

Abstract
The major obstacles for the efficacy of tumor immunotherapies are their immune-related systemic adverse events. Therefore, tumor tropism property and pro-inflammatory ability of mesenchymal stem cells (MSCs) could be utilized in combination to potentiate local immunity for cancer eradication. We previously observed that MSCs with the type III histone deacetylase silent information regulator 2 homologue 1 (Sirt1) overexpression displayed a pro-inflammatory capacity. However, the anti-tumor effect of Sirt1-overexpressing MSCs and the role of Sirt1 in regulating the pro-inflammatory capacity of MSCs still need to be clarified. In this study, utilizing the hepatic metastasis model of colorectal carcinoma, we demonstrated that Sirt1-overexpressing MSCs significantly exerted anti-tumor activity through increasing the number of CD8+ T cells. Furthermore, Sirt1 did not affect chemokine secretion in MSCs induced by inflammatory cytokines, but impaired the immunosuppressive ability of MSCs through suppressing inflammatory cytokine-stimulated inducible nitric oxide synthase (iNOS) production via deacetylating p65. iNOS overexpression negated the anti-tumor effect of Sirt1-overexpressing MSCs. Collectively, our data defined Sirt1 as the critical regulator for modulating the pro-inflammatory ability of MSCs, and they suggested that Sirt1-overexpressing MSCs secreting chemokines but little iNOS under the inflammatory milieu were capable of attracting immune cells to close proximity without suppressing their proliferation, thereby achieving a potent anti-tumor effect.
Keywords: mesenchymal stem cells, pro-inflammatory, Sirt1, iNOS, anti-tumor
Graphical Abstract
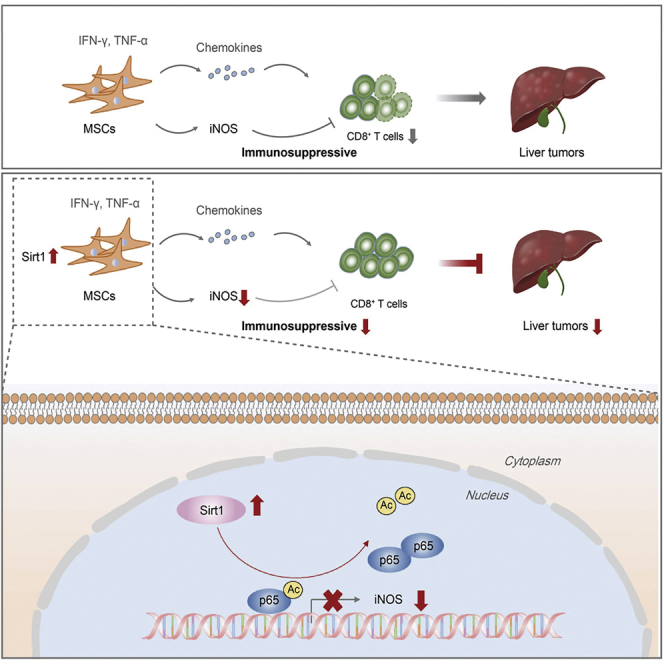
Ye et al. demonstrate that pro-inflammatory Sirt1-overexpressing MSCs exert anti-tumor activity through increasing the number of CD8+ T cells in an iNOS-dependent manner, providing a possible treatment option for cancer. Sirt1 does not affect chemokine secretion in inflammatory cytokine-induced MSCs, but it impairs the immunosuppressive ability of MSCs through suppressing iNOS via p65 deacetylation.
Introduction
The approaches to cancer treatment have made significant progress with outstanding advances in the clinical development of immunotherapy strategies.1 However, systemic immunotherapies still cause major side effects because they powerfully enhance the activity of the entire immune system.2,3 Thus, finding better options for cancer immunotherapy with less aggressive systemic immune responses and more enhanced specific anti-tumor immunity is therefore a highly sought-after goal. At present, mesenchymal stem cells (MSCs) have emerged as anti-tumor drug carriers for cancer treatment owing to their migration toward the malignant tumor site.4 MSCs genetically engineered with anti-cancer agents migrate to the tumor site and exhibit their local anti-tumor activity.5, 6, 7 Recently, with further investigation, the substantial data available have established that the immunoregulatory capacities of MSCs are not constitutive but are rather licensed by inflammation.8 MSCs can acquire a pro-inflammatory or anti-inflammatory phenotype depending on the inflammation type or concentration.9 Thus, the utilization of pro-inflammatory MSCs for the reversion of tumor-derived immunosuppression may elicit potent anti-tumor immunity and achieve tumor eradication or suppression. Their advantages of the tumor-tropic potential and no toxic systemic side effects, along with their pro-inflammatory properties, make MSCs suitable for efficient clinical applications in cancer immunotherapy.
Based on our previous findings, we found that MSCs with the type III histone deacetylase silent information regulator 2 homologue 1 (Sirt1) overexpression exhibited pro-inflammatory potential. These observations prompted us to hypothesize that MSCs with Sirt1 overexpression may be therapeutically effective against tumors through reversing the immunosuppressive microenvironment. Emerging evidence suggested that Sirt1 could produce a positive effect on regulating the activation and function of immune cells.10 For example, Sirt1 negatively regulated T cell activation and played a major role in clonal T cell anergy in mice.11 The myeloid deletion of Sirt1 impaired dendritic cell (DC) maturation and reduced T helper (Th)1 and Th17 differentiation.12 Additionally, studies have demonstrated that macrophage Sirt1 plays important roles in the regulation of M1/M2 polarization.13 So far, the critical role of Sirt1 in regulating the immunomodulatory properties of MSCs has not been unveiled. Therefore, elucidating the potential mechanism of Sirt1-mediated modulation of pro-inflammatory activity in MSCs could help us gain a better understanding of the dual immunomodulatory functions of MSCs.
The aims of the present study are to test the hypothesis that Sirt1-overexpressing MSCs with pro-inflammatory activity may enhance local immunity and reverse immunosuppressive conditions, thereby exerting anti-tumor effect, and to further explore the mechanism of Sirt1 in regulating the pro-inflammatory ability of MSCs. In this study, bone marrow-derived MSCs were transfected with adenovirus vector encoding Sirt1 gene to overexpress Sirt1 protein (AdSirt1-MSCs). Then, we set out to investigate the anti-tumor effect of AdSirt1-MSCs in the hepatic metastasis model of colorectal carcinoma and the pro-inflammatory role of AdSirt1-MSCs in carbon tetrachloride (CCl4)-induced acute liver injury. We subsequently explored the key immunoregulatory factor mediating the pro-inflammatory function of AdSirt1-MSCs. The molecular mechanism underlying the regulation of the key immunoregulatory factor via Sirt1 was also investigated. Our findings may reveal an essential role for Sirt1 in regulating the pro-inflammatory ability of MSCs and raise the possibility for the potential applications of pro-inflammatory Sirt1-overexpressing MSCs as a new therapeutic strategy for future battles against cancer.
