

SUMMARY
Acute cardiac hypoxia produces life-threatening elevations in late sodium current (ILATE) in the human heart. Here, we show the underlying mechanism: hypoxia induces rapid SUMOylation of NaV1.5 channels so they reopen when normally inactive, late in the action potential. NaV1.5 is SUMOylated only on lysine 442, and the mutation of that residue, or application of a deSUMOylating enzyme, prevents hypoxic reopenings. The time course of SUMOylation of single channels in response to hypoxia coincides with the increase in ILATE, a reaction that is complete in under 100 s. In human cardiac myocytes derived from pluripotent stem cells, hypoxia-induced ILATE is confirmed to be SUMO-dependent and to produce action potential prolongation, the pro-arrhythmic change observed in patients.
Graphical Abstract
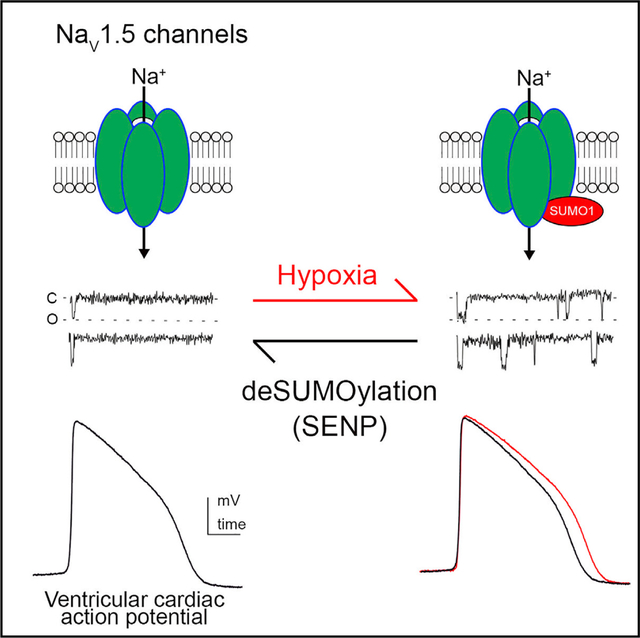
In Brief
The cardiac channel NaV1.5 passes pro-arrhythmic late sodium currents in response to hypoxia. Plant et al. demonstrate the pathophysiological mechanism to be rapid, hypoxia-induced monoSUMOylation of NaV1.5 channels. Blocking SUMOylation of lysine442 prevents hypoxia-induced late currents and attendant prolongation of the action potential in human cardiomyocytes derived from pluripotent stem cells.
INTRODUCTION
In the human heart, NaV1.5 voltage-gated sodium channels pass INa, a rapidly activating and inactivating Na+ current that determines the rise, duration, and timing of action potentials (Marbán, 2002; Catterall, 1986). In addition, NaV1.5 channels pass a small, persistent Na+ current, the late sodium current (ILATE), which contributes to maintaining the action potential plateau (Makielski, 2016). In healthy hearts, ILATE is less than 0.5% of the magnitude of peak INa and is passed by a small number of channels that do not stay closed after inactivation (Makielski, 2016). In patients with an ischemic and failing heart, sudden infant death syndrome (SIDS), and mutations in SCN5A, the gene that encodes the NaV1.5 channel, which produces long QT syndrome, the size of ILATE can increase to 4%–5% of peak INa (Bennett et al., 1995; Plant et al., 2006; Belardinelli et al., 2015). Furthermore, acute hypoxia and ischemia have been recognized to increase ILATE in cardiac myocytes (Saint et al., 1992; Ju et al., 1996; Carmeliet, 1999; Belardinelli et al., 2006) prior to slower processes like remodeling (West, 2017), and excess ILATE has been shown to be pro-arrhythmic because it prolongs action potential duration (APD), reducing repolarization reserve, increasing susceptibility to after-depolarizations, and causing a predisposition to torsades de pointes (TdP), a ventricular dysrhythmia that is lethal when sustained (Gaur et al., 2009; Shryock et al., 2013; Chadda et al., 2017). Thus, increases in ILATE above baseline by just 0.3% to 1% predisposition to sudden cardiac death (Bennett et al., 1995; Belardinelli et al., 2015). These observations underpin proposals that inhibiting ILATE has therapeutic potential (Belardinelli et al., 2006; Song et al., 2006).
We recently reported that the rapid influx of Na+ flux into neurons in response to acute hypoxic challenge is due to SUMOylation of NaV1.2, the major voltage-gated sodium channel in the brain; in that channel, SUMO modification of lysine 38 alters opening and closing with changes in voltage and does not effect ILATE (Plant et al., 2016). SUMOylation is the enzyme-mediated linkage of one of three SUMO isoforms to the ε amino group of specific lysine residues in a target protein (Henley et al., 2014). Present in all eukaryotic cells, the SUMO pathway was known to regulate the trafficking and activity of nuclear transcription factors when we described it to operate as well at the plasma membrane by direct SUMOylation of Na+ and K+ channels to regulate excitability (Rajan et al., 2005; Plant et al., 2010, 2011, 2012, 2016; Xiong et al., 2017). Given the important role of hypoxia-induced increases in ILATE in heart disease, we tested the hypothesis that SUMOylation of NaV1.5 channels was the underlying mechanism.
We demonstrate that hypoxia induces a rapid increase in ILATE in human cardiac myocytes derived from pluripotent stem cells (iPS-CMs). The response is reproduced by application of cytosolic SUMO1 at ambient levels of oxygen and suppressed by the deSUMOylating enzyme SENP1. SUMOylation of NaV1.5 on lysine 442, a residue located in the segment between channel domains I and II, is shown to be necessary and sufficient to explain hypoxia-induced changes in ILATE by reconstitution of the response in heterologous cells and by studies using whole-cell and single-channel patch-clamp recording and live cell Förster resonance energy transfer (FRET). The implied mechanism was confirmed using total internal reflection fluorescence microscopy (TIRFM) to study single particles on the surface of live cells in real-time; the time course was the same for hypoxic challenge, the recruitment of SUMO1 to NaV1.5 channels on cell surface, and the increase in ILATE. Furthermore, hypoxia-induced, SUMO-dependent increases in ILATE were observed to increase APD in human induced pluripotent stem cells (iPSCs) by recording spontaneous action potentials in current-clamp mode; and the changes were suppressed by ranolazine, a drug that inhibits NaV1.5 channel late current. Incorporating the measured hypoxia-induced increase in ILATE in iPS-CMs into the O’Hara-Rudy model for human cardiac action potentials (O’Hara et al., 2011) was sufficient to reproduce the observed pro-arrhythmic increase in APD. Given the role of ILATE in arrhythmogenesis, SUMOylation of NaV1.5 is identified as a mechanistically defined target for therapeutic intervention.
RESULTS
Hypoxia Rapidly Increases ILATE in Human iPS-CMs
At ambient O2 levels (21%), INa in human iPS-CMs activated rapidly to a mean peak of −40 ± 4 pA/pF when measured at −30 mV in whole-cell mode (Figure 1A; Table 1). INa then inactivated to produce a residual ILATE that was 0.46% ± 0.1% of the magnitude of peak current 50 to 100 ms after the onset of inactivation. When O2 was lowered from ambient levels to 1.5% by the perfusion of cells with a hypoxic solution (Plant et al., 2016), mean peak INa was unchanged (−42 ± 3 pA/pF), but ILATE increased ~10-fold to ~4.4% (−1.85 pA/pF) of peak INa after 100 s of hypoxia. The hypoxia-induced increase in ILATE rose to half its maximal level in 40 ± 2 s and remained stable for at least 3 min after iPS-CMs were restored to ambient O2. Cells were not studied beyond 600 s to maintain consistent voltage-clamp control of INa (Figure 1A). These changes are like those observed in isolated rat ventricular myocytes (Ju et al., 1996) and human cardiac muscle (Makielski, 2016; Wei et al., 2016).
Figure 1. Acute Hypoxia and SUMO1 Augment the ILATE in Human iPS-CMs.
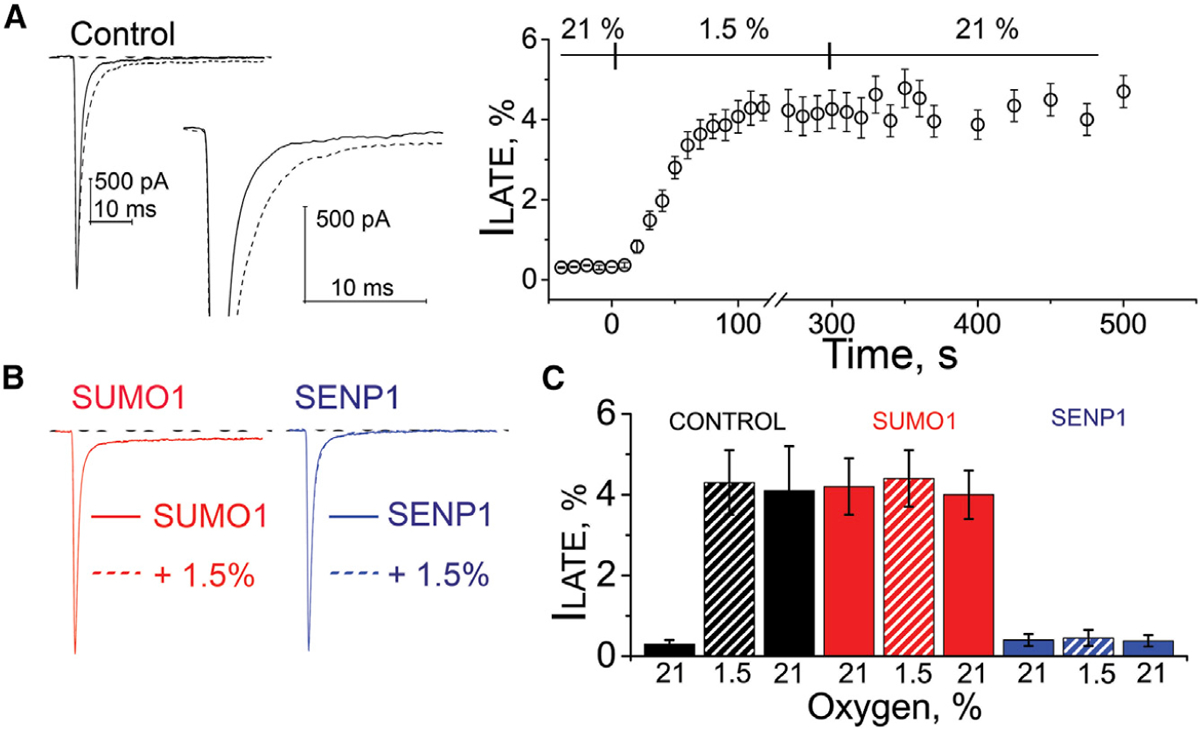
INa in human iPS-CMs was studied by whole-cell patch clamp. ILATE was measured at −30 mV and is shown as a mean percentage of the peak current remaining between 50 and 100 ms after the peak current. Hypoxia challenge was a drop in O2 from 21% to 1.5%. The O2 level was measured in real time in the recording chamber and fell from 21% to 1.5% O2 in 11 ± 3 s (not shown). The time course of hypoxic modulation of INa was studied by steps from −100 mV to −30 mV every 10 s. Whole-cell currents were not studied for more than 600 s to maintain consistent voltage clamp control of INa. Cells were studied with control solution (black), 1 nM SUMO1 (red), or 1 nM SENP1 (blue) in the recording pipette. Data are mean ± SEM for 8 to 10 cells per group. Measured values and statistical analyses are listed in Table 1.
(A) Left: example traces showing ILATE at −30 mV increased when control perfusate at 21% O2 (black) was exchanged with a hypoxic solution at 1.5% O2 (black dash). Inset: the increase in ILATE shown in response to hypoxia. Right: the time course for changes in ILATE in response to hypoxia and on return to normoxia (21%). Scale bars represent 500 pA and 10 ms.
