

Abstract
The PI3K/AKT pathway is a key target in oncology where most efforts are focussed on phenotypes such as cell proliferation and survival. Comparatively, little attention has been paid to PI3K in stemness regulation, despite the emerging link between acquisition of stem cell-like features and therapeutic failure in cancer. The aim of this review is to summarise current known and unknowns of PI3K-dependent stemness regulation, by integrating knowledge from the fields of developmental, signalling and cancer biology. Particular attention is given to the role of the PI3K pathway in pluripotent stem cells (PSCs) and the emerging parallels to dedifferentiated cancer cells with stem cell-like features. Compelling evidence suggests that PI3K/AKT signalling forms part of a ‘core molecular stemness programme’ in both mouse and human PSCs. In cancer, the oncogenic PIK3CAH1047R variant causes constitutive activation of the PI3K pathway and has recently been linked to increased stemness in a dose-dependent manner, similar to observations in mouse PSCs with heterozygous versus homozygous Pten loss. There is also evidence that the stemness phenotype may become ‘locked’ and thus independent of the original PI3K activation, posing limitations for the success of PI3K monotherapy in cancer. Ongoing therapeutic developments for PI3K-associated cancers may therefore benefit from a better understanding of the pathway's two-layered and highly context-dependent regulation of cell growth versus stemness.
Keywords: cancer, development, PI3K signalling, PIK3CA, pluripotent stem cells, stemness
An unsolved puzzle
Development and cancer can be described as two sides of the same coin, with cancer cells progressively co-opting and corrupting embryonic processes to support tumour growth and metastasis. The class IA phosphoinositide 3-kinase (PI3K) pathway is among the best studied in human biology, and its pathological hyperactivation is considered a ‘driver’ in numerous cancers as well as benign, developmental overgrowth [1]. The last two decades have provided a detailed mechanistic understanding of how this pathway regulates fundamental cellular processes such as survival, proliferation, migration and metabolism [2] (Figure 1). Accumulating evidence also suggests an important role for PI3K signalling in the regulation of stemness, yet the underlying mechanisms remain largely enigmatic.
Figure 1. An overview of class IA PI3K signalling.

The class IA PI3K heterodimer consists of one of three different catalytic subunits (p110α/β/γ) and one of five different regulatory subunits (p85α/β, p55α/γ, p50α). Its activation involves recruitment to the plasma membrane where its substrate, the phosphoinositide PI(4,5)P2 is located. A common mechanism of activation involves the binding of the regulatory p85 subunit to phosphotyrosine residues on receptor tyrosine kinases (RTKs) or their associated adaptor proteins. The catalytic subunits of PI3Kα and PI3Kδ can also interact and be activated by RAS. This is not the case for PI3Kβ which instead can be activated by other small GTPases downstream of G protein-coupled receptors (GPCRs). The immediate output of class IA PI3K activation is the generation of the second messenger PI(3,4,5)P3 and its derivative PI(3,4)P2. These are detected by proteins with specialised phosphoinositide-binding domains, with AKT representing one of the most studied examples. Through AKT-dependent and -independent effectors, the PI3K pathway orchestrates an array of diverse phenotypic modules whose execution is highly context-dependent [8]. Negative feedback loops and cross-talk with other pathways are omitted for clarity. For comprehensive PI3K signalling reviews, the reader is referred to Ref. [1,2,4,113].
Given the emerging link between cancer stemness and disease progression, a better mechanistic understanding of how the PI3K pathway impinges on critical developmental processes — either in forward (normal development) or reverse (cancer) mode — will be important for continued therapeutic development for PI3K-associated cancers. Collaterally, such research may also improve our understanding of key embryonic processes operating at early stages of developmental PI3K-related overgrowth disorders. Finally, insight into PI3K-dependent stemness regulation is likely to inform current efforts to establish improved stem cell culture protocols in developmental biology and regenerative medicine.
The aim of this review is to provide an overview of PI3K signalling in stemness regulation, with a focus on pluripotent stem cells (PSCs) and emerging parallels to cancer cells with stem cell-like properties. The need for a better mechanistic understanding of context-dependent PI3K-mediated stemness is highlighted, alongside the potential for systems biology and interdisciplinary approaches to gain insight into these important questions.
PI3K signalling: the pathway that seems to do it all
Class IA PI3Ks are heterodimers of a regulatory (p85) and a catalytic (p110) subunit, with the resulting complexes referred to as PI3Kα, PI3Kβ and PI3Kδ based on the identity of the catalytic subunit (Figure 1). Among these, the ubiquitously expressed PI3Kα is essential for organismal growth and survival, with pleiotropic functions ranging from control of tissue patterning, angiogenesis and insulin-dependent metabolic regulation. Activating mutations in PIK3CA, the gene encoding the catalytic p110α subunit of PI3Kα, are also considered disease-drivers in human cancers as well as developmental overgrowth disorders known as PROS (PIK3CA-related overgrowth spectrum) [3].
Irrespective of the exact enzymatic complex, the primary output of PI3K activation is the production of the second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3) and its dephosphorylated derivative PI(3,4)P2. Among their key effectors are the three serine/threonine kinase AKT isoforms, which control the activity of major cellular proteins, including the glycogen synthase kinase 3 isoforms (GSK3α/β), forkhead box O (FOXO) transcription factors and mechanistic target of rapamycin complex 1 (mTORC1) [4]. Through modulation of the actin cytoskeleton, PI3K activity also regulates multiple AKT-independent nodes, including those involved in membrane ruffling and cell migration [5] (Figure 1). Given this ability to impinge on critical cellular processes, the PI3K pathway is subject to exquisite control, including multiple negative feedback loops [1,4] and direct inactivation by several lipid phosphatases, most notably by the ubiquitously expressed tumour suppressor phosphatase and tensin homologue (PTEN) which 3-dephosphorylates both PIP3 and PI(3,4)P2 [6,7]. PI(3,4)P2 is generated by the 5-phosphatases SHIP1 and/or SHIP2, which are found both on the plasma membrane and the early endosomal compartments, thus contributing to the spatial regulation of phosphoinositide signalling [2].
While PI3K signalling might be seen as capable of regulating most major cellular processes (Figure 1), its output is usually rather specific and highly context-dependent — governed by cell-specific gene expression programmes, signalling thresholds and environmental context [8]. Considering PI3K signalling as a ‘pathway’ is itself a simplification used to conceptualise a complicated network of signalling components. In reality, PI3K signalling components cross-talk with effectors of other major pathways, including those of RAS/MAPK [9,10], WNT/β-catenin [11,12], NF-κB [13] and TGFβ [14–17]. The resulting complexity presents a significant challenge for conventional reductionist approaches and, consequently, remains poorly understood, with most studies focussing on isolated PI3K signalling effects.
Cancer: ‘reverse’ development
There are numerous mechanisms through which normal cells may acquire malignant features [18]. A common feature, however, is the convergence on a phenotypic programme with aberrant access to cellular functions with key roles in embryogenesis and tissue self-renewal [19]. Characteristics such as replicative immorality, lineage plasticity and the ability to undergo epithelial-to-mesenchymal transition (EMT) are shared between cancer cells and the PSCs that orchestrate early embryonic development [20,21]. Accordingly, embryonic markers such as NANOG, OCT3/4 and SOX2 are re-expressed across different human cancers and have been linked to poor clinical outcome [22]. Furthermore, a recent systems-level analysis of 17 major cancer types identified up-regulation of cell growth genes and the down-regulation of differentiation genes as a general pattern associated with shorter patient survival [23].
There is also ample evidence for a link between the acquisition of stemness properties and therapeutic resistance in cancer [24,25]. Stemness features in tumours are attributed to the presence of a subpopulation of cancer stem cells (CSCs), defined as cells with high self-renewal capacity and the ability to regenerate the heterogeneity of the primary tumour in functional experimental assays [25]. Understanding the molecular mechanisms that stabilise the CSC state, and that set it apart from the bulk of the remaining tumour cells, is therefore critical for effective therapeutic targeting [24].
PI3K signalling in cancer stemness
The PI3K pathway is frequently hyperactivated across multiple human cancers, either due to direct genetic and/or epigenetic dysregulation of pathway effectors or indirectly, due to aberrant signalling inputs (e.g. hyperactivation of upstream receptors, loss of negative feedback regulation). In particular, activating mutations in PIK3CA, the gene encoding the catalytic subunit of PI3Kα, are among the most common across multiple human cancer types and are also the cause of benign yet highly debilitating developmental overgrowth disorders [3].