Results
Sirt1-Overexpressing MSCs Exert a Dramatic Anti-tumor Effect by Increasing the Number of CD8+ T Cells
To test the hypothesis that Sirt1-overexpressing MSCs with a pro-inflammatory capacity exerted the anti-tumor effect via reversing tumor immunosuppressive conditions, we first successfully constructed Sirt1-overexpressing MSCs with the recombinant adenovirus vector encoding Sirt1 (referred to as AdSirt1-MSCs), and we transfected MSCs with the recombinant adenovirus vector encoding EGFP as the control (referred to as AdEGFP-MSCs). Our data showed that transfection with AdSirt1 markedly increased Sirt1 protein level (Figures S1A and S1B), and Sirt1 overexpression did not affect the migration and proliferative capacity of MSCs (Figures S1C–S1E). Next, we established the hepatic metastasis model of colorectal carcinoma by intrasplenic injection of CT26 cells into syngeneic BALB/c mice. The mice were distributed into four groups in our study: I, CT26; II, CT26 + MSCs; III, CT26 + AdEGFP-MSCs; and IV, CT26 + AdSirt1-MSCs (Figure 1A). Compared with the control CT26 group, we found that the co-injection of AdSirt1-MSCs with CT26 significantly suppressed tumor development in liver, and metastatic liver tumors appeared significantly smaller in the AdSirt1-MSC group than did those in the control CT26 group. However, the co-injection of MSCs or AdEGFP-MSCs, respectively, with CT26 promoted tumor progression in liver with more tumor nodules when compared with the CT26 group (Figure 1B). The number of metastatic liver surface tumors was lower and the maximal liver tumor volume was also reduced in the AdSirt1-MSC group as compared with the control CT26 group (**p < 0.01 versus CT26) (Figure 1C). Histologically, the AdSirt1-MSC group showed smaller areas of metastatic hepatic necrotizing tissue (Figure 1D). Importantly, note that CD8+ T cells are capable of evoking a potent anti-tumor immune response. Thus, we next examined the accumulation of CD8+ T cells in the liver. We found that there was a marked increase in the number of CD8+ T cells at the liver tumor site in the AdSirt1-MSC group (***p < 0.001 versus CT26), but the MSC or AdEGFP-MSC group had no statistical significance compared with the CT26 group in the difference of hepatic CD8+ T cell numbers (p > 0.05 versus CT26) (Figures 1E and 1F). We also detected CD4+ T cells in the liver metastasis tissue, but there was no significant difference in the number of CD4+ T cells between the above-defined four animal groups (Figures S2A and S2B).
Figure 1.

The Administration of Sirt1-Overexpressing MSCs Suppresses Tumor Development and Increases the Number of Hepatic CD8+ T Cells in Mice Suffering from Hepatic Metastasis of Colorectal Carcinoma
(A) Schematic representation depicting the experimental design to investigate the therapeutic efficacy of the AdSirt1-MSC transfusion during liver metastasis of colorectal carcinoma in mice. The mice were randomly divided into four experimental groups as described in Materials and Methods. (B) Liver surface metastatic nodules were detected macroscopically. The representative photographs show the hepatic metastases in mice from the above four groups (21 days post-treatment). (C) The surface tumor number and maximal tumor volume at the hepatic tumor site were counted and measured in mice (21 days post-treatment) from the above four groups. Values represent means and standard error of three independent experiments. **p < 0.01, ***p < 0.001 versus CT26 group. (D) H&E staining was used to evaluate liver tissue of mice (21 days post-treatment) from the above four groups. Scale bars, 100 μm. (E) Immunohistochemical analysis of CD8+ T cells at the liver tumor site in mice (21 days post-treatment) from the above four groups. Representative immunohistochemical staining of CD8 in liver samples from each group is shown. Scale bars, 100 μm. The black arrows point to CD8+ T cells. (F) Quantitation of CD8+ T cells at hepatic tumor site in mice (21 days post-treatment) in each group. At least five fields (magnification, ×200) were counted for each specimen. ***p < 0.001 versus CT26 group. p > 0.05 versus CT26 group; NS, not significant (p > 0.05).
To further verify the anti-tumor effect of AdSirt1-MSCs on the established liver metastasis model of colorectal carcinoma (Figure S3A), we first identified liver metastatic nodules in mice on day 11 after the intrasplenic injection of CT26 (Figure S3B). Consistent with the above results, we found that the administration of AdSirt1-MSCs at day 11 after intrasplenic injection of CT26 also significantly attenuated tumor progression with reduced areas of metastatic hepatic necrotizing tissue and increased the number of CD8+ T cells at the liver site (**p < 0.01 versus CT26) (Figures S3C–S3F).
Taken together, our data demonstrated that the administration of AdSirt1-MSCs significantly suppressed tumor development and promoted anti-tumor immunity via increasing the number of CD8+ T cells to reverse tumor immunosuppressive conditions.
Sirt1-Overexpressing MSCs Display Profound Pro-inflammatory Activity in CCl4-Induced Acute Liver Injury
Our data have already demonstrated that AdSirt1-MSCs promote anti-tumor immunity with an increased number of CD8+ T cells, implying that AdSirt1-MSCs may have the pro-inflammatory properties to overcome the immunosuppressive tumor microenvironment. To further verify the pro-inflammatory capacity of AdSirt1-MSCs in vivo, we established an acute liver injury model induced by CCl4. Mice were randomly divided into five groups as shown in the schematic diagram (Figure 2A): the control group (olive oil), CCl4 group, CCl4 + MSC group, CCl4 + AdEGFP-MSC group, and CCl4 + AdSirt1-MSC group. Histologically, when compared with the CCl4 group, the massive necrosis of hepatocytes occurred in the AdSirt1-MSC group (Figure 2B). AdSirt1-MSC infusion led to the significant upregulation in the serum concentrations of pro-inflammatory cytokines (interleukin [IL]-1β, IL-12(p40), IL-13, and tumor necrosis factor [TNF]-α) (**p < 0.01 versus CCl4 group) (Figure 2C).
Figure 2.

The Administration of AdSirt1-MSCs Promotes Liver Inflammation in Mice with CCl4-Induced Acute Liver Injury
(A) Schematic diagram depicting the experimental approach to evaluate the effect of the AdSirt1-MSC transfusion on mice with acute liver injury induced by CCl4. The mice were randomly divided into five experimental groups as described in Materials and Methods. (B) H&E-stained sections of liver tissues from the animals of the above five groups (48 h post-treatment). Representative images are shown for each sample. Scale bars, 100 μm. (C) Pro-inflammatory cytokine production of IL-1β, IL-12(p40), IL-13, TNF-α, IL-6, IL-17, and IL-1α was detected in serum of mice from the above five animal groups (48 h post-treatment) by Bio-Plex analysis. **p < 0.01, *p < 0.05 versus CCl4 group. p > 0.05 versus CCl4 group; NS, not significant (p > 0.05). (D) Representative samples of immunochemistry staining of CD8+ T cells in liver tissue of mice from the above five groups (48 h post-treatment). The black arrows indicate CD8+ T cells. (E) Quantification of the CD8+ T cell populations identified by CD8 immunostaining in liver sections of mice from the above five animal groups (48 h post-treatment). Scale bars, 100 μm. The counts of CD8+ T cells were measured by counting five randomly selected ×200 high-power fields (HPFs) per paraffin section under light microscopy. Scale bars, 100 μm. ***p < 0.001 versus CCl4 group. p > 0.05 versus CCl4 group; NS, not significant (p > 0.05).
We next detected the lymphocyte infiltration in the liver tissue of mice with acute liver injury induced by CCl4. The accumulated CD8+ T cells in the liver dramatically increased in mice treated with AdSirt1-MSCs when compared with those treated with CCl4 alone (***p < 0.001 versus CCl4 group) (Figures 2D and 2E). In addition, we found that there was no significant difference in the number of CD4+ T cells in liver specimens of mice from the above four groups (CCl4, CCl4 + MSC, CCl4 + AdEGFP-MSC, and CCl4 + AdSirt1-MSC) (Figures S4A and S4B). Taken together, we demonstrated that AdSirt1-MSCs effectively promoted inflammation in mice with CCl4-induced acute liver injury, which suggested that AdSirt1-MSCs exerted the potent pro-inflammatory effect in vivo.