(B) Left: SUMO1 in the pipette (red) increased ILATE. No further change in ILATE was observed when the control perfusate was exchanged with a hypoxic solution at 1.5% O2 (red dash). Right: ILATE was not observed with SENP1 in the pipette when cells were studied at 21% (blue) or 1.5% O2 (blue dash). In each case, the dashed lines overlap with the solid lines.
(C) A histogram showing the mean ILATE as a percentage of the peak current from cells studied at 21% O2 (solid color) after 200 s at 1.5% O2 (hatched color) and after 200 s following a return to 21% O2 (solid color) with control solution (black), 1 nM SUMO1 (red), or 1 nM SENP1 (blue) in the recording pipette.
Table 1.
Effects of Hypoxia, SUMO1, and SENP1 on INa in iPS-CMs and NaV1.5 Channels in CHO Cells
| INa-iPS-CMs | Nav1.5 CHO-K1 | Nav1.5-K442Q CHO-K1 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Condition | Activation | SSI | Current, pA/pF | Activation | SSI | Current, pA/pF | Activation | SSI | Current, pA/pF | |||||||||
| O2 | V1/2, mV | K | V1/2, mV | K | IPEAK | ILATE | V1/2, mV | K | V1/2, mV | K | IPEAK | ILATE | V1/2, mV | K | V1/2, mV | K | IPEAK | ILATE |
| 21% | −44.3 ± 2 | 7.1 ± 1 | −87.2 ± 3 | 6.2 ± 1 | −40 ± 4 | −0.22 ± 1 | −45.1 ± 2 | 7.8 ± 1 | −87.2 ± 2 | 6.5 ± 1 | −198 ± 6 | −0.96 ± 0.2 | −47.0 ± 3 | 7.4 ± 2 | −87.4 ± 2 | 6 ± 2 | −200 ± 4 | −0.93 ± 0.3 |
| 21 % to 1.5% | −50.5 ± 1* | 5 ± 1* | −81.7 ± 3* | 4.6 ± 1* | −42 ± 3 | −1.85 ± 0.5 | −50.3 ± 3* | 4.4 ± 2* | −81 ± 2* | 4.5 ± 2* | −201 ± 4 | −8.3 ± 0.6 | −46 ± 1 | 7.2 ± 2 | −87.2 ± 1 | 5.8 ± 3 | −197 ± 6 | −0.91 ± 0.4 |
| SENP1 21% | −44.6 ± 2 | 7 ± 2 | −89 ± 2.5 | 6.5 ± 2 | −40 ± 2 | −0.28 ± 0.7 | −44.7 ± 3 | 7.8 ± 2 | −85.2 ± 3 | 6.2 ± 1 | −196 ± 5 | −1.1 ± 0.4 | −48 ± 2.3 | 7.3 ± 2 | −83.7 ± 2.5 | 5.9 ± 2 | −194 ± 5 | −0.88 ± 0.4 |
| SENP1 1.5% | −43.7 ± 2 | 7.2 ± 1 | −90 ± 2 | 6.4 ± 2 | −39 ± 3 | 0.25 ± 0.8 | −45.4 ± 2 | 7.6 ± 1 | −85.3 ± 2 | 6.7 ± 3 | −197 ± 4 | −1.3 ± 0.3 | −46.7 ± 3.7 | 7.2 ± 3 | −86.2 ± 2 | 5.9 ± 1 | −199 ± 5 | −0.93 ± 0.5 |
| SUMO1 21% | −52 ± 2* | 5.2 ± 1* | −81.9 ± 2* | 4.3 ± 1* | −43 ± 2 | −2.18 ± 0.9 | −51.5 ± 4* | 4.5 ± 1* | −82 ± 2* | 4.6 ± 2* | −202 ± 5 | −8.7 ± 0.6 | −47.5 ± 2.4 | 7.2 ± 1 | −82.7 ± 3 | 5.7 ± 1 | −203 ± 5 | −0.98 ± 0.6 |
| SUMO1 1.5% | −51 ± 3* | 5.3 ± 1* | −82 ± 3* | 4.2 ± 2* | −42 ± 4 | −2.22 ± 1 | −50.6 ± 3* | 4.5 ± 2* | −82.5 ± 3* | 4.1 ± 1* | −198 ± 5 | −9.1 ± 0.5 | −46.5 ± 3 | 7.3 ± 2 | −83.5 ± 3.7 | 6 ± 3 | −201 ± 4 | −0.94 ± 0.5 |
Human cardiac myocytes derived from iPS cells (Figure 1) or cloned channels in CHO-K1 cells (Figure 3) were studied in whole-cell mode. Stimulation protocols are described in the Methods. V1/2, the voltage evoking half-maximal conductance and k, for activation or steady-state inactivation (SSI), were obtained by fitting the normalized current plotted against voltage to a Boltzmann function, I = Imax/(1+exp[−(V−V1/2)/k]), where Imax is maximum current. For comparison between groups, current densities were measured at −30 mV, the potential that gave peak current in all conditions studied. Data are means ± SEM for 8 to 12 cells per group; Changes from values measured from cells studied at 21% oxygen O2 under control conditions were assessed by ANOVA and are indicated with *p < 0.05.
SUMO1 Increases ILATE in Human iPS-CMs
We delivered 1 nM purified SUMO1 peptide into iPS-CMs by the patch pipette; a concentration that evoked maximal effects on NaV1.2 channels in cerebellar granule neurons (CGNs) and three different K+ channels in CGNs, hippocampal neurons, and rat cardiac myocytes (Xiong et al., 2017; Plant et al., 2011, 2012, 2016). SUMO1 induced changes in cardiac INa like those produced by hypoxia: ILATE increased to ~4.2% of peak INa without a change in the peak magnitude (Figures 1B and 1C; Table 1). When SUMO1 was in the recording pipette, changing the solution bathing iPS-CMs from 21% to 1.5% O2 did not further alter the parameters of INa peak or ILATE. The notion that hypoxic regulation of INa proceeds by SUMOylation was further supported by the observation that delivering 1 nM SENP1 deSUMOylase into the iPS-CMs by the recording pipette suppressed the increase in ILATE induced by hypoxia (Figures 1B and 1C; Table 1).
ILATE is observed in the “window” between the voltages where NaV channels activate and inactivate (Figure S1; Table 1). Thus, exposure of the iPS-CMs to 1.5% O2 caused a leftward shift in the half-maximal activation voltage of INa (V1/2-act) of −6 ± 4 mV and a rightward shift in the steady-state inactivation midpoint (V1/2-SSI) of 6 ± 3 mV, increasing the range of voltages over which INa channels are likely to be activated but not fully inactivated. In ambient O2, SUMO1 in the pipette increased the magnitude of the window current in a manner like that observed on exposure to 1.5% O2, and hypoxic challenge with SUMO1 in the pipette did not further alter the parameters. Furthermore, inclusion of 1 nM SENP1 prevented the effects of hypoxia, maintaining the window current at control levels. Of note, the kinetics of recovery of INa from the fast-inactivated state was insensitive to hypoxia, SUMO1, and SENP1 (Figure S1).
SUMOylation of NaV1.5 on Lys442 Is Necessary and Sufficient to Recapitulate the Hypoxic Response
In cardiomyocytes, NaV1.5 channel complexes contain the 2016 amino-acid-pore-forming subunit (Figure 2A) and an accessory subunit, NaVβ1 (Catterall, 2012). A combination of spectroscopic and electrophysiological assays in live CHO cells showed that SUMOylation in response to hypoxia occurred only on NaV1.5 K442 and that modification of the site mediated the response to acute hypoxia.
Figure 2. FRET between NaV1.5 and SUMO1 at the Cell Surface Requires K442.
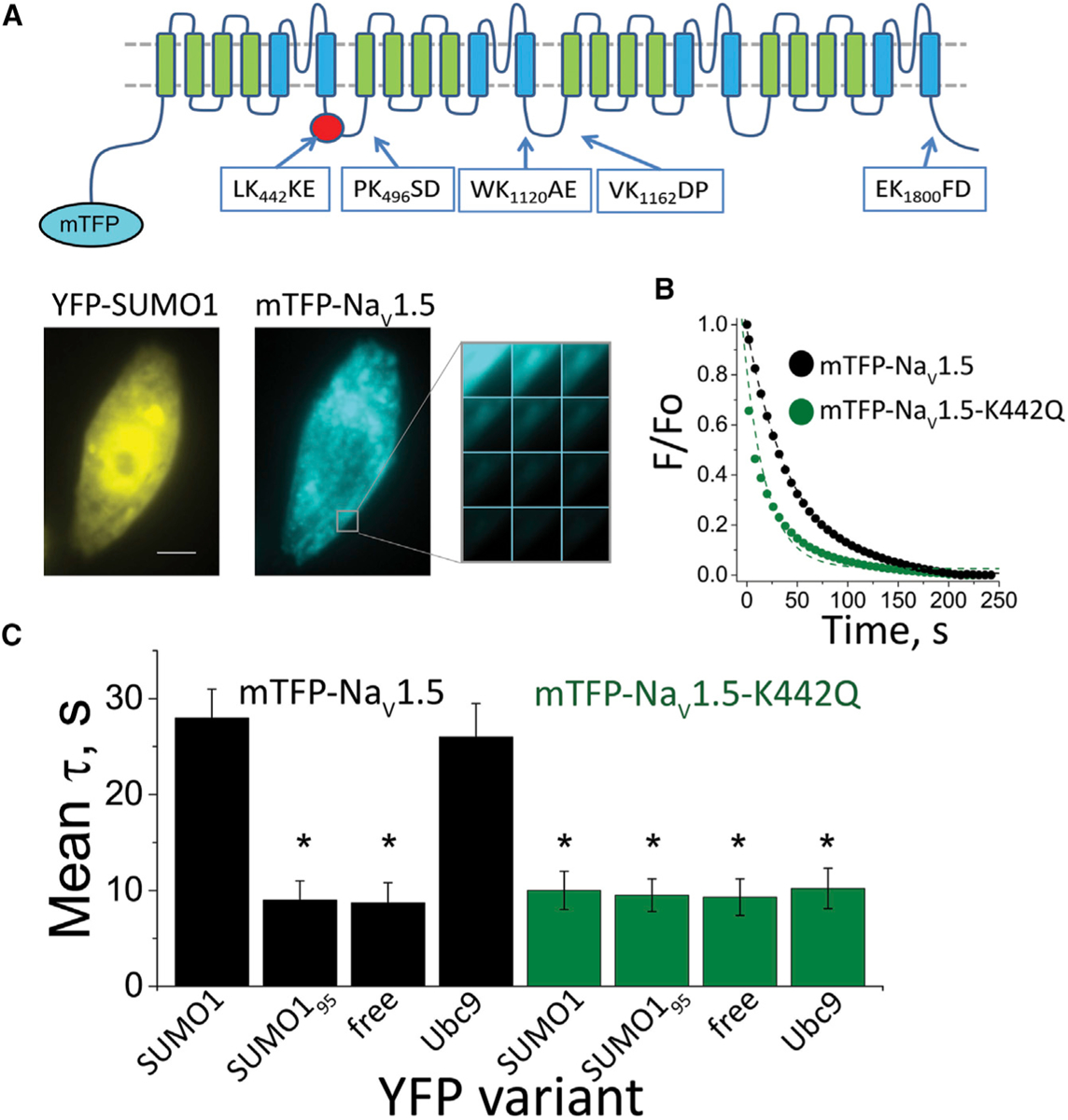
Human NaV1.5 was expressed with NaVβ1 and studied in live CHO-K1 cells per the STAR Methods. FRET was assessed by measuring the time constant (τ) for mTFP photobleaching (donor) in the presence of YFP (acceptor) from 3 regions of 5–7 cells per group. Data are mean τ ± SEM, and significant changes in τ compared to free YFP are indicated (*p < 0.001, two-way ANOVA).
