

Early-onset epileptic encephalopathy (EOEE) is associated with variants in the SCN2A gene encoding the voltage-gated sodium channel NaV1.2. Thompson et al. reveal that several of these variants have a greater effect on channel function and neuronal excitability when present in neonatal splice isoforms.
Abstract
Epileptic encephalopathies are severe forms of infantile-onset epilepsy often complicated by severe neurodevelopmental impairments. Some forms of early-onset epileptic encephalopathy (EOEE) have been associated with variants in SCN2A, which encodes the brain voltage-gated sodium channel NaV1.2. Many voltage-gated sodium channel genes, including SCN2A, undergo developmentally regulated mRNA splicing. The early onset of these disorders suggests that developmentally regulated alternative splicing of NaV1.2 may be an important consideration when elucidating the pathophysiological consequences of epilepsy-associated variants. We hypothesized that EOEE-associated NaV1.2 variants would exhibit greater dysfunction in a splice isoform that is prominently expressed during early development. We engineered five EOEE-associated NaV1.2 variants (T236S, E999K, S1336Y, T1623N, and R1882Q) into the adult and neonatal splice isoforms of NaV1.2 and performed whole-cell voltage clamp to elucidate their functional properties. All variants exhibited functional defects that could enhance neuronal excitability. Three of the five variants (T236S, E999K, and S1336Y) exhibited greater dysfunction in the neonatal isoform compared with those observed in the adult isoform. Computational modeling of a developing cortical pyramidal neuron indicated that T236S, E999K, S1336Y, and R1882Q showed hyperexcitability preferentially in immature neurons. These results suggest that both splice isoform and neuronal developmental stage influence how EOEE-associated NaV1.2 variants affect neuronal excitability.
Introduction
Variants in genes encoding voltage-gated sodium (NaV) channels are associated with epilepsy and neurodevelopmental disorders with a wide range of severity (George, 2005; Oliva et al., 2012). This is illustrated by variants in SCN2A encoding the NaV1.2 channel, which may be associated with relatively benign familial epilepsy (Heron et al., 2002; Berkovic et al., 2004; Herlenius et al., 2007; Misra et al., 2008), early-onset epileptic encephalopathy (EOEE; Beal et al., 2012; Nakamura et al., 2013; Touma et al., 2013; Howell et al., 2015; Wolff et al., 2017), autism spectrum disorder (O’Roak et al., 2011; Sanders et al., 2012; D’Gama et al., 2015; Ben-Shalom et al., 2017), or intellectual disability (Baasch et al., 2014; Sanders et al., 2018). One of the most severe conditions associated with SCN2A mutation is Ohtahara syndrome, which has onset within the first 2 wk of life and features intractable focal or generalized seizures sometimes accompanied by a suppression-burst pattern on electroencephalography (EEG), eventually manifesting with severe cognitive and neurodevelopmental impairment (Le Bouter et al., 2003; Nakamura et al., 2013; Touma et al., 2013; Zerem et al., 2014). The factors contributing to the very early onset and severity of Ohtahara syndrome and related disorders associated with NaV1.2 variants are not entirely clear (Ogiwara et al., 2009).
Previous reports have suggested that the majority of EOEE-associated NaV1.2 variants exert gain-of-function effects (Ben-Shalom et al., 2017; Berecki et al., 2018), although only a few variants have been studied. Because NaV1.2 is strongly expressed in neocortical glutamatergic pyramidal neurons, where it localizes to axon initial segments (AISs), nodes of Ranvier, and the somatodendritic compartment in the developing brain, gain-of-function variants are predicted to enhance network excitability (Hu et al., 2009; Liao et al., 2010; Tian et al., 2014; Yamagata et al., 2017; Spratt et al., 2019). During maturation, NaV1.2 is replaced at the distal AIS and nodes of Ranvier by NaV1.6, suggesting that gain-of-function variants in NaV1.2 may have less severe consequences on action potential initiation and propagation in mature neurons due to its diminished contribution to these processes (Hu et al., 2009; Liao et al., 2010; Tian et al., 2014). Consistent with this developmental switch in NaV channel expression, recent work has suggested that sodium channel–blocking antiepileptic drugs are most effective in the setting of EOEE associated with gain-of-function NaV1.2 variants (Wolff et al., 2017).
SCN2A undergoes developmentally regulated alternative mRNA splicing that leads to incorporation of an alternate exon encoding a portion of the domain I voltage-sensor (S3 and S4 helices). The two resulting protein isoforms (proteoforms) differ by one amino acid at position 209, encoding an asparagine in the splice proteoform expressed primarily during early development (neonatal proteoform, designated here as NaV1.2N) and an aspartic acid in the proteoform expressed mainly in adult brain (NaV1.2A). While this splicing event has been demonstrated in human brain tissue, the time course over which the transition from neonatal to adult proteoforms occurs is unknown (Kasai et al., 2001). In mouse cortex, the neonatal splice proteoform is present in approximately twofold excess compared with the canonical adult-expressed NaV1.2A transcript in early development, but drops to one third of the canonical splice proteoform within the first 2 wk of life (Kasai et al., 2001; Copley, 2004; Gazina et al., 2010). An analogous splicing event affecting NaV1.1 influences the pharmacological properties of that channel (Thompson et al., 2011). The contribution of alternative splicing to the pathogenesis of SCN2A-associated EOEE has not been systematically investigated, and prior studies determining the functional consequences of SCN2A variants were performed using the canonical splice proteoform expressed predominantly in adult brain (Misra et al., 2008; Ben-Shalom et al., 2017; Wolff et al., 2017; Berecki et al., 2018). However, some work suggests that variants in the neonatal splice proteoform negatively impact neuronal excitability (Xu et al., 2007; Gazina et al., 2015).
In this study, we examined the functional consequences of five NaV1.2 variants (T236S, E999K, S1336Y, T1623N, and R1882Q) in both the neonatal and adult proteoforms. Four of the five variants were not previously studied for their functional effects, while R1882Q was previously shown to exhibit a depolarized shift in voltage dependence of inactivation and slowed onset of inactivation compared with WT channels (Berecki et al., 2018; Mason et al., 2019). However, none of the variants have been studied in the two splice isoforms. We demonstrate that NaV1.2 variants associated with EOEE exhibit greater dysfunction when expressed in the neonatal splice proteoform. Our results indicate that both developmentally regulated alternative mRNA splicing of NaV1.2 and developmental stage are important factors promoting neuronal hyperexcitability. These observations have implications for understanding the pathophysiology of EOEE and for guiding experimental strategies to determine the functional consequences of NaV channel variants associated with these life-threatening, early-life neurodevelopmental disorders.
Materials and methods
Mutagenesis and heterologous expression of NaV1.2
Mutagenesis of recombinant human NaV1.2 was performed as described previously (Lossin et al., 2002; Rhodes et al., 2004; Kahlig et al., 2008; Thompson et al., 2011). To generate the neonatal splice isoform (NaV1.2N), a single substitution (D209N) was introduced. Five EOEE-associated variants (T236S, E999K, S1336Y, T1623N, and R1882Q) were introduced into both NaV1.2N and NaV1.2A splice isoforms. To minimize spontaneous mutagenesis of NaV1.2 cDNA in bacteria, recombinants were always propagated in Stbl2 cells (Invitrogen) at 30°C.
Heterologous expression of WT or NaV1.2 variants was performed in tsA201 cells. Heterologous expression in tsA201 cells was chosen for these experiments to enable precise measurement of biophysical parameters associated with WT or mutant NaV1.2 channels, without the contamination of other sodium currents that may be found in native neuronal cells. Cells were grown in 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, 2 mM l-glutamine, 50 units/ml penicillin, and 50 µg/ml streptomycin. Cells were transiently cotransfected with NaV1.2 and the accessory β1 and β2 subunits (2 µg total plasmid DNA was transfected with a cDNA ratio of 10:1:1 for NaV1.2:β1:β2 subunits) using SuperFect reagent (Qiagen). Human β1 and β2 cDNAs were cloned into plasmids encoding the CD8 receptor (CD8-IRES-hβ1) or enhanced GFP-IRES-hβ2, respectively, as transfection markers, as previously described.