Expression of the most frequently occurring PIK3CA cancer hotspot variant, H1047R, has been linked to dedifferentiation and stemness in mouse models of breast [26–28], lung [29] and colorectal [30] cancers. A similar phenotype has been observed upon wild-type PIK3CA overexpression in a murine head and neck cancer model [31]. Nevertheless, the exact molecular mechanism(s), including an often-reported requirement for additional oncogenic hits, have remained elusive. It is noteworthy that heterozygous PIK3CAH1047R on its own rarely suffices to induce cancer in mice [32], consistent with the benign disease in individuals with congenital PIK3CA-related overgrowth [3]. Interestingly, vascular malformations represent one of the most common and debilitating phenotypes in the PIK3CA-related overgrowth spectrum (PROS), and when modelled in mice, these lesions exhibit loss of arteriovenous identity markers, suggesting lineage identity loss and dedifferentiation even in some benign disease settings [33].
Oncogenic PI3Kα activation has also been linked with induction of EMT [28,30,34–36] — a process that is itself characterised by enormous plasticity and multiple intermediate states [24,37]. The connection between cancer stemness and EMT is suggested to hinge upon induction of autocrine signalling loops, including those involving the pro-tumorigenic action of the TGFβ pathway [24,38]. Given compelling evidence for a link between TGFβ and PI3K signalling in regulation of stemness in cancer-relevant cell models [31,35,39], as well as the involvement of both pathways in developmental stemness (see below), it will be important for future studies to determine whether cancer cells co-opt the developmental functions of the two pathways to acquire stemness properties that are associated with therapeutic resistance.
Such studies are inherently difficult to perform because CSCs are thought to represent rare cell populations in most tumours [25]. They may be enriched by using in vitro cancer spheroid models [40], but will still fall short of capturing the evolution of the stemness phenotype upon induction of oncogenic PI3K signalling in otherwise normal cells. While adult stem cells may represent an alternative option, hyperactivation of PI3K signalling in these cells is often associated with stem cell exhaustion and terminal differentiation (see section ‘A note on context'). For instance, homozygous Pten loss in mice leads to depletion of haematopoietic stem cells (HSCs), but promotes the generation of their transformed counterparts — leukemic stem cells [41]. This suggests that studies of PI3K-dependent stemness regulation in the context of cancer progression will benefit from availability of non-transformed cell lines that are nevertheless capable of unlimited self-renewal in the face of oncogenic pathway activation. Normal PSCs are characterised by a diploid genome and lack of oncogenic mutations, yet are naturally immortal and exhibit many phenotypic parallels to cancer cells [20,21]. This provides an opportunity to use PSCs as a model system to study PI3K pathway-dependent regulation of stemness, with subsequent testing of relevant findings in bona fide CSCs.
PI3K signalling in developmental stemness
PSC primer: the importance of species and developmental stage
PSCs such as mouse and human embryonic stem cells (mESCs and hESCs, respectively), or the corresponding induced pluripotent stem cells (iPSCs), are capable of multilineage differentiation and can theoretically give rise to any cell type in the adult organism (Box 1).
BOX 1: The many faces of stemness
‘Stemness’ is used to describe lack of differentiation or partial dedifferentiation and is typically applied in studies of stem cells, yet the definition of a stem cell is itself dependent on the particular context under study. Mammalian development starts with a fully undifferentiated single cell known as the totipotent zygote (see Figure below). This ultimate state of stemness is transient and quickly gives rise to the two cell lineages that define the developing blastocyst — the inner cell mass and the trophectoderm. The inner cell mass consolidates into the pluripotent epiblast from which all future embryonic lineages develop [130]. While short-lived in vivo, PSCs can be isolated and propagated indefinitely under the right conditions in vitro, thus forming the basis for the so-called human embryonic stem cells (hESCs). In this context, ‘stemness’ refers to the indefinite self-renewal of the undifferentiated cells, while pluripotency denotes their ability to differentiate to derivatives of the three embryonic germ layers (ectoderm, mesoderm, endoderm) [131].
At the other end of the spectrum, fully differentiated cells can acquire ‘stemness’ properties through the process of partial or complete dedifferentiation as seen in cancer or during the process of artificial reprogramming of somatic cells into induced pluripotent stem cells (iPSCs). Somewhere in between these two lie multipotent, bipotent and unipotent adult stem cells which are relatively differentiated yet capable of self-renewal and additional specification into tissue-specific cell types (Figure below). Neural stem cells (NSCs), mesenchymal stem cells (MSCs) and HSCs are examples of multipotent stem cell types, whereas more tissue-specific stem cells such as mammary stem cells or intestinal stem cells are more limited in their differentiation capacity.
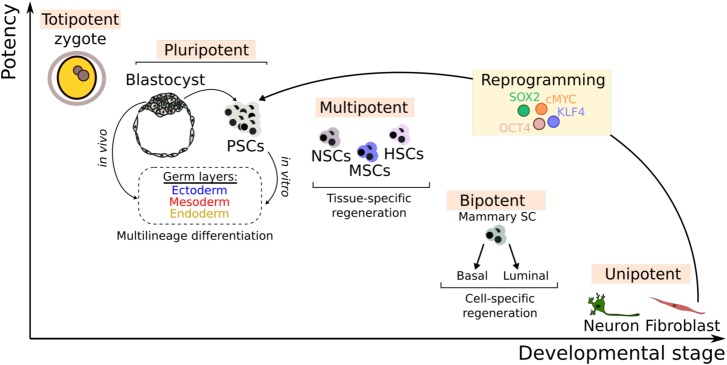
Comparisons of mouse and human PSCs have revealed critical differences, including the timing of transcriptional activation of the embryonic genome, distinct mechanisms to achieve X-chromosome dosage compensation in female lines, as well as differences in the configuration of signalling networks (reviewed in Ref. [42–45]). In addition to species divergence, these differences reflect the in vitro stabilisation of two distinct developmental states — the naïve pre-implantation state for mESCs and the primed post-implantation state for human PSCs (hPSCs) [43,46].
Differences between mouse and hPSCs are particularly important to consider when it comes to extrapolation of mechanistic insights from one system to the other. The core pluripotency gene regulatory network is a pertinent example. While co-ordinated by the same highly conserved transcription factors — NANOG, SOX2, OCT3/4 — in both mouse and hPSCs, downstream target gene regulation is poorly conserved [43,47,48]. More generally, this illustrates a recurrent point in this review — the notion that the same set of signalling effectors can be plugged into different regulatory layers in different cell types.
PI3K-induced stemness in mouse and human PSCs
Although several of the major cell signalling pathways, including MAPK/ERK and WNT, have opposing effects on mouse versus hPSCs [49,50], both cell systems exhibit a consistent reliance on PI3K signalling not only for survival but also for sustained stemness. This suggests that the PI3K pathway forms part of a ‘core molecular stemness programme’ in PSCs [51], with the underlying signalling network undergoing substantial remodelling in response to differentiation signals [52].
The strongest evidence for PI3K-induced stemness comes from genetic perturbations that result in constitutive activation of the pathway. This was initially achieved through Pten ablation in mESCs, resulting in their impaired differentiation both in vitro and in vivo[53]. The effect of Pten loss is allele dose-dependent, with Pten−/− mESCs giving rise to large, undifferentiated tumours in vivo, whereas their heterozygous counterparts generate well-differentiated tumours composed of tissues from all three embryonic germ layers [53]. Such tumours are known as teratomas and the capacity to form them is used to test for pluripotency. Therefore, homozygous but not heterozygous Pten−/− mESCs exhibit sustained stemness alongside impaired pluripotency. Although a subsequent study using a different mouse strain did not observe alternations in differentiation capacity between Pten−/− mESCs and wild-type counterparts, once differentiated, a subset of Pten-null mESC derivatives failed to down-regulate Nanog and Oct3/4 expression, resulting in a greater capacity for tumour formation [54]. These results are consistent with residual stemness and impaired pluripotency in a subset of Pten−/− mESCs. Similar to mESCs, hESCs with PTEN knock-down exhibit increased self-renewal and up-regulated expression of NANOG and OCT3/4, in conjunction with activation of canonical PI3K/AKT signalling and resistance to multilineage differentiation in three-dimensional (3D) embryoid body (EB) assays in vitro [55].
Given that PTEN can have PI3K-independent effects [56], other studies have investigated the link between PI3K pathway activation and stemness more directly by modulating key pathway effectors. In mESCs and primate PSCs, overexpression or constitutive activation of AKT results in self-sustained stemness, characterised by persistent expression of PSC markers and impaired differentiation in vitro [57,58]. More recently, an allelic series of isogenic hPSCs with endogenous heterozygous or homozygous expression of the PI3Kα-activating cancer-driver mutation PIK3CAH1047R were shown to exhibit a striking allele dose-dependent stemness phenotype [59] — similar to the aforementioned findings with heterozygous versus homozygous loss of Pten in mESCs. Thus, homozygous but not heterozygous PIK3CAH1047R mutants were characterised by self-sustained stemness both in vitro and in vivo, accompanied by graded activation of the PI3K pathway, partial loss of epithelial morphology and widespread transcriptional remodelling with up-regulated expression of multiple PSC markers, including NANOG and OCT3/4 [59]. The stemness phenotype of PIK3CAH1047R/H1047R hPSCs is similar to previous observations in mESCs with enhanced AKT activation [57,58] or GSK3α/β double knock-out [60], as well as to mouse and monkey PSCs expressing membrane-targeted and thus constitutively active PDK1 (3-phosphoinositide-dependent protein kinase 1; PDPK1) or AKT [57,61]. Combined, these studies suggest that above a certain threshold, constitutive PI3K activation leads to AKT-dependent self-sustained renewal of PSCs.