(A) Top: a cartoon showing the secondary structure of a mTFP-NaV1.5 subunit and the location of putative SUMOylation sites identified by the SUMOplot algorithm. The NaV1.5 α-subunit has four homologous domains (I–IV), each with six transmembrane segments that fold to form one central ion conduction pore. Domains I and II are critical to opening the pore (activation) (Catterall, 2017; Cestèle et al., 1998, 2001). Fast inactivation results from pore occlusion by an IFM triplet of residues in the linker between domains III and IV(Vassilev et al., 1988; West et al., 1992; Cha et al., 1999). Bottom: fluorescent photomicrographs showing that mTFP-NaV1.5 (blue) and YFP-SUMO1 (yellow) reach the cell surface. Scale bar represents 10 μm. The montage shows photobleaching of mTFP-NaV1.5 in response to continued excitation.
(B) Exemplar photobleaching studies show the decay of mTFP fluorescence intensity for single cells expressing mTFP-NaV1.5 (black circle) or mTFP-NaV1.5-K442Q (green circle) with YFP-SUMO1, fit by an exponential to give τ.
(C) A histogram of mean FRET τ values show the assembly of mTFP-NaV1.5 (black bars) with YFP-SUMO1 and YFP-Ubc9 (τ = 28 ± 3* and 26 ± 3.5*, respectively) but not with linkage-incompetent YFP-SUMO195 or free YFP (τ = 9 ± 2 and 9 ± 2, respectively). In contrast, mTFP-NaV1.5-K442Q (green bars) did not show FRET with YFP-SUMO1, YFP-SUMO195, YFP-Ubc9, or free YFP (τ = 10 ± 2; 9.5 ± 2; 10 ± 2; and 9 ± 2, respectively).
First, the association of heterologously expressed human NaV1.5 and SUMO proteins at the surface of cells was studied by measuring FRET between subunits tagged with the fluorescent proteins mTFP1 and YFP (Figure 2A). The NaVβ1 subunit was expressed in these studies but did not carry a fluorescent protein. Tagging the N terminus of NaV1.5 (mTFP-NaV1.5) did not alter the biophysical properties of the channel nor its responsiveness to hypoxia or SUMO1 or SENP1 in the recording pipette (Figures S2; Table S1). Similarly, tagging the N terminus of SUMO1 (YFP-SUMO1) did not alter its operation with NaV1.5, as previously observed with NaV1.2 and three K+ channels (Xiong et al., 2017; Plant et al., 2010, 2011, 2012, 2016).
Both mTFP1-NaV1.5 and YFP-SUMO1 were visualized at the plasma membrane when expressed in CHO cells (Figure 2A). Indicating FRET, and thus the intimate association of the two fluorophores consistent with SUMOylation, the time course of mTFP1 photobleaching under continuous illumination had a time constant (τ) that was prolonged from less than 10 s to 27 s when the channel was expressed with YFP-SUMO1 (Figures 2B and 2C). mTFP-NaV1.5 also showed FRET with YFP-Ubc9, the SUMO conjugating enzyme that binds to target proteins only when a lysine subject to modification is present (Xiong et al., 2017; Plant et al., 2016). In contrast, FRET was not observed when mTFP-NaV1.5 was expressed with linkage-incompetent YFP-SUMO195 (a variant that lacks the terminal Gly-Gly motif present in SUMO1 and the matured isoform SUMO197) or soluble YFP; in both cases, τ was less than 10 s.
To identify SUMOylation site(s) in NaV1.5 channels, we analyzed the primary sequence of the subunit with an algorithm that looks for variants of the canonical SUMO motif ψ-K-X-E/D, as before with NaV1.2 and the K+ channels (Xiong et al., 2017; Plant et al., 2010, 2011, 2012, 2016). We mutated each of five potential target lysines individually to glutamine in mTFP-NaV1.5 and used FRET with YFP-SUMO1 to assess interaction (Figure S2). Only K442, within the motif L-K442-K-E, was required to produce FRET. Thus, mTFP-NaV1.5-K442Q was expressed on the cell surface like wild-type mTFP1-NaV1.5 but did not show FRET with YFP-SUMO1 or YFP-Ubc9 (Figure 2C). The absence of FRET supported the conclusion that K442 was necessary for SUMOylation of the NaV1.5 channel complex and, moreover, that no other lysine residue in the α subunit or in the NaVβ1 was subject to SUMOylation.
To confirm that K442 in the NaV1.5 sequence motif was subject to SUMOylation, a mass spectrometry (MS) strategy was used that we had previously developed for K+ channels (Plant et al., 2010, 2011). SUMO197T95K, a variant that leaves a Gly-Gly remnant of SUMO1 on the ε amino group of the target Lys after enzyme cleavage, was expressed with NaV1.5 residues 353–502 bearing a His6 affinity tag and the mammalian E1 and E2 SUMOylation enzymes in bacteria. Affinity purification followed by SDS-PAGE revealed a band of the expected apparent mass for the NaV1.5-SUMO adduct (~35 kDa). The purified protein was treated with trypsin and subjected to MS to yield a sequence coverage for NaV1.5353–502 of 42% and was identified uniquely as the fragment carrying the Gly-Gly SUMO1 remnant (Figure S4).
Next, we confirmed that K442 controlled the response to hypoxia by studying human NaV1.5 channels expressed in CHO cells. With the wild-type channel, decreasing O2 from 21% to 1.5% was associated with a rapid increase in ILATE from ~0.5% (−0.96 ± 0.2 pA/pF) to ~4.1% (−8.3 ± 0.6 pA/pF), without a change in the peak current, ~200 pA/pF at −30 mV (Figure 3A; Table 1), as observed for ILATE in iPS-CMs. As predicted, NaV1.5-K442Q channels were insensitive to hypoxia; ILATE for the point mutant channels remained less than ~0.5% of the peak current when O2 was decreased to 1.5%. Furthermore, peak current measured at −30 mV was like wild-type channels (~ −200 pA/pF) and was also unaltered by hypoxia (Figures 3A and 3B). Of note, the time course for the change in ILATE for NaV1.5 channels heterologously expressed in the CHO cells was like that for native INa in iPS-CMs, increasing from 0.5 to 4.2% over ~100 s, with half the rise time of 39.7 ± 2 s, and the increase was stable after the cells were restored to ambient levels of O2 for at least 3 min (Figure 3C).
Figure 3. K442 Is Required for Hypoxia and SUMO1-Mediated ILATE.
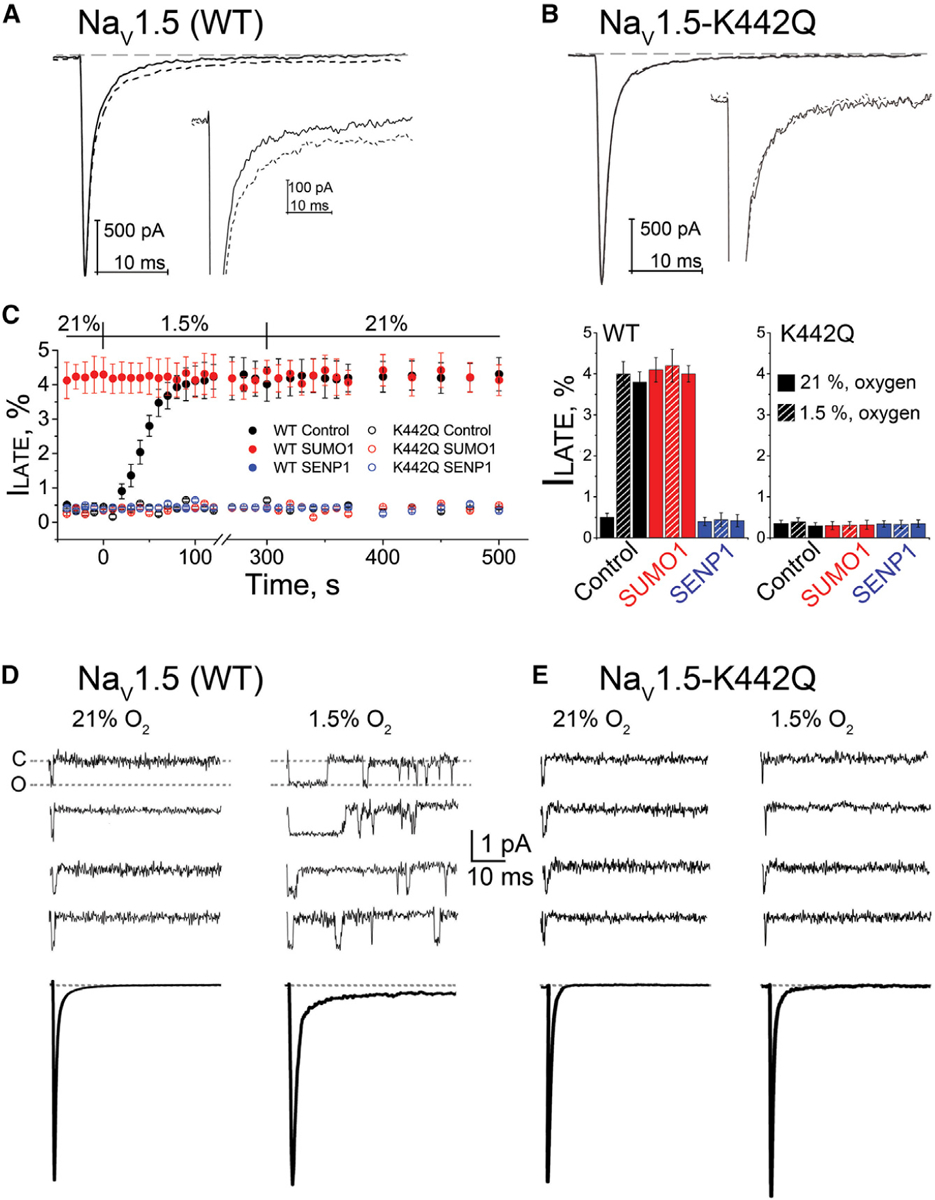
Human NaV1.5 wild-type (WT) or NaV1.5-K442Q was expressed in CHO-K1 cells with NaVβ1 and studied under normoxic (21%) and hypoxic (1.5%) O2 conditions with control solution (black), 1 nM SUMO1 (red), or 1 nM SENP1 (blue) in the recording pipette, as indicated. The late current (ILATE) was studied at −30 mV and is shown as a mean percentage of the peak current remaining between 50 and 100 ms after the peak current. The time course of hypoxic modulation of INa was studied by steps from −100 mV to −30 mV every 10 s. Data are mean ± SEM for 8 to 12 cells per group. Single-channel currents were studied in cell-attached mode and elicited every 5 s by a 50-ms depolarizing pulse to −30 mV from a holding potential of −120 mV. Data were recorded at filter and sampling frequencies of 5 and 50 kHz, respectively, and processed offline using a 1.2-kHz Bessel filter for display. Recording pipettes were filled with extracellular buffer. For each cell, null sweeps (with no channel activity) were identified, averaged offline, and subtracted from the data sweeps before analysis.
(A) Example traces from a cell expressing WT NaV1.5 channel activation at −30 mV; ILATE increased when control perfusate at 21% O2 (black) was exchanged with a hypoxic solution at 1.5% O2 (black dash). Inset: the increase in ILATE shown in response to hypoxia.
(B) Example traces showing that hypoxia does not induce an increase in ILATE in cells expressing NaV1.5-K442Q channels.
(C) Left: the time course for changes in ILATE in response to hypoxia and on return to normoxia for cells expressing WT NaV1.5 (closed circles) or NaV1.5-K442Q channels (open circles) with control solution (black), SUMO1 (red), or SENP1 (blue) in the recording pipette. Right: a histogram showing the mean ILATE as a percentage of the peak current from cells studied at 21% O2, after 200 s at 1.5% O2 and after 200 s following a return to 21% O2, with the pipette solutions indicated.