Sirt1 Does Not Affect the Chemoattractive Capacity of MSCs Induced by Inflammatory Cytokines
Recent studies have revealed that MSCs attract the immune cells in their proximity through the secretion of chemokines and then exert their potent immunosuppressive effect.14 Given that our above experimental data showed that AdSirt1-MSC infusion resulted in an increased number of CD8+ T cells in vivo, we first detected whether Sirt1 could affect the expression of T cell chemokines in MSCs. Our data showed that MSCs, AdEGFP-MSCs, and AdSirt1-MSCs produced small amounts of mRNA levels in T cell chemokines (Cxcl9, Cxcl10, Cxcl11, Icam1, and Vcam1) without interferon (IFN)-γ and TNF-α (IT) treatment (Figure 3A). When treated with IT, the mRNA expression of T cell chemokines (Cxcl9, Cxcl10, Cxcl11, Icam1, and Vcam1) in the three groups increased sharply (***p < 0.001 versus MSC), but no significant difference was observed among these three groups (p > 0.05 versus IT-MSC) (Figure 3A). As the chemoattractive property of MSCs has been reported to be induced by inflammatory cytokines,14 we used the chemotaxis assay described by Shi et al.15 to further examine the effect of Sirt1 on the chemoattractive capacity of inflammatory cytokine-induced MSCs. We found that IT pre-stimulated MSCs, AdEGFP-MSCs, or AdSirt1-MSCs could attract splenocytes to their vicinity (p > 0.05 versus IT-MSC) (Figures 3B and 3C). Therefore, our data suggested that Sirt1 had no impact on the induction of chemokines for T cell recruitment in inflammatory cytokine-stimulated MSCs.
Figure 3.

Sirt1 Has No Effect on Chemokine Secretion and Chemotactic Activity of Inflammatory Cytokine-Induced MSCs
(A) The expression of chemokine genes Cxcl9, Cxcl10, Cxcl11, Icam1, and Vcam1 was quantified by real-time PCR in MSCs, AdEGFP-MSCs, and AdSirt1-MSCs with or without stimulation of IFN-γ (10 ng/mL) and TNF-α (10 ng/mL) for 8 h. ***p < 0.001 versus MSC; p > 0.05 versus IT-MSC; NS, not significant (p > 0.05). (B) MSCs, AdEGFP-MSCs, or AdSirt1-MSCs pretreated with IFN-γ and TNF-α (10 ng/mL each) for 24 h were respectively seeded in the lower chamber, and splenocytes were added to the upper chamber. After 0 or 3 h of incubation, the extent of cell aggregation was screened under light microscopic observation. (C) Quantification of splenocyte aggregation was measured by counting the total number of migrated splenocytes in each group. p > 0.05 versus IT-MSC; NS, not significant (p > 0.05).
Sirt1 Blocks the Immunosuppressive Capacity of MSCs
To further determine the potential mechanisms involving the role of Sirt1 in regulating the pro-inflammatory properties of MSCs, as our above data showed that Sirt1 did not affect the T cell chemokine secretion in MSCs with inflammatory cytokine stimulation, we next investigated whether Sirt1 could affect the immunosuppressive ability of MSCs with a splenocyte proliferation assay. As expected, MSCs and AdEGFP-MSCs effectively suppressed the proliferation of concanavalin A (Con A)-activated splenocytes (**p < 0.01 versus spl + ConA), but surprisingly, no significant inhibition of splenocyte proliferation was observed in the AdSirt1-MSCs group (p > 0.05 versus spl + ConA) (Figures 4A and 4B). Under optical microscopic observation, MSCs and AdEGFP-MSCs strongly inhibited splenocyte proliferation, as indicated by a diminished number of splenocyte proliferation clones as compared to that of Con A-induced splenocyte proliferation. Conversely, AdSirt1-MSCs could not exert the immunosuppressive effect on Con A-activated splenocyte proliferation with an evidently increased number of splenocyte proliferation clones (Figure 4C). Taken together, our data demonstrated that AdSirt1-MSCs lost the immunosuppressive effect, and they suggested that Sirt1 hindered the immunosuppressive activity of MSCs.
Figure 4.

Sirt1 Impairs the Immunosuppressive Ability of MSCs and Inhibits Inflammatory Cytokine-Induced iNOS Production in MSCs
(A) MSCs, AdEGFP-MSCs, or AdSirt1-MSCs were respectively co-cultured with CFSE-labeled splenocytes at a ratio of 1:10 in the presence of Con A (5 μg/mL). The CFSE-diluted splenocytes were detected by flow cytometry after 72 h of incubation. A representative staining of three independent experiments is shown. (B) Quantitation data were determined by the percentage of proliferating CFSE-labeled splenocytes in each group. **p < 0.01 versus splenocytes (spl) + Con A. p > 0.05 versus spl + ConA; NS, not significant (p > 0.05). (C) Under the same treatment conditions as in (A), the splenocyte proliferation clones at 72 h were inspected by microscopy. Microphotographs show a representative image of splenocyte proliferation clones in each group. Scale bars, 100 μm. (D) The mRNA levels of Il6, Il10, Tnfaip6, Hgf, and Nos2 were detected by real-time PCR in MSCs, AdEGFP-MSCs, or AdSirt1-MSCs treated with or without IFN-γ and TNF-α (IT, 10 ng/mL each) for 8 h. ***p < 0.001, **p < 0.01, p > 0.05 versus MSC; **p < 0.01, p > 0.05 versus IT-MSC; NS, not significant (p > 0.05). (E) The protein levels of Sirt1 and iNOS were determined by western blotting in MSCs, AdEGFP-MSCs, or AdSirt1-MSCs treated with IFN-γ and TNF-α (IT, 10 ng/mL each) for 24 h. MSCs without IFN-γ and TNF-α treatment were used as the control. (F) The level of nitrate representing the iNOS activity was measured using Griess reagent in the supernatant of different groups of MSCs exposed to the treatments as described in (E). ***p < 0.001 versus MSC. p > 0.05 versus MSC. NS: not significant (p > 0.05).
Since our above data have shown that AdSirt1-MSCs could not exert immunosuppressive activity, as well as that the infusion of AdSirt1-MSCs resulted in an increased number of CD8+ T cells in vivo (Figures 1E and 2D), to further determine the proliferation of which subtype of T cells (CD4+ or CD8+ T cells) may be affected by AdSirt1-MSCs, we detected the percentage and total number of CD4+ or CD8+ T cells in the co-culture system of MSCs with Con A-activated splenocytes. Our data showed that the proliferation of both CD4+ and CD8+ T cells was not inhibited by the AdSirt1-MSCs. In addition, AdSirt1-MSCs did not selectively promote the proliferation of CD4+ or CD8+ T cell subsets in vitro (p > 0.05 versus spl + ConA) (Figures S5A–S5C). Collectively, under the inflammatory conditions, Sirt1 did not affect T cell chemokine secretion in MSCs, but it impaired the immunosuppressive function of MSCs, thereby resulting in the Sirt1-mediated pro-inflammatory capacity of MSCs.
Sirt1 Impairs the Immunosuppressive Capacity of MSCs by Inhibiting iNOS Expression
Our data clearly showed that Sirt1 blocked the immunosuppressive effect of MSCs, and it is well known that MSC-mediated immunosuppression is licensed by inflammation and mainly acts through the secretion of various immunosuppressive factors;16 therefore, we next sought to examine which immunosuppressive factor was affected by Sirt1 in MSCs under inflammatory stimulation. The results showed that Sirt1 did not affect the gene expression of Il6, Il10, Tnfaip6, or Hgf in MSCs with or without inflammatory stimulation (Figure 4D). However, we found that IT stimulation increased inducible nitric oxide synthase (iNOS) expression at both the mRNA and protein levels in MSCs and AdEGFP-MSCs (***p < 0.001 versus MSC), but IT-stimulated AdSirt1-MSCs displayed the drastically lower mRNA and protein levels of iNOS (**p < 0.01 versus IT-MSC) (Figures 4D and 4E). Furthermore, NO is generated by iNOS,17 so we then detected the stable reaction product derived from NO, nitrate, as the good indicator for iNOS activity by using the Griess test. We found dramatically increased amounts of nitrate in the supernatants of IT-induced MSCs and AdEGFP-MSCs compared to that of the MSCs without IT stimulation (***p < 0.001 versus MSC), but there was no significant difference in nitrate concentrations of the supernatants between IT-induced AdSirt1-MSCs and MSCs without IT treatment (p > 0.05 versus MSC) (Figure 4F).