A different question is whether physiological levels of PI3K activation are required for continuous maintenance of PSCs. Work in this area has primarily been carried out in mESCs, and caution is warranted before extrapolating the proposed mechanisms to hPSCs (see section ‘Pluripotent stem cell (PSC) primer: the importance of species and developmental stage'). Such limitations notwithstanding, genetic and pharmacological studies by the Welham group have demonstrated that PI3K signalling is required for maintenance of the undifferentiated state in mESCs [62–64]. Conversely, knock-down of Akt1 leads to loss of mESC self-renewal [65,66]. Pharmacological PI3K inhibition in hPSCs has also been linked to increased differentiation [67–71], yet the evidence is mainly based on the use of the pan-PI3K inhibitors, LY294002 and wortmannin, which are known to be promiscuous towards multiple other kinases — including mTOR — at the applied concentrations [72–76]. The use of these inhibitors is strongly discouraged by experts in the PI3K signalling field [4].
Mechanistic insights: the known unknowns
Despite substantial evidence that PI3K signalling promotes stemness in PSCs, the underlying mechanisms have yet to be defined [77]. From receptor activation to the specific PI3K isoform(s) and its downstream effectors, the exact sequence of events and their contribution to PSC phenotypes warrant more systematic studies. The following is an attempt to summarise the known unknowns and thus facilitate the generation of novel hypotheses for future studies in this area.
Receptor-mediated PI3K activation
Advances in regenerative medicine have long called for more defined culture conditions for hPSCs, including coating substrate and growth medium. At present, the most widely used media solutions in the field are the commercially available mTESR1 and Essential 8/E8, with the latter allowing cells to be cultured in DMEM/F12 supplemented with only eight components [78]. Three of these eight components — insulin, FGF2 and TGFβ (or its alternative, NODAL) — represent growth factors/cytokines that are critical for hPSC survival and continued self-renewal (Figure 2) [78–86]. Insulin is well known to act in a PI3K-dependent manner, but is also able to induce activation of the mitogenic MAPK/ERK pathway [87]. FGF2 is a potent inducer of MAPK/ERK and can also activate PI3K. Further complexity emerges from the context-dependent cross-talk between the two pathways [88], either directly or indirectly. It is, however, unclear to what extent each growth factor leads to activation of one pathway over the other, and whether a specific balance needs to be attained for continuous PSC self-renewal — as observed in endothelial regulation of HSC maintenance [89]. Beyond FGF2 and insulin, whether TGFβ pathway activation promotes PI3K signalling in hPSCs — as reported in other contexts [14,17,90–92] — requires further and more systematic investigation. The available evidence remains inconclusive and may reflect differences in culture conditions and examined signalling time points [69,93].
Figure 2. Core signalling pathways maintaining human pluripotent stem cells (hPSCs).

The pluripotent state is inherently unstable and minor perturbations disrupting the balance within the signalling network may lead to the initiation of differentiation to either one of the three germ layers or to extraembryonic derivatives. As a consequence, the shown pathways may act both to promote stemness in one setting and differentiation in another, all depending on the microenvironmental context, as well as the subcellular localisation and signalling dynamics of individual pathway components. Note that several of the displayed effectors exist in multiple isoforms and are currently omitted for clarity, although there are cases in which two isoforms may have different or even opposing effects on PSC biology [143]. The red question mark is used to denote existing uncertainty about the ability of PI3K and WNT signalling to access the same GSK3 pool. Dashed lines are used to indicate indirect regulatory relationships. Positive regulation is shown in green and negative regulation in black.
Additional inputs into the PI3K pathway are also known to arise from autocrine and paracrine signals such as the endogenously secreted peptide ELABELA [94]. Finally, mechanotransduction is closely intertwined with both PI3K and MAPK/ERK signalling [95], and it is conceivable that differences in coating substrate may alter the dependencies on one or several of the aforementioned growth factors when it comes to PI3K activation. PI3K pathway activation is itself linked to altered expression of extracellular matrix components, at least in mESCs and their derivatives [96,97].
The specific PI3K isoform
At the level of the PI3K heterodimer itself, very few studies have attempted to determine the identity of the main PI3K catalytic isoform(s) responsible for pathway activation in PSCs. Treatment of hPSCs with a relatively low-dose (100 nM) of the PI3Kα-specific inhibitor BYL719 reduces AKT phosphorylation both at baseline and in response to different growth factors, whereas treatment with TGX221 at a dose (500 nM) that would inhibit both PI3Kβ and PI3Kδ has no effect [58,98,99]. These findings agree with the identification of PIK3CA as an essential gene in a recent knock-out CRISPR screen in haploid hPSCs [100]. In contrast with findings in mESCs [64], however, PI3Kα and not PI3Kβ so far appears to be the main isoform responsible not only for promoting survival but also stemness in hPSCs [58,98,99].
Downstream effectors
The exact signalling mechanisms whereby activation of PI3K signalling leads to increased expression of stemness markers are the least well understood, particularly when it comes to hPSCs (for an mESC-focussed review on PI3K signalling, see Ref. [101]). Well-studied AKT-dependent PI3K pathway effectors with known roles in stemness include GSK3 and MYC. MYC represents a central hub in stemness regulation via its pleiotropic roles on the transcriptome and epigenome of PSCs [102], and is also one of the four Yamanaka factors used to reprogramme somatic cells to iPSCs [103]. GSK3-mediated phosphorylation of MYC primes this transcription factor for degradation [104], thus AKT-dependent inhibition of GSK3 downstream of PI3K activation would be expected to have the opposite effect. While this mechanism appears to operate in mESCs [105], its importance in hPSCs has been disputed [93,106]. PI3K signalling can also lead to increased MYC levels through a translational mechanism that relies on mTORC1 activation [107], but this has yet to be studied in a stem cell context.
PI3K-dependent GSK3 inhibition may also lead to direct stabilisation of β-catenin, although the extent of this cross-talk remains subject to debate and may reflect indirect transcriptional changes through MYC or other effectors, as opposed to rapid post-translational regulation [108]. Among its many transcriptional targets, β-catenin — a downstream effector of canonical WNT signalling — promotes NODAL expression and thus TGFβ signalling, with both WNT and TGFβ pathways known to function in a dose- and time-dependent manner in developmental biology. It remains to be determined whether the recent discovery of dose-dependent stemness regulation downstream of oncogenic PI3K activation in hPSCs features an initial β-catenin-driven enhancement of NODAL expression and subsequent induction of self-sustained stemness, in a manner that is strictly dependent on a particular threshold of PI3K pathway activation (Box 2).
BOX 2: PIK3CAH1047R dose-dependent effects on stemness
Mainly performed in mouse ESCs treated with LY294002 or expressing dominant-negative PI3K regulators, some early studies linked PI3K activity to increased Nanog expression through a PI3Kβ-dependent but AKT-independent mechanism [64,132,133]. More recently, specific perturbation of PI3Kα by both genetic and pharmacological means revealed a previously unknown link between activation of this enzyme and acute dose- as well as time-dependent regulation of NODAL expression, prior to any changes in NANOG [58,99] — a well-known transcriptional target of TGFβ/NODAL signalling [134]. Furthermore, homozygous PIK3CAH1047R hPSCs no longer require continuous PI3K pathway activation to sustain the enhanced stemness gene signature, consistent with their autocrine activation of TGFβ/NODAL signalling [99]. Evidence from bona fide cancer models of oncogenic PI3K pathway activation also suggests that the stemness phenotype can become uncoupled from the original trigger and thus no longer reversible simply through PI3K inhibition [99,135,136].
Exactly how the PI3K pathway controls NODAL expression in a dose-dependent manner remains unknown. One candidate worthy of further investigation is β-catenin due to its ability to activate transcription of NODAL, with NODAL subsequently sustaining its own expression through an autoregulatory positive feedback loop [137]. There is some evidence for an interaction between WNT/β-catenin and oncogenic PI3K pathway activation in promoting intestinal [30], mammary [138] and leukemic [135] stem cell maintenance, yet further studies will be required to determine the nature of this cross-talk and whether it operates in response to PIK3CAH1047R expression in hPSCs.