(D) Top: upon depolarization, single NaV1.5 channels opened, inactivated rapidly, and did not reopen. NaV1.5 channels reopened upon exposure to 1.5% O2. Null traces in normoxia and hypoxia were 55% ± 5% (n = 20 patches; 2,000 sweeps) and 53% ± 4% (n = 20 patches; 2,000 sweeps), respectively. Bottom: ensemble average traces (n = 200–220 sweeps) for the condition indicated.
(E) Top: single NaV1.5-K442Q channels did not reopen in ambient or hypoxic conditions. Null traces were 51% ± 3% (n = 20 patches; 2,000 sweeps) and 50% ± 2% (n = 20 patches; 2,000 sweeps), respectively. Bottom: ensemble average traces (n = 200–225 sweeps) for the condition indicated.
NaV1.5 channels heterologously expressed in tissue culture cells also responded to SUMO1 and SENP1 like INa in iPS-CMs. Thus, NaV1.5 channels with SUMO1 in the recording pipette showed ILATE of ~4% of the peak current at both 21% and 1.5% O2 (Figure 3C; Table 1). Furthermore, the window-current voltages increased in 1.5% O2, or with 1 nM SUMO1, by −6 mV for V1/2-act and +6 mV for V1/2-SSI (Figure S3), as observed for INa in iPS-CMs. Also like INa in iPS-CMs, ILATE for NaV1.5 channels was below 0.5%, with 1 nM SENP1 in the pipette at both 21% and 1.5% O2, with no change in the window current, V1/2-act, or V1/2-SSI. Finally, the biophysical properties of NaV1.5-K442Q channels were insensitive to hypoxia and were not altered by SUMO1 or SENP1, showing less than 0.5% ILATE and without changes in V1/2-act and V1/2-SSI.
Single NaV1.5 Channels Are Reopened from the Inactive State by Hypoxia
Single NaV1.5 channels are closed at rest, open briefly upon depolarization, and then inactivate rapidly; the channels remain in the inactive state until the membrane returns to the resting potential and the channels move back to the closed state from which they are available to open once again in response to the next heartbeat. Thus, single NaV1.5 channels in cell-attached patches open just once in response to depolarization and almost always remain inactive (Figure 3D). As predicted by their behavior in whole-cell mode, hypoxia induced abnormal single-channel reopenings during sustained depolarization. Ensemble currents generated by the cumulative averaging of 300 repetitive cycles of depolarization demonstrated that the reopening of inactivated NaV1.5 channels in response to hypoxia was sufficient to increase ILATE from 0.4% ± 0.1% to 4.8% ±0.3%. In contrast, NaV1.5-K442Q channels opened only once and remained inactive when cells were studied in either ambient levels of O2 or at 1.5% O2 (Figure 3E).
Single NaV1.5 Channels Are Reopened from the Inactive State by SUMOylation
To confirm the mechanistic role of SUMO, single NaV1.5 channels were studied in inside-out membrane patches excised from CHO cells, allowing perfusion of the intracellular face of the channel with experimental solutions. In ambient O2, single NaV1.5 channels opened once and remained inactive for the duration of the depolarizing step, and in this configuration reducing O2 from 21% to 1.5% did not produce channel reopening (Figure S5). This suggested that hypoxia-induced reopening in whole-cell mode depended on the presence of cytoplasmic SUMO, a hypothesis confirmed when patches were perfused with 1 nM SUMO1, and reopening was observed at both 21% and 1.5% O2.
Like the changes observed in whole-cell mode with hypoxia, single channels in the patches reopened within 150 s of exposure to SUMO1, with 50% of the patches showing channel reopening at 65 ± 10 s (Figure S5). In agreement with the cell-attached recordings, ensemble currents generated from single NaV1.5 channels by averaging 500 cycles of depolarization in off-cell patches showed that SUMO1 increased ILATE from 0.4% ±0.2% to 4.1% ± 0.3% (Figure S5). As anticipated, single NaV1.5-K442Q channels in inside-out patches opened once and did not reopen even when exposed to SUMO1 or hypoxia (Figure S5). When considered in conjunction with the cell-attached studies, these findings support three conclusions: SUMOylation is necessary and sufficient for aberrant reopening of single NaV1.5 channels, K442 is required to mediate the effect of SUMO1 on NaV1.5, and hypoxia-evoked changes in ILATE are mediated by the SUMO pathway.
Hypoxia Induces Rapid monoSUMOylation of NaV1.5 Channels
To determine the stoichiometry of SUMOylation, we directly counted the number of SUMO1 monomers on individual NaV1.5 channels at the surface of live CHO cells by using TIRFM photobleaching, as before for NaV1.2 and IKs channels (Xiong et al., 2017; Plant et al., 2014, 2016). When mTFP-NaV1.5 and SUMO1 tagged with mCherry (m-SUMO1) were co-expressed, particles containing only mTFP-NaV1.5 and others with both mTFP-NaV1.5 and m-SUMO1 were observed at the cell membrane (Figures 4A and 4B; Table 2). Approximately 10% of mTFP-NaV1.5 channels were co-localized with one m-SUMO1 in ambient oxygen: continuous, two-color excitation bleached each fluorophore in the complexes in a single step consistent with one subunit of each type. On exposure to hypoxia, m-SUMO1 was recruited to the cell surface but only at sites with mTFP-NaV1.5, increasing the number of pixels that contained both mTFP-NaV1.5 and m-SUMO1 channels from 10% to 87% within ~100 s (Figure 4B). Increased co-localization did not alter the 1:1 stoichiometry of NaV1.5 channel and m-SUMO1 complexes, as judged by bleaching.
Figure 4. Hypoxia Recruits One SUMO1 Monomer to Each Cell Surface NaV1.5 Channel.
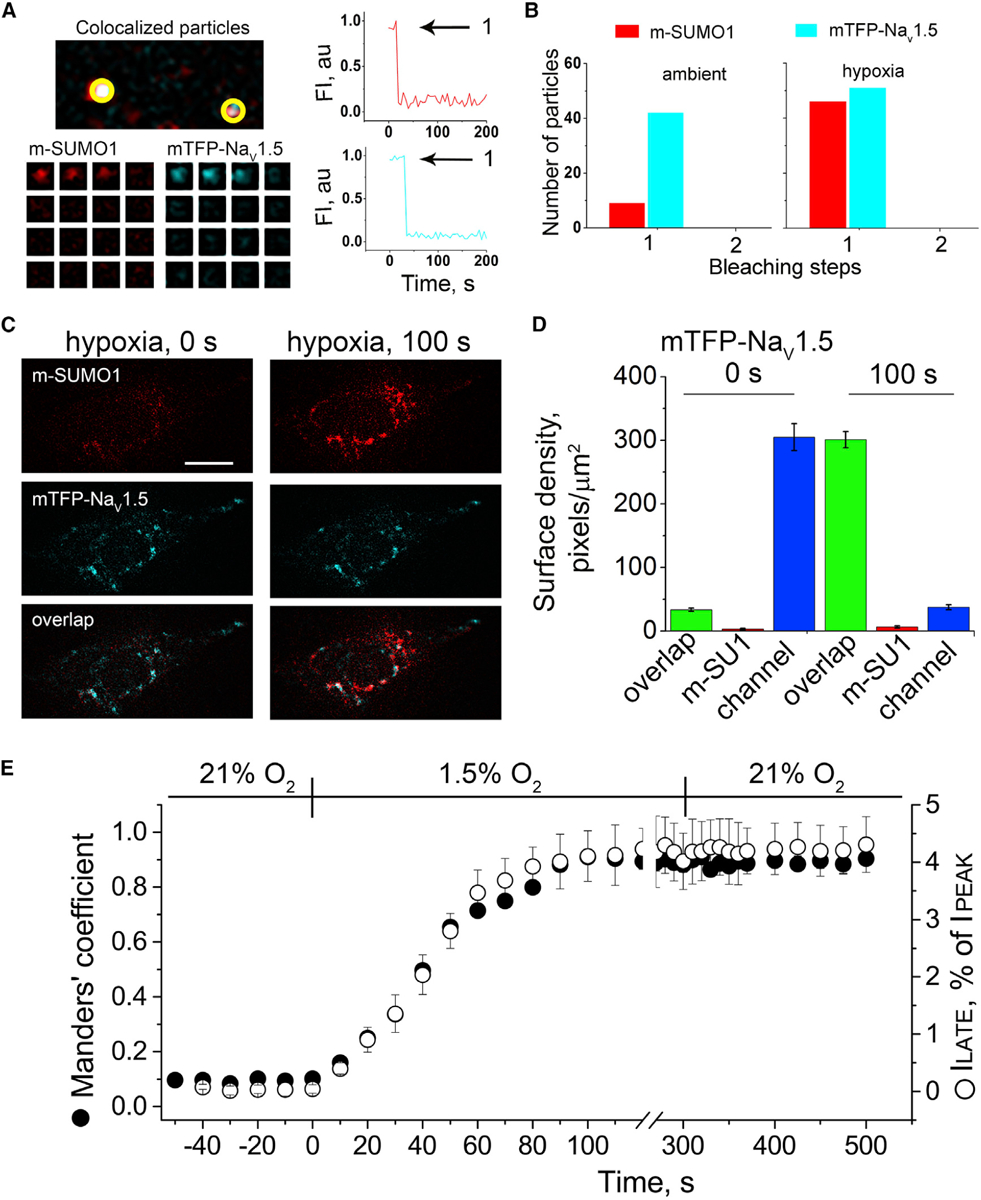
Single mTFP-NaV1.5 channels (blue) and SUMO1 tagged with mCherry (m-SUMO1, red) were studied in CHO-K1 cells by TIRFM. Stoichiometric (photobleaching) and pixel-by-pixel analysis for subunit density and co-localization were performed as described in the STAR Methods. Manders’ coefficients were assessed post hoc for 3–5 regions per cell, and co-localization was defined as the presence of both fluorophores at more than 30% of the maximum fluorescence level recorded in that stack (and their overlap is represented in the images as white pixels). The time course of hypoxic modulation of NaV1.5 ILATE was studied with steps from −100 mV to −30 mV every 10 s and normalized to the peak current. Data represent 5–8 cells, and biophysical parameters and single particle statistical analyses are summarized in Tables S1 and S3.
(A) Left: single co-localized particles with mCherry and TFP fluorescence were observed at the surface of cells expressing NaV1.5 and SUMO1. The time courses for simultaneous photobleaching of the fluorophores revealed that complexes have one subunit of each type.
(B) Histogram of photobleaching steps showing that hypoxia increased single m-SUMO1 (red) subunits at the cell surface co-localized with mTFP-NaV1.5 channels (blue), without a change in subunit stoichiometry.
(C) Left: the images show that in ambient O2, the surface density of m-SUMO1 (top) is low compared to NaV1.5 (middle) with little co-localization (bottom). Right: hypoxia recruits m-SUMO1 to the cell surface within ~100 s (top), where it is co-localized with NaV1.5 channels (bottom); surface levels of NaV1.5 were not observed to change (middle). The scale bar represents 10 μm.
(D) Histogram of surface density summarizing six cells studied as described in (A). In ambient O2, the density of pixels per μm2 with SUMO1 alone (red) was 3 ± 1, and 305 ± 21 for NaV1.5 (blue). The density of pixels with both fluorophores (green) was 33 ± 3 per μm2. Hypoxia increased co-localization to 301 ± 13 pixels per μm2 and decreased the density of free NaV1.5 channels (37 ± 4) without altering the density of free SUMO1 (6 ± 2).
(E) The time course for the hypoxia-induced increase in the co-localization of mTFP-NaV1.5 and m-SUMO1 (Manders’ coefficient, black circle) and the magnitude of ILATE, as a percentage of the peak current (open circle) were coincident. A mean Manders’ coefficient of 0.10 ± 0.01 measured in ambient O2 increased to 0.91 ± 0.03 in ~100 s of exposure to hypoxia. The mean ILATE rose from 0.46% ± 0.1% to 4.4% ± 0.8%. Increases in the Manders’ coefficient and late current versus peak current ratio were unchanged for at least 3 min after cells were restored to ambient O2.