Next, we examined whether Sirt1-mediated downregulation of iNOS expression may drive AdSirt1-MSCs to become immunosuppressive incompetent. To experimentally address this point, iNOS was overexpressed in the AdSirt1-MSCs (iNOS-AdSirt1-MSCs) with plasmid encoding mouse iNOS cDNA driven under the cytomegalovirus promoter (pCMV-iNOS), and MSCs transfected with the empty pCMV vector (vector-AdSirt1-MSCs) were used as the control (Figure 5A). Clearly, a high level of nitrate was detected in the culture supernatant of IT-stimulated iNOS-AdSirt1-MSCs (***p < 0.001 versus IT + AdSirt1-MSC) (Figure 5B). We next performed the co-culture experiments of MSCs with Con A-activated splenocytes and found that the iNOS-AdSirt1-MSCs suppressed the proliferation of activated splenocytes more efficiently than did AdSirt1-MSCs or vector-AdSirt1-MSCs (***p < 0.001 versus spl + ConA) (Figures 5C and 5D). Taken together, our data clearly showed that Sirt1 significantly inhibited inflammatory cytokine-induced iNOS expression in MSCs, and that iNOS overexpression dramatically restored the immunosuppressive capacity of Sirt1-overexpressing MSCs. We demonstrated that Sirt1 impaired the immunosuppressive function of MSCs in an iNOS-dependent manner.
Figure 5.

iNOS Overexpression Promotes the Recovery of Immunosuppressive Function in AdSirt1-MSCs
(A) AdSirt1-MSCs were transfected with control empty pCMV vector (vector-AdSirt1-MSCs) or pCMV plasmid containing cDNA encoding mouse iNOS (iNOS-AdSirt1-MSCs), and then the AdSirt1-MSCs, vector-AdSirt1-MSCs, or iNOS-AdSirt1-MSCs were stimulated respectively with IFN-γ and TNF-α (IT, 10 ng/mL each) for 24 h, and iNOS expression was measured by immunoblotting analysis. (B) Under the same treatment conditions as in (A), the nitrite level in cell culture supernatant was detected using Griess reagent. ***p < 0.001 versus IT + AdSirt1-MSC. p > 0.05 versus IT + AdSirt1-MSC; NS, not significant (p > 0.05). (C) MSCs, AdSirt1-MSCs, vector-AdSirt1-MSCs, or iNOS-AdSirt1-MSCs were co-cultured respectively with CFSE-labeled splenocytes at a ratio of 1:10 for 72 h in the presence of Con A (5 μg/mL). The splenocytes were collected for proliferation analysis and assessed by the decrease in CFSE fluorescence intensity for cell division via flow cytometry after 72 h of co-culture. (D) Quantitation of splenocyte proliferation was determined by the percentage of CFSE-diluted splenocytes among total CFSE-labeled splenocytes in each group. ***p < 0.001 versus splenocytes (spl) + Con A. p > 0.05 versus spl + ConA; NS, not significant (p > 0.05).
Sirt1 Inhibits iNOS Expression in Inflammatory Cytokine-Induced MSCs through Deacetylating p65
It has been reported that iNOS expression is elicited by inflammatory stimuli mainly through activation of the nuclear factor κB (NF-κB) signaling pathway.18 We therefore determined whether the NF-κB pathway was involved in the induction of iNOS expression in MSCs treated with inflammatory cytokines. For this purpose, we used SN50 (an NF-κB inhibitor) to inhibit the NF-κB signaling pathway. We observed that iNOS expression was significantly inhibited in IT-treated MSCs after adding SN50 (Figure 6A), and lower amounts of nitrate were also detected in IT-stimulated MSCs treated with SN50 compared with those of IT-stimulated MSCs (**p < 0.01 versus IT + MSC) (Figure 6B). These results indicated that inflammatory cytokine-induced iNOS expression was mainly mediated by the NF-κB signaling pathway in MSCs.
Figure 6.

Sirt1 Inhibits iNOS Expression in Inflammatory Cytokine-Induced MSCs through Deacetylating p65
(A) MSCs were pretreated with or without NF-κB inhibitor SN50 (18 μM) for 2 h, and then MSCs were stimulated with TNF-α and IFN-γ (IT, 10 ng/mL, each) for 24 h. MSCs treated without SN50 and IT were used as the control. After 24 h, cells were subjected to western blot for analyzing iNOS protein level. GAPDH was used as the internal control. (B) Under the same treatment conditions as in (A), the nitrite level in cell culture supernatant was measured using Griess reagent.. **p < 0.01 versus IT + MSC; ***p < 0.001 versus control MSC. (C) AdEGFP-MSCs or AdSirt1-MSCs were treated respectively with IT (10 ng/mL, each) for 30 min, and MSCs treated with IT (10 ng/mL, each) for 30 min were used as the control. The cells were then subjected to western blot for analyzing protein levels of acetyl-(Lys310)-p65, p65, and Sirt1. GAPDH was used as the internal control. (D) MSCs, AdEGFP-MSCs, or AdSirt1-MSCs were treated respectively with IT (10 ng/mL, each); after 30 min, proteins were extracted for immunoprecipitation with anti-p65, and the immunoprecipitate was subjected to immunoblotting (IB) with antibodies against acetyl-NF-κB p65 (Lys310) antibody.
Having demonstrated that NF-κB was responsible for iNOS expression in IT-induced MSCs, we further examined the role of Sirt1 in the regulation of NF-κB signaling pathway. Since the well-characterized target of Sirt1 deacetylation was the acetylation of Lys310 in the p65 subunit of NF-κB,19 this led us to investigate the effect of Sirt1 on acetylation of the p65 subunit in inflammatory cytokine-induced MSCs. We found that the acetylation of p65 (Lys310) markedly decreased in AdSirt1-MSCs compared with that in MSCs or AdEGFP-MSCs upon IT treatment (Figure 6C). The immunoprecipitation with p65 antibody revealed that Sirt1 overexpression dramatically decreased acetylation level of p65 in IT-induced MSCs (Figure 6D). Collectively, we provided evidence that Sirt1 impeded the activation of NF-κB signaling pathways through deacetylating p65, leading to the suppression of NF-κB-mediated iNOS expression in pro-inflammatory cytokine-stimulated MSCs.
iNOS Overexpression Impairs the Anti-tumor Ability of AdSirt1-MSCs in the Hepatic Metastasis Model of Colorectal Carcinoma
Because we demonstrated that decreased iNOS expression is essential for the pro-inflammatory properties of AdSirt1-MSCs, we next determined whether iNOS overexpression could alter the anti-tumor effect of AdSirt1-MSCs in vivo. The liver metastasis model of colorectal carcinoma was established by intrasplenic inoculation of CT26 cells in BALB/c mice, and treatment with different groups of MSCs was done in the experimental procedures as presented in the schematic diagram (Figure 7A; Figure S6A). We found that the co-injection of iNOS-overexpressing AdSirt1-MSCs with CT26 or injection of iNOS-overexpressing AdSirt1-MSCs at day 11 after the inoculation of CT26 led to significant tumor promotion with increased liver metastatic nodules as compared to the other three groups (Figure 7B; Figure S6B). The extensive tumor necrosis detected by histopathological examination (Figure 7C; Figure S6C) and the presence of dramatically decreased hepatic CD8+ T cells (Figures 7D and 7E; Figures S6D and S6E) were also confirmed in mice with administration of iNOS-overexpressing AdSirt1-MSCs.
Figure 7.

iNOS Overexpression Negates the Anti-tumor Effect of Sirt1-Overexpressing MSCs In Vivo
(A) Schematic representation depicting the experimental design to investigate the efficacy of the iNOS-overexpressing AdSirt1-MSC transfusion during liver metastasis of colorectal carcinoma in mice. (B) Liver surface metastatic nodules were detected macroscopically. Representative photographs show the hepatic tumor metastases in mice from the above four groups (21 days post-treatment). (C) H&E staining was used to evaluate liver samples of mice (21 days post-treatment) from the above four groups. Scale bars, 100 μm. (D) Representative immunohistochemical staining of CD8 in liver samples in mice (21 days post-treatment) from the above four groups are shown. Scale bars, 100 μm. The black arrows point to CD8+ T cells. (E) Quantitation of CD8+ T cells at hepatic tumor site in mice (21 days post-treatment) from the above four groups. At least five fields (magnification, ×200) were counted for each specimen. **p < 0.01 versus CT26 group. p > 0.05 versus CT26 group; NS, not significant (p > 0.05).
