

Abstract
Bis-3-chloropiperidines are a new class of DNA-active compounds capable of alkylating nucleobases and inducing strand cleavage. In this report, we investigated the reactivity of these mustard-based agents with both single- and double-stranded DNA constructs. Polyacrylamide gel electrophoresis (PAGE) and electrospray ionization mass spectrometry (ESI-MS) were employed to obtain valuable insights into their mechanism at the molecular level and to investigate their time- and concentration-dependence. The results revealed the preferential formation of mono- and bifunctional adducts at nucleophilic guanine sites. In stepwise fashion, alkylation was followed by depurination and subsequent strand scission at the ensuing apurinic site. We demonstrated that the covalent modifications introduced by this new class of compounds can inhibit the activity of essential DNA-processing proteins, such as topoisomerase IIα, thus suggesting that bis-3-chloropiperidines may have excellent anticancer potential.
Keywords: bis-3-chloropiperidines, DNA alkylation, DNA depurination, topoisomerase IIα inhibition, anticancer agents
Graphical Abstract
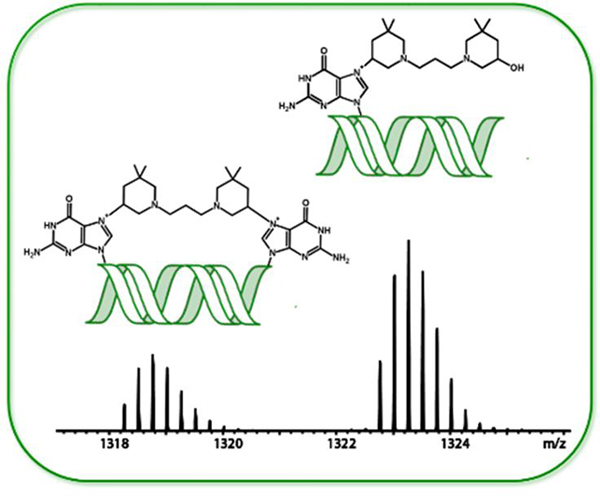
The assault on guanines! Exploring the molecular details of bis-3-chloropiperidines’s reactivity with DNA, our results demonstrate that the initial attack is directed towards the N7 position of guanine, generating both mono- and bi-functional adducts. The DNA damage inflicted by bis-3-chloropiperidines possesses meaningful properties on the activity of human topoisomerases IIα to develop this class of DNA-active compounds as anticancer agents.
Introduction
Nucleic acids are the molecular targets of different classes of small molecule drugs, which find broad clinical application in anticancer and antiviral therapy. DNA alkylating agents, such as nitrogen mustards, represent the oldest class of anticancer drugs, which is still widely used in cancer chemotherapy due to its ability to interfere with a wide range of biological processes.[1] These types of agents react covalently with nucleophilic sites on DNA bases, leading to alkylation at specific positions. Base alkylation prevents DNA replication and induces DNA cleavage, which can ultimately lead to cell death. Toxic side effects and the rapid emergence of drug resistance have typically limited the scope of their therapeutic applications [2] and spurred the search for more effective analogues. In this context, we recently developed nitrogen-linked bis-3-chloropiperidines as a new class of DNA alkylating agents.[3] The synthetic procedures were described in a recent report, together with a preliminary analysis of their structure-activity relationship (SAR).[3] Our library of bis-3-chloropiperidines included derivatives with different linker structures (i.e. flexible, rigid, lysine ester linkers), which we have shown capable of modulating the DNA-alkylating properties.[3] Figure 1A depicts the chemical structure of compound 1, which exemplifies the series of derivatives with flexible hydrocarbon linkers.[3b] Preliminary studies have shown that the reaction of bis-3-chloropiperidines with DNA proceeds through a bicyclic aziridinium intermediate formed from intramolecular nucleophilic displacement of chloride by nitrogen, followed by attack of nucleophilic centers present on the DNA substrate (Figure 1B).[4] In this report, we completed a systematic study with both single- and double-stranded oligodeoxyribonucleotides to gain a greater understanding of their mechanism of action and to unambiguously determine the molecular details of their reaction with DNA.
Figure 1.
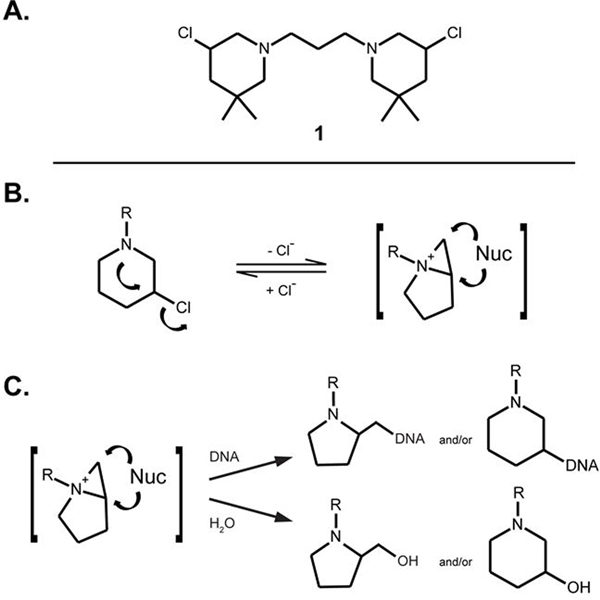
A. Chemical structure of representative bis-3-chloropiperidine derivative 1.[3] B. Chemical structure of the bicyclic aziridinium intermediate, which can be readily attacked by nucleophiles (Nuc). C. The electrophilic bicyclic aziridinium intermediate can be attacked by nucleophilic sites present on either DNA or solvent to give 5- and/or 6-membered ring adducts.
Based on the earlier study,[3b] bis-3-chloropiperidine 1 (Figure 1A) was selected as representative for the entire class of compounds. Established sequencing techniques were employed to confirm the adduct position, whereas mass spectrometric (MS) analysis was used to identify the reaction products. Additionally, we examined the effect of 1 on vital DNA-protein interactions to obtain an assessment on possible therapeutic applications. We specifically tested its ability to inhibit the activity of topoisomerases involved in the regulation of the topological state of DNA during replication, transcription, recombination and chromatin remodeling.[5] The fact that these enzymes are essential to maintain genome integrity and that their expression and activity increase at the onset of tumors[6] make them into excellent indicators of putative pathological states. Correlating the degree of alkylation and depurination with corresponding effects on the activity of human topoisomerase II allowed us to assess the potential of bis-3-chloropiperidines as possible candidates for the development of new anticancer therapeutics.