Table 2.
Co-localization of SUMO1 with Nav1.5 in Response to Hypoxia
| Subunits Expressed | mTFP-NaV1.5 + m-SUMO1 | mTFP-NaV1.5-K442Q + m-SUMO1 | ||||||
|---|---|---|---|---|---|---|---|---|
| Condition | Ambient O2 | Hypoxia 50 s | Hypoxia 100 s | Recovery 150 s | Ambient O2 | Hypoxia 50 s | Hypoxia 100 s | Recovery 150 s |
| Single particle stoichiometry SUMO1: NaV1.5 | 1: 1 | 1: 1 | 1: 1 | 1: 1 | 0: 1 | 0: 1 | 0: 1 | 0: 1 |
| Free mTFP-NaV1.5, pixels/μm2 | 305 ± 21 | 166 ± 18* | 37 ± 4** | 39 ± 5** | 328 ± 26 | 333 ± 29 | 326 ± 25 | 308 ± 19 |
| Free m-SUMO1, pixels/μm2 | 3 ± 1 | 4 ± 2 | 7 ± 2 | 5 ± 2 | 3 ± 1 | 4 ± 1 | 4 ± 2 | 6 ± 2 |
| Co-localized pixels/μm2 | 33 ± 3 | 179 ± 15** | 301 ± 13** | 302 ± 15** | 4 ± 1 | 4 ± 2 | 3 ± 1 | 4 ± 1 |
NaV1.5 or NaV1.5-K442Q subunits tagged with mTFP1 were expressed in CHO-K1 cells with mCherry-SUMO1 (m-SUMO1) and studied by TIRFM and whole-cell patch clamp (Figures 3 and 5). The number of photobleaching steps observed for each fluorophore in each single fluorescent spot reports on the stoichiometry of the channel complex. NaV1.5 channels are monomers and show no more than one bleaching step when tagged with mTFP1 (Figure 5). No more than one bleaching steps was observed for mCherry-tagged SUMO1 subunits (free or co-localized with the channel). A 1:1 stoichiometry is maintained when cells are exposed to hypoxia (1.5% O2). SUMO1 was not observed to co-localize with NaV1.5-K442Q channels. The surface density of subunits was quantified as the mean of four 100- by 100-pixel regions for 6–10 cells per group. Exposure to hypoxia increased the number of SUMO1 monomers observed at the cell surface within 100 s, and almost all were co-localized with NaV1.5. Data are mean ± SEM for 5 to 8 cells per group;
p < 0.05,
p < 0.01 compared with cells studied in ambient O2 for each channel type.
Evidence that the co-localization represented a covalent interaction of the channel and SUMO1 was provided by two additional experiments (Figure S6). First, m-SUMO1 was not observed at the membrane in cells expressing mTFP-NaV1.5-K442Q and rarely yielded co-localization of the fluorophores in 21% or 1.5% O2. Second, hypoxia did not increase the number of particles at the membrane with m-SUMO195, the SUMO1 variant that is unable to covalently attach to target lysines. Because NaV1.5 channels contain just one α-subunit, the observed 1:1 stoichiometry with SUMO1 supports three additional conclusions: NaV1.5 subunits are SUMOylated only on K442, the channel is monoSUMOylated and polySUMO1 chains were not observed, and the NaVβ1 subunit is not SUMOylated.
Although hypoxia recruited m-SUMO1 to the cell surface, it did not change the number of NaV1.5 channels at the plasma membrane (Figures 4C and 4D; Table 2). This was shown by studying the surface density and localization of mTFP-NaV1.5 and m-SUMO1 subunits by using pixel-by-pixel analysis, a method we have applied before (Xiong et al., 2017; Plant et al., 2014, 2016). In ambient O2, about 10% of mTFP-NaV1.5 fluorescent pixels were localized with m-SUMO1 (33 ± 3/μm2) and 90% of the channels were unmodified (305 ± 21/μm2). After 100 s of hypoxia, the ratio was reversed; the number of mTFP-NaV1.5 pixels co-localized with m-SUMO1 increased to about 90% (301 ± 13/μm2), and 10% of the channels remained unmodified by m-SUMO1 (37 ± 4/μm2). Thus, hypoxia increased the surface density of m-SUMO1 ~33-fold (from 10/μm2 to 334/μm2), whereas the density of mTFP-NaV1.5 did not change (338/μm2).
Consistent with cause and effect, the appearance of m-SUMO1 at the plasma membrane with mTFP-NaV1.5, demonstrated above to require covalent modification of the channel on K442, matched the time course of hypoxia-induced increase in ILATE observed in iPS-CMs and NaV1.5 channels studied in CHO cells (Figures 1 and 3; Table 1). These rates also matched the increase in the mean Manders’ coefficient of co-localization of m-SUMO1 and mTFP-NaV1.5 (Figure 4E) as well as Pearson’s correlation coefficient (Table S2). Also, as observed for ILATE in iPS-CMs and NaV1.5 channels in CHO cells, the number of mTFP-NaV1.5 surface particles and the fraction of channels localized with m-SUMO1 were stable on restoration to ambient O2 for 3 min after hypoxia (Figure 4E; Table 2). Supporting the notion that heterologous expression of m-SUMO1 increased basal SUMOylation of mTFP-NaV1.5, about 10% of mTFP-NaV1.5 channels co-localized with m-SUMO1 in these cells and ILATE was 2.5-fold higher (~1% of peak current) than the control in ambient O2, rising to 4.5% with hypoxia (Tables 1 and S1).
Hypoxia-Induced ILATE in iPS-CMs Yields SUMO-Dependent APD Prolongation
To evaluate if the increase in ILATE we observed was sufficient to produce pro-arrhythmic APD prolongation, iPS-CMs were studied in current-clamp mode and the time to repolarize the membrane to resting values during spontaneous action potentials was measured. Under control conditions, the cells had a resting potential of −45 ± 3 mV, and the phase 1 action potential up-stroke mediated by INa peaked at 43 ± 4 mV (n = 6). Hypoxia increased the time for 50% restoration (APD50) by 22% without changing the resting potential or the action potential height (Figure 5A). To confirm that action potential prolongation was due to an increase in ILATE, we applied ranolazine, an open-state blocker of NaV1.5 channels in clinical use that preferentially inhibits ILATE rather than peak INa (Maier and Sossalla, 2013). Ranolazine reversed hypoxia-induced lengthening of APD50 to within 5% of control values without altering other action potential biophysical parameters (Table S3).
Figure 5. Hypoxia-Induced Increase in APD Mediated by SUMO Modulation of ILATE.
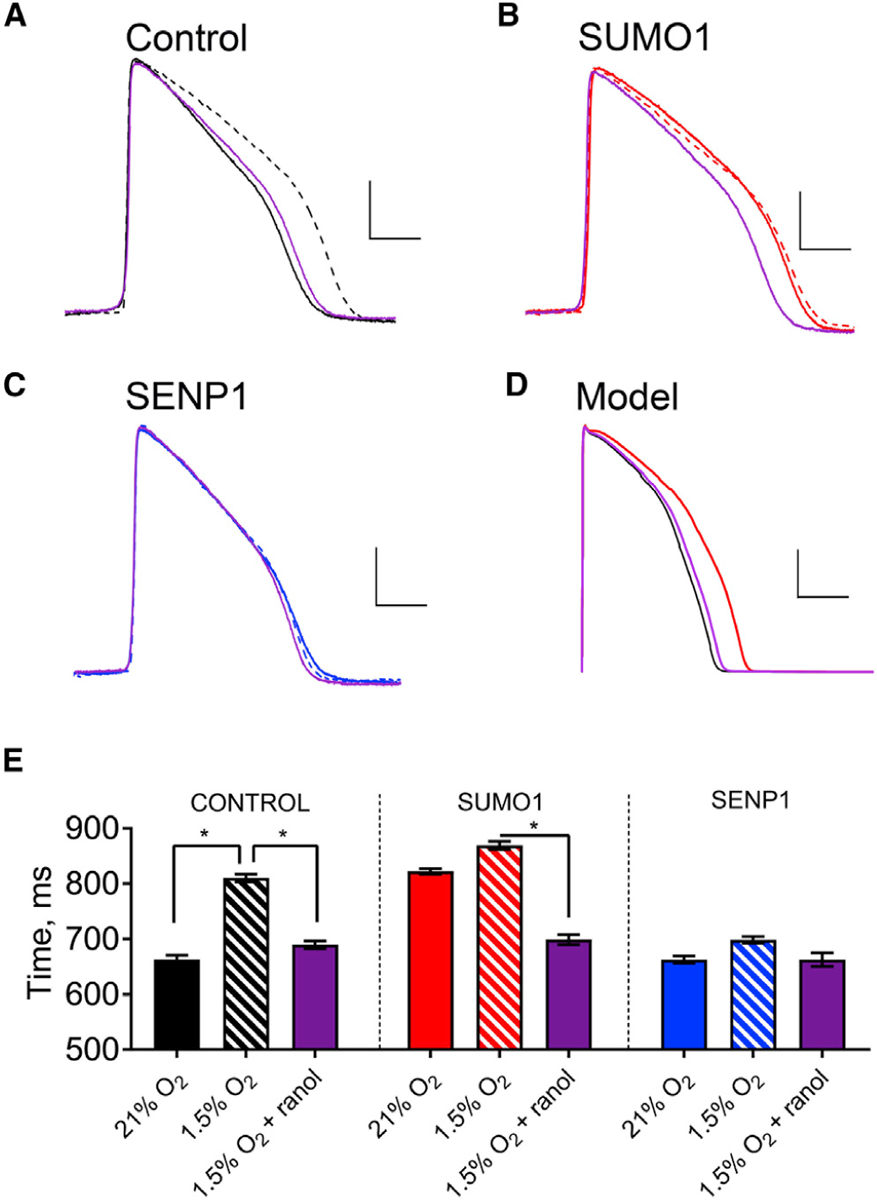
Spontaneous action potentials from human iPS-CMs were recorded in current-clamp mode with the same sequence of perfusates: normoxic (21% O2, solid line), hypoxic (1.5% O2, dashed line), and then hypoxia with 1 μM ranolazine (purple). Action potentials were studied with control intracellular solution (black) or with 1 nM SUMO1 (red) or SENP1 (blue) in the recording pipette. APD50 was determined by calculating the time required for the membrane potential to return to 50% of the resting value from the peak deflection of the action potential. Hypoxia was induced as described in the STAR Methods. Data are means ± SEM for 6–9 cells studied per group (*p < 0.01, two-way paired t test). The O’Hara-Rudy model for human action potentials was applied as described in the STAR Methods and Table S3. Scale bars represent 25 mV and 250 ms in (A)–(C) and 30 mV and 150 ms in panel (D).
(A) Examplar spontaneous action potentials recorded from iPS-CMs studied with control pipette solution under normoxic conditions and hypoxia with and without ranolazine. The APD50 increased with hypoxia by 22%, from 663 ± 2 ms to 810 ± 2 ms, and application of ranolazine restored the APD50 to 689 ± 2 ms.
(B) When SUMO1 was included in the pipette solution, APD50 was 822 ± 2 ms, and this increased to 870 ± 2 ms with hypoxia; ranolazine with hypoxia produced an APD50 of 699 ± 3 ms like that observed without SUMO1 in the pipette under normoxic conditions.
(C) APD50 was 663 ± 2 ms when cells were studied with SENP1 in the recording pipette, and the APD50 increased only to 698 ± 2 ms with hypoxia and was returned to 663 ± 4 ms with ranolazine.
(D) When a 5-fold increase in ILATE (half the maximal effect observed with hypoxia/SUMOylation), was simulated using the O’Hara-Rudy model, the APD50 increased by 27%, from 205 ms in normoxia to 260 ms.
(E) Histograms summarizing the mean APD50 of iPS-CMs under the conditions described in (A)–(C).