Voltage-clamp recording and data analysis
Whole-cell voltage clamp experiments of heterologous cells were performed as previously described (Thompson et al., 2011, 2012). Whole-cell voltage-clamp recordings were made at room temperature using an Axopatch 200B amplifier (Molecular Devices). Patch pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus) with a multistage P-1000 Flaming-Brown micropipette puller (Sutter Instruments) and fire-polished using a microforge (Narashige MF-830) to a resistance of 1.5–2.5 MΩ. The pipette solution consisted of (in mM): 10 NaF, 105 CsF, 20 CsCl, 2 EGTA, and 10 HEPES, with pH adjusted to 7.35 with CsOH and osmolality adjusted to 300 mOsmol/kg with sucrose. The recording solution was continuously perfused with bath solution containing (in mM): 145 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, with pH 7.35 and osmolality 310 mOsmol/kg. The reference electrode consisted of a 2% agar bridge with composition similar to the bath solution. All chemicals were purchased from Sigma-Aldrich.
Voltage-clamp pulse generation and data collection were done using Clampex 10.4 (Molecular Devices). Whole-cell capacitance was determined by integrating capacitive transients generated by a voltage step from −120 mV to −110 mV filtered at 100 kHz low-pass Bessel filtering. Series resistance was compensated with prediction >70% and correction >90% to ensure that the command potential was reached within microseconds with a voltage error <3 mV. Leak currents were subtracted by using an online P/4 procedure. All whole-cell currents were filtered at 5 kHz low-pass Bessel filtering and digitized at 50 kHz. All voltage-clamp experiments were conducted at room temperature (20–25°C).
Data were analyzed using a combination of Clampfit 10.4 (Molecular Devices), Excel 2013 (Microsoft), and SigmaPlot 12.5 (Systat Software). Peak current was normalized for cell capacitance and plotted against step voltage to generate a peak current density–voltage relationship. Whole-cell conductance (GNa) was calculated as GNa = I/(V − Erev), where I is the measured peak current, V is the step potential, and Erev is the calculated sodium reversal potential. GNa at each voltage step was normalized to the maximum conductance between −80 mV and 20 mV. To calculate voltage dependence of activation (protocol described in figure insets), normalized GNa was plotted against voltage and fitted with the Boltzmann function G/Gmax = (1 + exp[(V – V1/2)/k])−1, where V1/2 indicates the voltage at half-maximal activation, and k is a slope factor describing voltage sensitivity of the channel. Voltage dependence of steady-state inactivation was assessed by plotting currents generated by the −10-mV postpulse voltage step normalized to the maximum current against prepulse voltage step from −140 to −10 mV in 10-mV increments. The plot was fitted with the Boltzmann function. Time-dependent recovery from inactivation was evaluated by fitting peak current recovery with a two-exponential function. Chord conductance at 0 mV (20 trials from a holding potential of −120 mV) was determined using non-stationary fluctuation analysis where σ2(I) − iI − (I2/N), where σ2 is variance, i is single-channel current, I is average current, and N is number of channels.
Results are presented as mean ± SEM. The statistical tests performed are specified in the figure legends. P < 0.05 was considered statistically significant.
Computational modeling
Simulations were performed using a pyramidal cell model in the NEURON environment, as previously described (Schmidt-Hieber and Bischofberger, 2010; Hallermann et al., 2012; Ben-Shalom et al., 2017; Spratt et al., 2019). All EOEE syndrome variants were modeled as changes relative to this baseline. All NaV channel simulations were performed at 25°C to match recording conditions. All neuronal simulations were performed at 33°C, with kinetics scaled by the Q10 factor within the model. Because changes in whole-cell current densities may be a byproduct of expression system, we only modeled changes in sodium channel biophysical parameters. All neuronal simulations assumed heterozygosity. Model files will be provided upon request to the author.
Results
Developmentally regulated alternative mRNA splicing is a conserved feature of human NaV channels. In human NaV1.2, alternative splicing involves a pair of mutually exclusive exons encoding part of S3 and S4 in domain I and generates two channel proteoforms (designated NaV1.2-neonatal [NaV1.2N] and NaV1.2-adult [NaV1.2A]) that differ by a single amino acid residue (adult to neonatal: D209N, Fig. 1 A). We compared the functional properties of heterologously expressed WT NaV1.2N and NaV1.2A proteoforms. Total whole-cell current density, voltage dependence of channel inactivation, and recovery from fast inactivation were not significantly different between the two WT NaV1.2 proteoforms (Fig. 1). However, NaV1.2N exhibited a significantly depolarized voltage dependence of activation compared with NaV1.2A (activation V1/2: WT-NaV1.2N, −19.6 ± 0.8 mV; NaV1.2A, −23.5 ± 1.3 mV, n = 15; P = 0.0172; Fig. 1 D). This depolarized voltage dependence of activation may limit neuronal excitability during early development (Xu et al., 2007; Gazina et al., 2015).
Figure 1.
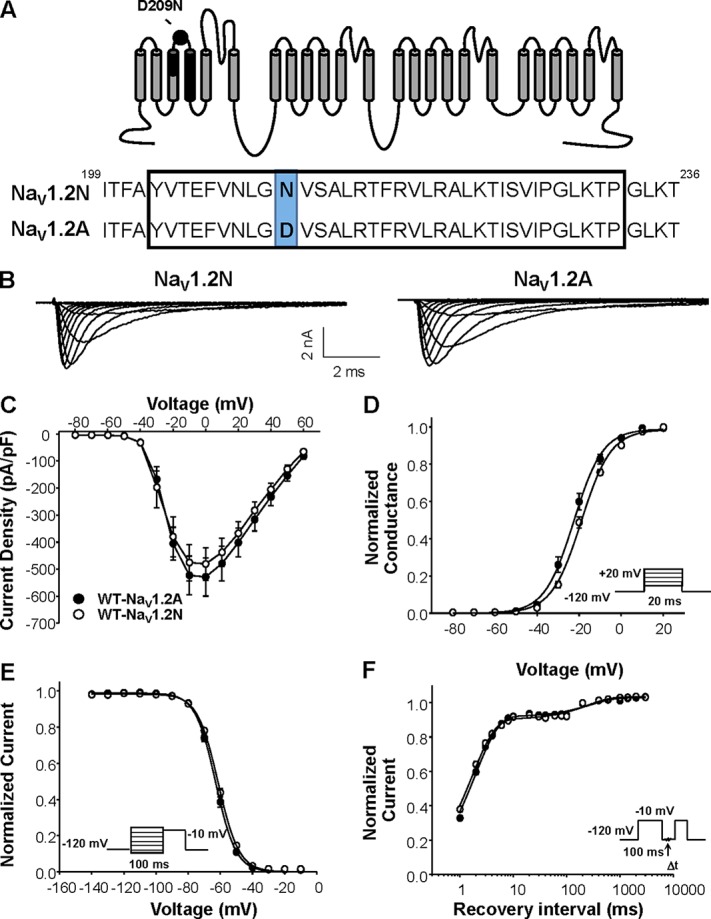
Biophysical properties of NaV1.2 splice variants. (A) Top: Predicted transmembrane topology of NaV1.2, highlighting the location of the exon 5 coding region (black shaded) and amino acid change associated with alternative exon splicing (black dot). Bottom: Amino acid alignment of NaV1.2N and NaV1.2A. (B) Representative whole-cell sodium currents recorded from tsA201 cells coexpressing either NaV1.2N (left) or NaV1.2A (right) with β1 and β2 subunits. (C) Current density elicited by test pulses to various membrane potentials and normalized to cell capacitance. (D) Voltage dependence of channel activation. (E) Voltage dependence of channel inactivation. (F) Time-dependent recovery from inactivation. Closed symbols represent NaV1.2A, and open symbols represent NaV1.2N. All data are expressed as mean ± SEM for 15 measurements. Statistical comparisons were made using an unpaired Student’s t test.
Biophysical properties of epileptic encephalopathy–associated NaV1.2 variants
We investigated the functional consequences of five EOEE-associated NaV1.2 variants. Four variants, reported originally by Nakamura et al. (2013), were identified in neonates having seizures associated with a suppression-burst EEG pattern beginning within the first week of life (age 0–1 d for three variants, age 6 d for E999K). Three of the subjects were described as having medically intractable epilepsy (an exception was the carrier of T236S, who responded to a four-antiepileptic-drug regimen), and all had severe developmental delay. The initial diagnosis was Ohtahara syndrome in all cases, but three were later reclassified as West syndrome due to emergence of hypsarrhythmia on EEG. The other variant, R1882Q, is recurrent and presents with focal seizures on the first day of life that evolves to severe developmental delay (Howell et al., 2015). These variants were specifically chosen because the associated clinical phenotypes had very early onset, and they were also chosen to represent each domain of the sodium channel.