Results and Discussion
Bis-3-chloropiperidines react with the N7 of guanine
Earlier studies of the biological activity of bis-3-chloropiperidines demonstrated their ability to react very efficiently with nucleic acid molecules, leading to predominant cleavage of double-stranded DNA at guanine (dG) nucleotides.[3] Our hypothesis envisaged the N7-position of dG, which constitutes the most nucleophilic site present on DNA nucleobases,[7] as the alkylation site preferred by these novel mustard-based agents. N7-alkyation is typically followed by loss of the covalently modified nucleobase, which can lead to facile strand scission.[8] Consistent with these fundamental properties, our bis-3-chloropiperidines were shown capable of inducing cleavage of model oligodeoxyribonucleotides and plasmid DNA under a broad range of conditions. In this report, we employed different oligodeoxyribonucleotide constructs to obtain direct experimental evidence that bis-3-chloropiperidines attack predominantly the N7-position of dG nucleotides. For instance, a 22-mer double-stranded construct (ds-7DG-scr, Table 1) was assembled by annealing complementary strands, one of which contained a 7-deaza-2’-deoxyguanosine (7DG in Figure 2) that was expected to be impervious to alkylating agents. Save for this 7DG nucleotide placed in position 5, the construct replicated the actual sequence of a substrate used in our previous electrophoretic studies.[3] Compound 1 (Figure 1A) was selected as the representative member of this new class of alkylating agents by virtue of its excellent reactivity towards DNA substrates.[3b] The reaction was performed in accordance with experimental conditions described earlier,[3] which included mixing 2 μM of ds-7DG-scr with either 5 or 50 μM of compound 1 in BPE buffer at 37°C (see Experimental). Different incubation times ranging from 1 to 15 h were employed to monitor the reaction progress. After incubation, analysis by high-resolution poly-acrylamide gel electrophoresis (PAGE) was used to evaluate the effects of compound 1 on strand integrity.
Table 1.
Name, chemical modification, description, and sequence of oligodeoxynucleotides used in the study.
| Name | 5’-end modifier | Description | Sequence |
|---|---|---|---|
| 7DG-scr | 6-FAM | single-stranded DNA | 5’-FAM-GGAT(7DG)TGAGTGTGAGTGTGAGG-3’ |
| co-scr | None | single-stranded DNA | 5’-CCTCACACTCACACTCACATCC-3’ |
| ds-7DG-scr | see 7DG-scr see co-scr |
double-stranded DNA | 5’-FAM-GGAT(7DG)TGAGTGTGAGTGTGAGG-3’ 3’-CCTA C ACTCACACTCACACTCC-5’ |
| ODN1 | None | single-stranded DNA | 5’-TAGGGGGAAGCTTTGG-3’ |
| ODN2 | None | single-stranded DNA | 5’-ATTCCAAAGCTTCCCCCTA-3’ |
| dsDNA | see ODN2 see ODN1 |
double-stranded DNA | 5’-ATTCCAAAGCTTCCCCCTA-3’ 3’-GGTTTCGAAGGGGGAT-5’ |
Figure 2.
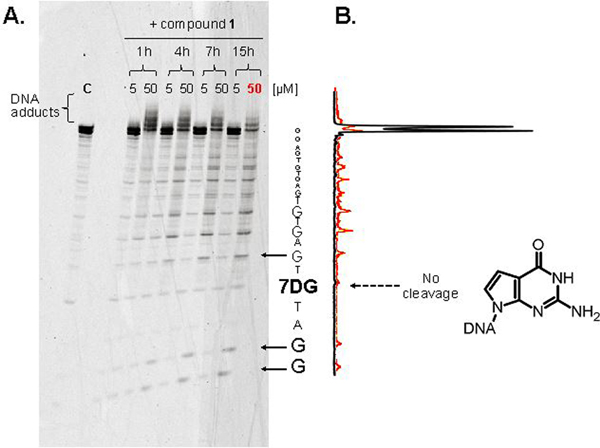
A. Time- and concentration-dependence of the reaction between bis-3-chloropiperidine 1 and ds-7DG-scr. The duplex construct included a strand carrying a 5’-FAM label to enable visualization and a 7-deaza-2’-deoxyguanosine nucleotide (7DG, chemical structure shown in figure) in position 5. 2 μM aliquots of ds-7DG-scr were treated with either 5 or 50 μM final concentrations of compound 1 at 37 °C in BPE buffer, pH 7.4, and incubated for the time indicated (see Experimental). The reaction mixtures were analyzed by denaturing polyacrylamide gel electrophoresis (PAA 20 %, 7M urea, TBE 1X). The 50 μM sample incubated for 15 h is labeled “50” and highlighted in red. Arrows indicate the position of cleavage products. C (control) indicates an untreated sample of oligodeoxynucleotide duplex. B. Densitograms used to quantify the intensities of cleavage products. The black line was obtained from the control lane C of panel 2A, while the red line was from the “50” sample, corresponding to the reaction of ds-7DG-scr with 50 μM 1 for 15 h.
The data shown in Figure 2 provide a comprehensive view of the time- and concentration-dependent features of the reaction between 1 and ds-7DG-scr. Consistent with substrate alkylation, bands migrating with lower mobility than the untreated sample (C, control) were already detectable after 1 h incubation (panel 2A). Numerous slower bands observed in the 50 μM lane suggested the formation of a range of adducts induced by the higher reagent concentration. Faster migrating bands were instead ascribed to the products of strand cleavage, which became progressively more intense at longer incubation times. As highlighted by the densitogram in panel 2B, which quantifies the observed electrophoretic bands, the dG nucleotides in the ds-7DG-scr strand were affected by cleavage, whereas the 7DG variant was not. This observation was confirmed by comparing the densitograms obtained from the control and 15 h incubation sample (e.g., black and red line in panel 2B, respectively). These data supported the working hypothesis that the N7 position of dG may represent the predominant site of bis-3-chloropiperidine alkylation.
Bis-3-chloropiperidines form both mono- and bi-functional adducts
To elucidate the molecular details of the products observed by denaturing PAGE, the characterization of reaction mixtures was carried out by electrospray ionization-mass spectrometry (ESI-MS). In this case, the selected substrate consisted of a single-stranded oligodeoxynucleotide containing a stretch of five adjacent dG nucleotides to provide multiple nucleophilic centers (ODN1, Table 1). At the same time, a 5’-FAM-labeled version of this substrate was used in parallel PAGE experiments to verify the ability of compound 1 to alkylate and cleave single-stranded oligodeoxynucleotides. Concentrations, incubation times, and other reaction conditions replicated those described earlier (see Experimental). The analysis confirmed the formation of both slow and fast migrating bands analogous to those observed for the double-stranded substrate, which were respectively ascribed to alkylation and cleavage processes (Figure S1). Based on these results, we performed an experiment by reacting 2 μM of ODN1 with 5 μM of compound 1 for 2 h at 37°C. The reaction was then quenched by ethanol precipitation, which enabled direct infusion ESI-MS analysis by accomplishing sample desalting (see Experimental). A representative ESI-MS spectrum is provided in Figure 3, which shows only the region containing the 4- charge state for the sake of clarity. An intense signal corresponding to unreacted substrate was readily recognized (labeled [ODN1-4H]4-). Additional signals corresponding to different bis-3-chloropiperidine adducts were also detected, which were assigned by matching experimental masses with values calculated from the ODN1 sequence and the structure of compound 1. Table 2 summarizes experimental and calculated masses and provides the assignment of the various species detected in Figure 3.
