations of cardiolipin interactions and the organization of the adenine nucleotide translocase in model mitochondrial membranes. Biochemistry 55, 6238–6249 10.1021/acs.biochem.6b00751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duncan A.L., Ruprecht J.J., Kunji E.R.S. and Robinson A.J. (2018) Cardiolipin dynamics and binding to conserved residues in the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta 1860, 1035–1045 10.1016/j.bbamem.2018.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yen H.Y., Hoi K.K., Liko I., Hedger G., Horrell M.R., Song W.L. et al. (2018) Ptdins(4,5)P-2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 559, 424–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q., Corey R.A., Hedger G., Aryal P., Grieben M., Nazrallah C. et al. (2019) Lipid interactions of a ciliary membrane TRP channel: simulation and structural studies of polycystin-2 (PC2). Structure (in press) 10.1016/j.str.2019.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corradi V., Mendez-Villuendas E., Ingólfsson H.I., Gu R.-X., Siuda I., Melo M.N. et al. (2018) Lipid–protein interactions are unique fingerprints for membrane proteins. ACS Cent. Sci. 4, 709–717 10.1021/acscentsci.8b00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dupuy A.D. and Engelman D.M. (2008) Protein area occupancy at the center of the red blood cell membrane. Proc. Natl Acad. Sci. U.S.A. 105, 2848–2852 10.1073/pnas.0712379105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Javanainen M., Hammaren H., Monticelli L., Jeon J.-H., Miettinen M.S., Martinez-Seara H. et al. (2013) Anomalous and normal diffusion of proteins and lipids in crowded lipid membranes. Faraday Disc. 161, 397–417 10.1039/C2FD20085F [DOI] [PubMed] [Google Scholar]
- 36.Goose J.E. and Sansom M.S.P. (2013) Reduced lateral mobility of lipids and proteins in crowded membranes. PLoS Comput. Biol. 9, e1003033 10.1371/journal.pcbi.1003033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu E.L., Cheng X., Jo S., Rui H., Song K.C., Davila-Contreras E.M. et al. (2014) CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004 10.1002/jcc.23702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wassenaar T.A., Ingólfsson H.I., Böckmann R.A., Tieleman D.P. and Marrink S.J. (2015) Computational lipidomics with insane: a versatile tool for generating custom membranes for molecular simulations. J. Chem. Theor. Comput. 11, 2144–2155 10.1021/acs.jctc.5b00209 [DOI] [PubMed] [Google Scholar]
- 39.Stansfeld P.J., Goose J.E., Caffrey M., Carpenter E.P., Parker J.L., Newstead N.. et al. (2015) MemProtMD: automated insertion of membrane protein structures into explicit lipid membranes. Structure 23, 1350–1361 10.1016/j.str.2015.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newport T.D., Sansom M.S.P. and Stansfeld P.J. (2019) The MemProtMD database: a resource for membrane-embedded protein structures and their lipid interactions Nucl. Acids Res. 47, D390-D397 10.1093/nar/gky1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Husic B.E. and Pande V.S. (2018) Markov state models: from an art to a science. J. Am. Chem. Soc. 140, 2386–2396 10.1021/jacs.7b12191 [DOI] [PubMed] [Google Scholar]
- 42.Limongelli V., Bonomi M. and Parrinello M. (2013) Funnel metadynamics as accurate binding free-energy method. Proc. Natl Acad. Sci. U.S.A. 110, 6358–6363 10.1073/pnas.1303186110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiwary P., Limongelli V., Salvalaglio M. and Parrinello M. (2015) Kinetics of protein–ligand unbinding: predicting pathways, rates, and rate-limiting steps. Proc. Natl Acad. Sci. U.S.A. 112, E386-E391 10.1073/pnas.1424461112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song W.L., Yen H.Y., Robinson C.V. and Sansom M.S.P. (2019) State-dependent lipid interactions with the A2a receptor revealed by MD simulations using in vivo-mimetic membranes. Structure 27, 392–403 10.1016/j.str.2018.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Souaille M. and Roux B. (2001) Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 135, 40–57 10.1016/S0010-4655(00)00215-0 [DOI] [Google Scholar]