To verify that SUMOylation mediates hypoxia-induced APD prolongation, 1 nM SUMO1 or SENP1 was included in the recording pipette. As expected, SUMO1 lengthened APD50 to 123% of control values without causing a significant shift in the resting membrane potential or the height of spontaneous action potentials in 21% O2, and hypoxia evoked only a further 5% increase in the APD50 (Figure 5B). Subsequent application of ranolazine reversed the effects of SUMOylation as it had the effects of hypoxia with control solution in the pipette, reducing APD50 to within 5% of its control value. In contrast, 1 nM SENP1 in the recording pipette yielded APD50, resting membrane potential, and action potential height like control cells studied in 21% O2, and neither hypoxia nor ranolazine altered the three parameters of excitability (Figure 5C). These observations support the conclusion that hypoxia-induced changes in APD are primarily due to the SUMO-mediated increase in ILATE passed by NaV1.5 channels. Of note, baseline APD in iPS-CMs is longer than that in native cardiomyocytes, apparently due to a lower expression of K+ channels (Yang et al., 2014).
The O’Hara-Rudy model for human cardiac action potentials was used to demonstrate that the increase in ILATE due to SUMOylation of NaV1.5 channels observed in reconstituted CHO cells and iPS-CMs was sufficient to meet the theoretical requirements for pro-arrhythmic prolongation of APD. As anticipated, the time to repolarize the membrane from peak to rest by 50% (APD50) was prolonged by 27% when ILATE was increased in the model by 5-fold, half the observed maximal effect in the cells, from 205 ms under control conditions to 260 ms (Figure 5D); also as observed in iPS-CMs, there was no appreciable change in the resting membrane potential or the action potential peak current in the model (Table S3).
DISCUSSION
Myocardial hypoxia increases ILATE leading to increased APD, a common setting for dangerous arrhythmias. Here, we show that hypoxia induces SUMOylation of NaV1.5 channels on K442 and this is necessary and sufficient to increase ILATE and APD in human cardiomyocytes. Supporting a direct mechanism, SUMOylation of NaV1.5 produces ILATE in the absence of hypoxia in membrane patches excised from cells, basal levels of NaV1.5 SUMOylation are low under control conditions in iPS-CMs, and hypoxia leads to SUMOylation of the channels with the same time course as the increase in ILATE. Furthermore, SUMOylation produces changes in the gating of single NaV1.5 ion channels, macroscopic INa, and APDs like those described in human heart and in animal models with hypoxia (Chadda et al., 2017; Austen et al., 1963; Thung et al., 1962; Brown et al., 2014); and the effects are suppressed by the mutation of NaV1.5 K442, the application of SENP1 deSUMOylase, or the application of ranolazine, an inhibitor of ILATE.
Increased ILATE and AP prolongation predispose to arrhythmia because they increase the influx of Na+, raising the intracellular Na+ concentration and depolarizing the membrane potential. These changes impact the activity of other ion channels and transporters, including the Na/K-ATPase, leading to Ca2+ loading by L-type Ca2+ channel reactivation and reverse operation of the sodium-calcium exchanger (NCX). These excitatory effects increase the likelihood of early after-depolarizations (EADs), delayed after-depolarizations (DADs), triggered activity, and spatiotemporal changes in repolarization that promote reentrant arrhythmias, including ventricular TdP (Gaur et al., 2009; Shryock et al., 2013; Chadda et al., 2017). Medications like ranolazine that suppress ILATE minimize disruption of Ca2+ homeostasis, reducing EADs and DADs (Maier and Sossalla, 2013). The hypoxic increase in ILATE we observe with iPS-CMs modeled in the O’Hara-Rudy algorithm are found sufficient to underlie the increase in APD that induces these downstream effects (Figure 5). In addition to hypoxia and ischemia (Saint, 2006; Hammarström and Gage, 2002), cardiac ILATE is reported to increase with oxidative stress (Ward and Giles, 1997), acidosis (Plant et al., 2006), heart failure (Valdivia et al., 2005; Undrovinas et al., 2006), and inflammation (Ward et al., 2006), suggesting a role for SUMOylation in the transduction of these stimuli that merits study.
Rapid SUMOylation of NaV1.5 channels on the cardiac plasma membrane in response to hypoxia is reminiscent of our finding that hypoxia mediates SUMOylation of NaV1.2 channels on the surface of central neurons in under 60 s on K38, leading to an increase peak current (without changes in ILATE) due to changes in the voltage dependence of activation, consistent with early, rapid Na+ flux in cerebral ischemia, a prelude to downstream pathology (Plant et al., 2016). In both cases, rapid recruitment of SUMO in the first few minutes of hypoxia initiates deleterious sequelae. In contrast, chronic hypoxia has been associated with cardioprotection by increased SUMOylation of the ATPase SERCA2A in heart failure and ischemia-reperfusion injury (Du et al., 2018) and with neuroprotection by a global increase in SUMOylation, changes in gene expression, and remodeling of protein expression in ground squirrel hibernation torpor (Kho et al., 2011, 2015; Lee et al., 2007) and chronic oxygen-glucose deprivation (Lee et al., 2009).
NaV1.5 is recognized as the preeminent sodium channel in the heart, although other voltage-gated sodium channels are expressed at lower levels and in specific cardiac regions (Maier et al., 2004), notably purkinje fibers (Haufe et al., 2005). Although non-NaV1.5 channels (Maier et al., 2004) have been described as contributing only 5%–10% of peak current, a role for hypoxia-induced ILATE is possible because non-NaV1.5 currents have been implicated in the late currents measured with ambient oxygen in isolated canine and murine myocytes (Biet et al., 2012; Yang et al., 2012). We do not infer roles for NaV1.2, NaV1.1, or NaV1.6 in SUMO-mediated hypoxic late current because we have demonstrated that the stimuli alter peak current and do not induce ILATE with NaV1.2 in rat CGN and CHO cells and that NaV1.1 and NaV1.6 channels are insensitive to SUMOylation in rat hippocampal neurons and rat CGN, respectively (Plant et al., 2016). We cannot rule out contributions from NaV1.3, NaV1.4, NaV1.7, NaV1.8, or NaV1.9.
At the single-channel level, the abnormal increases in ILATE we observe with hypoxia are like those recorded with mutations in NaV1.5 that cause LQT3 that destabilize internal pore occlusion by the IFM inactivation “lid” that resides between channel domains III and IV (Bennett et al., 1995; West et al., 1992). Rather than a single opening followed by inactivation, the LQT3 channels re-open with maintained depolarization, suggesting a lowered energetic barrier for return to the open state. Consistent with the idea that SUMOylated NaV1.5 channels also experience potentials where the energetic barriers to moving between the open and inactive states are lowered, we observe hypoxia to increase the window between activation and steady-state inactivation voltage curves (Figure S3). Furthermore, our failure to observe long first openings suggests that the barrier to inactivation is not increased by hypoxia. Of note, destabilization of the inactivated state can rationalize ILATE in association with channels that recover from inactivation too rapidly during repolarization (Belardinelli et al., 2006, 2015; Shryock et al., 2013). It is feasible that hypoxia-induced SUMOylation of NaV1.5 on K442, a site on the linker between domains I and II, may interfere with the stability of the interaction of the IFM lid and its pore receptor.
Here, we apply SUMO1101 to the inner surface of excised membrane patches to SUMOylate NaV1.5 channels, as we observed before for K2P1, KV2.1, and NaV1.2 channels; this indicates that enzymes stably associated with the cytoplasmic face of the plasma membrane mature SUMO1 to SUMO97 and mediate coupling to target lysine (Xiong et al., 2017; Plant et al., 2010, 2011, 2012, 2016). In contrast, when SUMOylation of NaV1.5 channels is induced by hypoxia, the membrane must be in continuity with the cytosol, indicating cellular components have a role in transducing the signal that oxygen tension is low to the SUMO pathway. Thus far, exogenous SENP1 must be applied to deSUMOylate the ion channels in off-cell mode in all cases we have studied.
It is notable that changes in Ca2+ homeostasis are not required for hypoxia-induced SUMOylation of NaV1.5 in whole cells as the reaction proceeds despite 10 mM EGTA in the recording pipette to suppress changes in cellular Ca2+ and 200 μM CdCl2 in the extracellular bath solution to suppress conductance through voltage-gated calcium channels (Figure 1A). The similar attributes of SUMOylated NaV1.5 currents in intact cells and single channels in excised patches also support the conclusion that changes in Ca2+ at the cytoplasmic face of the channel are not necessary for SUMO-induced increases in ILATE (Figure S5). Those points do not rule out a role for variation in cytoplasmic Ca2+ when the ionic concentration is not clamped by chelation and blockade.
Ion channel SUMOylation appears to enhance excitability by decreasing potassium currents (K2P1, KV2.1, and Kv7.1/KCNE1) (Rajan et al., 2005; Plant et al., 2010, 2012) or increasing currents passed by NaV1.2 (Plant et al., 2016) and NaV1.5 (this work). Our prior study showing that acute hypoxia leads to the rapid SUMOylation of NaV1.2 in CGN and influx of Na+ flux due to altered voltage-dependent gating (but not a change in ILATE) (Plant et al., 2016) and this study of NaV1.5 suggest that hypoxia-mediated SUMOylation is an emerging common mechanism shared by some, but not all, ion channels.
We observed previously that acidosis increases ILATE in NaV1.5 channels bearing a common polymorphism associated with SIDS and arrhythmia in African American adults, presenting a model for the genetic predisposition to dysrhythmia in the face of an environmental challenge (Plant et al., 2006). Inherited ion channel mutations that predispose to arrhythmia by increasing APD also yield life-threatening events in response to specific stimuli, for example, concurrent drug blockade (Sesti and Goldstein, 1998). Here, we present the mechanism for the sensitivity of wild-type NaV1.5 channels to the environmental challenge of hypoxia. Identifying the role of the SUMO pathway in the increase in ILATE in response to hypoxia supports targeting the pathway for therapeutic intervention.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further Information and requests for resources and reagents should be directed to the Lead Contact, S.A.N. Goldstein (sgoldst2@uci.edu). Materials generated through this work are available from the Lead Contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
CHO-K1 cells (RRID: CVCL_0214) were obtained from ATCC and used for heterologous expression, as detailed below. Human induced pluripotent stem cell cardiomyocytes2 were purchased from Cellular Dynamics and cultured on #1.5 glass coverslips coated with gelatin (0.1% w/v in water) according to the manufacturer’s instructions. Cells were maintained in iCell maintenance medium (Cellular Dynamics) and incubated at 37°C in a humidified atmosphere containing 5% CO2 / 95% air (21% O2). The iPS cells were studied between days 5 – 10 in culture at which time both isolated cells and cells in syncytia exhibited rhythmic beating behavior, a criterium for mature myocyte attributes shown to correspond with expression of troponin-T and sarcomeric α-actinin (Ma et al., 2011). To further verify that the currents under study in iPS cells were mediated by NaV1.5 channels, we employed protocols and solutions to minimize conductance through potassium and calcium channels (per Figures 1, S1, and 5); showed that the currents had biophysical hallmarks like those of human NaV1.5 in tissue culture cells (per Table 1 and Figures 3 and S3); and demonstrated blockade with ranolazine, a drug that inhibits ILATE mediated by NaV1.5 (per Table S3 and Figure 5).
METHOD DETAILS
Molecular Biology and Reagents
Human NaV1.5 (NM_198056.1) was handled in pcDNA1, as previously described (Plant et al., 2006) and was transiently cotransfected with GFP-tagged SCN1B (the β subunit of cardiac SCN5A channel, isoform b; GenBank: NM_001037). Human SUMO1101 (GenBank: NM_003352.8) and Ubc9 (GenBank: NM_003345.5) were amplified from a brain cDNA library (Clontech) and inserted into pMAX, as described before (Rajan et al., 2005). Sequences encoding mCherry or mTFP1 were inserted as described (Plant et al., 2010) at the N terminus of SUMO1 or NaV1.5 respectively. SUMO1 and Ubc9 were tagged with eYFP using the same strategy. Mutations were introduced with Pfu Quikchange PCR (Agilent). Purified SUMO197, SUMO293, SENP1 and SENP2 were purchased from Boston Biochemical.