Figure 3.
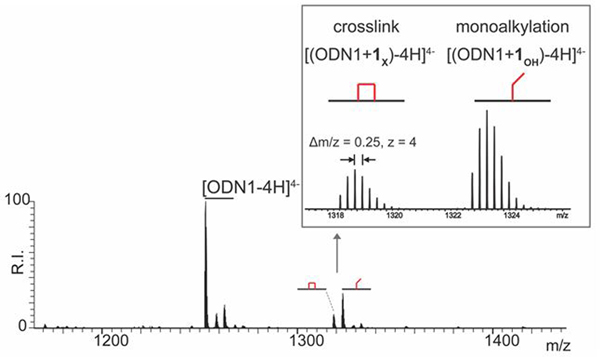
ESI-MS spectrum of reaction mixture obtained by incubating 2 μM of oligodeoxynucleotide ODN1 with 5 μM of bis-3-chloropiperidine 1 at 37 °C for 2 h. Spectra were recorded in 150 mM ammonium acetate (see Experimental Section for conditions). Lower intensity signals near free/bound species consist of typical sodium and ammonium adducts. The inset shows an expanded view of the 4- charge state of the crosslinked ODN1+1X ( ) and mono-alkylated ODN1+1OH (
) and mono-alkylated ODN1+1OH ( ) adducts.
) adducts.
Table 2.
Name, description, symbol, experimental and calculated mass for the species detected in Figure 3. These products were obtained by reacting 2 μM of oligodeoxynucleotide ODN1 with 5 μM of bis-3-chloropiperidine 1 at 37 °C for 2 h (see Experimental). Monoisotopic masses are reported in mass units (u).
| Name | Description | Symbol | Experimental mass (u) | Calculated mass (u) |
|---|---|---|---|---|
| ODN1 | single-stranded oligodeoxynucleotide | 5014.86 | 5014.86 | |
| ODN1+1X | crosslinked adduct of 1 on ODN1 | 5277.10 | 5277.10 | |
| ODN1+1OH | mono-alkylation adduct of 1 on ODN1 | 5295.10 | 5295.11 | |
The masses of the species observed by ESI-MS matched those of products generated by either mono- or bis-alkylation reactions. For instance, the species with experimental mass 5295.10 u (labeled  ) displayed a 280.24 u increment over the mass of the initial ODN1 construct (i.e., 5014.86 u, Table 2). This increment was consistent with the formation of a distinctive mono-alkylation adduct in which the remaining 3-chloropiperidine function was hydrolyzed to a 3-hydroxyl counterpart. The latter is due to the ability of an aziridinium intermediate (Figure 1B) to alternatively react with nucleophilic groups present on either the nucleic acid substrate or solvent components (Figure 1C). In contrast, the species with experimental mass 5277.10 u displayed a 262.24 u increment over that of ODN1, which corresponded to a characteristic bi-functional adduct formed by reaction of both 3-chloropiperidine functions with the nucleic acid substrate (labeled
) displayed a 280.24 u increment over the mass of the initial ODN1 construct (i.e., 5014.86 u, Table 2). This increment was consistent with the formation of a distinctive mono-alkylation adduct in which the remaining 3-chloropiperidine function was hydrolyzed to a 3-hydroxyl counterpart. The latter is due to the ability of an aziridinium intermediate (Figure 1B) to alternatively react with nucleophilic groups present on either the nucleic acid substrate or solvent components (Figure 1C). In contrast, the species with experimental mass 5277.10 u displayed a 262.24 u increment over that of ODN1, which corresponded to a characteristic bi-functional adduct formed by reaction of both 3-chloropiperidine functions with the nucleic acid substrate (labeled  , Table 2). These observations are consistent with the previously proposed mechanism of DNA alkylation by bis-3-chloropiperidines,[3] which involves the nucleophilic displacement of chloride by nitrogen to afford the rapid formation of the aziridinium ion, followed by either nucleic acid alkylation or solvolysis. These results provided an excellent explanation for the slow-migrating bands observed by denaturing PAGE. The various bands labeled as DNA adducts in Figure 2 were consistent with multiple alkylation hits allowed by the presence of several susceptible sites on the ds-7DG-scr substrate. The resolution of typical PAGE analysis, however, was not sufficient to discriminate between mono- and bi-functional products.
, Table 2). These observations are consistent with the previously proposed mechanism of DNA alkylation by bis-3-chloropiperidines,[3] which involves the nucleophilic displacement of chloride by nitrogen to afford the rapid formation of the aziridinium ion, followed by either nucleic acid alkylation or solvolysis. These results provided an excellent explanation for the slow-migrating bands observed by denaturing PAGE. The various bands labeled as DNA adducts in Figure 2 were consistent with multiple alkylation hits allowed by the presence of several susceptible sites on the ds-7DG-scr substrate. The resolution of typical PAGE analysis, however, was not sufficient to discriminate between mono- and bi-functional products.
DNA adducts induced by bis-3-chloropiperidines occur predominantly at guanine residues
The identity of the nucleobases involved in the bis-3-chloropiperidine reactions was investigated by tandem mass spectrometry (MS/MS).[9] As previously described, nucleobase alkylation weakens the N-glycosidic bond and facilitates characteristic base losses upon gas-phase activation.[10] The products generated by this process are more readily evident in positive ion mode, owing to the relatively basic character of the nucleobase leaving group. [11] For this reason, an abundant fragment with m/z 432.31 was readily observed upon activation of the 3+ charge state of mono-functional adduct ODN1+1OH (Figure S2), which corresponded to a G nucleobase modified by the 3-hydroxyl version of compound 1 (G+1OH, Figure 4A). In analogous fashion, gas-phase activation of the 3+ charge state of the bi-functional ODN1+1X generated products detected at m/z 565.35 and m/z 549.36 (Figure S2), which corresponded to different combinations of purine nucleobases crosslinked by compound 1 (G+1X+G and G+1X+A, respectively, Figure 4B). These results clearly confirmed the ability of bis-3-chloropiperidine to bridge across contiguous nucleotides. It was also noted that the signal provided by G+1X+G was significantly more intense than that of G+1X+A (Figure S2).
Figure 4.
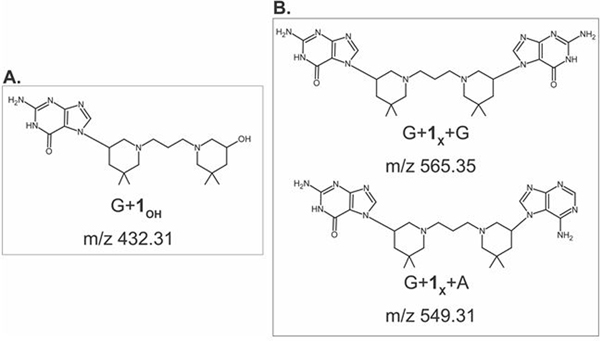
Fragments obtained by gas-phase activation of the 3+ charge states of A) ODN1+1OH and B) ODN1+1X.