Investigation of a potential link to the MYC oncogene is also warranted given its prominent role as a hub gene in computational network analyses of homozygous PIK3CAH1047R hPSCs [99]. Conditional MYC activation in mESCs has been shown to establish a self-sustained stemness phenotype which ultimately becomes independent of the presence of MYC activation [139], similar to the inability of PI3Kα inhibition to reverse the self-sustained stemness phenotype in homozygous PIK3CAH1047R hPSCs [99]. It is also noteworthy that MYC's biological effects have been linked to distinct thresholds of abundance [140]. Finally, PIK3CAH1047R was recently shown to co-operate with oncogenic KRAS in promoting MYC activity and tumorigenesis in mammary breast epithelial cells [141], a cellular system in which a link between oncogenic PI3K signalling and stemness has been demonstrated [35,142].
Others have suggested an alternative model linking PI3K, GSK3 and TGFβ signalling to explain the stemness-promoting ability of the PI3K pathway in hPSCs [93]. According to this model, PI3K activation indirectly promotes GSK3 activity and thus inhibits WNT/β-catenin signalling, which serves to keep TGFβ signalling below the activity threshold required for mesendodermal differentiation [93]. This indirect mechanism relies on PI3K-dependent inhibition of ERK, thus relieving ERK's inhibitory phosphorylation of GSK3 on the same Serine residue that is also known to be phosphorylated by AKT [93]. However, the supporting evidence is based on the use of the relatively non-specific inhibitors, LY294002 and PI-103, thus warranting additional confirmation. Arguing against this mechanism, GSK3 phosphorylation is ablated in response to PI3Kα-specific inhibition in wild-type as well as PIK3CAH1047R hPSCs, irrespective of the elevated ERK phosphorylation seen in PIK3CA mutant cells [99].
Another potential mechanism whereby PI3K signalling may promote stemness involves alteration of cellular metabolism and ‘knock-on’ effects on epigenetic regulation. The cancer field has contributed tremendous insight into how PI3K pathway activation alters major metabolic fluxes, most notably those associated with glycolysis, the citric acid cycle and lipid synthesis. This is closely linked with altered levels of key metabolites acting as substrates for chromatin- and DNA-modifying enzymes [109]. One well-studied example is the AKT-dependent increase in Acetyl-CoA levels in cells with hyperactive PI3K signalling, which in turn results in enhanced histone acetylation [110]. Conversely, a recent study demonstrated that Acetyl-CoA and the associated increase in histone acetylation sustains the stemness phenotype of hPSCs [111]. Moreover, MYC has been suggested to orchestrate the metabolic phenotype of hPSCs [112], although this has not been studied specifically in the context of PI3K activation and epigenetic changes. Thus, future studies are warranted to determine to what extent PI3K-induced stemness reflects metabolic regulation of the epigenome.
A note on context
An important point about the aforementioned AKT-regulated PI3K pathway effectors is that they are all subject to regulation by other components beyond those involved in PI3K signalling. The relative contribution of individual inputs is likely dependent on the exact cellular state. In the case of mTOR, additional complexity arises from its incorporation into two different complexes (mTORC1 and mTORC2) and their involvement in an array of cellular signalling pathways [113]. Moreover, mTORC1 and its effector ribosomal S6 kinase comprise a negative feedback loop that limits upstream PI3K activity [113], thus resulting in a non-linear relationship between PI3K and mTORC1 activation. Currently, knowledge about the effect of mTOR activity on stemness in hPSCs remains unclear [114,115], though recent data suggest the existence of dose-dependent regulation [116]. The context-dependent wiring of the signalling network and its dynamic properties should also be considered when assessing the mechanisms behind the dose-dependent effects on stemness following PTEN inactivation or oncogenic PIK3CA expression in PSCs. The similarity of the cellular phenotypes, notwithstanding, PI3K pathway activation in PTEN-null cells remains dependent on external ligand stimulation, whereas strongly activating PIK3CA mutations (e.g. H1047R) also promote constitutive ligand-independent PI3K signalling [3]. Such differences could give rise to altered signalling dynamics and feedback regulation [8], which in turn may impinge on stemness regulation through distinct mechanisms.
While PI3K pathway activation seems to promote stemness in PSCs, this ability is not universal when it comes to adult stem cells, reflecting not only different stem cell niches and developmental timings, but also differences in differentiation stage among stem cells residing within the same niche. Given multiple reports of adult stem cell exhaustion or differentiation in response to oncogenic activation of the PI3K pathway [41,117–121], and in particular mTORC1 activation (reviewed in Ref. [122]), it will be important for future studies to determine the exact factors that allow the same set of pathway components to promote stemness in one setting but not in another. Similarly, continued studies of PSCs and their derivatives may also help explain the apparent lineage skewing and relative lack of increased cancer risk in overgrowth patients with the embryonic acquisition of otherwise highly oncogenic PIK3CA mutations [3]. This also calls for a better understanding of the potential contribution from non-cell-autonomous mechanisms, driven by interactions between PIK3CA mutant and wild-type cells in a mosaic context [3]. Precedents for such mechanisms already exist, with previous work demonstrating that the enhanced stemness of HSCs in mice with germline Ship1 loss is a non-cell-autonomous consequence of an altered haematopoietic niche [123].
Future directions
The medieval proverb ‘All roads lead to Rome' can conveniently be superimposed on to the current picture of PI3K-dependent stemness regulation. While the puzzle remains unsolved, efforts are made to approach it from multiple, perhaps even diametrically opposite, ways. This, in turn, can result in confusion and give the impression of inconsistent findings. Although true inconsistencies do occur – often owing to the use of non-specific approaches (e.g. the widespread application of non-specific PI3K inhibitors in the PSC field) — the vast majority are likely to reflect the true complexity of the phenomenon under study and the limited ability of conventional approaches to capture the cellular system in its entirety.
Recent technological advances in high content imaging and -omics technologies are offering novel ways in which future studies may address the complexity of PI3K-mediated stemness, through a combination of conventional mechanistic studies and emerging systems biology strategies applied successfully in other areas [124–127]. To succeed in providing a unifying picture of PI3K-mediated stemness in development and cancer, such systems biology approaches will necessitate better interdisciplinary ‘cross-talk’ to combine the multifaceted mechanistic data on this pathway already available into comprehensive computational models. The power of these models lies in their ability to handle the complexity of temporal parameters, signalling thresholds and combinatorial pathway interactions. This, in turn, allows for the generation of mechanistic predictions for otherwise poorly understood signalling phenomena. These predictions can subsequently undergo formal testing by conventional approaches and the results used to refine and improve the original models in what may be considered a cycle of continuous reiteration.
A mathematical model of PI3K signalling in hPSCs has been developed and used to study the pathway's information transmission principles in this particular context [128,129]. This model may serve as a starting point for further refinement based on prior and future data, potentially enabling previously intractable questions to be addressed: How are different doses and patterns of PI3K activation sensed and decoded by hPSCs as a function of genetic background and environmental context? Are similar decoding principles shared by CSCs, thereby allowing hPSCs to be used as a valid in vitro model system for an otherwise rare subpopulation of therapeutically relevant cancer cells? Are there selective vulnerabilities whereby inhibition of PI3K-induced stemness can be achieved without knock-on effects on essential functions such as metabolic regulation?
Answering these fundamental questions is valuable in its own right and may also inform further therapeutic development for PI3K-associated disorders. Undoubtedly, solving the ‘PI3K-stemness puzzle’ will be an investment with many returns.
Perspectives
Given the link between cancer stemness and therapeutic relapse, understanding the PI3K pathway's two-layered regulation of growth versus stemness is an important task for the future. Beyond its direct translational value, this understanding may further efforts to develop improved PSC culturing protocols in developmental biology and regenerative medicine.
Oncogenic PI3K pathway activation has been linked to enhanced stemness in both cancer models and PSCs. At least in some contexts, this link appears to be exquisitely dependent on the dose of oncogenic PI3K signalling, yet the underlying mechanisms remain obscure.
Solving the ‘PI3K-stemness puzzle’ will hinge upon adoption of emerging systems biology approaches, including computational models capable of handling the context-dependent regulation of the phenomenon under study. For such approaches to succeed, there is a need for greater cross-talk between the fields of cancer, signalling and developmental biology.
Acknowledgements
I would like to thank Prof Bart Vanhaesebroeck and Dr Benoit Bilanges for critical review and feedback on the manuscript.
Abbreviations
- CSCs
cancer stem cells
- EB
embryoid body
- EMT
epithelial-to-mesenchymal transition
- FOXO
forkhead box O
- HSCs
haematopoietic stem cells
- iPSCs
induced pluripotent stem cells
- MSCs
mesenchymal stem cells
- NSCs
neural stem cells
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- PROS
PIK3CA-related overgrowth spectrum
- PSCs
pluripotent stem cells
- PTEN
phosphatase and tensin homologue
Author Contributions
R.R.M. conceptualised and wrote the review, including figure design.