We first examined whole-cell sodium current density for WT and mutant NaV1.2N. Current density for NaV1.2N-E999K, NaV1.2N-T1623N, and NaV1.2N-R1882Q was not statistically different from WT. However, T236S expressed in NaV1.2N had approximately twofold larger currents than the WT channel (NaV1.2N-WT: −480.9 ± 60.9 pA/pF, n = 15; NaV1.2N-T236S: −985.9 ± 184.8 pA/pF, n = 15, P = 0.0037; Fig. 2, B and C; and Fig. 3). We calculated chord conductance at 0 mV using non-stationary fluctuation analysis and found no difference between NaV1.2N-WT and NaV1.2N-T236S (19.9 ± 4.4 vs. 23.4 ± 3.4 pS, respectively, n = 5 each, P = 0.75). Also, we observed a strong trend toward smaller current density compared with WT NaV1.2N for NaV1.2N-S1336Y (−113.7 ± 26.9 pA/pF, n = 10, P = 0.0549). Similar to that of the neonatal isoform, neither NaV1.2A-E999K, NaV1.2A-NaV1.2A-T1623N, nor NaV1.2A-R1882Q showed any alterations in current density. The NaV1.2A-S1336Y variant showed similarly small currents compared with the neonatal isoform of the channel (−104.7 ± 14.7 pA/pF, n = 11, P = 0.0045; Fig. 2, B and C; and Fig. 3). However, the larger current density observed for NaV1.2N-T236S was not evident in the adult isoform (WT NaV1.2A: −518.4 ± 50.9 pA/pF, n = 15; NaV1.2A-T236S: −405.2 ± 83.9 pA/pF, n = 9, P = 0.87; Fig. 2, B and C; and Fig. 3).
Figure 2.
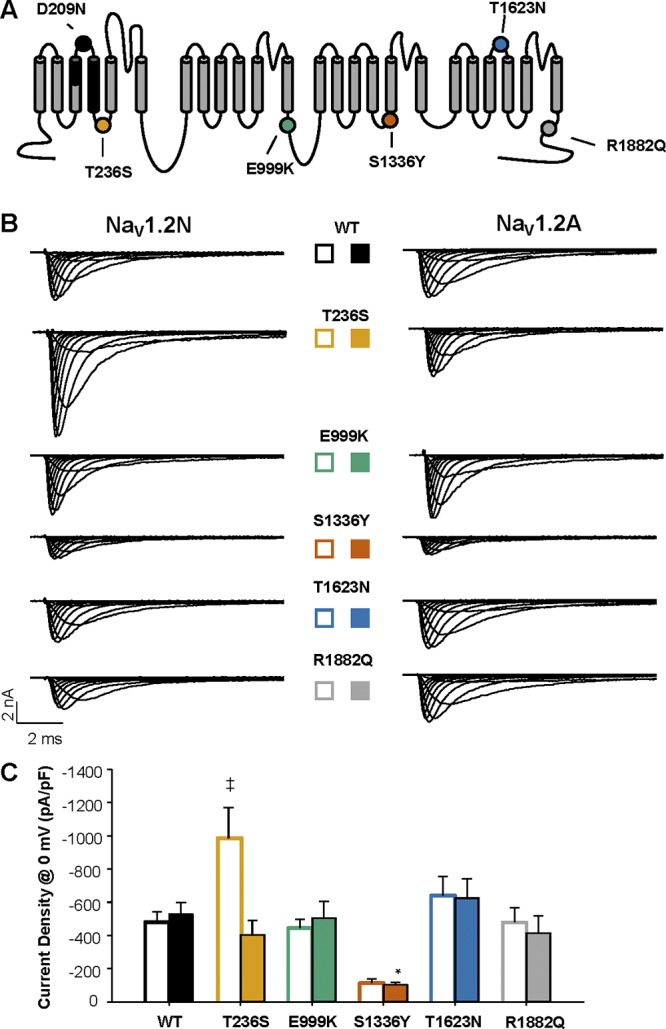
Function of EOEE-associated variants. (A) Predicted transmembrane topology of NaV1.2, highlighting the location of the exon 5 coding region (black shaded), amino acid change associated with alternative exon splicing (black dot), and EOEE-associated variants (colored dots). (B) Whole-cell sodium currents of EOEE-associated NaV1.2 variants in either the adult (NaV1.2N; left) or neonatal (NaV1.2A; right) splice isoforms. (C) Peak current density elicited by test pulses to 0 mV from a holding potential of −120 mV for NaV1.2N (open bars) and NaV1.2A (closed bars) proteoforms. All data are expressed as mean ± SEM for 8–15 measurements. *, P < 0.05 compared with NaV1.2A; ‡, P < 0.05 NaV1.2N. EOEE-associated variants were compared with WT NaV1.2 of the same proteoform using a one-way ANOVA, followed by Dunnett’s post hoc test.
Figure 3.
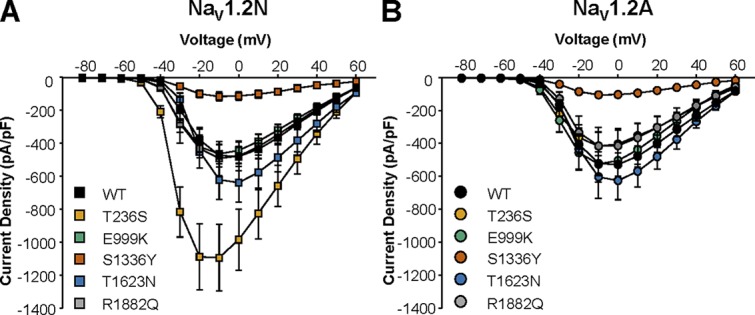
The impact of NaV1.2 alternative splicing on sodium current density. (A and B) Current–voltage relationships for WT and EOEE-associated variants expressed in the neonatal (A) or adult (B) NaV1.2 splice isoforms. All data are expressed as mean ± SEM for 8–15 measurements.
We determined if the variants altered the voltage dependence of channel activation. We observed that NaV1.2N-T236S, NaV1.2N-E999K, and NaV1.2N-S1336Y all had hyperpolarized voltage dependence of activation compared with WT NaV1.2N (NaV1.2N: −19.6 ± 0.8 mV, n = 15; NaV1.2N-T236S: −29.9 ± 1.1 mV, n = 15, P < 0.0001; NaV1.2N-E999K: −26.7 ± 1.7 mV, n = 14, P = 0.003; NaV1.2N-S1336Y: −25.3 ± 1.9 mV, n = 10, P = 0.0278; Fig. 4 and Table 1). However, when voltage dependence of activation was measured in the adult isoform, we found that the mutants were indistinguishable from WT NaV1.2A (WT-NaV1.2A: −23.5 ± 1.3 mV, n = 15; NaV1.2A-T236S: −25.6 ± 2.0 mV, n = 9, P = 0.78; NaV1.2A-E999K: −24.6 ± 1.4 mV, n = 10, P = 0.96; NaV1.2A-S1336Y: −23.2 ± 1.4 mV, n = 11, P = 0.99; Fig. 4 and Table 1). Interestingly, in NaV1.2A, we observed a depolarized shift in the voltage dependence of activation for T1623N (−18.4 ± 1.4 mV, n = 8, P = 0.0316; Fig. 4 and Table 1). These results suggest that three variants, T236S, E999K, and S1336Y, may confer neuronal hyperexcitability, but only when expressed in NaV1.2N.
Figure 4.