Collectively, we demonstrated that iNOS overexpression negated the anti-tumor effect of AdSirt1-MSCs in vivo, and Sirt1-overexpressing MSCs relied on their decreased iNOS expression to reverse the local immunosuppressive environment and boost the potent anti-tumor immunity.
Sirt1 Decreases the Production of IDO in Human MSCs
Given that the key molecule mediating immunosuppression by MSCs was species-dependent, that is, indoleamine 2,3-dioxygenase (IDO) in humans and iNOS in mice,20 we next examined whether Sirt1 could affect the IDO expression in human umbilical cord-derived MSCs (hUC-MSCs). The recombinant adenovirus vector encoding Sirt1 (AdSirt1) was transfected into hUC-MSCs to construct the Sirt1-overexpressing hUC-MSCs (AdSirt1-hUC-MSCs), and hUC-MSCs transfected with recombinant adenovirus vector encoding EGFP was used as the control (AdEGFP-hUC-MSCs). Our data showed that transfection with AdSirt1 markedly increased the Sirt1 protein level in hUC-MSCs (Figures S7A and S7B). When incubated with no inflammatory cytokines, western blotting revealed that hUC-MSCs, AdEGFP-MSCs, or AdSirt1-hUC-MSCs did not produce IDO (Figure S7B). When stimulated with inflammatory cytokines, IDO increased sharply in hUC-MSCs and AdEGFP-hUC-MSCs, but there was a significant decrease of IDO in AdSirt1-hUC-MSCs as compared with that in hUC-MSCs or AdEGFP-hUC-MSCs (Figures S7C).
Thus, our results demonstrated that Sirt1 overexpression profoundly suppressed IDO expression in inflammatory cytokine-stimulated human MSCs, and they suggested Sirt1 as the critical regulator for modulating IDO production in human MSCs.
Discussion
The immunosuppressive microenvironment facilitated tumor immune escape and promoted tumor progression.21 Thus, reversing immunosuppression in the tumor microenvironment could achieve therapeutic efficacy in cancer. In this study, to overcome the tumor immunosuppressive conditions, we exploited this new approach to apply MSCs with pro-inflammatory properties to improve anti-tumor treatment by enhancing local immunity. In the hepatic metastasis model of colorectal carcinoma, we found that Sirt1-overexpressing MSCs with the pro-inflammatory abilities strongly inhibited tumor development via enhancing local immunity with increased numbers of anti-tumor CD8+ T cells. We also defined Sirt1 as the critical regulator for the pro-inflammatory function of MSCs in an iNOS-dependent manner. Moreover, iNOS overexpression negated the anti-tumor effect of Sirt1-overexpressing MSCs.
Previous studies revealed that Sirt1 played a critical regulatory role in the immune system.10 SIRT1 has been reported to deacetylate retinoic acid-related orphan receptor (ROR)γt and enhance Th17 cell generation.22 van Loosdregt et al.23 demonstrated that the inhibition of SIRT1 resulted in functionally improved regulatory T (Treg) cells. During respiratory syncytial virus infection, a study showed that SIRT1 promoted DC activation and autophagy-mediated processes.24 Substantial evidence has shown that Sirt1 regulates the fates and functions of immune cells as well as controlling their activation and maturation. However, the effect of Sirt1 activity on the immunomodulatory ability of MSCs remains unknown. In this study, we characterized Sirt1 as the critical factor in regulating the immunomodulatory ability of MSCs. We found that Sirt1 did not affect the chemokines in inflammatory cytokine-induced MSCs, but it impaired the immunosuppressive function of MSCs through decreasing iNOS production. Sirt1-overexpressing MSCs can attract immune cells to their proximity without suppressing the viability of these immune cells, thereby resulting in a potent anti-tumor effect via enhancing local immunity in the hepatic metastasis model of colorectal carcinoma. Accordingly, Sirt1-overexpressing MSCs exhibited pro-inflammatory activity in CCl4-induced acute liver injury. However, there are some conflicting reports stating that Sirt1 deficiency leads to the pro-inflammatory ability of certain immune cells. Zhang et al.13 pointed out that SIRT1-deficient macrophages displayed a significant increase in basal and IFN-γ/lipopolysaccharide (LPS)-stimulated expression of the M1 macrophage marker iNOS, suggesting that SIRT1 deletion promoted activation of pro-inflammatory M1 macrophages. However, our results showed that Sirt1-overexpressing MSCs displayed the potent pro-inflammatory capacity through decreasing the expression of the immunosuppressive factor iNOS. There are some explanations for these conflicts. For M1 pro-inflammatory macrophages, iNOS was involved in generating the microbicidal NO to combat invading pathogens.25 For immunosuppressive MSCs, IDO mediated tryptophan degradation to inhibit allogeneic T cell responses in human MSCs,26 whereas NO produced by iNOS played a critical role in suppression of T cell proliferation in mouse MSCs.27 We think that diverse activity of iNOS may partially explain why Sirt1 contributed to the discordant effects on regulating pro-inflammatory or anti-inflammatory activities in distinct cell types.
Our study also clarified the molecular mechanisms underlying Sirt1-regulated iNOS expression in MSCs. Detailed studies have regarded the inflammatory transcription factor NF-κB as a strong inducer of iNOS expression.18 It is also well known that Sirt1 deacetylates the RelA/p65 subunit of NF-κB at Lys310 and suppresses NF-κB-associated transcription, further resulting in a reduction in inflammatory responses.28,29 In this regard, we first found that the NF-κB signaling pathway was mainly involved in the induction of inflammatory cytokine-induced iNOS expression in MSCs. In support of our data, the regulation of iNOS via the NF-κB pathway has also been reported in various cell types, such as murine macrophages,30 murine fibroblasts,31 and human lung adenocarcinoma cell.32 Moreover, our data revealed that Sirt1 overexpression significantly reduced the acetylation level of NF-kB p65, leading to suppression of NF-κB-regulated iNOS production in MSCs treated with inflammatory cytokines. Consistent with our findings, Park et al.33 demonstrated that SIRT1 activation downregulated LPS/IFN-γ-mediated NF-κB activity by inhibiting p65 acetylation and it suppressed the expression of M1 macrophage-related genes, such as CCL2, iNOS, IL-12 p35, and IL-12 p40. Other experimental evidence indicated that Sirt1 overexpression protected osteoblasts against TNF-α-induced cell injury partly by repressing NF-κB activity and NF-κB downstream genes, including iNOS.34 Research has shown that SIRT1 overexpression completely inhibited iNOS expression through inhibition of the NF-κB signaling pathway via deacetylation of p65 in pancreatic β cells.35 However, in addition to NF-κB, other transcription factors have also been reported to be involved in the regulation of iNOS transcription, such as STAT1 homodimers,36 STAT3,37 IRF-1,38 AP-1,39 and C/EBPβ.40 It will be of interest to further evaluate the other related signaling pathways for iNOS induction that are affected by Sirt1 in inflammatory cytokine-treated MSCs.