Funding
R.R.M. is supported by funding provided to Prof Bart Vanhaesebroeck (B.V.). Work in the laboratory of B.V. is supported by PTEN Research, Cancer Research U.K. (C23338/A25722), the U.K. Biotechnology and Biological Sciences Research Council (BB/I007806/1, BB/M013278/1, BB/R017972/1) and the U.K. NIHR University College London Hospitals Biomedical Research Centre.
Open Access
Open access for this article was enabled by the participation of University College London in an all-inclusive Read & Publish pilot with Portland Press and the Biochemical Society under a transformative agreement with JISC.
Competing Interests
The author declares that there are no competing interests associated with this manuscript.
References
- 1.Fruman D.A., Chiu H., Hopkins B.D., Bagrodia S., Cantley L.C. and Abraham R.T. (2017) The PI3K pathway in human disease. Cell 170, 605–635 10.1016/j.cell.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bilanges B., Posor Y. and Vanhaesebroeck B. (2019) PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 20, 515–534 10.1038/s41580-019-0129-z [DOI] [PubMed] [Google Scholar]
- 3.Madsen R.R., Vanhaesebroeck B. and Semple R.K. (2018) Cancer-associated PIK3CA mutations in overgrowth disorders. Trends Mol. Med. 24, 856–870 10.1016/j.molmed.2018.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manning B.D. and Toker A. (2017) AKT/PKB signaling: navigating the network. Cell 169, 381–405 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hawkins P.T. and Stephens L.R. (2016) Emerging evidence of signalling roles for PI(3,4)P2 in Class I and II PI3K-regulated pathways. Biochem. Soc. Trans. 44, 307–314 10.1042/BST20150248 [DOI] [PubMed] [Google Scholar]
- 6.Malek M., Kielkowska A., Chessa T., Anderson K.E., Barneda D., Pir P., et al. (2017) PTEN regulates PI(3,4)P2 signaling downstream of class I PI3K. Mol. Cell, 566–580 10.1016/j.molcel.2017.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goulden B.D., Pacheco J., Dull A., Zewe J.P., Deiters A. and Hammond G.R.V. (2019) A high-avidity biosensor reveals plasma membrane PI(3,4)P2 is predominantly a class I PI3K signaling product. J. Cell Biol. 218, 1066–1079 10.1083/jcb.201809026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madsen R.R. and Vanhaesebroeck B. (2020) Cracking the context-specific PI3K signaling code. Sci. Signal. 13, eaay2940 10.1126/scisignal.aay2940 [DOI] [PubMed] [Google Scholar]
- 9.Mendoza M.C., Er E.E. and Blenis J. (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 36, 320–328 10.1016/j.tibs.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castellano E., Molina-Arcas M., Krygowska A.A., East P., Warne P., Nicol A. et al. (2016) RAS signalling through PI3-Kinase controls cell migration via modulation of Reelin expression. Nat. Commun. 7, 11245 10.1038/ncomms11245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inoki K., Ouyang H., Zhu T., Lindvall C., Wang Y., Zhang X., et al. (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126, 955–968 10.1016/j.cell.2006.06.055 [DOI] [PubMed] [Google Scholar]
- 12.Fang D., Hawke D., Zheng Y., Xia Y., Meisenhelder J., Nika H. et al. (2007) Phosphorylation of β-catenin by AKT promotes β-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229 10.1074/jbc.M611871200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oeckinghaus A., Hayden M.S. and Ghosh S. (2011) Crosstalk in NF-κB signaling pathways. Nat. Immunol. 12, 695–708 10.1038/ni.2065 [DOI] [PubMed] [Google Scholar]
- 14.Zhang L., Zhou F. and ten Dijke P. (2013) Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 38, 612–620 10.1016/j.tibs.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 15.Hamidi A., Song J., Thakur N., Itoh S., Marcusson A., Bergh A. et al. (2017) TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci. Signal. 10, eaal4186 10.1126/scisignal.aal4186 [DOI] [PubMed] [Google Scholar]
- 16.Budi E.H., Muthusamy B.P. and Derynck R. (2015) The insulin response integrates increased TGF-β signaling through Akt-induced enhancement of cell surface delivery of TGF-β receptors. Sci. Signal. 8, ra96 10.1126/scisignal.aaa9432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L., Zhou F., Drabsch Y., Gao R., Snaar-Jagalska B.E., Mickanin C., et al. (2012) USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nat. Cell Biol. 14, 717–726 10.1038/ncb2522 [DOI] [PubMed] [Google Scholar]
- 18.Hanahan D. and Weinberg R.A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 19.Lineweaver C.H., Davies P.C.W. and Vincent M.D. (2014) Targeting cancer's weaknesses (not its strengths): therapeutic strategies suggested by the atavistic model. Bioessays 36, 827–835 10.1002/bies.201400070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ben-David U. and Benvenisty N. (2011) The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 11, 268–277 10.1038/nrc3034 [DOI] [PubMed] [Google Scholar]
- 21.Hadjimichael C., Chanoumidou K., Papadopoulou N., Arampatzi P., Papamatheakis J. and Kretsovali A. (2015) Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells 7, 1150–1184 10.4252/wjsc.v7.i9.1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hepburn A.C., Steele R.E., Veeratterapillay R., Wilson L., Kounatidou E.E., Barnard A., et al. (2019) The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene 38, 4425 10.1038/s41388-019-0826-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uhlen M., Zhang C., Lee S., Sjöstedt E., Fagerberg L., Bidkhori G., et al. (2017) A pathology atlas of the human cancer transcriptome. Science 357, eaan2507 10.1126/science.aan2507 [DOI] [PubMed] [Google Scholar]
- 24.Shibue T. and Weinberg R.A. (2017) EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 14, 611–629 10.1038/nrclinonc.2017.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nassar D. and Blanpain C. (2016) Cancer stem cells: basic concepts and therapeutic implications. Annu. Rev. Pathol. Mech. Dis. 11, 47–76 10.1146/annurev-pathol-012615-044438 [DOI] [PubMed] [Google Scholar]
- 26.Van Keymeulen A., Lee M.Y., Ousset M., Brohée S., Rorive S., Giraddi R.R., et al. (2015) Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 525, 119–123 10.1038/nature14665 [DOI] [PubMed] [Google Scholar]
- 27.Koren S., Reavie L., Couto J.P., De Silva D., Stadler M.B., Roloff T., et al. (2015) PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 525, 114–118 10.1038/nature14669 [DOI] [PubMed] [Google Scholar]
- 28.Hanker A.B., Pfefferle A.D., Balko J.M., Kuba M.G., Young C.D., Sánchez V., et al. (2013) Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc. Natl Acad. Sci. U.S.A. 110, 14372–14377 10.1073/pnas.1303204110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Veen J.E., Scherzer M., Boshuizen J., Chu M., Liu A., Landman A. et al. (2019) Mutationally-activated PI3′-kinase-α promotes de-differentiation of lung tumors initiated by the BRAFV600E oncoprotein kinase. eLife 8, e43668 10.7554/eLife.43668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riemer P., Rydenfelt M., Marks M., van Eunen K., Thedieck K., Herrmann B.G. et al. (2017) Oncogenic β-catenin and PIK3CA instruct network states and cancer phenotypes in intestinal organoids. J. Cell Biol. 216, 1567–1577 10.1083/jcb.201610058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du L., Chen X., Cao Y., Lu L., Zhang F., Bornstein S., et al. (2016) Overexpression of PIK3CA in murine head and neck epithelium drives tumor invasion and metastasis through PDK1 and enhanced TGFβ signaling. Oncogene 35, 4641–4652 10.1038/onc.2016.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell C.B. and Phillips W.A. (2019) Mouse models for exploring the biological consequences and clinical significance of PIK3CA mutations. Biomolecules 9, 158 10.3390/biom9040158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castillo S.D., Tzouanacou E., Zaw-Thin M., Berenjeno I.M., Parker V.E.R., Chivite I., et al. (2016) Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans. Sci. Transl. Med. 8, 332ra43 10.1126/scitranslmed.aad9982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallin J.J., Guan J., Edgar K.A., Zhou W., Francis R., Torres A.C., et al. (2012) Active PI3K pathway causes an invasive phenotype which can be reversed or promoted by blocking the pathway at divergent nodes. PLoS ONE 7, e36402 10.1371/journal.pone.0036402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chakrabarty A., Surendran S., Bhola N.E., Mishra V.S., Wani T.H., Baghel K.S. et al. (2019) The H1047R PIK3CA oncogene induces a senescence-like state, pleiotropy and acute HSP90 dependency in HER2+ mammary epithelial cells. Carcinogenesis 40, 1179–1190 10.1093/carcin/bgz118 [DOI] [PubMed] [Google Scholar]