- 46.Li P.C., Miyashita N., Im W., Ishido S. and Sugita Y. (2013) Multidimensional umbrella sampling and replica-exchange molecular dynamics simulations for structure prediction of transmembrane helix dimers. J. Comput. Chem. 35, 300–308 10.1002/jcc.23494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Domański J., Hedger G., Best R., Stansfeld P.J. and Sansom M.S.P. (2017) Convergence and sampling in determining free energy landscapes for membrane protein association. J. Phys. Chem. B. 121, 3364–3375 10.1021/acs.jpcb.6b08445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shirts M.R. and Mobley D.L. (2013). An introduction to best practices in free energy calculations In Biomolecular Simulations: Methods and Protocols (Monticelli L. and Salonen E. eds), pp. 271–311. Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 49.Aldeghi M., Heifetz A., Bodkin M.J., Knappcd S. and Biggin P.C. (2016) Accurate calculation of the absolute free energy of binding for drug molecules. Chem. Sci. 7, 207–218 10.1039/C5SC02678D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klimovich P.V., Shirts M.R. and Mobley D.L. (2015) Guidelines for the analysis of free energy calculations. J. Comput. Aided Mol. Des. 29, 397–411 10.1007/s10822-015-9840-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corey R.A., Vickery O.N., Sansom M.S.P. and Stansfeld P.J. (2019) Insights into membrane protein–lipid interactions from free energy calculations. J. Chem. Theor. Comput. 15, 5727–5736 10.1021/acs.jctc.9b00548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barducci A., Bussi G. and Parrinello M. (2008) Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 100, 020603 10.1103/PhysRevLett.100.020603 [DOI] [PubMed] [Google Scholar]
- 53.Bussi G., Laio A. and Parrinello M. (2006) Equilibrium free energies from nonequilibrium metadynamics. Phys. Rev. Lett. 96, 090601 10.1103/PhysRevLett.96.090601 [DOI] [PubMed] [Google Scholar]
- 54.Planas-Iglesias J., Dwarakanath H., Mohammadyani D., Yanamala N., Kagan V.E. and Klein-Seetharaman J. (2015) Cardiolipin interactions with proteins. Biophys. J. 109, 1282–1294 10.1016/j.bpj.2015.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stahelin R.V., Scott J.L. and Frick C.T. (2014) Cellular and molecular interactions of phosphoinositides and peripheral proteins. Chem. Phys. Lipids 182, 3–18 10.1016/j.chemphyslip.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hansen S.B. (2015) Lipid agonism: the PIP2 paradigm of ligand-gated ion channels. Biochim. Biophys. Acta 1851, 620–628 10.1016/j.bbalip.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sampaio J.L., Gerl M.J., Klose C., Ejsing C.S., Beug H., Simons K. et al. (2011) Membrane lipidome of an epithelial cell line. Proc. Natl Acad. Sci. U.S.A. 108, 1903–1907 10.1073/pnas.1019267108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sriram K. and Insel P.A. (2018) G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258 10.1124/mol.117.111062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hanson M.A., Cherezov V., Griffith M.T., Roth C.B., Jaakola V.-P., Chien E.Y.T. et al. (2008) A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 16, 897–905 10.1016/j.str.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gimpl G. (2016) Interaction of G protein coupled receptors and cholesterol. Chem. Phys. Lipids 199, 61–73 10.1016/j.chemphyslip.2016.04.006 [DOI] [PubMed] [Google Scholar]
- 61.Oates J. and Watts A. (2011) Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr. Opin. Struct. Biol. 21, 802–807 10.1016/j.sbi.2011.09.007 [DOI] [PubMed] [Google Scholar]