At first sight, it would be tempting to attribute this observation to a greater reactivity of bis-3-chloropiperidine towards dG than dA. However, one must consider also that the sequence of ODN1 includes more instances of contiguous dG-dG nucleotides than dG-dA or dA-dG (Table 1). The greater number of susceptible sites could alone explain the different abundances displayed by the crosslinked products. In this context, it is significant that no fragment corresponding to A+1OH was detected upon activation of mono-functional ODN1+1OH, which could imply an initial attack to dA (Figure S2). The absence of this product suggests that dA alkylation may be more likely to occur as an intramolecular bridging reaction within an initial dG mono-functional adduct, which is entropically favored over the competing solvolysis. These considerations indicate that dG remains the most likely site for initial bis-3-chloropiperidine alkylation.
Bis-3-chloropiperidines alkylation leads to DNA depurination and strand cleavage in solution
The observation of multiple adducts by PAGE analysis and the almost complete loss of intact DNA detected at the higher doses indicated that both alkylation and strand cleavage were dose dependent.[3] We investigated the effects of dose on product distribution by analyzing mixtures that contained 2 μM of ODN1 and 50 μM of compound 1, followed by 2 h incubation at 37 °C (see Experimental). ESI-MS analysis showed signals corresponding to stable 1:1 bi- and mono-functional covalent adducts (labelled  and
and  in Figure 5), which matched those observed at lower concentrations (compare with spectrum in Figure 3). In this case however, no signal was detected for the initial unmodified substrate, whereas intense signals were observed for additional alkylation products. These data confirmed that, at this concentration, the initial ODN1 oligodeoxynucleotide had been completely converted into different reaction products after 2 h of incubation. Based on matching experimental and calculated masses (Table S1, Supporting Information), the new signals were assigned to species containing combinations of mono and bi-functional adducts (labelled
in Figure 5), which matched those observed at lower concentrations (compare with spectrum in Figure 3). In this case however, no signal was detected for the initial unmodified substrate, whereas intense signals were observed for additional alkylation products. These data confirmed that, at this concentration, the initial ODN1 oligodeoxynucleotide had been completely converted into different reaction products after 2 h of incubation. Based on matching experimental and calculated masses (Table S1, Supporting Information), the new signals were assigned to species containing combinations of mono and bi-functional adducts (labelled  ,
,  and
and  in Figure 5). Additional products were also observed with masses that were 151.05 u or 133.04 u lower than the corresponding adducts, which were consistent with the formal loss of G nucleobase to leave an abasic site produced by base elimination or hydrolytic attack (labelled respectively
in Figure 5). Additional products were also observed with masses that were 151.05 u or 133.04 u lower than the corresponding adducts, which were consistent with the formal loss of G nucleobase to leave an abasic site produced by base elimination or hydrolytic attack (labelled respectively  and
and  in Figure 5).
in Figure 5).
Figure 5.
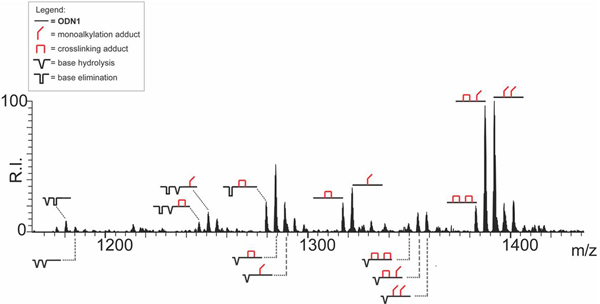
ESI-MS spectrum of reaction mixture obtained by incubating 2 μM of oligodeoxynucleotide ODN1 with 50 μM of bis-3-chloropiperidine 1 at 37 °C for 2 h. The analysis was performed in 150 mM ammonium acetate (see Experimental for conditions). Lower intensity signals near free/bound species consist of typical sodium and ammonium adducts. The spectrum shows the concentration-dependent reactions induced by compound 1 on ODN1. Only the region containing the 4- charge state is shown. To facilitate the interpretation, we included in the spectrum the graphical representation of identified reaction products (see inset for symbols legend). The symbol  corresponds to unmodified oligodeoxynucleotide ODN1;
corresponds to unmodified oligodeoxynucleotide ODN1;  mono-alkylation adduct in which the remaining 3-chloropiperidine function was hydrolyzed to a 3-hydroxyl;
mono-alkylation adduct in which the remaining 3-chloropiperidine function was hydrolyzed to a 3-hydroxyl;  crosslinking adduct;
crosslinking adduct;  base hydrolysis (modified G formally replaced by OH);
base hydrolysis (modified G formally replaced by OH);  base elimination (elimination of alkylated G nucleobase).
base elimination (elimination of alkylated G nucleobase).
The formation of abasic products is ascribable to a putative stepwise process triggered by the initial reaction with bis-3-chloropiperidine (Scheme 1). Alkylation of the N7 position of dG nucleotides places a formal positive charge on the ring system, which makes the nucleobase into an excellent leaving group for a nucleophilic substitution reaction. In contrast, alkylation of the six-member pyrimidine ring does not increase significantly the reactivity of the purine system in substitution reactions.[12] In aqueous solution, the oxocarbenium intermediate (c in Scheme 1) can readily react with water to produce an abasic cyclic acetal ( in Figure 5 and d in Scheme 1). In Figure 5, the possibility of multiple bis-piperidine hits on the same substrate explains the complexity of the data obtained from these reaction mixtures. The rather fast nature of the depurination process can account for the simultaneous detection of species containing all possible combinations of abasic sites, mono-functional 3-hydroxyl adducts, and bi-functional crosslinks (Figure 5).
in Figure 5 and d in Scheme 1). In Figure 5, the possibility of multiple bis-piperidine hits on the same substrate explains the complexity of the data obtained from these reaction mixtures. The rather fast nature of the depurination process can account for the simultaneous detection of species containing all possible combinations of abasic sites, mono-functional 3-hydroxyl adducts, and bi-functional crosslinks (Figure 5).
Scheme 1.