In our study, we demonstrated that Sirt1 affected the pro-inflammatory ability and the anti-tumor effect of MSCs through downregulating iNOS expression. However, well-documented studies have noted that immunosuppressive factors secreted by MSCs are species-dependent: human MSCs utilize IDO whereas rodent MSCs express iNOS in response to inflammatory cytokines.20 Therefore, we also performed additional experiments to demonstrate the effect of Sirt1 overexpression on the key immunosuppressive molecule IDO in human umbilical cord-derived MSCs (hUC-MSCs). We found that Sirt1 overexpression significantly decreased IDO production in inflammatory cytokine-treated hUC-MSCs, which suggested that Sirt1 overexpression could also play an important role in reversing the immunosuppressive properties of human MSCs. Furthermore, the effect of Sirt1-overexpressing human MSCs on clinical anti-tumor therapy needs intense exploration. Studies have already shown that the interplay of the STAT1 and phosphatidylinositol 3-kinase (PI3Kα) pathways regulates IFN-γ‐induced IDO production in MSCs.41 Therefore, the potential of Sirt1 to modulate the key molecule in the signaling pathway of IDO expression in human MSCs deserves our further investigation, and the new insight into the critical role of Sirt1 for the regulation of immunomodulatory activity in human MSCs will help to optimize their clinical application.
In summary, our study demonstrated that the Sirt1-overexpressing MSCs that produced chemokines but little iNOS under the inflammatory status were effective in attracting immune cells to their proximity without suppressing immune cell proliferation, thereby exerting the anti-tumor effect through reversing tumor immunosuppressive conditions and boosting local immunity. We also found that Sirt1 modulated the pro-inflammatory properties of MSCs by decreasing iNOS production. Thus, modulation of Sirt1 may be a strategy for harnessing the pro-inflammatory properties of MSCs. Additionally, considering the pro-inflammatory effect of Sirt1-overexpressing MSCs, they are worthy of further clinical applications to alter the immunosuppressive state and enhance the potent immune response, especially in cancer patients.
Materials and Methods
Cell Lines and Culture Conditions
The murine colorectal carcinoma cell line CT26 (syngeneic to BALB/c) was cultured in RPMI 1640 culture medium (Gibco, Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS; Gibco, Invitrogen, Carlsbad, CA, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin and was cultured in a CO2 incubator under saturated humidity (5% CO2 and 95% air) at 37°C.
Isolation and Culture of MSCs from Mouse Bone Marrow
The mouse bone marrow MSCs were isolated and characterized according to the protocols of our previously published studies.42,43 The bone marrow cells were isolated from BALB/c or C57BL/6 male mice aged 6–8 weeks and were collected by flushing the medullary cavities from the femurs and tibias, and then the derived cells were cultured in low-glucose DMEM (L-DMEM) medium containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. After 72 h, culture medium was replaced and the non-adherent cells were removed. The fresh medium was replaced every 3 days. At day 7 after isolation, the attached cells at 80%–90% confluency were trypsinized and seeded into culture flasks for further expansion. The adherent cells were passaged three times and then were used for the following experiments as purified MSCs.
Animals
Male BALB/c or C57BL/6 mice, 6–8 weeks of age, were purchased from the Shanghai Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China) and were housed under standard conditions. Animal experiments were done in accordance with the Institutional Animal Welfare Guidelines of the Eastern Hepatobiliary Surgery Hospital of the Second Military Medical University, Shanghai, China.
Experimental Design of the Hepatic Metastasis Model of Colorectal Carcinoma
The 6- to 8-week-old male BALB/c mice were randomly divided into four experimental groups (n = 5 per group), including group I (CT26 group), group II (CT26 + MSC group), group III (CT26 + AdEGFP-MSC group), and group IV (CT26 + AdSirt1-MSC group). Mice in group I were inoculated intrasplenically with CT26 cells (2 × 105) to induce the hepatic metastasis model of colorectal carcinoma in the liver. In groups II–IV, mice were treated with co-injection of CT26 cells (2 × 105) mixed with MSCs, AdEGFP-MSCs, or AdSirt1-MSCs respectively at a ratio of 1:1 via intrasplenic injection. The hepatic metastasis model of colorectal carcinoma was also established to investigate the effect of iNOS-AdSirt1-MSCs. Animals were sacrificed on day 21 after treatment, and murine blood and liver were removed and processed for histology assessment. Maximum tumor volume (V) was measured with the following equation: V = ½(a × b2), where a and b represent maximum and minimum tumor diameters, respectively.
Experimental Design of CCl4-Induced Acute Liver Injury
The male C57BL/6 mice (8–10 weeks of age) were randomly divided into five groups (n = 5 per group) including group I (olive oil group), group II (CCl4 group), group III (CCl4 + MSC group), group IV (CCl4 + AdEGFP-MSC group), and group V (CCl4 + AdSirt1-MSC group). CCl4 (Sinopharm, Shanghai, China) was used to induce acute liver injury in mice. The mice were injected intraperitoneally (i.p.) with 0.3% CCl4 dissolved in olive oil (10 mL/kg body weight) in groups II–V, whereas mice that were injected i.p. with olive oil represented the control (group I). Then, MSCs, AdEGFP-MSCs, or AdSirt1-MSCs (5 × 105) were intravenously injected into mice 4 h after CCl4 administration in groups III, IV, or V, respectively. 48 h after CCl4 challenge, the animals were anesthetized to collect the blood sample and then sacrificed to obtain the livers. Blood and liver samples were collected for further analysis as described below.
Bio-Plex Assay
A Bio-Plex assay was performed according to the manufacturer’s instruction using the Bio-Plex cytokine assay kit from Bio-Rad. Briefly, samples were thawed at room temperature and incubated with antibody microbeads for 30 min. After washing, the beads were incubated with the detection antibody cocktail; after another wash step, the beads were incubated with streptavidin phycoerythrin for 10 min and washed. Then, the concentration of each cytokine was measured with the Bio‐Plex suspension array reader.
Hematoxylin and Eosin Staining
Mouse liver samples were rinsed with PBS, fixed in 4% paraformaldehyde, and then embedded in paraffin. The 5-μm-thick paraffin sections prepared for the experiments were stained with hematoxylin and eosin (H&E) according to the staining protocol.
Immunohistochemistry Staining
The anti-CD8 antibody (1:500; Abcam, Cambridge, UK) and anti-CD4 antibody (1:1,000; Abcam, Cambridge, UK) were used as primary antibodies. The anti-mouse and anti-rabbit horseradish peroxidase-conjugated secondary antibodies were used correspondingly.
Quantitative Real-Time PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen/Life Technologies). 1 μg of total RNA sample was reverse transcribed into cDNA using a PrimeScript RT reagent kit (Takara, Kyoto, Japan) for cDNA synthesis. Quantitative PCR was performed using a SYBR Green PCR kit (Applied Biosystems) according to the manufacturer’s protocols. The primers are presented in Table 1.
Table 1.
Primer Sequences
| Target Gene | Sequence (5′→3′) | |
|---|---|---|
| Mouse Cxcl9 | forward | GGAGTTCGAGGAACCCTAGTG |
| reverse | GGGATTTGTAGTGGATCGTGC | |
| Mouse Cxcl10 | forward | CCAAGTGCTGCCGTCATTTTC |
| reverse | TCCCTATGGCCCTCATTCTCA | |
| Mouse Cxcl11 | forward | TGTAATTTACCCGAGTAACGGC |
| reverse | CACCTTTGTCGTTTATGAGCCTT | |
| Mouse Icam1 | forward | TGCCTCTGAAGCTCGGATATAC |
| reverse | TCTGTCGAACTCCTCAGTCAC | |
| Mouse Vcam1 | forward | TTGGGAGCCTCAACGGTACT |
| reverse | GCAATCGTTTTGTATTCAGGGGA | |
| Mouse Sirt1 | forward | ATGACGCTGTGGCAGATTGTT |
| reverse | CCGCAAGGCGAGCATAGAT | |
| Mouse Nos2 | forward | ACATCGACCCGTCCACAGTAT |
| reverse | CAGAGGGGTAGGCTTGTCTC | |
| Mouse Tnfaip6 | forward | GTGAGCGATGGGATGCCTATT |
| reverse | AGCCGAATGTGCCAGTAGC | |
| Mouse Il10 | forward | AGCCTTATCGGAAATGATCCAGT |
| reverse | GGCCTTGTAGACACCTTGGT | |
| Mouse Il6 | forward | CTGCAAGAGACTTCCATCCAG |
| reverse | AGTGGTATAGACAGGTCTGTTGG | |
| Mouse Hgf | forward | AACAGGGGCTTTACGTTCACT |
| reverse | CGTCCCTTTATAGCTGCCTCC | |
| Mouse Gapdh | forward | TGGCCTTCCGTGTTCCTAC |
| reverse | GAGTTGCTGTTGAAGTCGCA | |
Western Blot Assay
The anti-Sirt1 (1:1,000; CST, Danvers, MA, USA), anti-iNOS (1:1,000; Abcam, Cambridge, UK), NF-κB-p65 (1:1,000; CST, Danvers, MA, USA), and acetyl-NF-κB p65 (Lys310) antibodies (1:1,000, CST, Danvers, MA, USA) and the anti-GAPDH antibody (1:1,000; Bioworld Technology, St. Louis Park, MN, USA) were used as primary antibodies. Western blot experiments were repeated three times for each protein sample.