- 62.Manna M., Niemelä M., Tynkkynen J., Javanainen M., Kulig W., Müller D.J. et al. (2016) Mechanism of allosteric regulation of β2-adrenergic receptor by cholesterol. eLife 5, e18432 10.7554/eLife.18432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lovera S., Cuzzolin A., Kelm S., De Fabritiis G. and Sands Z.A. (2019) Reconstruction of apo A2A receptor activation pathways reveal ligand-competent intermediates and state-dependent cholesterol hotspots. Sci. Rep. 9, 14199 10.1038/s41598-019-50752-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee J.Y. and Lyman E. (2012) Predictions for cholesterol interaction sites on the A2A adenosine receptor. J. Am. Chem. Soc. 134, 16512–16515 10.1021/ja307532d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Genheden S., Essex J.W. and Lee A.G. (2017) G protein coupled receptor interactions with cholesterol deep in the membrane. Biochim. Biophys. Acta 1859, 268–281 10.1016/j.bbamem.2016.12.001 [DOI] [PubMed] [Google Scholar]
- 66.Salari R., Joseph T., Lohia R., Henin J. and Brannigan G. (2018) A streamlined, general approach for computing ligand binding free energies and its application to GPCR-bound cholesterol. J. Chem. Theor. Comput. 14, 6560–6573 10.1021/acs.jctc.8b00447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hedger G., Koldsø H., Chavent M., Siebold C., Rohatgi R. and Sansom M.S.P. (2019) Cholesterol interaction sites on the transmembrane domain of the hedgehog signal transducer and class F G protein-coupled receptor smoothened. Structure 27, 549–559 10.1016/j.str.2018.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Domicevica L., Koldso H. and Biggin P.C. (2018) Multiscale molecular dynamics simulations of lipid interactions with P-glycoprotein in a complex membrane. J. Mol. Graph. Model. 80, 147–156 10.1016/j.jmgm.2017.12.022 [DOI] [PubMed] [Google Scholar]
- 69.Rosenhouse-Dantsker A., Noskov S., Durdagi S., Logothetis D.E. and Levitan I. (2013) Identification of novel cholesterol-binding regions in Kir2 channels. J. Biol. Chem. 288, 31154–31164 10.1074/jbc.M113.496117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee A.G. (2019) Interfacial binding sites for cholesterol on G protein-coupled receptors. Biophys. J. 116, 1586–1597 10.1016/j.bpj.2019.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gater D.L., Saurel O., Iordanov I., Liu W., Cherezov V. and Milon A. (2014) Two classes of cholesterol binding sites for the β2AR revealed by thermostability and NMR. Biophys. J. 107, 2305–2312 10.1016/j.bpj.2014.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gambin Y., Lopez-Esparza R., Reffay M., Sierecki E., Gov N.S., Genest M. et al. (2006) Lateral mobility of proteins in liquid membranes revisited. Proc. Natl Acad. Sci. U.S.A. 103, 2098–2102 10.1073/pnas.0511026103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie L.H., John S.A., Ribalet B. and Weiss J.N. (2008) Phosphatidylinositol-4,5-bisphosphate (PIP(2)) regulation of strong inward rectifier Kir2.1 channels: multilevel positive cooperativity. J. Physiol. 586, 1833–1848 10.1113/jphysiol.2007.147868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dawaliby R., Trubbia C., Delporte C., Masureel M., Van Antwerpen P., Kobilka B.K. et al. (2016) Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat. Chem. Biol. 12, 35–39 10.1038/nchembio.1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hedger G., Shorthouse D., Koldso H. and Sansom M.S. (2016) Free energy landscape of lipid interactions with regulatory binding sites on the transmembrane domain of the EGF receptor. J. Phys. Chem. B 120, 8154–8163 10.1021/acs.jpcb.6b01387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nury H., Dahout-Gonzalez C., Trezeguet V., Lauquin C.J.M., Brandolin G. and Pebay-Peyroula E. (2005) Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett. 579, 6031–6036 10.1016/j.febslet.2005.09.061 [DOI] [PubMed] [Google Scholar]
- 77.Ruprecht J.J., King M.S., Zogg T., Aleksandrova A.A., Pardon E., Crichton P.G. et al. (2019) The molecular mechanism of transport by the mitochondrial ADP/ATP carrier. Cell 176, 435–447 10.1016/j.cell.2018.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arnarez C., Marrink S.J. and Periole X. (2016) Molecular mechanism of cardiolipin-mediated assembly of respiratory chain supercomplexes. Chem. Sci. 7, 4435–4443 10.1039/C5SC04664E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kalli A.C. and Sansom M.S.P. (2014) Interactions of peripheral proteins with model membranes as viewed by molecular dynamics simulations. Biochem. Soc. Trans. 42, 1418–1424 10.1042/BST20140144 [DOI] [PubMed] [Google Scholar]