- 36.Yamaguchi H., Yoshida S., Muroi E., Yoshida N., Kawamura M., Kouchi Z. et al. (2011) Phosphoinositide 3-kinase signaling pathway mediated by p110α regulates invadopodia formation. J. Cell Biol. 193, 1275–1288 10.1083/jcb.201009126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pastushenko I., Brisebarre A., Sifrim A., Fioramonti M., Revenco T., Boumahdi S., et al. (2018) Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 10.1038/s41586-018-0040-3 [DOI] [PubMed] [Google Scholar]
- 38.Scheel C., Eaton E.N., Li S.H.J., Chaffer C.L., Reinhardt F., Kah K.J., et al. (2011) Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145, 926–940 10.1016/j.cell.2011.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katsuno Y., Meyer D.S., Zhang Z., Shokat K.M., Akhurst R.J., Miyazono K. et al. (2019) Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal. 12, eaau8544 10.1126/scisignal.aau8544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mehta P., Novak C., Raghavan S., Ward M. and Mehta G. (2018) Self-renewal and CSCs in vitro enrichment: growth as floating spheres. Methods Mol. Biol. 1692, 61–75 10.1007/978-1-4939-7401-6_6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yilmaz Ö.H, Valdez R., Theisen B.K., Guo W., Ferguson D.O., Wu H. et al. (2006) Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482 10.1038/nature04703 [DOI] [PubMed] [Google Scholar]
- 42.Boroviak T. and Nichols J. (2017) Primate embryogenesis predicts the hallmarks of human naïve pluripotency. Development 144, 175–186 10.1242/dev.145177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Theunissen T.W. and Jaenisch R. (2017) Mechanisms of gene regulation in human embryos and pluripotent stem cells. Development 144, 4496–4509 10.1242/dev.157404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossant J. and Tam P.P.L. (2017) New insights into early human development: lessons for stem cell derivation and differentiation. Cell Stem Cell 20, 18–28 10.1016/j.stem.2016.12.004 [DOI] [PubMed] [Google Scholar]
- 45.Wu J., Carlos J. and Belmonte I. (2016) Stem cells: a renaissance in human biology research. Cell 165, 1572–1585 10.1016/j.cell.2016.05.043 [DOI] [PubMed] [Google Scholar]
- 46.Kinoshita M. and Smith A. (2018) Pluripotency deconstructed. Dev. Growth Differ. 60, 44–52 10.1111/dgd.12419 [DOI] [PubMed] [Google Scholar]
- 47.Loh Y.H., Wu Q., Chew J.L., Vega V.B., Zhang W., Chen X., et al. (2006) The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 38, 431–440 10.1038/ng1760 [DOI] [PubMed] [Google Scholar]
- 48.Boyer L., Lee T.I., Cole M., Johnstone S., Levine S., Zucker J., et al. (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956 10.1016/j.cell.2005.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dalton S. (2013) Signaling networks in human pluripotent stem cells. Curr. Opin. Cell Biol. 25, 241–246 10.1016/j.ceb.2012.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu J. and Izpisua Belmonte J.C. (2015) Dynamic pluripotent stem cell states and their applications. Cell Stem Cell 17, 509–525 10.1016/j.stem.2015.10.009 [DOI] [PubMed] [Google Scholar]
- 51.Sato N., Munoz Sanjuan I., Heke M., Uchida M., Naef F. and Brivanlou A.H. (2003) Molecular signature of human embryonic stem cells and its comparison with the mouse. Dev. Biol. 260, 404–413 10.1016/S0012-1606(03)00256-2 [DOI] [PubMed] [Google Scholar]
- 52.Van Hoof D., Muñoz J., Braam S.R., Pinkse M.W.H., Linding R., Heck A.J.R. et al. (2009) Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell 5, 214–226 10.1016/j.stem.2009.05.021 [DOI] [PubMed] [Google Scholar]
- 53.Di Cristofano A., Pesce B., Cordon-Cardo C. and Pandolfi P.P. (1998) Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355 10.1038/1235 [DOI] [PubMed] [Google Scholar]
- 54.Lindgren A.G., Natsuhara K., Tian E., Vincent J.J., Li X., Jiao J. et al. (2011) Loss of Pten causes tumor initiation following differentiation of murine pluripotent stem cells due to failed repression of nanog. PLoS ONE 6, e16478 10.1371/journal.pone.0016478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alva J.A., Lee G.E., Escobar E.E. and Pyle A.D. (2011) Phosphatase and tensin homolog regulates the pluripotent state and lineage fate choice in human embryonic stem cells. Stem Cells 29, 1952–1962 10.1002/stem.748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee Y.-R., Chen M. and Pandolfi P.P. (2018) The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol. 19, 547–562 10.1038/s41580-018-0015-0 [DOI] [PubMed] [Google Scholar]
- 57.Watanabe S., Umehara H., Murayama K., Okabe M., Kimura T. and Nakano T. (2006) Activation of Akt signaling is sufficient to maintain pluripotency in mouse and primate embryonic stem cells. Oncogene 25, 2697–2707 10.1038/sj.onc.1209307 [DOI] [PubMed] [Google Scholar]
- 58.Pritsker M., Ford N.R., Jenq H.T. and Lemischka I.R. (2006) Genomewide gain-of-function genetic screen identifies functionally active genes in mouse embryonic stem cells. Proc. Natl Acad. Sci. U.S.A. 103, 6946–6951 10.1073/pnas.0509861103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Madsen R.R., Knox R.G., Pearce W., Lopez S., Mahler-Araujo B., McGranahan N. et al. (2019) Oncogenic PIK3CA promotes cellular stemness in an allele dose-dependent manner. Proc. Natl Acad. Sci. U.S.A. 116, 8380–8389 10.1073/pnas.1821093116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doble B.W., Patel S., Wood G.A., Kockeritz L.K. and Woodgett J.R. (2007) Functional redundancy of GSK-3α and GSK-3β in Wnt/β-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 12, 957–971 10.1016/j.devcel.2007.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ling L.S., Voskas D. and Woodgett J.R. (2013) Activation of PDK-1 maintains mouse embryonic stem cell self-renewal in a PKB-dependent manner. Oncogene 32, 5397–5408 10.1038/onc.2013.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Welham M.J., Storm M.P., Kingham E. and Bone H.K. (2007) Phosphoinositide 3-kinases and regulation of embryonic stem cell fate. Biochem. Soc. Trans. 35, 225–228 10.1042/BST0350225 [DOI] [PubMed] [Google Scholar]
- 63.Paling N.R.D., Wheadon H., Bone H.K. and Welham M.J. (2004) Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J. Biol. Chem. 279, 48063–48070 10.1074/jbc.M406467200 [DOI] [PubMed] [Google Scholar]
- 64.Kingham E. and Welham M. (2009) Distinct roles for isoforms of the catalytic subunit of class-IA PI3K in the regulation of behaviour of murine embryonic stem cells. J. Cell Sci. 122, 2311–2321 10.1242/jcs.046557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jeong C.H., Cho Y.Y., Kim M.O., Kim S.H., Cho E.J., Lee S.Y., et al. (2010) Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells 28, 2141–2150 10.1002/stem.540 [DOI] [PubMed] [Google Scholar]
- 66.Fang L., Zhang L., Wei W., Jin X., Wang P., Tong Y. et al. (2014) A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol. Cell 55, 537–551 10.1016/j.molcel.2014.06.018 [DOI] [PubMed] [Google Scholar]
- 67.Armstrong L., Hughes O., Yung S., Hyslop L., Stewart R., Wappler I., et al. (2006) The role of PI3K/AKT, MAPK/ERK and NFκβ signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum. Mol. Genet. 15, 1894–1913 10.1093/hmg/ddl112 [DOI] [PubMed] [Google Scholar]
- 68.Na J., Furue M.K. and Andrews P.W. (2010) Inhibition of ERK1/2 prevents neural and mesendodermal differentiation and promotes human embryonic stem cell self-renewal. Stem Cell Res. 5, 157–169 10.1016/j.scr.2010.06.002 [DOI] [PubMed] [Google Scholar]
- 69.Yu J.S.L., Ramasamy T.S., Murphy N., Holt M.K., Czapiewski R., Wei S.-K. et al. (2015) PI3K/mTORC2 regulates TGF-β/activin signalling by modulating Smad2/3 activity via linker phosphorylation. Nat. Commun. 6, 7212 10.1038/ncomms8212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McLean A.B., D'Amour K.A., Jones K.L., Krishnamoorthy M., Kulik M.J., Reynolds D.M., et al. (2007) Activin a efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells 25, 29–38 10.1634/stemcells.2006-0219 [DOI] [PubMed] [Google Scholar]