Transient Transfection Assay
The iNOS overexpression plasmid was provided by Heyuan Biotechnology (Shanghai, China). Cells were transfected with the iNOS overexpression plasmid using Lipofectamine 3000 (Invitrogen). Cells were transfected in parallel with the empty vector as the control.
Coimmunoprecipitation Assay
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing a cocktail of protease inhibitors (Thermo Fisher Scientific) and PMSF (Sigma) for 30 min on ice. Each sample of cell protein lysates was coimmunoprecipitated using anti-p65 antibody (1:100; CST, Danvers, MA, USA) overnight at 4°C. Normal immunoglobulin G (IgG) was used as the control. The immunoprecipitates were added to the protein A/G agarose beads (Thermo Fisher Scientific) and rotated for 2 h. The immunoprecipitates attached to the bead resins were eluted in elution buffer and subjected to western blotting analysis.
Measurement of NO Production
The Griess method was applied to evaluate NO concentration in MSCs. A Griess reagent kit (Beyotime Biotech, Hangzhou, China) was used according to the manufacturer’s instructions. Briefly, supernatant from each group was mixed with Griess reagents in a 96-well microtiter plate. The optical density (OD) values of the mixtures were detected at 540 nm.
Splenocyte Proliferation Assay In Vitro
The extent of splenocyte proliferation was measured by carboxyfluorescein diacetate succinimidyl ester (CFSE) staining detected by flow cytometric analysis. Freshly isolated splenocytes from the BALB/c mice were incubated with 5 μM CFSE (eBioscience) for 10 min at 37°C and then were subsequently washed twice with ice-cold FBS, after which the splenocytes labeled with CFSE were co-cultured respectively with MSCs, AdEGFP-MSCs, and Adirt1-MSCs at a ratio of 10:1 for 72 h in the presence of 5 μg/mL Con A (eBioscience), and then they were collected for flow cytometric measurement of CFSE dilution to detect splenocyte proliferation.
Chemotaxis Assay
Chemotaxis was tested with the NeuroProbe ChemoTx chemotaxis system (NeuroProbe, Gaithersburg, MD, USA) as described previously by Shi et al.15 MSCs, AdEGFP-MSCs, or AdSirt1-MSCs pre-treated with IFN-γ plus TNF-α (10 ng/mL each) for 24 h were respectively seeded into the lower chambers. The control group was added with L-DMEM in the lower chamber. Then, the polyvinylpyrrolidone-free polycarbonate membrane with 5-μm pores was overlaid. The freshly isolated splenocytes from mice were added to the top chambers. After a 3-h incubation, cells that had migrated through pores and into bottom wells were observed under the microscope.
Statistical Analysis
All statistical data were analyzed with t tests using GraphPad Prism software v6.0. Data are presented as the mean ± standard deviation (SD). Differences with p < 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001; NS: not significant).
Author Contributions
F.Y. conducted and designed the experiments, analyzed and interpreted the data, and wrote the paper. L.W. and Z.H. designed the study and interpreted the data. J.J. and C.Z. conducted experiments and analyzed the data. X.Y. performed the animal experiments. L.G. performed immunohistochemistry staining. Y.M. drew the schematic figure. R.L. performed measurement of nitric oxide production. Q.Z. performed qRT-PCR experiments.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This project was supported by National Key R&D Program of China Grant 2018YFA0107500; National Natural Science Foundation of China Grants 81630070, 81872243, 81802395, 31700788, 81702320, 81972254, 81401308, and 81472737; Special Funds for National Key Sci-Tech Special Project of China Grant 2018ZX10723204-005-004; Shanghai Science and Technology Committee Grant, China 16JC1405200; and Science Fund for Creative Research Groups, NSFC, China Grant 81521091.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.01.018.
Contributor Information
Zhipeng Han, Email: hanzhipeng0311@126.com.
Lixin Wei, Email: weilixin_smmu@163.com.
Supplemental Information
References
- 1.Bender E. Cancer immunotherapy. Nature. 2017;552:S61. doi: 10.1038/d41586-017-08699-z. [DOI] [PubMed] [Google Scholar]
- 2.Postow M.A., Sidlow R., Hellmann M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018;378:158–168. doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- 3.June C.H., O’Connor R.S., Kawalekar O.U., Ghassemi S., Milone M.C. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 4.Wu H.H., Zhou Y., Tabata Y., Gao J.Q. Mesenchymal stem cell-based drug delivery strategy: from cells to biomimetic. J. Control. Release. 2019;294:102–113. doi: 10.1016/j.jconrel.2018.12.019. [DOI] [PubMed] [Google Scholar]
- 5.Malik Y.S., Sheikh M.A., Xing Z., Guo Z., Zhu X., Tian H., Chen X. Polylysine-modified polyethylenimine polymer can generate genetically engineered mesenchymal stem cells for combinational suicidal gene therapy in glioblastoma. Acta Biomater. 2018;80:144–153. doi: 10.1016/j.actbio.2018.09.015. [DOI] [PubMed] [Google Scholar]
- 6.Grisendi G., Spano C., D’souza N., Rasini V., Veronesi E., Prapa M., Petrachi T., Piccinno S., Rossignoli F., Burns J.S. Mesenchymal progenitors expressing TRAIL induce apoptosis in sarcomas. Stem Cells. 2015;33:859–869. doi: 10.1002/stem.1903. [DOI] [PubMed] [Google Scholar]
- 7.Sage E.K., Thakrar R.M., Janes S.M. Genetically modified mesenchymal stromal cells in cancer therapy. Cytotherapy. 2016;18:1435–1445. doi: 10.1016/j.jcyt.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y., Chen X., Cao W., Shi Y. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat. Immunol. 2014;15:1009–1016. doi: 10.1038/ni.3002. [DOI] [PubMed] [Google Scholar]
- 9.Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell. 2012;10:709–716. doi: 10.1016/j.stem.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Chen X., Lu Y., Zhang Z., Wang J., Yang H., Liu G. Intercellular interplay between Sirt1 signalling and cell metabolism in immune cell biology. Immunology. 2015;145:455–467. doi: 10.1111/imm.12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J., Lee S.M., Shannon S., Gao B., Chen W., Chen A., Divekar R., McBurney M.W., Braley-Mullen H., Zaghouani H., Fang D. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J. Clin. Invest. 2009;119:3048–3058. doi: 10.1172/JCI38902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woo S.J., Lee S.M., Lim H.S., Hah Y.S., Jung I.D., Park Y.M., Kim H.O., Cheon Y.H., Jeon M.G., Jang K.Y. Myeloid deletion of SIRT1 suppresses collagen-induced arthritis in mice by modulating dendritic cell maturation. Exp. Mol. Med. 2016;48:e221. doi: 10.1038/emm.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z., Xu J., Liu Y., Wang T., Pei J., Cheng L., Hao D., Zhao X., Chen H.Z., Liu D.P. Mouse macrophage specific knockout of SIRT1 influences macrophage polarization and promotes angiotensin II-induced abdominal aortic aneurysm formation. J. Genet. Genomics. 2018;45:25–32. doi: 10.1016/j.jgg.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Ren G., Zhang L., Zhao X., Xu G., Zhang Y., Roberts A.I., Zhao R.C., Shi Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–150. doi: 10.1016/j.stem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 15.Shi Y., Kornovski B.S., Savani R., Turley E.A. A rapid, multiwell colorimetric assay for chemotaxis. J. Immunol. Methods. 1993;164:149–154. doi: 10.1016/0022-1759(93)90307-s. [DOI] [PubMed] [Google Scholar]