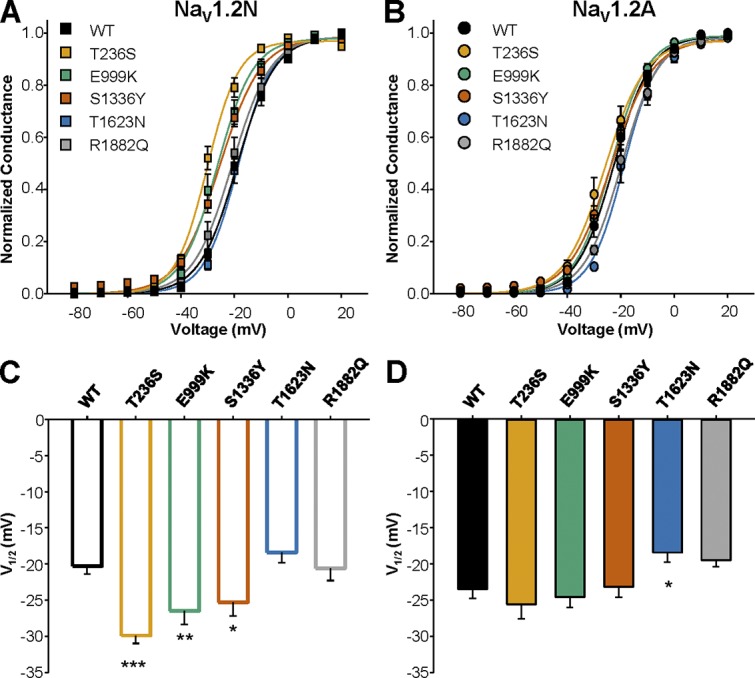
Alternative splicing of NaV1.2 differentially modulates the effect of EOEE-associated variants on channel activation. (A and B) Voltage dependence of activation for WT and EOEE-associated NaV1.2 variants in the neonatal (A) or adult (B) proteoform. (C and D) Values for V1/2 of activation for WT and EOEE-associated NaV1.2 variants in the neonatal (C) or adult (D) proteoform. All data are expressed as mean ± SEM for 8–15 measurements. EOEE-associated variants were compared with WT NaV1.2 of the same proteoform using one-way ANOVA, followed by Dunnett’s post hoc test. ***, P < 0.0001; **, P < 0.01; *, P < 0.05.
Table 1. Biophysical properties of NaV1.2 variants.
| Variant | Voltage dependence of activation | Voltage dependence of fast inactivation | Recovery from fast inactivation (−10 mV) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| V1/2 (mV) | k (mV) | n | V1/2 (mV) | k (mV) | n | τf (ms) (amplitude) | τs (ms) (amplitude) | n | |
| NaV1.2A | −23.5 ± 1.3 | 6.3 ± 0.3 | 15 | −63.5 ± 0.9 | −6.2 ± 0.1 | 15 | 1.9 ± 0.1 (0.90 ± 0.01) | 275.6 ± 16.4 (0.12 ± 0.01) | 15 |
| NaV1.2A-T236S | −25.6 ± 2.0 | 7.0 ± 0.3 | 9 | −65.3 ± 1.8 | −5.6 ± 0.1 | 9 | 2.1 ± 0.1 (0.87 ± 0.01) | 270.1 ± 16.8 (0.14 ± 0.01) | 8 |
| NaV1.2A-E999K | −4.6 ± 1.4 | 6.5 ± 0.3 | 10 | −63.1 ± 1.2 | −6.2 ± 0.1 | 10 | 1.9 ± 0.2 (0.90 ± 0.01) | 269.3 ± 21.2 (0.14 ± 0.01) | 10 |
| NaV1.2A-S1336Y | −23.2 ± 1.4 | 7.9 ± 0.4 | 11 | −57.7 ± 1.3a | −6.6 ± 0.3 | 11 | 1.3 ± 0.2 (0.84 ± 0.03) | 210.3 ± 20.4 (0.21 ± 0.02) | 11 |
| NaV1.2A-T1623N | −18.4 ± 1.4a | 6.4 ± 0.6 | 8 | −56.7 ± 0.8a | −6.0 ± 0.1 | 8 | 2.1 ± 0.1 (0.92 ± 0.01) | 240.4 ± 17.4 (0.09 ± 0.01) | 8 |
| NaV1.2A-R1882Q | −19.5 ± 0.9 | 6.7 ± 0.3 | 15 | −51.2 ± 1.5a | −7.6 ± 0.3a | 15 | 1.6 ± 0.1 (0.84 ± 0.02) | 248.2 ± 22.0 (0.16 ± 0.02) | 15 |
| NaV1.2N | −19.5 ± 0.8b | 7.0 ± 0.3 | 15 | −62.4 ± 1.2 | −6.4 ± 0.1 | 15 | 1.7± 0.1 (0.90 ± 0.01) | 267.3 ± 28.8 (0.13 ± 0.01) | 15 |
| NaV1.2N-T236S | −29.9 ± 1.1a | 5.7 ± 0.4a | 15 | −62.3 ± 0.8 | 6.1 ± 0.4 | 8 | 1.9 ± 0.1 (0.87 ± 0.01) | 245.3 ± 7.7 (0.14 ± 0.01) | 7 |
| NaV1.2N-E999K | −26.2 ± 1.7a | 6.3 ± 0.4 | 14 | −63.1 ± 1.3 | −5.8 ± 0.1 | 13 | 1.8 ± 0.1 (0.89 ± 0.01) | 263.2 ± 22.9 (0.12 ± 0.01) | 13 |
| NaV1.2N-S1336Y | −25.3 ± 1.9a | 7.3 ± 0.7 | 10 | −56.9 ± 1.0a | −7.9 ± 0.8a | 10 | 1.0 ± 0.1 (0.89 ± 0.02) | 261.0 ± 38.8 (0.16 ± 0.03) | 10 |
| NaV1.2N-T1623N | −18.4 ± 1.4 | 6.2 ± 0.3 | 9 | −56.3 ± 0.7a | −6.3 ± 0.4 | 9 | 2.0 ± 0.1 (0.87 ± 0.02) | 213.6 ± 23.9 (0.14 ± 0.02) | 9 |
| NaV1.2N-R1882Q | −20.6 ± 1.7 | 6.2 ± 0.4 | 14 | −53.4 ± 1.3a | −7.0 ± 0.2a | 14 | 1.6 ± 0.1 (0.88 ± 0.02) | 258.2 ± 27.8 (0.12 ± 0.02) | 14 |
P < 0.05 between WT and variant in same splice isoform.
P < 0.05 between neonatal and adult isoforms.
We investigated whether the variants exhibited a compensatory hyperpolarization in the voltage dependence of inactivation when expressed in NaV1.2N. We found that both T236S and E999K were indistinguishable from WT NaV1.2N (WT-NaV1.2N: −62.4 ± 1.2 mV, n = 15; NaV1.2N-T236S: −62.3 ± 0.8 mV, n = 8, P = 0.999; NaV1.2N-E999K: −63.1 ± 1.3 mV, n = 13, P = 0.992; Fig. 5 and Table 1). However, NaV1.2N-S1336Y, NaV1.2N-T1623N, and NaV1.2N-R1882Q all showed a depolarized shift in the voltage dependence of inactivation (NaV1.2N-S1336Y: −56.9 ± 1.0 mV, n = 10, P = 0.0075; NaV1.2N-T1623N: −56.3 ± 0.7 mV, n = 9, P = 0.0049; NaV1.2N-R1882Q: −53.4 ± 1.3 mV, n = 14, P < 0.0001; Fig. 5, A and C; and Table 1). These differences in voltage dependence of inactivation were proteoform independent, with similar depolarized shifts observed in NaV1.2A (WT-NaV1.2A: −63.5 ± 0.9 mV, n = 15; NaV1.2A-S1336Y: −57.7 ± 1.3 mV, n = 11, P = 0.0011; NaV1.2A-T1623N: −56.7 ± 0.8 mV, n = 8, P = 0.0055; NaV1.2A-R1882Q: −51.2 ± 1.5 mV, n = 14, P < 0.0001; Fig. 5, B and D; and Table 1).
Figure 5.