- 71.Shcherbina A., Li J., Narayanan C., Greenleaf W., Kundaje A. and Chetty S. (2019) Brief report: cell cycle dynamics of human pluripotent stem cells primed for differentiation. Stem Cells 37, 1151–1157 10.1002/stem.3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gharbi S.I., Zvelebil M.J., Shuttleworth S.J., Hancox T., Saghir N., Timms J.F. et al. (2007) Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 404, 15–21 10.1042/BJ20061489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brunn G.J., Williams J., Sabers C., Wiederrecht G., Lawrence J.C. and Abraham R.T. (1996) Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J. 15, 5256–5267 10.1002/j.1460-2075.1996.tb00911.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davies S.P., Reddy H., Caivano M. and Cohen P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 10.1042/bj3510095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cross M.J., Stewart A., Hodgkin M.N., Kerr D.J. and Wakelam M.J.O. (1995) Wortmannin and its structural analogue demethoxyviridin inhibit stimulated phospholipase A2 activity in Swiss 3T3 cells. Wortmannin is not a specific inhibitor of phosphatidylinositol 3-kinase. J. Biol. Chem. 270, 25352–25355 10.1074/jbc.270.43.25352 [DOI] [PubMed] [Google Scholar]
- 76.Nakanishi S., Catt K.J. and Balla T. (1995) A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc. Natl Acad. Sci. U.S.A. 92, 5317–5321 10.1073/pnas.92.12.5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu J.S. and Cui W. (2016) Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 143, 3050–3060 10.1242/dev.137075 [DOI] [PubMed] [Google Scholar]
- 78.Chen G., Gulbranson D.R., Hou Z., Bolin J.M., Ruotti V., Probasco M.D., et al. (2011) Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 8, 424–429 10.1038/nmeth.1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.James D., Levine A.J., Besser D. and Hemmati-Brivanlou A. (2005) TGFβ/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 132, 1273–1282 10.1242/dev.01706 [DOI] [PubMed] [Google Scholar]
- 80.Vallier L., Alexander M. and Pedersen R.A. (2005) Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J. Cell Sci. 118, 4495–4509 10.1242/jcs.02553 [DOI] [PubMed] [Google Scholar]
- 81.Xu R.H., Peck R.M., Li D.S., Feng X., Ludwig T. and Thomson J.A. (2005) Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat. Methods 2, 185–190 10.1038/nmeth744 [DOI] [PubMed] [Google Scholar]
- 82.Xu C., Rosler E., Jiang J., Lebkowski J.S., Gold J.D., O'Sullivan C. et al. (2005) Basic fibroblast growth factor supports undifferentiated human embryonic stem cell growth without conditioned medium. Stem Cells 23, 315–323 10.1634/stemcells.2004-0211 [DOI] [PubMed] [Google Scholar]
- 83.Xu R.H., Sampsell-Barron T.L., Gu F., Root S., Peck R.M., Pan G., et al. (2008) NANOG is a direct target of TGFβ/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell 3, 196–206 10.1016/j.stem.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang L., Schulz T.C., Sherrer E.S., Dauphin D.S., Shin S., Nelson A.M., et al. (2007) Self-renewal of human embryonic stem cells requires insulin-like growth factor-1 receptor and ERBB2 receptor signaling. Blood 110, 4111–4119 10.1182/blood-2007-03-082586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bendall S.C., Stewart M.H., Menendez P., George D., Vijayaragavan K., Werbowetski-Ogilvie T., et al. (2007) IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature 448, 1015–1021 10.1038/nature06027 [DOI] [PubMed] [Google Scholar]
- 86.Shahbazi M., Cundiff P., Zhou W., Lee P., Patel A., D'Souza S.L. et al. (2019) The role of insulin as a key regulator of seeding, proliferation, and mRNA transcription of human pluripotent stem cells. Stem Cell Res. Ther. 10, 228 10.1186/s13287-019-1319-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Belfiore A., Malaguarnera R., Vella V., Lawrence M.C., Sciacca L., Frasca F. et al. (2017) Insulin receptor isoforms in physiology and disease: An updated view. Endocr. Rev. 38, 379–431 10.1210/er.2017-00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fey D., Matallanas D., Rauch J., Rukhlenko O.S. and Kholodenko B.N. (2016) The complexities and versatility of the RAS-to-ERK signalling system in normal and cancer cells. Semin. Cell Dev. Biol. 58, 96–107 10.1016/j.semcdb.2016.06.011 [DOI] [PubMed] [Google Scholar]
- 89.Kobayashi H., Butler J.M., O'Donnell R., Kobayashi M., Ding B.-S., Bonner B., et al. (2010) Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat. Cell Biol. 12, 1046–1056 10.1038/ncb2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lamouille S. and Derynck R. (2007) Cell size and invasion in TGF-b-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178, 437–451 10.1083/jcb.200611146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu W.-T., Huang K.-Y., Lu M., Huang H.-L., Chen C., Cheng Y. et al. (2017) TGF-β upregulates the translation of USP15 via the PI3K/AKT pathway to promote p53 stability. Oncogene 36, 2715–2723 10.1038/onc.2016.424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xue G., Restuccia D.F., Lan Q., Hynx D., Dirnhofer S., Hess D. et al. (2012) Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov. 2, 248–259 10.1158/2159-8290.CD-11-0270 [DOI] [PubMed] [Google Scholar]
- 93.Singh A.M., Reynolds D., Cliff T., Ohtsuka S., Mattheyses A.L., Sun Y. et al. (2012) Signaling network crosstalk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell 10, 312–326 10.1016/j.stem.2012.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ho L., Tan S.Y.X., Wee S., Wu Y., Tan S.J.C., Ramakrishna N.B., et al. (2015) ELABELA is an endogenous growth factor that sustains hESC self-renewal via the PI3K/AKT pathway. Cell Stem Cell 17, 435–447 10.1016/j.stem.2015.08.010 [DOI] [PubMed] [Google Scholar]
- 95.Sun Y., Chen C.S. and Fu J. (2012) Forcing stem cells to behave: a biophysical perspective of the cellular microenvironment. Annu. Rev. Biophys. 41, 519–542 10.1146/annurev-biophys-042910-155306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li X., Talts U., Talts J.F., Arman E., Ekblom P. and Lonai P. (2001) Akt/PKB regulates laminin and collagen IV isotypes of the basement membrane. Proc. Natl Acad. Sci. U.S.A. 98, 14416–14421 10.1073/pnas.251547198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nahuel Villegas S., Rothová M., Barrios-Llerena M.E., Pulina M., Hadjantonakis A.K., Le Bihan T. et al. (2013) PI3K/Akt1 signalling specifies foregut precursors by generating regionalized extra-cellular matrix. eLife 2, e00806 10.7554/eLife.00806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Madsen R.R. (2019) Investigation of Genetic PIK3CA Activation in Genome-Edited Human Pluripotent Stem Cells, University of Cambridge [Google Scholar]
- 99.Madsen R. R., Longden J., Knox R. G., Xavier R., Völlmy F., Macleod K. G., et al. (2019) High-dose oncogenic PIK3CA drives constitutive cellular stemness through self-sustained TGFβ pathway activation. bioRxiv (preprint) 10.1101/753830 [DOI]
- 100.Yilmaz A., Peretz M., Aharony A., Sagi I. and Benvenisty N. (2018) Defining essential genes for human pluripotent stem cells by CRISPR–Cas9 screening in haploid cells. Nat. Cell Biol. 20, 610–619 10.1038/s41556-018-0088-1 [DOI] [PubMed] [Google Scholar]
- 101.Voskas D., Ling L.S. and Woodgett J.R. (2014) Signals controlling un-differentiated states in embryonic stem and cancer cells: role of the phosphatidylinositol 3′ kinase pathway. J. Cell. Physiol. 229, 1312–1322 10.1002/jcp.24603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fagnocchi L. and Zippo A. (2017) Multiple roles of MYC in integrating regulatory networks of pluripotent stem cells. Front. Cell Dev. Biol. 5, 1–19 10.3389/fcell.2017.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K. et al. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- 104.Gregory M.A., Qi Y. and Hann S.R. (2003) Phosphorylation by glycogen synthase kinase-3 controls c-Myc proteolysis and subnuclear localization. J. Biol. Chem. 278, 51606–51612 10.1074/jbc.M310722200 [DOI] [PubMed] [Google Scholar]