- 80.Baylon J.L., Vermaas J.V., Muller M.P., Arcario M.J., Pogorelov T.V. and Tajkhorshid E. (2016) Atomic-level description of protein–lipid interactions using an accelerated membrane model. Biochim. Biophys. Acta Biomembr. 1858, 1573–1583 10.1016/j.bbamem.2016.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khan H.M., He T., Fuglebakk E., Grauffel C., Yang B.Q., Roberts M.F. et al. (2016) A role for weak electrostatic interactions in peripheral membrane protein binding. Biophys. J. 110, 1367–1378 10.1016/j.bpj.2016.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fuglebakk E. and Reuter N. (2018) A model for hydrophobic protrusions on peripheral membrane proteins. PLoS Comp. Biol. 14, e1006325 10.1371/journal.pcbi.1006325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alwarawrah M. and Wereszczynski J. (2017) Investigation of the effect of bilayer composition on PKCα-C2 domain docking using molecular dynamics simulations. J. Phys. Chem. B 121, 78–88 10.1021/acs.jpcb.6b10188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kutateladze T.G. (2010) Translation of the phosphoinositide code by PI effectors. Nat. Chem. Biol. 6, 507–513 10.1038/nchembio.390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vonkova I., Saliba A.E., Deghou S., Anand K., Ceschia S., Doerks T. et al. (2015) Lipid cooperativity as a general membrane-recruitment principle for PH domains. Cell Rep. 12, 1519–1530 10.1016/j.celrep.2015.07.054 [DOI] [PubMed] [Google Scholar]
- 86.Lai C.-L., Landgraf K.E., Voth G.A. and Falke J.J. (2010) Membrane docking geometry and target lipid stoichiometry of membrane-bound PKCα C2 domain: a combined molecular dynamics and experimental study. J. Mol. Biol. 402, 301–310 10.1016/j.jmb.2010.07.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lai C.-L., Srivastava A., Pilling C., Chase A.R., Falke J.J. and Voth G.A. (2013) Molecular mechanism of membrane binding of the GRP1 PH domain. J. Mol. Biol. 425, 3073–3090 10.1016/j.jmb.2013.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yamamoto E., Kalli A.C., Akimoto T., Yasuoka K. and Sansom M.S.P. (2015) Anomalous dynamics of a lipid recognition protein on a lipid membrane surface. Sci. Rep. 5, 18245 10.1038/srep18245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yamamoto E., Kalli A.C., Yasuoka K. and Sansom M.S.P. (2016) Interactions of pleckstrin homology domains with membranes: adding back the bilayer via high throughput molecular dynamics. Structure 24, 1421–1431 10.1016/j.str.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamamoto E., Akimoto T., Kalli A.C., Yasuoka K. and Sansom M.S.P. (2017) Dynamic interactions between a membrane binding protein and lipids induce fluctuating diffusivity. Sci. Adv. 3, e1601871 10.1126/sciadv.1601871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Herzog F.A., Braun L., Schoen I. and Vogel V. (2016) Improved side chain dynamics in MARTINI simulations of protein lipid interfaces. J. Chem. Theor. Comput. 12, 2446–2458 10.1021/acs.jctc.6b00122 [DOI] [PubMed] [Google Scholar]
- 92.Wang Q., Pechersky Y., Sagawa S., Pan A.C. and Shaw D.E. (2019) Structural mechanism for Bruton's tyrosine kinase activation at the cell membrane. Proc. Natl Acad. Sci. U.S.A. 116, 9390–9399 10.1073/pnas.1819301116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Naughton F.B., Kalli A.C. and Sansom M.S.P. (2016) Association of peripheral membrane proteins with membranes: free energy of binding of GRP1 PH domain with PIP-containing model bilayers. J. Phys. Chem. Lett. 7, 1219–1224 10.1021/acs.jpclett.6b00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Naughton F., Kalli A.C. and Sansom M.S.P. (2018) Modes of interaction of pleckstrin homology domains with membranes: toward a computational biochemistry of membrane recognition. J. Mol. Biol. 430, 372–388 10.1016/j.jmb.2017.12.011 [DOI] [PubMed] [Google Scholar]