Heterologous Expression
CHO-K1 cells (RRID: CVCL_0214) were maintained in F12K medium supplemented with 10% FBS. Plasmids were transfected into cells with Lipofectamine 2000 according to the manufacturer’s instructions (Life Technologies). Experiments were performed 24 to 48 hours post transfection at room temperature. The human β1 accessory subunit was co-expressed with NaV1.5 subunits unless otherwise indicated.
Electrophysiology
INa and action potentials in iPS-CMs, and whole-cell NaV1.5 channel currents in heterologous cells were recorded using an Axopatch 200B amplifier and pCLAMP software (Molecular Devices) at filter and sampling frequencies of 10 and 50 kHz respectively. For voltage-clamp studies, cells were superfused with a solution comprising, in mM: NaCl 130, CsCl 4, CaCl2 2, MgCl2 1.2, glucose 5.5, HEPES 10 and, 200 μM CdCl2, a concentration previously shown to block >98% of voltage-gated calcium channel current in primary cells (Pearson et al., 1993). The pH was adjusted to 7.4 with NaOH and HCl. Cells were studied at room temperature with borosilicate glass pipettes (Clark Kent) with a resistance of 2–3 MΩ when filled with a solution comprising, in mM: CsCl 60, CsF 80, CaCl2 1, MgCl2 1, Na2ATP 5, EGTA 10, HEPES 10, pH 7.4 with CsOH. Pipettes were coated with Sylgard (Dow Corning) prior to use. Capacitance artifacts were subtracted online, series resistance was compensated to 70% and cells with a series resistance of less than 10 MΩ were studied. Once whole-cell mode was established, cells were not studied for more than 600 s in order to maintain consistent membrane seals and voltage-clamp control of INa. Current-voltage relationships were evoked from a holding potential of −100 mV by 100 ms test pulses between −100 and 0 mV, in 10 mV increments. Steady-state inactivation was studied by holding cells at −140 mV and then comparing currents evoked by 50 ms test pulses between −140 mV and −20 mV to those evoked by a 100 ms prepulse to 0 ms. A 10 s interpulse interval was used in both cases. Normalized peak current values are plotted against prepulse potential (mV). A Boltzmann function, I = Imax/(1 + exp[V – V1/2/k]), where Imax is the maximum current and k is slope factor, was used to fit normalized activation-voltage relationships. Recovery from fast inactivation was studied by holding cells at −100 mV and comparing currents evoked by a pair of 50 ms test step to −30 mV separated by an interpulse interval that increased in duration by 5 ms increments per sweep. The time constant for recovery from inactivation (τ) was obtained from mono-exponential fits of the normalized current amplitude to the recovery time using It = Imax +A[e−t/τ], where A is the amplitude of components and t is time. Whole-cell currents were normalized to cell capacitance. Mean ± SEM capacitance values were 28 ± 5 pF for iPS cells and 10 ± 2 pF for CHO cells.
For current-clamp studies, iPS-CMs were superfused with a solution comprising, in mM: NaCl 135, KCl 3.5, CaCl2 2, MgCl2 1.2, glucose 8, HEPES 10 and the pH was adjusted to 7.4. The intracellular solution contained, in mM: KCl 140, MgCl2 2, Na2ATP 5, EGTA 1, HEPES 5 and the pH was adjusted to 7.4.
Acute Hypoxia for Electrophysiology
Acute hypoxia was achieved as described previously (Plant et al., 2002). Briefly, the cells were made hypoxic (1.5% O2) by switching the perfusate with one that had been bubbled with nitrogen for at least 30 minutes prior to perfusion. Oxygen tension was measured at the cell by a polarized carbon fiber electrode; solution exchange occurred in less than 10 s. Ambient O2, or normoxia recording conditions, were achieved by allowing recording solutions to equilibrate with room air for at least 30 minutes prior to perfusion. The ambient O2 solution was measured to be 21% O2 at the cell. All solutions were equilibrated, measured, and perfused at room temperature (18°C).
Two-Color TIRFM
Single protein complexes at the surface of live CHO-K1 cells were identified using TIRFM as previously described (Plant et al., 2014). Cells were studied in a solution comprising (in mM): NaCl 130, KCl 4, MgCl2 1.2, CaCl2 2, HEPES 10, pH was adjusted to 7.4 with NaOH. The critical angle for TIRF was adjusted using a CellTIRF illuminator (Olympus) and a high numerical aperture apochromat objective (150x, 1.45 NA) mounted on an automated fluorescence microscope (IX81) controlled by Metamorph software (Molecular Devices). For simultaneous illumination of two fluorophores, CellTIRF software (Olympus) was used to adjust the critical angle for each excitation wavelength to generate evanescent waves of equivalent depth (100 nm). mCherry was excited by a 561-nm laser line and mTFP1 was excited a 445-nm laser line. When mTFP1 was studied with mCherry the emitted light signals were split using a 520-nm dichroic mirror mounted in a DualView adaptor (Photometrics) and each wavelength was directed to one half of an EM-CCD. The dichroic mirror was disengaged when single fluorophores were studied.
To assess stoichiometry, fluorophores were photobleached by continual excitation and data were captured as movies of 100–400 frames acquired at 1 Hz using an EM-CCD camera (Hamamatsu). When mTFP1 was studied with mCherry in the same cell, the data for each fluorophore were saved as separate stacks and processed in an identical manner. Images were background corrected by subtracting the mean of 5 fully bleached frames from the end of each stack analyzed. Misalignment of the data between stacks was corrected in ImageJ using StackReg. Fluorescent spots were defined as a discrete 3 × 3-pixel region around a pixel of maximum intensity, as before (Plant et al., 2010, 2014). Fluorescence is reported as the change in fluorescence intensity normalized by the initial fluorescence for each trace. The density of co-localized and single fluor-fluorescent particles was determined following thresholding and watershed separation in ImageJ. The particle number was counted in 4, separate 100 × 100-pixel regions of interest for 6–10 cells per group using the Analyze particles plugin.
Manders’ coefficient of colocalization was assessed from live-cell simultaneous two-color TIRF images captured at 5 s intervals to minimize photobleaching. Data stacks were background subtracted and aligned prepared for each fluorophore post hoc, as above. Co-localization of partner pixels from the two stacks of images was defined as the presence of both fluorophores with at least 30% of maximum fluorescence levels recorded in that region of interest. Mean Manders’ coefficients were calculated for 3–5 separate 100 × 100-pixel regions of interest per cell.
Live Cell FRET Microscopy
Donor-decay time-course was studied as before (Plant et al., 2010), using an Olympus inverted epi-fluorescence microscope. Cells were studied in a solution comprising (in mM): NaCl 130, KCl 4, MgCl2 1.2, CaCl2 2, HEPES 10, pH was adjusted to 7.4 with NaOH. mTFP1 was excited at 445 nm and the emission collected through a 470–500 nm bandpass filter, YFP was excited at 514 nm and the emission collected through a 525–575 nm filter. Images were captured using a scientific camera controlled by μmanager or Metamorph software (Molecular Devices) and were analyzed with ImageJ.
Mass Spectrometry
Human NaV1.5 residues 353–502 were cloned into pET28a vector with six-His residues and a tobacco etch virus (TEV) cleavage site replacing the thrombin site and expressed in BL21(DE3) E. Coli with a vector carrying mouse E1 (as a linear fusion product of Aos1 and Uba2), E2 (Ubc-9) and SUMO11–97 with a T95K mutation so that trypsin digestion before MS leaves a Gly-Gly tag on the ε-amino group of target lysines as before (Plant et al., 2010). Protein was purified with Ni-NTA affinity columns. The eluate was reduced with TCEP and alkylated using iodoacetamide prior to tryptic digestion via filter-assisted digestion. LC MS/MS analysis was performed using a Thermo Scientific Dionex UltiMate 3000 system coupled in-line to an Orbitrap Fusion Lumos MS. A 25 cm × 75 μm PepMap EASY-Spray Acclaim PepMap C18 Column was used to separate peptides over acetonitrile gradients of 4% to 28% at a flow rate of 300 nL/min. Each duty cycle consists of one MS scan in FT mode (350–1500 m/z, resolution of 120,000 at m/z 400) followed by data-dependent tandem mass scans in IT mode for 3 s at top speed, utilizing HCD with NCE 30% on the most intense ions with charge states +2 or higher. The RAW data file acquired was extracted to MGF format using MSConvert (http://proteowizard.sourceforge.net/tools.shtml) and subjected to database searching using Batch-Tag within a developmental version of Protein Prospector (v. 6.1.0) against a decoy database consisting of a normal Homo sapiens Swissprot database concatenated with a randomized version (SwissProt.2019.4.8.random.concat, total 20,418 protein entries). The mass accuracy for parent ions and fragment ions were set as ± 10 ppm and 0.2 Da, respectively. Trypsin was set as the enzyme and maximum of two missed cleavages were allowed. Protein N-terminal acetylation, methionine oxidation, Gly-Gly, and N-terminal conversion of glutamine to pyroglutamic acid were selected as variable modifications. Minimum protein and peptide scores were set as 22.0 and 5.0 respectively, while maximum E values for proteins and peptides were set to 0.01 and 0.1. A list of identified precursor ions corresponding to peptides carrying GlyGly modifications were generated for targeted sequencing for further validation. All MS/MS spectra for potential SUMOylated peptides were further inspected manually and evaluated based on spectral quality.
Simulations
Action potentials were simulated using the O’Hara Rudy (ORD) model (O’Hara et al., 2011) under normal condition or by scaling the conductance of ILate. To simulate the effect of SUMO (or hypoxia-induced SUMOylation) a scaling factor named “SUMO” that varies from 1 (No SUMO) to 10 (maximum effect at 1 nM SUMO1) was added to the model to increase the conductance of Ilate:GNalate SUMO = GNaLate * SUMO, were GNaLate is the nominal conductance value from the O’Hara-Rudy ventricular myocyte model and GNalate SUMO is the conductance used for simulation in the modified ORD. Other variables and all equations of the model were otherwise as described in the published model, except that the external ionic concentrations were set to match the experimental conditions used here to study the iPS-CMs (in mM): [Na+]o = 135; [Ca2+]o = 2.0; and [K+]o = 3.5. A pacing cycle length of 800 ms was used and the numbers of beats was set to 1000. The duration of the APD50 was measured from the time of maximum dV/dt to the time that membrane voltage reached 50% of complete repolarization; resting potential was measured immediately before each beat.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using pClamp, Origin, GraphPad and Excel software. Quantification and analysis specific in each experimental technique is described in the Method Details section, above. Data were assessed for statistical differences between groups by one-way analysis of variance with Bonferroni post hoc analysis to test differences within pairs of group means for all dataset with an F-value of p < 0.05. Data are presented, where indicated as the mean ± standard error of the mean (SEM). The number of replicates for each study are described in the legends.