- 105.Bechard M. and Dalton S. (2009) Subcellular localization of glycogen synthase kinase 3 controls embryonic stem cell self-renewal. Mol. Cell. Biol. 29, 2092–2104 10.1128/MCB.01405-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Singh A.M., Bechard M., Smith K. and Dalton S. (2012) Reconciling the different roles of Gsk3β in ‘naïve’ and ‘primed’ pluripotent stem cells. Cell Cycle 11, 2991–2996 10.4161/cc.21110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Csibi A., Lee G., Yoon S.O., Tong H., Ilter D., Elia I. et al. (2014) The mTORC1/S6K1 pathway regulates glutamine metabolism through the eif4b-dependent control of c-Myc translation. Curr. Biol. 24, 2274–2280 10.1016/j.cub.2014.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 108.Voskas D., Ling L.S. and Woodgett J.R. (2010) Does GSK-3 provide a shortcut for PI3K activation of Wnt signalling? F1000 Biol. Rep. 2, 82 10.3410/B2-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reid M.A., Dai Z. and Locasale J.W. (2017) The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 19, 1298–1306 10.1038/ncb3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee J.V., Carrer A., Shah S., Snyder N.W., Wei S., Venneti S., et al. (2014) Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306–319 10.1016/j.cmet.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moussaieff A., Rouleau M., Kitsberg D., Cohen M., Levy G., Barasch D., et al. (2015) Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21, 392–402 10.1016/j.cmet.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 112.Cliff T.S., Wu T., Boward B.R., Yin A., Yin H., Glushka J.N. et al. (2017) MYC controls human pluripotent stem cell fate decisions through regulation of metabolic flux. Cell Stem Cell 21, 502–516.e9 10.1016/j.stem.2017.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Saxton R.A. and Sabatini D.M. (2017) mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 10.1016/j.cell.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou J., Su P., Wang L., Chen J., Zimmermann M., Genbacev O., et al. (2009) mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc. Natl Acad. Sci. U.S.A. 106, 7840–7845 10.1073/pnas.0901854106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Easley C.A., Ben-Yehudah A., Redinger C.J., Oliver S.L., Varum S.T., Eisinger V.M. et al. (2010) mTOR-mediated activation of p70 S6K induces differentiation of pluripotent human embryonic stem cells. Cell 12, 263–273 10.1089/cell.2010.0011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nazareth E.J.P., Rahman N., Yin T. and Zandstra P.W. (2016) A multi-lineage screen reveals mTORC1 inhibition enhances human pluripotent stem cell mesendoderm and blood progenitor production. Stem Cell Rep. 6, 679–691 10.1016/j.stemcr.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yue F., Bi P., Wang C., Li J., Liu X. and Kuang S. (2016) Conditional loss of pten in myogenic progenitors leads to postnatal skeletal muscle hypertrophy but age-dependent exhaustion of satellite cells. Cell Rep. 17, 2340–2353 10.1016/j.celrep.2016.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Castilho R.M., Squarize C.H., Chodosh L.A., Williams B.O. and Gutkind J.S. (2009) mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell 5, 279–289 10.1016/j.stem.2009.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Magri L., Cambiaghi M., Cominelli M., Alfaro-Cervello C., Cursi M., Pala M., et al. (2011) Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 9, 447–462 10.1016/j.stem.2011.09.008 [DOI] [PubMed] [Google Scholar]
- 120.Kharas M.G., Okabe R., Ganis J.J., Gozo M., Khandan T., Paktinat M. et al. (2010) Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 115, 1406–1415 10.1182/blood-2009-06-229443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ying Z., Sandoval M. and Beronja S. (2018) Oncogenic activation of PI3K induces progenitor cell differentiation to suppress epidermal growth. Nat. Cell Biol. 20, 1256–1266 10.1038/s41556-018-0218-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Meng D., Frank A.R. and Jewell J.L. (2018) mTOR signaling in stem and progenitor cells. Development 145, dev152595 10.1242/dev.152595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hazen A.L., Smith M.J., Desponts C., Winter O., Moser K. and Kerr W.G. (2009) SHIP is required for a functional hematopoietic stem cell niche. Blood 113, 2924–2933 10.1182/blood-2008-02-138008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gross S.M. and Rotwein P. (2016) Mapping growth-factor-modulated Akt signaling dynamics. J. Cell Sci. 129, 2052–2063 10.1242/jcs.183764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sulaimanov N., Klose M., Busch H. and Boerries M. (2017) Understanding the mTOR signaling pathway via mathematical modeling. Wiley Interdiscip. Rev. Syst. Biol. Med. 9, 1–18 10.1002/wsbm.1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sampattavanich S., Steiert B., Kramer B.A., Gyori B.M., Albeck J.G. and Sorger P.K. (2018) Encoding growth factor identity in the temporal dynamics of FOXO3 under the combinatorial control of ERK and AKT kinases. Cell Syst. 6, 664–678.e9 10.1016/j.cels.2018.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gerosa L., Chidley C., Froehlich F., Sanchez G., Lim S. K., Muhlich J., et al. (2019) Sporadic ERK pulses drive non-genetic resistance in drug-adapted BRAF V600E melanoma cells. bioRxiv (preprint) 1–44 10.1101/762294 [DOI] [Google Scholar]
- 128.Mathew S., Sundararaj S., Mamiya H. and Banerjee I. (2014) Regulatory interactions maintaining self-renewal of human embryonic stem cells as revealed through a systems analysis of PI3K/AKT pathway. Bioinformatics 30, 2334–2342 10.1093/bioinformatics/btu209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mathew S. and Banerjee I. (2014) Quantitative analysis of robustness of dynamic response and signal transfer in insulin mediated PI3K/AKT pathway. Comput. Chem. Eng. 71, 715–727 10.1016/j.compchemeng.2014.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hackett J.A. and Azim Surani M. (2014) Regulatory principles of pluripotency: from the ground state up. Cell Stem Cell 15, 416–430 10.1016/j.stem.2014.09.015 [DOI] [PubMed] [Google Scholar]
- 131.Cahan P. and Daley G.Q. (2013) Origins and implications of pluripotent stem cell variability and heterogeneity. Nat. Rev. Mol. Cell Biol. 14, 357–368 10.1038/nrm3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Storm M.P., Kumpfmueller B., Thompson B., Kolde R., Vilo J., Hummel O. et al. (2009) Characterization of the phosphoinositide 3-kinase-dependent transcriptome in murine embryonic stem cells: Identification of novel regulators of pluripotency. Stem Cells 27, 764–775 10.1002/stem.3 [DOI] [PubMed] [Google Scholar]
- 133.Storm M.P., Bone H.K., Beck C.G., Bourillot P.Y., Schreiber V., Damiano T. et al. (2007) Regulation of nanog expression by phosphoinositide 3-kinase-dependent signaling in murine embryonic stem cells. J. Biol. Chem. 282, 6265–6273 10.1074/jbc.M610906200 [DOI] [PubMed] [Google Scholar]
- 134.Vallier L., Mendjan S., Brown S., Chng Z., Teo A., Smithers L.E., et al. (2009) Activin/Nodal signalling maintains pluripotency by controlling nanog expression. Development 136, 1339–1349 10.1242/dev.033951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhu H., Zhang L., Wu Y., Dong B., Guo W., Wang M., et al. (2018) T-ALL leukemia stem cell ‘stemness’ is epigenetically controlled by the master regulator SPI1. eLife 7, e38314 10.7554/eLife.38314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen X., Cao Y., Sedhom W., Lu L., Liu Y., Wang H., et al. (2019) Distinct roles of PIK3CA in the enrichment and maintenance of cancer stem cells in head and neck squamous cell carcinoma. Mol. Oncol. 14, 139–158 10.1002/1878-0261.12584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hill C.S. (2018) Spatial and temporal control of NODAL signaling. Curr. Opin. Cell Biol. 51, 50–57 10.1016/j.ceb.2017.10.005 [DOI] [PubMed] [Google Scholar]
- 138.Korkaya H., Paulson A., Charafe-Jauffret E., Ginestier C., Brown M., Dutcher J. et al. (2009) Regulation of mammary stem/progenitor cells by PTEN/Akt/β-catenin signaling. PLoS Biol. 7, e1000121 10.1371/journal.pbio.1000121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fagnocchi L., Cherubini A., Hatsuda H., Fasciani A., Mazzoleni S., Poli V., et al. (2016) A Myc-driven self-reinforcing regulatory network maintains mouse embryonic stem cell identity. Nat. Commun. 7, 11903 10.1038/ncomms11903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Murphy D.J., Junttila M.R., Pouyet L., Karnezis A., Shchors K., Bui D.A. et al. (2008) Distinct thresholds govern myc's biological output in vivo. Cancer Cell 14, 447–457 10.1016/j.ccr.2008.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bell C.M., Raffeiner P., Hart J.R. and Vogt P.K. (2019) PIK3CA cooperates with KRAS to promote MYC activity and tumorigenesis via the bromodomain protein BRD9. Cancers (Basel) 11, 1634 10.3390/cancers11111634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thomas C., Henry W., Cuiffo B.G., Collmann A.Y., Marangoni E., Benhamo V., et al. (2017) Pentraxin-3 is a PI3K signaling target that promotes stem cell–like traits in basal-like breast cancers. Sci. Signal. 10, eaah4674 10.1126/scisignal.aah4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen X., Wang R., Liu X., Wu Y., Zhou T., Yang Y., et al. (2017) A chemical-genetic approach reveals the distinct roles of GSK3α and GSK3β in regulating embryonic stem cell fate. Dev. Cell 43, 563–576.e4 10.1016/j.devcel.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]