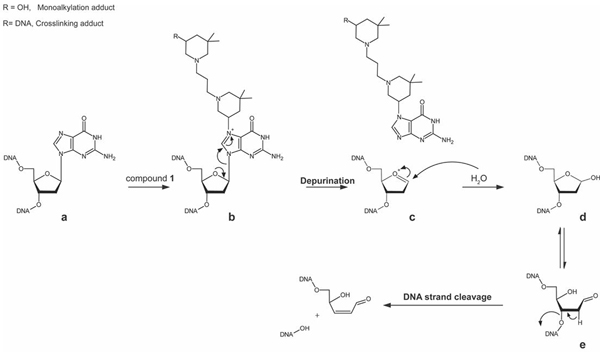
Established mechanism of DNA depurination of N7-alkylguanines residues by bis-3-chloropiperidine 1 and strand cleavage. Guanine residues in DNA (a) can be attacked by reactive bis-3-chloropiperidine 1. The alkylation of the N7 position of guanine places a formal positive charge on the guanine ring system (b), leading to the loss of the alkylguanine. The carbocation intermediate (c) react with the solvent (i.e. water) to form the abasic cyclic acetal (d), which is in equilibrium with small amounts of the ring-opened aldehyde form (e). The loss of the acidic proton adjacent to the carbonyl residue in the aldehydic form of the abasic site can lead to a strand break via elimination of the 3’-phosphate group.[13]
The same type of reaction was monitored for up to 15 h to assess the effects of incubation time on product distribution. The ESI-MS data obtained from this sample showed that the oligodeoxynucleotide ODN1 was completely transformed over time into products with lower molecular masses, whereas adducts of the initial substrate were not detected. Additional species were detected with masses corresponding to the possible loss of the 5’-tail TAG, TAGG, TAGGG, etc. from the initial ODN1 substrate (Figure S3). Each 5’-tail counterpart was also identified. These data are consistent with putative strand-cleavage events taking place at specific locations of the substrate sequence. Indeed, the phosphodiester bonds of abasic sites are well known for their susceptibility to hydrolysis, which results in strand cleavage (Scheme 1).[13] The “ladder” series observed here suggest that hydrolysis may take place at any possible dG nucleotide in the construct. The fact that these nucleotides were also the preferred targets of bis-3-chloropiperidine reactivity confirmed a direct correlation between alkylation, depurination and strand cleavage.
Bis-3-chloropiperidines reactivity with double-stranded DNA
The characterization of adducts with double-stranded constructs was carried out to evaluate the possible formation of inter-strand crosslinks. For this purpose, a duplex sample named dsDNA (Table 1) was prepared by annealing ODN1 with its complementary oligodeoxynucleotide ODN2. The sequences of these components were chosen to strategically place two adjacent dG nucleotides on opposing strands (i.e., G10 on ODN1 and G9 on ODN2, Table 2) to create a putative inter-strand reactive site. Alkylation reactions were carried out by incubating 2 μM of dsDNA with either 5 of 50 μM compound 1, followed by 2 h incubation at 37 °C. Figure 6 displays the ESI-MS spectrum obtained after desalting the reaction mixture (see Experimental). Experimental and calculated masses for all detected species are provided in Table S2 (Supporting Information). The results were very similar to those obtained upon reaction with single-stranded substrates. For instance, signals corresponding to initial unreacted duplex were observed together with those corresponding to 1:1 mono-alkylation and crosslinked adducts (labeled  and
and  , respectively).
, respectively).
Figure 6.
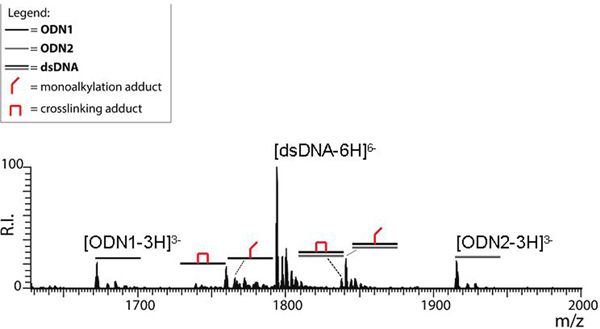
ESI-MS spectrum of a sample obtained by incubating 2 μM of the duplex dsDNA with 5 μM of bis-3-chloropiperidine 1 at 37 °C for 2 h. The analysis was performed in 150 mM ammonium acetate (see Experimental for conditions). Lower intensity signals near free/bound species consist of typical sodium and ammonium adducts. For the sake of clarity, the spectrum shows only the region containing the 6- charge state of the duplex and the 3- charge states of its single-stranded components. The symbol  corresponds to unmodified single-stranded oligodeoxynucleotide ODN1;
corresponds to unmodified single-stranded oligodeoxynucleotide ODN1;  single-stranded oligodeoxynucleotide ODN2;
single-stranded oligodeoxynucleotide ODN2;  double-stranded oligodeoxynucleotide dsDNA;
double-stranded oligodeoxynucleotide dsDNA;  mono-alkylation adduct in which the remaining 3-chloropiperidine function was hydrolyzed to a 3-hydroxyl;
mono-alkylation adduct in which the remaining 3-chloropiperidine function was hydrolyzed to a 3-hydroxyl;  crosslinking adduct.
crosslinking adduct.
A close examination of the spectrum in Figure 6 revealed signals corresponding to the single-stranded components of dsDNA, which were detected in both unmodified and alkylated form. Consistent with its dG-rich nature, ODN1 displayed mono-alkylation and crosslinking adducts, whereas the dG-poor ODN2 did not. These results suggested that also in double-stranded substrates, the availability of susceptible sites remained a major determinant of the extent of DNA damage introduced by bis-3-chloropiperidine.
The soft desolvation conditions employed in this determination ensure that a duplex construct of this size and base composition can be readily observed intact with no strand dissociation (see Figure S4 in Supporting Information).[14] Therefore, the detection of single-stranded products could be readily attributed to possible destablization of base-pairing interactions introduced by bis-3-chloropiperidine adducts. This hypothesis was supported by the data obtained at the higher reagent concentration, in which single-stranded products displayed the most intense signals (see Figure S5 and Table S3 in Supporting Information). In fact, 1:1 mono- and bi-functional adducts were observed not only for ODN1, but also for its dG-poor counterpart ODN2, which was not modified at lower reagent concentration (Figure 6). Further, additional signals corresponding to different combinations of 2:1 adducts were unambiguously recognized. Failure to detect any intact duplex, either in unmodified or alkylated form, confirmed the ability of bis-3-chloropiperidine adducts to destabilize single-stranded structures. This observation ruled out also the formation of detectable inter-strand crosslinks under the selected conditions.
Effects of bis-3-chloropiperidine adduction on human topoisomerase IIα activity
The ability of bis-3-chloropiperidines to induce depurination and strand-cleavage of small model substrates was consistent with the plasmid-nicking properties described in our previous report.[3b] At the cellular level, the formation of adducted bases, abasic sites and strand breaks can interfere with fundamental nuclear processes, such as DNA replication and transcription, which can adversely affect cell survival. To evaluate the possible biological effects of DNA lesions induced by bis-3-chloropiperidines, we utilized an assay based on the activity of human topoisomerase IIα (topo IIα), an essential enzyme overexpressed in proliferating cells, which is capable of both cleaving and resealing double-stranded nucleic acids.[15] The kinetoplast DNA (kDNA) of C. fasciculate, which consists of a network of catenated DNA rings, was used as a substrate that can be readily resolved into minicircle monomers by topo IIα.[16] Readout was obtained by electrophoretic analysis on agarose gel, which can visualize the decatenated minicircles produced by the enzymatic cleaving/resealing process (see Experimental). The assay was performed in the absence and presence of bis-3-chloropiperidines to compare their intrinsic strand-cleavage properties with those of topo IIα, and to explore the possible effects of their covalent adducts on DNA processing.