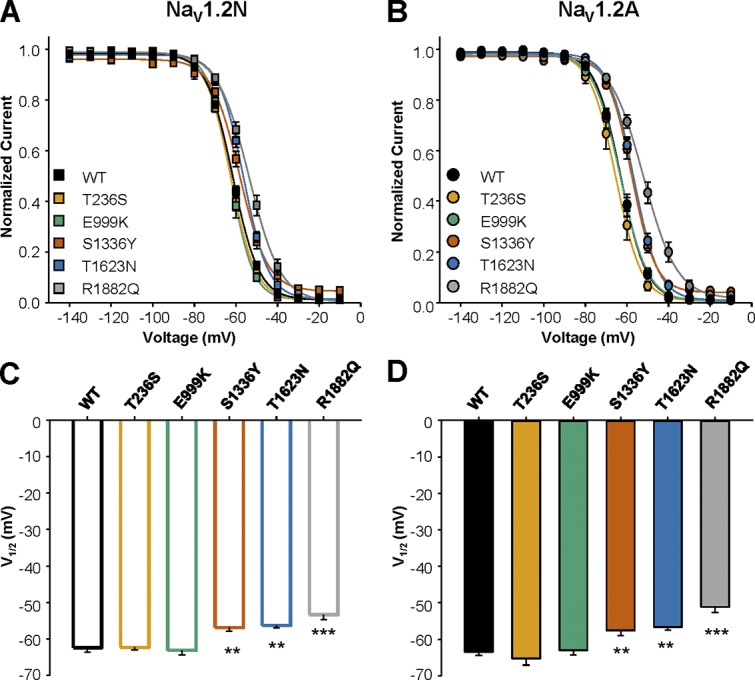
Voltage dependence of inactivation of EOEE-associated NaV1.2 variants. (A and B) Voltage dependence of inactivation for WT and EOEE-associated NaV1.2 variants in the neonatal (A) or adult (B) proteoform. (C and D) Values for V1/2 of inactivation for WT and EOEE-associated NaV1.2 variants in the neonatal (C) or adult (D) proteoform. All data are expressed as mean ± SEM for 8–15 measurements. EOEE-associated variants were compared with WT NaV1.2 of the same proteoform using one-way ANOVA, followed by Dunnett’s post hoc test. ***, P < 0.0001; **, P < 0.01.
The kinetics of inactivation (Fig. 6, A and B) and recovery from inactivation (Fig. 6, C and D) were indistinguishable between WT and most of the mutant channels regardless of the channel proteoform (Fig. 6 and Table 1). However, both T1623N and R1882Q exhibited a significantly slower entry into the fast inactivated state, as evidenced by the larger time constant for inactivation across a range of voltages in both NaV1.2N and NaV1.2A (Fig. 6, A and B).
Figure 6.
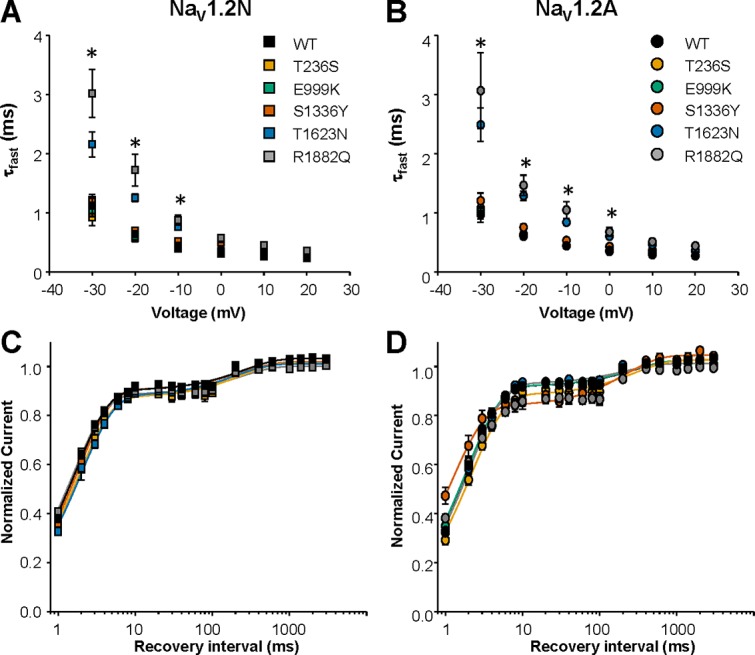
Alternative NaV1.2 splicing does not impact the effect of EOEE-associated variants on the kinetics of fast inactivation. (A and B) Voltage dependence of time constants for the onset of fast inactivation for WT and EOEE associated NaV1.2 variants in the NaV1.2N (A) and NaV1.2A (B) isoforms. All data are expressed as mean ± SEM for 8–15 measurements. (C and D) Recovery from fast inactivation for WT and EOEE-associated NaV1.2 variants in the neonatal (C) and adult (D) isoforms. All data are expressed as mean ± SEM for 8–15 measurements. EOEE-associated variants were compared with WT NaV1.2 of the same proteoform using one-way ANOVA, followed by Dunnett’s post hoc test. *, P < 0.05.
Integrating and interpreting functional effects of NaV1.2 variants
Missense variation in NaV1.2 can result in complex changes to channel functional properties. As a result, it can be difficult to understand how these changes sum to a net gain or loss of channel function. To better summarize and integrate the functional differences among channel variants and alternatively spliced proteoforms, we constructed radar plots incorporating peak sodium current density, voltage dependence of activation and inactivation, and the kinetics of inactivation, relative to the WT channel. By plotting the data together, we could better interpret the net effect of a variant on overall channel function. We configured each radar spoke such that points lying outside of the WT plot would be indicative of a gain of function, while points lying within the WT plot would indicate loss of function.
As illustrated in Fig. 7, all five EOEE-associated mutations showed significant deviations from WT channels in NaV1.2N. Most effects observed in the NaV1.2N background were consistent with gain of function, whereas one mutation (S1336Y) exhibited a mixture of gain- and loss-of-function defects. However, in the NaV1.2A background, the differences in voltage dependence of activation were absent for T236S, E999K, and S13336Y, but the overt differences in voltage dependence of inactivation were present for S1336Y, T1623N, and R1882Q.
Figure 7.
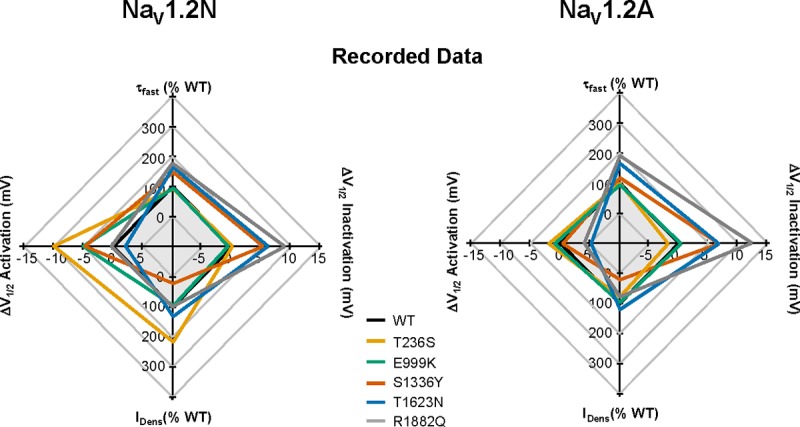
Radar plots illustrating differences in functional consequences of NaV1.2 variants. Plots were constructed with each spoke arranged such that points lying outside of the WT plot would be indicative of a gain of function, while points lying within the WT plot would indicate a loss of function. The effects of each mutant channel relative to WT are illustrated for NaV1.2N (left) and NaV1.2A (right) from experimentally collected data.
Computational modeling of EOEE-associated NaV1.2 variants
To evaluate how NaV1.2 dysfunction affects neuronal excitability, we incorporated the functional properties of each mutation into an established computational model of a cortical pyramidal neuron (Hallermann et al., 2012; Ben-Shalom et al., 2017). We created computational models to simulate sodium currents conducted by NaV1.2A or NaV1.2N, as well as each mutant channel, in both proteoforms. Our models successfully recapitulated the functional differences observed between NaV1.2A and NaV1.2N (Fig. 8, A and B), as well the biophysical properties of all mutations (Fig. 8 C). Importantly, because whole-cell current density is highly variable and may be dependent on cell type, we only modeled changes in voltage-dependent activation and inactivation along with kinetic parameters. For comparison, we modeled the benign familial neonatal-infantile seizures (BFNIS) associated NaV1.2 mutation L1563V, using previously published data (Fig. 8 C; Xu et al., 2007). This mutation has been shown to exhibit biophysical properties that are dependent on splice isoform.
Figure 8.

Computational modeling of WT NaV1.2 splice isoforms. (A) Simulated whole-cell sodium current for NaV1.2A (top) and NaV1.2N (bottom). (B) Voltage dependence of activation and inactivation for simulated sodium currents mediated by the NaV1.2N (open circles) and NaV1.2A (closed circles). Curves for voltage dependence of inactivation for both isoforms are superimposed. (C) Radar plots comparing changes in biophysical parameters of whole-cell sodium current from simulated data for NaV1.2N (left) and NaV1.2A (right).