- 95.Yamamoto E., Domański J., Naughton F.B., Best R.B., Kalli A.C., Stansfeld P.J. et al. (2019) Multiple lipid binding sites determine the affinity of PH domains for phosphoinositide-containing membranes Sci. Adv. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chan K.C., Lu L.Y., Sun F. and Fan J. (2017) Molecular details of the PH domain of ACAP1 (BAR-PH) protein binding to PIP-containing membrane. J. Phys. Chem. B 121, 3586–3596 10.1021/acs.jpcb.6b09563 [DOI] [PubMed] [Google Scholar]
- 97.Kalli A.C., Devaney I. and Sansom M.S.P. (2014) Interactions of phosphatase and tensin homologue (PTEN) proteins with phosphatidylinositol phosphates: insights from molecular dynamics simulations of PTEN and voltage sensitive phosphatase. Biochem. 53, 1724–1732 10.1021/bi5000299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Amos S.-B.T.A., Kalli A.C., Shi J. and Sansom M.S.P. (2019) Membrane recognition and binding by the phosphatidylinositol phosphate kinase PIP5K1A: a multiscale simulation study. Structure 27, 1336–1346.e1332 10.1016/j.str.2019.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karandur D., Nawrotek A., Kuriyan J. and Cherfils J. (2017) Multiple interactions between an Arf/GEF complex and charged lipids determine activation kinetics on the membrane. Proc. Natl Acad. Sci. U.S.A. 114, 11416–11421 10.1073/pnas.1707970114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Melcr J., Martinez-Seara H., Nencini R., Kolafa J., Jungwirth P. and Ollila O.H.S. (2018) Accurate binding of sodium and calcium to a POPC bilayer by effective inclusion of electronic polarization. J. Phys. Chem. B 122, 4546–4557 10.1021/acs.jpcb.7b12510 [DOI] [PubMed] [Google Scholar]
- 101.Chu H.Y., Peng X.D., Li Y., Zhang Y.B. and Li G.H. (2018) A polarizable atomic multipole-based force field for molecular dynamics simulations of anionic lipids. Molecules 23, 77 10.3390/molecules23010077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cabanos C., Wang M., Han X.L. and Hansen S.B. (2017) A soluble fluorescent binding assay reveals PIP2 antagonism of TREK-1 channels. Cell Rep. 20, 1287–1294 10.1016/j.celrep.2017.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gupta K., Li J., Liko I., Gault J., Bechara C., Wu D. et al. (2018) Identifying key membrane protein lipid interactions using mass spectrometry. Nat. Protoc. 13, 1106 10.1038/nprot.2018.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Inada M., Kinoshita M., Sumino A., Oiki S. and Matsumori N. (2019) A concise method for quantitative analysis of interactions between lipids and membrane proteins. Anal. Chim. Acta 1059, 103–112 10.1016/j.aca.2019.01.042 [DOI] [PubMed] [Google Scholar]
- 105.Robinson C.V., Rohacs T. and Hansen S.B. (2019) Tools for understanding nanoscale lipid regulation of ion channels. Trends Biochem. Sci. 44, 795–806 10.1016/j.tibs.2019.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Slochower D.R., Huwe P.J., Radhakrishnan R. and Janmey P.A. (2013) Quantum and all-atom molecular dynamics simulations of protonation and divalent ion binding to phosphatidylinositol 4,5-bisphosphate (PIP2). J. Phys. Chem. B 117, 8322–8329 10.1021/jp401414y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Slochower D.R., Wang Y.H., Radhakrishnan R. and Janmey P.A. (2015) Physical chemistry and membrane properties of two phosphatidylinositol bisphosphate isomers. Phys. Chem. Chem. Phys. 17, 12608–12615 10.1039/C5CP00862J [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bilkova E., Pleskot R., Rissanen S., Sun S., Czogalla A., Cwiklik L. et al. (2017) Calcium directly regulates phosphatidylinositol 4,5-bisphosphate headgroup conformation and recognition. J. Am. Chem. Soc. 139, 4019–4024 10.1021/jacs.6b11760 [DOI] [PMC free article] [PubMed] [Google Scholar]