The ability of bis-3-chloropiperidine to cause strand-cleavage in this type of substrate was confirmed at the highest concentration of compound 1 (i.e., 50 μM) by the detection not only of the initial catenated substrate, but also of monomeric decatenated minicircles (lane 3 of Figure 6). Consistent with the large number of DNA adducts detected by ESI-MS analysis, the compound-induced decatenated monomers displayed a slower electrophoretic mobility (dashed arrow) than the decatenated monomers, which were barely evident already in the untreated kDNA control (lane B). In the presence of topo IIα, decatenated minicircles were already detectable at the lower concentrations used in this series of experiments (i.e., lanes 1t and 2t). However, the conversion to decatenated species was much lower than that observed under the same conditions in the absence of bis-3-chloropiperidine treatment (lane C), thus indicating that this compound possessed extensive inhibitory properties on topo IIα activity. This observation contrasts with the results of previous studies in which alkylators, such as clerocidin, stimulated DNA cleavage by topoisomerase II.[17] The dose-dependent character of such properties was clearly evident at the highest concentration of bis-3-chloropiperidine in the series, which resulted in distinct inhibition of enzymatic decatenation (lane 3t).
The incubation interval was subsequently increased from 2 to 18 h to investigate the time-dependent aspects of these processes. Extending the reaction time intensified the effects of bis-3-chloropiperidine on kDNA (Figure 6). In the absence of topo IIα, two bands corresponding to distinct minicircle topologies were detected at the lower concentrations of compound 1 (lanes 4 and 5), whereas more widespread cleavage was observed at the highest one (i.e., diffused smearing in lane 6). These unresolved, fast-migrating species were consistent with the broad distribution of alkylation, depurination, and backbone cleavage products observed by ESI-MS analysis of smaller DNA models. Also in this case, the presence of DNA damage induced by bis-3-chloropiperidine seemed to hamper the activity of topo IIα, which resulted in the detection of partially resolved catenanes (lanes 4t and 5t). In contrast, the outcome observed at higher compound 1 concentration (lane 6t) could be traced to the well-known ability of topoisomerases to establish specific interactions with damaged DNA.[18] In particular, it has been shown that apurinic sites located within the four-base overhang generated by topo IIα can stimulate enzymatic DNA scission and lead to irreversible “suicide” cleavage.[19] Therefore, the complete degradation of initial kDNA substrate into unresolved fast-migrating products (lane 6t) could be ascribed to the compounded effects of a higher concentration of bis-3-chloropiperidine and the presence of topo IIα in solution.
Conclusions
This study aimed at obtaining a comprehensive view of the mechanism of action of bis-3-chloropiperidines with different types of DNA substrates. The concerted application of electrophoretic and mass spectrometric approaches to analyze reaction mixtures obtained under a broad range of experimental conditions enabled the characterization of prominent products that are typical of mustard-based alkylating agents.[13, 20] The results confirmed that the initial attack was directed towards the N7 position of guanine, which constitutes the most nucleophilic site present in nucleic acids and represents the alkylation site preferred by nitrogen mustards.[7] The presence of two 3-chloropiperidine moieties in the same molecule ensures that, upon initial attack carried out by the first function, the second one can alternatively react with the aqueous solvent, or additional nucleophilic sites in adjacent nucleotides. Indeed, the former generated a mono-functional adduct with a “dangling” 3-hydroxyl-piperidine moiety, whereas the latter led to actual bi-functional crosslinks, which provided characteristic mass and fragmentation signatures that enabled unambiguous identification. The pattern of adduct formation was found to be significantly affected by both reagent concentration and reaction interval. In particular, higher concentrations produced broader adduct distributions with greater modification stoichiometries. Longer incubation intervals promoted instead the formation of abasic lesions by hydrolysis of alkylated guanine nucleobases. Depurination was followed by effective backbone cleavage, consistent with the outcome of many other alkylation reactions capable of producing abasic lesions. The substrates employed in the study led to the exclusive detection of intra-strand crosslinks between adjacent purines, rather than inter-strand bridges possible in double-stranded structures. Additional studies are currently underway to investigate how the length and flexibility of the linker between 3-chloropiperidines moieties may affect the yield of inter-strand crosslinks within either B- or A-form helical structures.
The decatenation experiments provided valuable insights into the possible effects induced by bis-3-chloropiperidines on vital cellular processes. In particular, the results showed that the DNA damage inflicted by lower reagent concentrations possessed unanticipated inhibitory properties on the activity of human topoisomerase IIα. At higher reagent concentration, the balance between the strand cleaving and resealing components of such activity was disproportionally tipped towards the former. For this reason, the incidence of strand breaks induced by bis-3-chloropiperidine was significantly enhanced by enzymatic catalysis. This observation suggests that bis-3-chloropiperidines could potentially represent a new class of therapeutic agents that act by indirectly modifying the activity of topo IIα. Indeed, these compounds could transform this essential protein into a lethal cellular toxin capable of increasing the levels of DNA breaks in the genome of treated cells. For this reason, additional studies are currently underway to further investigate the significance of combining bis-3-chloropiperidines with topo IIα. We are specifically targeting a series of human cancer lines, which are expected to constitute excellent susceptible targets by virtue of their rapid proliferation. Evaluating the cytotoxic effects of our extensive library of derivatives will provide the foundations for the development of bis-3-chloropiperidines as possible anticancer agents.
Experimental Section
Nucleic acid substrates.
Synthetic oligodeoxynucleotides were obtained in lyophilized form from commercial sources. In particular, the DNA oligonucleotides used for gel electrophoresis analysis were chemically synthesized by Metabion International AG (Martinsried, Germany); those used for mass spectrometry analysis were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA). All oligonucleotides were employed as received with no further purification. The sample were stored at −20°C in TE 1X buffer (Tris-HCl 10 mM, EDTA 1 mM, pH 8). Names, chemical modifications, and sequences are reported in Table 1. Double-stranded duplexes, such as dsDNA and ds-7DG-scr, were obtained by annealing the corresponding single-stranded components. In each case, equimolar amounts of the complementary strands were mixed in BPE 1X buffer (BPE 5X: NaH2PO4·2H2O 2 mM, Na2HPO4·12H2O 6 mM, Na2EDTA·2H2O 1 mM, pH 7.4). The mixture was denatured at 95 °C for 5 min, and then left to cool to room temperature to obtain the desired DNA duplex (Table 1). Each strand possessed specific features explained in the text. Strands labelled with 6-carboxyfluorescein (FAM) at the 5’-end were employed to enable fluorescence detection upon gel electrophoretic analysis.
Chemical reagents.