To recapitulate the neuronal environment at different developmental stages, we performed simulations incorporating NaV1.2A and NaV1.2N at specific ratios based on published data from mouse brain (Gazina et al., 2010). To simulate an immature neuron, we developed a model expressing 67% NaV1.2N and 33% NaV1.2A. Simulations of the WT channels in this model showed that expression of 100% WT NaV1.2N or a mixture of NaV1.2A and NaV1.2N was associated with lower neuronal excitability compared with neurons expressing 100% WT NaV1.2A (Fig. 9). Therefore, while the ∼3-mV difference in voltage dependence of activation between NaV1.2N and NaV1.2A may appear small, it is sufficient to affect excitability. This is similar to predictions made by other neuronal models, which demonstrate that NaV1.2N-expressing neurons have lower excitability compared with neurons expressing WT channels (Xu et al., 2007). However, when we incorporated each EOEE-associated NaV1.2 mutation into the immature neuron model with a mixture of NaV1.2A and NaV1.2N, all but one (T1623N) exhibited overtly greater neuronal excitability (Fig. 10, A and B). We additionally modeled NaV1.2-L1563V, a mutation associated with BFNIS. This mutation evoked a modest enhancement of excitability compared with WT neurons but was not as excitable as most EOEE-associated mutations (Fig. 10, A and B). Interestingly, although R1882Q is associated with EOEE, it also exhibited a small enhancement of excitability in immature neurons (Fig. 10, A and B). This suggests that five of these six mutations exert gain-of-function effects strong enough to overcome the normally dampening effect of the NaV1.2N on neuronal excitability.
Figure 9.
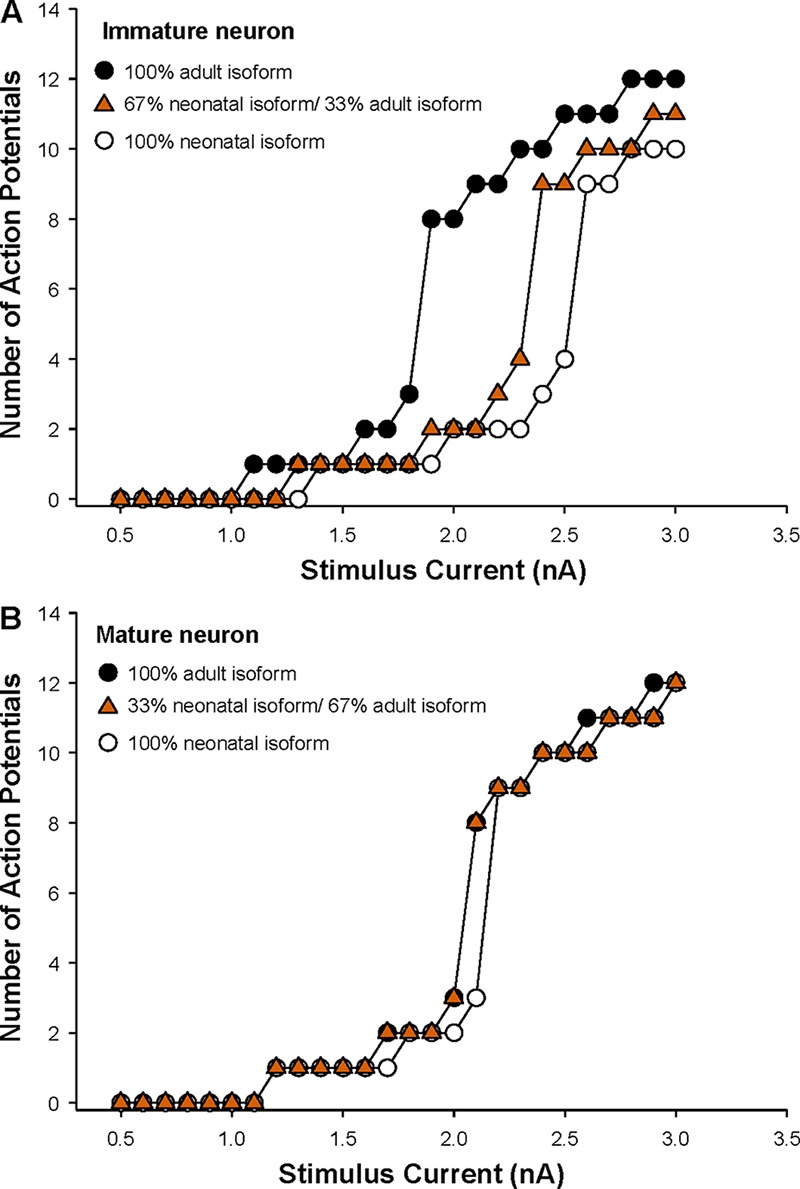
Alternative splicing of NaV1.2 modulates neuronal excitability. (A) Summary data plotting the number of action potentials against stimulation current for an immature neuron with specific ratios of neonatal/adult NaV1.2 isoforms. (B) Summary data plotting the number of action potentials against stimulation current for a mature neuron with specific ratios of neonatal/adult NaV1.2 isoforms.
Figure 10.
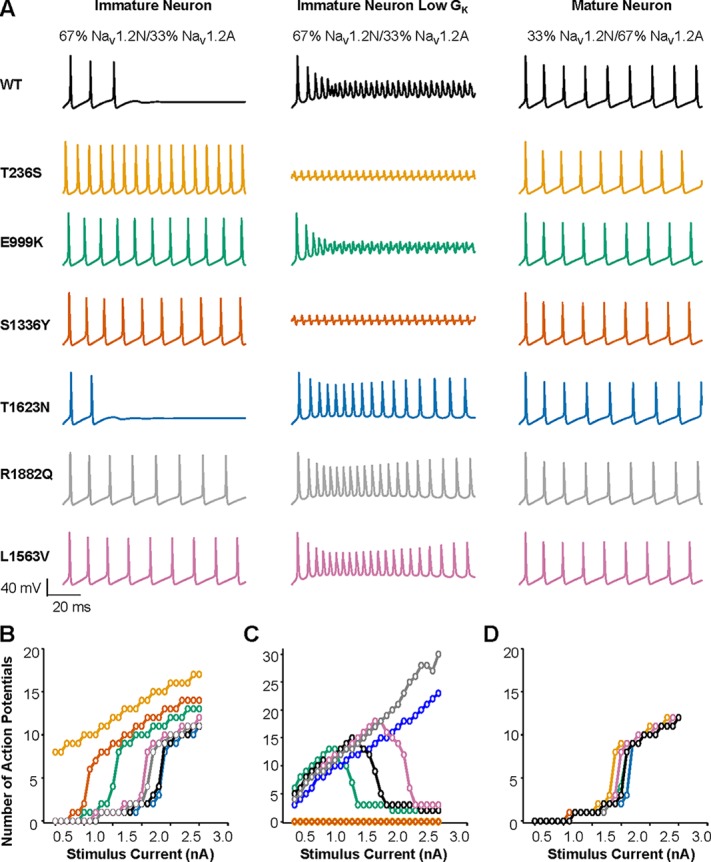
Neuronal maturity influences hyperexcitability associated with NaV1.2 variants. (A) Action potentials generated in response to a 2.2-nA somatic current stimulation for an immature neuron expressing 67% NaV1.2N and 33% NaV1.2A throughout the AIS (left), an immature neuron expressing 67% NaV1.2N and 33% NaV1.2A throughout the AIS, and lower potassium conductance (middle), and a mature neuron expressing 33% NaV1.2N and 67% NaV1.2A restricted to the proximal AIS (right). (B) Summary data plotting the number of action potentials against stimulation current for the immature neuron defined in A. (C) Summary data plotting the number of action potentials against stimulation current for the immature neuron with low potassium conductance defined in A. (D) Summary data plotting the number of action potentials against stimulation current for the mature neuron defined in A. All simulations assumed heterozygosity.
As discussed above, patients with NaV1.2-associated EOEE often present symptoms within the first days of life (Nakamura et al., 2013). Previous work has shown that potassium conductance is significantly smaller in mouse neocortical pyramidal neurons immediately after birth and steadily rises during the first weeks of life (Guan et al., 2011). Because we observed relatively mild phenotypes for two EE-associated mutations, T1623N and R1882Q, we hypothesized that a more overt phenotype may be observed at an earlier developmental time point when lower potassium conductance may predispose to hyperexcitability.