- 16.Lee D.K., Song S.U. Immunomodulatory mechanisms of mesenchymal stem cells and their therapeutic applications. Cell. Immunol. 2018;326:68–76. doi: 10.1016/j.cellimm.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Cinelli M.A., Do H.T., Miley G.P., Silverman R.B. Inducible nitric oxide synthase: regulation, structure, and inhibition. Med. Res. Rev. 2020;40:158–189. doi: 10.1002/med.21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004;75:639–653. doi: 10.1016/j.lfs.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 19.Yeung F., Hoberg J.E., Ramsey C.S., Keller M.D., Jones D.R., Frye R.A., Mayo M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su J., Chen X., Huang Y., Li W., Li J., Cao K., Cao G., Zhang L., Li F., Roberts A.I. Phylogenetic distinction of iNOS and IDO function in mesenchymal stem cell-mediated immunosuppression in mammalian species. Cell Death Differ. 2014;21:388–396. doi: 10.1038/cdd.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabinovich G.A., Gabrilovich D., Sotomayor E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim H.W., Kang S.G., Ryu J.K., Schilling B., Fei M., Lee I.S., Kehasse A., Shirakawa K., Yokoyama M., Schnölzer M. SIRT1 deacetylates RORγt and enhances Th17 cell generation. J. Exp. Med. 2015;212:607–617. doi: 10.1084/jem.20132378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Loosdregt J., Vercoulen Y., Guichelaar T., Gent Y.Y., Beekman J.M., van Beekum O., Brenkman A.B., Hijnen D.J., Mutis T., Kalkhoven E. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood. 2010;115:965–974. doi: 10.1182/blood-2009-02-207118. [DOI] [PubMed] [Google Scholar]
- 24.Owczarczyk A.B., Schaller M.A., Reed M., Rasky A.J., Lombard D.B., Lukacs N.W. Sirtuin 1 regulates dendritic cell activation and autophagy during respiratory syncytial virus-induced immune responses. J. Immunol. 2015;195:1637–1646. doi: 10.4049/jimmunol.1500326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murray P.J., Wynn T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meisel R., Zibert A., Laryea M., Göbel U., Däubener W., Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619–4621. doi: 10.1182/blood-2003-11-3909. [DOI] [PubMed] [Google Scholar]
- 27.Sato K., Ozaki K., Oh I., Meguro A., Hatanaka K., Nagai T., Muroi K., Ozawa K. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood. 2007;109:228–234. doi: 10.1182/blood-2006-02-002246. [DOI] [PubMed] [Google Scholar]
- 28.Nopparat C., Sinjanakhom P., Govitrapong P. Melatonin reverses H2O2-induced senescence in SH-SY5Y cells by enhancing autophagy via sirtuin 1 deacetylation of the RelA/p65 subunit of NF-κB. J. Pineal Res. 2017;63:e12407. doi: 10.1111/jpi.12407. [DOI] [PubMed] [Google Scholar]
- 29.Kauppinen A., Suuronen T., Ojala J., Kaarniranta K., Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013;25:1939–1948. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Xie Q.W., Kashiwabara Y., Nathan C. Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J. Biol. Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- 31.Kleinert H., Euchenhofer C., Ihrig-Biedert I., Förstermann U. In murine 3T3 fibroblasts, different second messenger pathways resulting in the induction of NO synthase II (iNOS) converge in the activation of transcription factor NF-κB. J. Biol. Chem. 1996;271:6039–6044. doi: 10.1074/jbc.271.11.6039. [DOI] [PubMed] [Google Scholar]
- 32.Marks-Konczalik J., Chu S.C., Moss J. Cytokine-mediated transcriptional induction of the human inducible nitric oxide synthase gene requires both activator protein 1 and nuclear factor κB-binding sites. J. Biol. Chem. 1998;273:22201–22208. doi: 10.1074/jbc.273.35.22201. [DOI] [PubMed] [Google Scholar]
- 33.Park S.Y., Lee S.W., Lee S.Y., Hong K.W., Bae S.S., Kim K., Kim C.D. SIRT1/adenosine monophosphate-activated protein kinase α signaling enhances macrophage polarization to an anti-inflammatory phenotype in rheumatoid arthritis. Front. Immunol. 2017;8:1135. doi: 10.3389/fimmu.2017.01135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang W., Shang W.L., Wang H.D., Wu W.W., Hou S.X. Sirt1 overexpression protects murine osteoblasts against TNF-α-induced injury in vitro by suppressing the NF-κB signaling pathway. Acta Pharmacol. Sin. 2012;33:668–674. doi: 10.1038/aps.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J.H., Song M.Y., Song E.K., Kim E.K., Moon W.S., Han M.K., Park J.W., Kwon K.B., Park B.H. Overexpression of SIRT1 protects pancreatic β-cells against cytokine toxicity by suppressing the nuclear factor-κB signaling pathway. Diabetes. 2009;58:344–351. doi: 10.2337/db07-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howard M., Roux J., Lee H., Miyazawa B., Lee J.W., Gartland B., Howard A.J., Matthay M.A., Carles M., Pittet J.F. Activation of the stress protein response inhibits the STAT1 signalling pathway and iNOS function in alveolar macrophages: role of Hsp90 and Hsp70. Thorax. 2010;65:346–353. doi: 10.1136/thx.2008.101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jahani-Asl A., Bonni A. iNOS: a potential therapeutic target for malignant glioma. Curr. Mol. Med. 2013;13:1241–1249. doi: 10.2174/1566524011313080002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamijo R., Harada H., Matsuyama T., Bosland M., Gerecitano J., Shapiro D., Le J., Koh S.I., Kimura T., Green S.J. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 39.Do H., Pyo S., Sohn E.H. Suppression of iNOS expression by fucoidan is mediated by regulation of p38 MAPK, JAK/STAT, AP-1 and IRF-1, and depends on up-regulation of scavenger receptor B1 expression in TNF-α- and IFN-γ-stimulated C6 glioma cells. J. Nutr. Biochem. 2010;21:671–679. doi: 10.1016/j.jnutbio.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 40.Guo Z., Shao L., Feng X., Reid K., Marderstein E., Nakao A., Geller D.A. A critical role for C/EBPβ binding to the AABS promoter response element in the human iNOS gene. FASEB J. 2003;17:1718–1720. doi: 10.1096/fj.02-1172fje. [DOI] [PubMed] [Google Scholar]
- 41.Mounayar M., Kefaloyianni E., Smith B., Solhjou Z., Maarouf O.H., Azzi J., Chabtini L., Fiorina P., Kraus M., Briddell R. PI3kα and STAT1 interplay regulates human mesenchymal stem cell immune polarization. Stem Cells. 2015;33:1892–1901. doi: 10.1002/stem.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han Z., Tian Z., Lv G., Zhang L., Jiang G., Sun K., Wang C., Bu X., Li R., Shi Y. Immunosuppressive effect of bone marrow-derived mesenchymal stem cells in inflammatory microenvironment favours the growth of B16 melanoma cells. J. Cell. Mol. Med. 2011;15:2343–2352. doi: 10.1111/j.1582-4934.2010.01215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y., Yang X., Jing Y., Zhang S., Zong C., Jiang J., Sun K., Li R., Gao L., Zhao X. Contribution and mobilization of mesenchymal stem cells in a mouse model of carbon tetrachloride-induced liver fibrosis. Sci. Rep. 2015;5:17762. doi: 10.1038/srep17762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.