Bis-3-chloropiperidine 1 was synthesized in house as previously described.[3] Aliquots were freshly prepared by diluting an 8 mM DMSO stock in MilliQ water and were instantly reacted with the DNA substrate to avoid the typical quenching effects of the aqueous environment. All the other chemical reagents, including salts and solvents, were purchased from Sigma-Aldrich (Milan, Italy).
Reaction mixtures.
Samples containing a final 2 μM concentration of substrate in BPE buffer were added with compound 1 at final concentrations ranging from 0.5 to 50 μM, depending on the purpose of the experiment. Reactions mixtures were incubated at 37°C for 1, 2, 4, 7 and 15h, as indicated in the text.
Gel electrophoretic analysis.
Samples were dried in a vacuum centrifuge, re-suspended in 5 μL of denaturing buffer (10 mM Tris-HCl, 80% formamide, 0.025% bromophenol blue), and then loaded onto a 20% denaturing polyacrylamide gel (7 M urea) in TBE 1X buffer (Tris-HCl 89 mM, borate 89 mM, EDTA 2 mM). Acrylamide/bis-acrylamide (19:1) 40% solution was purchased from VWR International PBI Srl (Milan, Italy). Oligonucleotide products containing the FAM label were detected by using a STORM B40 Phosphorimager (GE Healthcare, Italy).
Mass spectrometric analysis.
Samples prepared in BPE were buffer-exchanged by performing ethanol precipitation in the presence of 1 M ammonium acetate. In case of reaction mixtures, the treatment served also to achieve reaction quenching. Samples were re-dissolved and diluted in 150 mM ammonium acetate (pH adjusted to 7.0) to achieve a final 2 μM concentration of total DNA. All samples were analyzed by direct infusion electrospray ionization (ESI) on Thermo Fisher Scientific (West Palm Beach, CA, USA) LTQ-Orbitrap Velos mass spectrometer. The analyses were performed in nanoflow mode by using quartz emitters produced in house by using a Sutter Instruments Co. (Novato, CA, USA) P2000 laser pipette puller. Up to 5 μL samples were typically loaded onto each emitter by using a gel-loader pipette tip. A stainless steel wire was inserted in the back-end of the emitter to supply an ionizing voltage that ranged between 0.8 and 1.2 kV. Source temperature and desolvation conditions were adjusted by closely monitoring the incidence of ammonium adducts and water clusters. Control determinations were completed to verify the experimental masses of either DNA substrate.
Full-fledged tandem mass spectrometry (MS/MS) determinations were accomplished by isolating the precursor ion of interest in the LTQ element of the instrument, which was then collided with N2 to activate fragmentation. The resulting products were mass analyzed in the orbitrap region of the instrument. Data were processed by using Xcalibur 2.1 software (Thermo Scientific).
Decatenation assay.
The activity of human topoisomerase IIα was determined by ATP-dependent decatenation of kinetoplast DNA (kDNA). Both enzyme and kDNA substrate were purchased from Inspiralis Ltd (UK). The decatenation assay is based on the different electrophoretic mobility of catenated kDNA, unable to migrate in the agarose gel, and of the monomeric decatenated products obtained by the topo IIα–mediated cleavage, which are visualized as discrete bands with higher electrophoretic mobility. 200 nanograms of substrate in MilliQ water were first added with 0.5, 5 and 50 μM final concentrations of compound 1, and then incubated at 37°C for either 2 or 18 h. The samples were subsequently added with 1 U of topoisomerase IIα at 37°C for 1 h. The environment consisted of Assay Buffer 1X (supplied as 10X stock: 50 mM Tris-HCl pH 7.5, 125 mM NaCl, 5 mM DTT, 100 μg/ml BSA, 10 mM MgCl2) and contained a final 1 mM concentration of ATP, as recommended by the supplier. In the samples without the enzyme, the volume of topoisomerase IIα solution was replaced with an equivalent volume of Dilution Buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM DTT, 50 μg/ml BSA, 0.5 mM EDTA, 50% v/v glycerol), which is the normal storage buffer for the enzyme. The final volume of each reaction was adjusted to 20 μL. A blank sample containing only kDNA and a control containing both kDNA and the enzyme (lane B and C in Figure 8) were also prepared to verify the activity of the enzyme.
Reactions were stopped by addition of 2 μL of denaturing gel loading buffer, which consisted of 49% TE (10 mM Tris-HCl pH 7.5, 20 mM NaCl, 1 mM EDTA), 49% glycerol, 2% SDS, 0.025% bromophenol blue, and 0.025% xylene cyanol. Samples were analyzed by electrophoresis on a 1% agarose gel in TBE buffer (0.5 X: 45 mM Tris-HCl, 45 mM Boric Acid, 10 mM EDTA). Electrophoresis was performed at 80 V for 90–120 minutes. Each gel was stained by using a 0.5 μg/mL solution of ethidium bromide. DNA bands were visualized on a Geliance 600 imaging system (Perkin Elmer).
Supplementary Material
Figure 7.
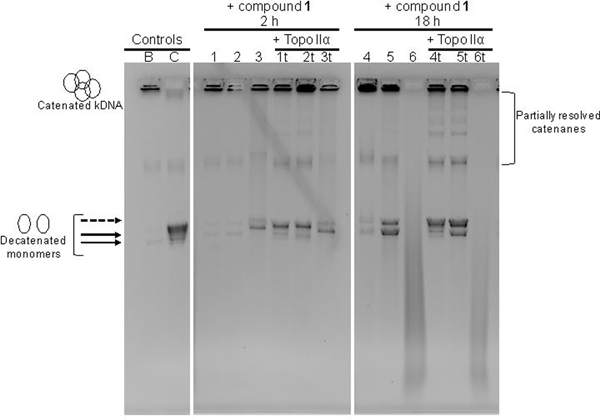
Effects of bis-3-chloropiperidine 1 on DNA integrity and decatenation by human topoisomerase IIα. Lane B contains kinetoplast DNA (kDNA) used as initial nucleic acid substrate. Lane C contains a mixture of kDNA and topoisomerase IIα (topo IIα) incubated for 1 h at 37°C (see Experimental Section for details). Lane 1, 2, and 3 contain kDNA samples treated respectively with 0.5, 5, and 50 μM of bis-3-chloropiperidine 1, which were incubated for 2 h at 37°C. Lane 1t, 2t, and 3t contain samples with the same compositions of those analyzed in Lane 1–3, which were further incubated with topo IIα for 1 h at 37°C. Lane 4–6 and 4t-6t contain samples analogous to those analyzed in Lane 1–3 and 1t-3t, which were instead reacted for 18 h prior to addition of topo IIα. To facilitate the interpretation, we included arrows to indicate the position of detected decatenated monomers; dashed arrow indicates the position of decatenated monomers migrating more slowly.