To determine the impact of EOEE-associated NaV1.2 variants on neuronal excitability in simulated neonatal neurons, we reduced the potassium channel conductance to one-third of that used for the immature neuron described above, while maintaining the sodium channel ratio of 67% NaV1.2N and 33% NaV1.2A (Fig. 10). Interestingly, neurons bearing either T236S or E999K, which exhibit large hyperpolarized shifts in the voltage dependence of activation, and S1336Y, which has hyperpolarized activation and depolarized inactivation, were either in a perpetual state of depolarization block (T236S and S1336Y) or entered into depolarization block more readily than WT neurons (E999K; Fig. 10, A and C). The mutations T1623N and R1882Q, which showed only modest effects on excitability with the full potassium current, showed greater excitability in the neonatal neuron model, being resistant to depolarization block compared with a WT neuron, while the BFNIS mutation exhibited only a modest enhancement of excitability (Fig. 10 C).
To simulate a mature neuron, we used a published model that restricts NaV1.2 expression to the proximal AIS and incorporates NaV1.6 into the distal AIS and nodes of Ranvier (Ben-Shalom et al., 2017). In the mature neuron model, NaV1.2 was represented as 33% NaV1.2N and 67% NaV1.2A. Simulations with WT channels showed that neuronal excitability was not different when 100% NaV1.2N, 100% NaV1.2A, or a mixture were present (Fig. 10 B). Further, mature neurons expressing each of the six variants exhibited minor differences in neuronal excitability (Fig. 10, A and D). Thus, differences in excitability among WT and mutant neurons depend greatly on the developmental context in which those variants are expressed, with hyperexcitability being exacerbated in immature developing neurons (Fig. 10). Thus, these results predict that the functional effects of EOEE-associated NaV1.2 variants will have the greatest impact on neuronal excitability in immature or neonatal neurons, which is consistent with the very early onset of these epilepsies.
Discussion
Variants in multiple NaV channel genes are associated with clinically diverse genetic epilepsies and neurodevelopmental disorders. Investigations of the functional consequences of various NaV channel variants have revealed fundamental mechanistic information implicating NaV1.1 loss of function in the pathogenesis of Dravet syndrome, and NaV1.6 gain of function as a cause for many cases of SCN8A epileptic encephalopathy (Yu et al., 2006; Ogiwara et al., 2007; Estacion et al., 2014; Wagnon et al., 2015; Lopez-Santiago et al., 2017). Functional studies have demonstrated a bifurcated pathophysiology of epilepsy and neurodevelopmental disorders associated with SCN2A variants. Specifically, NaV1.2 gain of function appears typical of EOEE, whereas loss of function seems more prevalent among variants associated with autism spectrum disorder or later-onset epilepsy, although there is significant comorbidity associated with autism spectrum disorders and epilepsy (Ben-Shalom et al., 2017). However, the fundamental reason for early-onset epilepsy in the setting of SCN2A mutation is not completely understood.
In this study, we investigated the contribution of a developmentally regulated alternative mRNA splicing event to the pathogenesis of SCN2A-associated epileptic encephalopathy. Previous studies have demonstrated that alternative splicing of SCN1A, SCN2A, SCN5A, and SCN9A have important effects on the functional and pharmacological properties of WT and mutant channels encoded by these genes (Kasai et al., 2001; Copley, 2004; Diss et al., 2004; Onkal et al., 2008; Thompson et al., 2011). With respect to SCN2A, there are functional differences between the two developmentally regulated alternatively spliced NaV1.2 isoforms, which may have physiological relevance. The neonatal-expressed NaV1.2N exhibits a depolarized voltage dependence of activation (Fig. 1), and this may dampen excitability in developing neurons.
Our results contrast with previous work that examined functional difference for NaV1.2N and NaV1.2A proteoforms (Xu et al., 2007). The prior work showed that NaV1.2N exhibited faster onset of inactivation and hyperpolarized voltage-dependent inactivation compared with NaV1.2A, while we observed only a depolarized shift in activation for NaV1.2N. One factor that may explain these differences is the coexpression of β1 and β2 subunits in our study. The β subunits have been shown to alter sodium channel biophysical properties and trafficking (Isom et al., 1992, 1995; Chen et al., 2002; Uebachs et al., 2010). However, both studies concur that the net effect of NaV1.2N biophysical properties predicts blunted neuronal excitability. This notion is supported by a previous study of mice engineered to express only NaV1.2A that demonstrated greater cortical pyramidal neuronal excitability beginning at postnatal day 3 (P3), and greater susceptibility to pentylenetetrazole-induced seizures beginning at P3 that persisted into adulthood (P53–P75; Gazina et al., 2015). Further, our demonstration that incorporating NaV1.2N into a compartmental model of a developing cortical pyramidal neuron results in lower firing frequency than neuron models incorporating only the adult proteoform (Fig. 9) also supports the relevance of SCN2A alternative splicing.
Given its physiological importance during early development, we investigated the impact of alternative splicing on a set of EOEE-associated SCN2A variants. We tested the hypothesis that the functional consequences of EOEE-associated NaV1.2 variants are potentiated in NaV1.2N, a phenomenon that may contribute to the early age of onset. The five NaV1.2 variants we studied exhibited mostly gain-of-function effects in NaV1.2N, but the functional consequences of these variants were more muted when expressed in the context of canonical NaV1.2A (adult expressed). Specifically, when expressed in NaV1.2N, three of the variants showed greater hyperpolarizing shifts in voltage dependence of activation, while one variant was protected from a depolarizing shift in voltage dependence of inactivation. Given that the splicing event involves a switch of the domain 1 voltage sensor, it is possible that variants with significant shifts in voltage dependence of activation may be disproportionately affected by this alternative splicing event. For NaV1.2N-T236S, given no change in single-channel conductance, the increased current density likely results from a hyperpolarized shift in open probability. These observations were supported by computational neuron modeling in which four of the five variants we examined, in which voltage dependence of activation was either hyperpolarized or unchanged, promoted overt hyperexcitability in an immature neuron where NaV1.2N is the sole sodium channel in the AIS and the nodes of Ranvier. Furthermore, recent work has suggested that shifts in voltage dependence of activation more strongly influence neuronal excitability than shifts in voltage dependence of inactivation (Liu et al., 2019). Work by Hu and Bean (2018) showed that a 3-mV shift in voltage dependence of channel activation may lead to up to twice as many sodium channels being recruited due to the steep slope of voltage-dependent channel activation. Thus, even small shifts in voltage dependence of activation may lead to greater recruitment of sodium channels in response to subthreshold events. Consistent with this, T1623N and R1882Q, which show depolarized voltage dependence of inactivation, show only modestly increased excitability.
One variant in our study, R1882Q, has been studied previously by two groups (Berecki et al., 2018; Mason et al., 2019). Our primary finding for this variant was a depolarized shift in voltage dependence of inactivation, as well as slower onset of inactivation. The previous studies showed similar effects on these parameters. However, Berecki et al. (2018) also observed larger whole-cell sodium current compared with the WT channel. Neither our results nor the results of Mason et al. (2019) showed larger currents mediated by the R1882Q mutation. Considering that Berecki et al. (2018) performed experiments in Chinese hamster ovary cells, while our work and the study by Mason et al. (2019) used human embryonic kidney–derived cell lines, the most likely explanation for discrepancies between these studies is different cell types.
Other human NaV1.2 variants are associated with BFNIS, which exhibits an early onset of pharmacoresponsive epilepsy that typically remits within the first year of life (Xu et al., 2007; Misra et al., 2008). Functional evaluation of BFNIS-associated variants have mostly demonstrated subtle alterations of channel properties, such as small hyperpolarized shifts in voltage dependence of activation or enhanced persistent current, which are consistent with mild gain of function. Two specific SCN2A variants, M252V and L1563V, exhibit biophysical defects only when studied in NaV1.2N (Xu et al., 2007; Liao et al., 2010). By contrast, one variant (V261M) exhibited functional abnormalities only when expressed in NaV1.2A (Liao et al., 2010). These findings were interpreted as evidence against the hypothesis that remission of epilepsy in BFNIS occurs following the developmental switch in SCN2A alternative splicing, but that remission is due to higher expression of NaV1.6 in these cells, which limits the functional contribution of NaV1.2. Consistent with this notion, computational simulations of NaV1.2-L1563V demonstrated only modestly increased excitability in immature neurons that normalized in the mature neuron (Xu et al., 2007).