DATA AND CODE AVAILABILITY
The study did not generate any unique datasets or codes.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| BL21(DE3) E. Coli | Thermo Fisher | Cat#: EC0114 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human: SUMO | Boston Biochem | Cat #: UL-712 |
| Human: His6-SENP1 catalytic domain | Boston Biochem | Cat#: E-700 |
| Ranolazine dihydrochloride | Sigma-Aldrich | Cat#: R6152 |
| Experimental Models: Cell Lines | ||
| Hamster: CHO-K1 cells | ATCC | Cat#: CCL-61; RRID: CVCL_0214 |
| Human: iPS-ventricular cardiomyocytes-2 | Cellular Dynamics | Cat# CMC-100-012-000.5 |
| Recombinant DNA | ||
| hNaV1.5-pcDNA1 | Plant et al., 2006 | DOI: 10.1172/JCI25618 |
| GFP-SCN1B-pcDNA1 | Plant et al., 2006 | DOI: 10.1172/JCI25618 |
| mCherry-SUMO1-pMAX | Plant et al., 2016 | https://doi.org/10.7554/eLife.20054 |
| mTFP1-hNav1.5-pMAX | This paper | N/A |
| eYFP-SUMO1-pMAX | Plant et al., 2010 | https://doi.org/10.1073/pnas.1004712107 |
| eYFP-Ubc9-pMAX | Plant et al., 2010 | https://doi.org/10.1073/pnas.1004712107 |
| Plasmid for MS with SUMO1(T95K)1–97 | Plant et al., 2010 | https://doi.org/10.1073/pnas.1004712107 |
| Plasmid for MS with Human NaV1.5 residues 353–502 | Plant et al., 2010, modified as described | https://doi.org/10.1073/pnas.1004712107 |
| Software and Algorithms | ||
| pClamp version 10 | Molecular Devices | N/A |
| Origin version 6 | Microcal | N/A |
| Prism version 8 | GraphPad | N/A |
| ImageJ-FIJI | Schindelin et al., 2012; https://imagej.net/Fiji | https://doi.org/10.1038/nmeth.2019 |
| MSConvert | http://proteowizard.sourceforge.net/tools.shtml | |
| Batch-Tag in Protein Prospector v. 6.1.0 | http://prospector.ucsf.edu/ | |
| O’Hara Rudy Model (ORD) | O’Hara et al., 2011 | https://doi.org/10.1371/journal.pcbi.1002061 |
Highlights.
Acute cardiac hypoxia elevates late sodium current (ILATE) to pro-arrhythmic levels
Increased ILATE is due to rapid SUMOylation of NaV1.5 channels at the cell membrane
SUMOylation of lysine442 reopens NaV1.5 channels when they are normally inactive
Blocking SUMOylation prevents increased ILATE and action potential prolongation
ACKNOWLEDGMENTS
The authors thank Anthony F. Arena, Jordie M. Kamuene, and Meghan Masotti for technical support. We are grateful to the late R.Y. Tsien (UCSD) for the mCherry clone and A. Periasamy (University of Virginia) for mTFP1. The work was funded by National Institutes of Health grants R01HL10549 (to S.A.N.G.) and R01HL144615 (to L.D.P.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.025.
DECLARATION OF INTERESTS
The authors report no competing financial interests.
REFERENCES
- Austen WG, Ebert PA, and Greenfield LJ (1963). Mechanism of cardiac arrest in acute hypoxia. Surgery 53, 784–791. [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC, and Fraser H (2006). Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92, iv6–iv14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli L, Giles WR, Rajamani S, Karagueuzian HS, and Shryock JC (2015). Cardiac late Na+ current: proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm 12, 440–448. [DOI] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, and George AL Jr. (1995). Molecular mechanism for an inherited cardiac arrhythmia. Nature 376, 683–685. [DOI] [PubMed] [Google Scholar]
- Biet M, Barajas-Martínez H, Ton AT, Delabre JF, Morin N, and Dumaine R (2012). About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na(+) channels. J. Mol. Cell. Cardiol 53, 593–598. [DOI] [PubMed] [Google Scholar]
- Brown SJ, Barnes MJ, and Mündel T (2014). Effects of hypoxia and hypercapnia on human HRV and respiratory sinus arrhythmia. Acta Physiol. Hung 101, 263–272. [DOI] [PubMed] [Google Scholar]
- Carmeliet E (1999). Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol. Rev 79, 917–1017. [DOI] [PubMed] [Google Scholar]
- Catterall WA (1986). Molecular properties of voltage-sensitive sodium channels. Annu. Rev. Biochem 55, 953–985. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2012). Voltage-gated sodium channels at 60: structure, function and pathophysiology. J. Physiol 590, 2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2017). Forty Years of Sodium Channels: Structure, Function, Pharmacology, and Epilepsy. Neurochem. Res 42, 2495–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestèle S, Qu Y, Rogers JC, Rochat H, Scheuer T, and Catterall WA (1998). Voltage sensor-trapping: enhanced activation of sodium channels by beta-scorpion toxin bound to the S3–S4 loop in domain II. Neuron 21, 919–931. [DOI] [PubMed] [Google Scholar]
- Cestèle S, Scheuer T, Mantegazza M, Rochat H, and Catterall WA (2001). Neutralization of gating charges in domain II of the sodium channel alpha subunit enhances voltage-sensor trapping by a beta-scorpion toxin. J. Gen. Physiol 118, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha A, Ruben PC, George AL Jr., Fujimoto E, and Bezanilla F (1999). Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron 22, 73–87. [DOI] [PubMed] [Google Scholar]
- Chadda KR, Jeevaratnam K, Lei M, and Huang CL (2017). Sodium channel biophysics, late sodium current and genetic arrhythmic syndromes. Pflugers Arch 469, 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Liu P, Xu T, Pan D, Zhu H, Zhai N, Zhang Y, and Li D (2018). Luteolin Modulates SERCA2a Leading to Attenuation of Myocardial Ischemia/ Reperfusion Injury via Sumoylation at Lysine 585 in Mice. Cell. Physiol. Biochem 45, 883–898. [DOI] [PubMed] [Google Scholar]
- Gaur N, Rudy Y, and Hool L (2009). Contributions of ion channel currents to ventricular action potential changes and induction of early afterdepolarizations during acute hypoxia. Circ. Res 105, 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström AK, and Gage PW (2002). Hypoxia and persistent sodium current. Eur. Biophys. J 31, 323–330. [DOI] [PubMed] [Google Scholar]
- Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S, Benndorf K, and Dumaine R (2005). Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc. Res 65, 117–127. [DOI] [PubMed] [Google Scholar]
- Henley JM, Craig TJ, and Wilkinson KA (2014). Neuronal SUMOylation: mechanisms, physiology, and roles in neuronal dysfunction. Physiol. Rev 94, 1249–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YK, Saint DA, and Gage PW (1996). Hypoxia increases persistent sodium current in rat ventricular myocytes. J. Physiol 497, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, and Hajjar RJ (2011). SUMO1-dependent modulation of SERCA2a in heart failure. Nature 477, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho C, Lee A, Jeong D, Oh JG, Gorski PA, Fish K, Sanchez R, De-Vita RJ, Christensen G, Dahl R, and Hajjar RJ (2015). Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun 6, 7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Miyake S, Wakita H, McMullen DC, Azuma Y, Auh S, and Hallenbeck JM (2007). Protein SUMOylation is massively increased in hibernation torpor and is critical for the cytoprotection provided by ischemic preconditioning and hypothermia in SHSY5Y cells. J. Cereb. Blood Flow Metab 27, 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Castri P, Bembry J, Maric D, Auh S, and Hallenbeck JM (2009). SUMOylation participates in induction of ischemic tolerance. J. Neurochem 109, 257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ, and January CT (2011). High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol 301, H2006–H2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier LS, and Sossalla S (2013). The late Na current as a therapeutic target: where are we? J. Mol. Cell. Cardiol 61, 44–50. [DOI] [PubMed] [Google Scholar]
- Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T, and Catterall WA (2004). Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation 109, 1421–1427. [DOI] [PubMed] [Google Scholar]
- Makielski JC (2016). Late sodium current: A mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc. Med 26, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbán E (2002). Cardiac channelopathies. Nature 415, 213–218. [DOI] [PubMed] [Google Scholar]
- O’Hara T, Virág L, Varró A, and Rudy Y (2011). Simulation of the undis-eased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput. Biol 7, e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson HA, Sutton KG, Scott RH, and Dolphin AC (1993). Ca2+ currents in cerebellar granule neurones: role of internal Mg2+ in altering characteristics and antagonist effects. Neuropharmacology 32, 1171–1183. [DOI] [PubMed] [Google Scholar]
- Plant LD, Kemp PJ, Peers C, Henderson Z, and Pearson HA (2002). Hypoxic depolarization of cerebellar granule neurons by specific inhibition of TASK-1. Stroke 33, 2324–2328. [DOI] [PubMed] [Google Scholar]
- Plant LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, Chen W, Kittles RA, and Goldstein SA (2006). A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J. Clin. Invest 116, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Dementieva IS, Kollewe A, Olikara S, Marks JD, and Goldstein SA (2010). One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc. Natl. Acad. Sci. USA 107, 10743–10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Dowdell EJ, Dementieva IS, Marks JD, and Goldstein SA (2011). SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J. Gen. Physiol 137, 441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Zuniga L, Araki D, Marks JD, and Goldstein SA (2012). SUMOylation silences heterodimeric TASK potassium channels containing K2P1 subunits in cerebellar granule neurons. Sci. Signal 5, ra84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Xiong D, Dai H, and Goldstein SA (2014). Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proc. Natl. Acad. Sci. USA 111, E1438–E1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Marks JD, and Goldstein SA (2016). SUMOylation of NaV1.2 channels mediates the early response to acute hypoxia in central neurons. eLife 5, e20054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S, Plant LD, Rabin ML, Butler MH, and Goldstein SA (2005). Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 121, 37–47. [DOI] [PubMed] [Google Scholar]
- Saint DA (2006). The role of the persistent Na(+) current during cardiac ischemia and hypoxia. J. Cardiovasc. Electrophysiol 17, S96–S103. [DOI] [PubMed] [Google Scholar]
- Saint KM, Abraham S, MacLeod BA, McGough J, Yoshida N, and Walker MJ (1992). Ischemic but not reperfusion arrhythmias depend upon serum potassium concentration. J. Mol. Cell. Cardiol 24, 701–709. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Argana-Carreras I, Frise E, Kaynig V, Longair M, Pietzsc T, Preibish S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti F, and Goldstein SA (1998). Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol 112, 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shryock JC, Song Y, Rajamani S, Antzelevitch C, and Belardinelli L (2013). The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc. Res 99, 600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Wagner S, Maier LS, and Belardinelli L (2006). Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J. Pharmacol. Exp. Ther 318, 214–222. [DOI] [PubMed] [Google Scholar]
- Thung N, Dammann JF Jr., Diaz-Perez R, Thompson WM Jr., San-marco M, and Mehegan C (1962). Hypoxia as the cause of hemorrhage into the cardiac conduction system, arrhythmia, and sudden death. J. Thorac. Cardiovasc. Surg 44, 687–695. [PubMed] [Google Scholar]
- Undrovinas AI, Belardinelli L, Undrovinas NA, and Sabbah HN (2006). Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J. Cardiovasc. Electrophysiol 17, S169–S177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, and Makielski JC (2005). Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol 38, 475–483. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, and Catterall WA (1988). Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 241, 1658–1661. [DOI] [PubMed] [Google Scholar]
- Ward CA, and Giles WR (1997). Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J. Physiol 500, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CA, Bazzazi H, Clark RB, Nygren A, and Giles WR (2006). Actions of emigrated neutrophils on Na(+) and K(+) currents in rat ventricular myocytes. Prog. Biophys. Mol. Biol 90, 249–269. [DOI] [PubMed] [Google Scholar]
- Wei X, Zhu A, Zhang Y, Yao S, and Mao W (2016). Pre- and Delayed Treatments With Ranolazine Ameliorate Ventricular Arrhythmias and Nav1.5 Downregulation in Ischemic/Reperfused Rat Hearts. J. Cardiovasc. Pharmacol 68, 269–279. [DOI] [PubMed] [Google Scholar]
- West JB (2017). Physiological Effects of Chronic Hypoxia. N. Engl. J. Med 376, 1965–1971. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, and Catterall WA (1992). A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc. Natl. Acad. Sci. USA 89, 10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong D, Li T, Dai H, Arena AF, Plant LD, and Goldstein SAN (2017). SUMOylation determines the voltage required to activate cardiac IKs channels. Proc. Natl. Acad. Sci. USA 114, E6686–E6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Atack TC, Stroud DM, Zhang W, Hall L, and Roden DM (2012). Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res 111, 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Pabon L, and Murry CE (2014). Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res 114, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The study did not generate any unique datasets or codes.