Acknowledgements
BG thanks the financial support by Ministero degli Affari Esteri e Cooperazione Internazionale (DRPG, Grant: PGR00171).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Avendaño C, Menendez JC, Medicinal chemistry of anticancer drugs, Elsevier, 2015; [Google Scholar]; b) Hurley LH, Nat. Rev. Cancer 2002, 2, 188–200; [DOI] [PubMed] [Google Scholar]; c) Kohn KW, Cancer Res. 1996, 56, 5533–5546. [PubMed] [Google Scholar]
- [2].a) Noll DM, Mason TM, Miller PS, Chem. Rev. 2006, 106, 277–301; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Panasci L, Xu ZY, Bello V, Aloyz R, Anticancer drugs 2002, 13, 211–220. [DOI] [PubMed] [Google Scholar]
- [3].a) Zuravka I, Roesmann R, Sosic A, Gottlich R, Gatto B, Bioorg. Med. Chem. 2015, 23, 1241–1250; [DOI] [PubMed] [Google Scholar]; b) Zuravka I, Roesmann R, Sosic A, Wende W, Pingoud A, Gatto B, Gottlich R, ChemMedChem 2014, 9, 2178–2185; [DOI] [PubMed] [Google Scholar]; c) Zuravka I, Sosic A, Gatto B, Gottlich R, Bioorg. Med. Chem. Lett. 2015, 25, 4606–4609. [DOI] [PubMed] [Google Scholar]
- [4].Henderson ND, Lacy SM, O’Hare CC, Hartley JA, McClean S, Wakelin LP, Kelland LR, Robins DJ, Anticancer Drug Des. 1998, 13, 749–768. [PubMed] [Google Scholar]
- [5].Brill SJ, DiNardo S, Voelkel-Meiman K, Sternglanz R, NCI Monographs 1987, 11–15. [PubMed] [Google Scholar]
- [6].Larsen AK, Skladanowski A, Biochim. Biophys. Acta 1998, 1400, 257–274. [DOI] [PubMed] [Google Scholar]
- [7].Mattes WB, Hartley JA, Kohn KW, Nucleic Acids Res. 1986, 14, 2971–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hara M, Yoshida M, Nakano H, Biochemistry 1990, 29, 10449–10455. [DOI] [PubMed] [Google Scholar]
- [9].Nordhoff E, Kirpekar F, Roepstorff P, Mass Spectrom. Rev. 1996, 15, 67–138. [DOI] [PubMed] [Google Scholar]
- [10].a) Kellersberger KA, Tan PV, Laiko VV, Doroshenko VM, Fabris D, Anal. Chem. 2004, 76, 3930–3934; [DOI] [PubMed] [Google Scholar]; b) Zhang Q, Yu ET, Kellersberger KA, Crosland E, Fabris D, J. Am. Soc. Mass Spectrom. 2006, 17, 1570–1581. [DOI] [PubMed] [Google Scholar]
- [11].Quinn R, Basanta-Sanchez M, Rose RE, Fabris D, J. Mass Spectrom. 2013, 48, 703–712; and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gates KS, Nooner T, Dutta S, Chem. Res. Toxicol. 2004, 17, 839–856. [DOI] [PubMed] [Google Scholar]
- [13].a) Gates KS, Chem. Res. Toxicol. 2009, 22, 1747–1760; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gates KS, Nooner T, Dutta S, Chem. Res. Toxicol. 2004, 17, 839–856; and references therein. [DOI] [PubMed] [Google Scholar]
- [14].a) Turner KB, Hagan NA, Fabris D, J. Mol. Biol. 2007, 369, 812–828; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stephenson W, Asare-Okai PN, Chen AA, Keller S, Santiago R, Tenenbaum SA, Garcia AE, Fabris D, Li PT, J. Am. Chem. Soc. 2013, 135, 5602–5611; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gabelica V, Pauw ED, J. Mass Spectrom. 2001, 36, 397–402. [DOI] [PubMed] [Google Scholar]
- [15].a) Nitiss JL, Nat. Rev. Cancer 2009, 9, 338–350; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Christensen MO, Larsen MK, Barthelmes HU, Hock R, Andersen CL, Kjeldsen E, Knudsen BR, Westergaard O, Boege F, Mielke C, J. Cell Biol. 2002, 157, 31–44; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang JC, Annu. Rev. Biochem. 1996, 65, 635–692. [DOI] [PubMed] [Google Scholar]
- [16].a) Furlanetto V, Zagotto G, Pasquale R, Moro S, Gatto B, Agric J. Food Chem. 2012, 60, 9162–9170; [DOI] [PubMed] [Google Scholar]; b) Cho KH, Pezzuto JM, Bolton JL, Steele VE, Kelloff GJ, Lee SK, Constantinou A, Eur. J. Cancer 2000, 36, 2146–2156; [DOI] [PubMed] [Google Scholar]; c) Haldane A, Sullivan DM, Methods Mol. Biol. 2001, 95, 13–23. [DOI] [PubMed] [Google Scholar]
- [17].a) Gatto B, Richter S, Moro S, Capranico G, Palumbo M, Nucleic Acids Res. 2001, 29, 4224–4230; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kwok Y, Hurley LH, J. Biol. Chem. 1998, 273, 33020–33026. [DOI] [PubMed] [Google Scholar]
- [18].Kingma PS, Osheroff N, Biochim. Biophys. Acta 1998, 1400, 223–232. [DOI] [PubMed] [Google Scholar]
- [19].a) Kingma PS, Corbett AH, Burcham PC, Marnett LJ, Osheroff N, J. Biol. Chem. 1995, 270, 21441–21444; [DOI] [PubMed] [Google Scholar]; b) Kingma PS, Greider CA, Osheroff N, Biochemistry 1997, 36, 5934–5939; [DOI] [PubMed] [Google Scholar]; c) Kingma PS, Osheroff N, J. Biol. Chem. 1997, 272, 1148–1155; [DOI] [PubMed] [Google Scholar]; d) Sabourin M, Osheroff N, Nucleic acids Res. 2000, 28, 1947–1954; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kingma PS, Osheroff N, J. Biol. Chem. 1998, 273, 17999–18002. [DOI] [PubMed] [Google Scholar]
- [20].a) Balcome S, Park S, Quirk Dorr DR, Hafner L, Phillips L, Tretyakova N, Chem. Res. Toxicol. 2004, 17, 950–962; [DOI] [PubMed] [Google Scholar]; b) Haapala E, Hakala K, Jokipelto E, Vilpo J, Hovinen J, Chem. Res. Toxicol. 2001, 14, 988–995; [DOI] [PubMed] [Google Scholar]; c) Mohamed D, Mowaka S, Thomale J, Linscheid MW, Chem. Res. Toxicol. 2009, 22, 1435–1446; [DOI] [PubMed] [Google Scholar]; d) Osborne MR, Wilman DE, Lawley PD, Chem. Res. Toxicol. 1995, 8, 316–320; [DOI] [PubMed] [Google Scholar]; e) Povirk LF, Shuker DE, Mutat. Res. 1994, 318, 205–226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.