Different functional consequences of mutant NaV1.2 channels due to alternative splicing is unlikely to be the only factor accounting for early-onset epilepsy and the subsequent severe neurodevelopmental impairments seen in epileptic encephalopathies. Early neuronal injury and subsequent tissue responses (e.g., reactive gliosis, inflammation) likely contribute to the perpetuation of the seizure-prone state (Loewen et al., 2016). A greater proclivity to seizure initiation during early development may be the primary contributing factor of enhanced channel dysfunction of mutant NaV1.2N. Indeed, our computational models with reduced potassium conductance show that EOEE-associated variants are prone to hyperexcitability very early in development, likely causing lasting changes to network development.
Many functional and genetic changes occur during the course of neuronal development. As stated above, there is a developmentally regulated switch between the neonatal and adult isoforms of NaV1.2 and escalating potassium conductance. However, it is apparent from previous studies that the switch is not complete, with some neonatal isoform persisting into adulthood (Gazina et al., 2010). Further, during the course of neuronal development, NaV1.2 is replaced in the distal AIS and the nodes of Ranvier by NaV1.6 (Liao et al., 2010). Our neuron simulations demonstrated that when EOEE-associated variants are present in both NaV1.2N and NaV1.2A in a developing neuron, the presence of the less severely affected adult isoform dampens the effect of the neonatal isoform, rendering the neuron less excitable than when the neonatal isoform is the sole NaV channel. Interestingly, when these mutant channels are incorporated into an adult neuron model where NaV1.6 is largely responsible for action potential initiation and propagation, we see that no variant dramatically affects excitability. These results suggest that EOEE-associated NaV1.2 variants (T236S, E999K, S1336Y, T1623N, and R1882Q) exert their effects very early in development, as is typical for other epileptic encephalopathies such as Dravet syndrome. While replacement of NaV1.2 at the AIS by NaV1.6 later in development may modulate seizures later in life, it is reasonable to predict that maladaptive changes in neuronal physiology occur in response to dysfunction of NaV1.2 early in development.
An important consideration of our work is that we are examining the effect of channel variants in a model of a cortical pyramidal neuron, due to the high expression of NaV1.2 throughout the neocortex during development and persistent expression throughout adulthood (Spratt et al., 2019). However, there is some evidence that NaV1.2 is expressed at the AIS of some interneurons, where it either mediates spontaneous action potential firing associated with basal network inhibition or is critical for network disinhibition (Ye et al., 2018). Ye et al. (2018) showed that epileptiform activity induced by magnesium deprivation was exacerbated by selective inhibition of NaV1.2 with low concentrations of phrixotoxin-3 in cortical slices. This abnormal activity could be normalized by inhibition of GABA-mediated neurotransmission (Ye et al., 2018). Additionally, NaV1.2 is found in unmyelinated axons of neurons from the hippocampal dentate, cerebellar granule cells, and medium spiny neurons of the striatum (Westenbroek et al., 1989; Gong et al., 1999; Miyazaki et al., 2014). Simulations of neuronal excitability suggested that the adult isoforms of two variants we studied, S1336Y and T1623N, are associated with lower neuronal excitability where NaV1.2 is the only channel at the AIS, while the neonatal isoform showed either dramatic (S1336Y) or modest (T1623N) hyperexcitability. Because these mutant channels do not influence neuronal excitability in an adult cortical pyramidal neuron, it is possible that complex network effects are set in place by having hyperexcitability during network development. Alternatively, loss of function of small populations of inhibitory interneurons may significantly disrupt excitation/inhibition balance.
In summary, our work has demonstrated that SCN2A-associated EOEE may result from channel gain of function that is potentiated in the neonatal NaV1.2N proteoform. Further, the developmental stage of the neuron, including NaV1.2 splice proteoform ratio, total NaV channel composition, and total potassium conductance, are critical determinants of neuronal excitability and are important considerations for functional evaluation of NaV1.2 variants associated with early-onset epilepsy.
Acknowledgments
Merritt C. Maduke served as editor.
We thank Jennifer D. Kunic for technical assistance with the project.
This work was funded in part by grants from the National Institutes of Health (U54-NS108874), grants from the Simons Foundation Autism Research Initiative to A.L. George Jr. and K.J. Bender (513133 to K.J. Bender), and a generous gift from the Davee Foundation.
The authors declare no competing financial interests.
Author contributions: C.H. Thompson, R. Ben-Shalom, K.J. Bender, and A.L. George Jr. designed the research; C.H. Thompson and R. Ben-Shalom performed experiments; C.H. Thompson and R. Ben-Shalom analyzed data; and C.H. Thompson, R. Ben-Shalom, K.J. Bender, and A.L. George Jr. wrote the paper.
References
- Baasch A.L., Hüning I., Gilissen C., Klepper J., Veltman J.A., Gillessen-Kaesbach G., Hoischen A., and Lohmann K.. 2014. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia. 55:e25–e29. 10.1111/epi.12554 [DOI] [PubMed] [Google Scholar]
- Beal J.C., Cherian K., and Moshe S.L.. 2012. Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatr. Neurol. 47:317–323. 10.1016/j.pediatrneurol.2012.06.002 [DOI] [PubMed] [Google Scholar]
- Ben-Shalom R., Keeshen C.M., Berrios K.N., An J.Y., Sanders S.J., and Bender K.J.. 2017. Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol. Psychiatry. 82:224–232. 10.1016/j.biopsych.2017.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berecki G., Howell K.B., Deerasooriya Y.H., Cilio M.R., Oliva M.K., Kaplan D., Scheffer I.E., Berkovic S.F., and Petrou S.. 2018. Dynamic action potential clamp predicts functional separation in mild familial and severe de novo forms of SCN2A epilepsy. Proc. Natl. Acad. Sci. USA. 115:E5516–E5525. 10.1073/pnas.1800077115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovic S.F., Heron S.E., Giordano L., Marini C., Guerrini R., Kaplan R.E., Gambardella A., Steinlein O.K., Grinton B.E., Dean J.T., et al. . 2004. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann. Neurol. 55:550–557. 10.1002/ana.20029 [DOI] [PubMed] [Google Scholar]
- Chen C., Bharucha V., Chen Y., Westenbroek R.E., Brown A., Malhotra J.D., Jones D., Avery C., Gillespie P.J. III, Kazen-Gillespie K.A., et al. . 2002. Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc. Natl. Acad. Sci. USA. 99:17072–17077. 10.1073/pnas.212638099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley R.R. 2004. Evolutionary convergence of alternative splicing in ion channels. Trends Genet. 20:171–176. 10.1016/j.tig.2004.02.001 [DOI] [PubMed] [Google Scholar]
- D’Gama A.M., Pochareddy S., Li M., Jamuar S.S., Reiff R.E., Lam A.N., Sestan N., and Walsh C.A.. 2015. Targeted DNA Sequencing from Autism Spectrum Disorder Brains Implicates Multiple Genetic Mechanisms. Neuron. 88:910–917. 10.1016/j.neuron.2015.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diss J.K., Fraser S.P., and Djamgoz M.B.. 2004. Voltage-gated Na+ channels: multiplicity of expression, plasticity, functional implications and pathophysiological aspects. Eur. Biophys. J. 33:180–193. 10.1007/s00249-004-0389-0 [DOI] [PubMed] [Google Scholar]
- Estacion M., O’Brien J.E., Conravey A., Hammer M.F., Waxman S.G., Dib-Hajj S.D., and Meisler M.H.. 2014. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol. Dis. 69:117–123. 10.1016/j.nbd.2014.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazina E.V., Leaw B.T., Richards K.L., Wimmer V.C., Kim T.H., Aumann T.D., Featherby T.J., Churilov L., Hammond V.E., Reid C.A., and Petrou S.. 2015. ‘Neonatal’ Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Hum. Mol. Genet. 24:1457–1468. 10.1093/hmg/ddu562 [DOI] [PubMed] [Google Scholar]
- Gazina E.V., Richards K.L., Mokhtar M.B., Thomas E.A., Reid C.A., and Petrou S.. 2010. Differential expression of exon 5 splice variants of sodium channel alpha subunit mRNAs in the developing mouse brain. Neuroscience. 166:195–200. 10.1016/j.neuroscience.2009.12.011 [DOI] [PubMed] [Google Scholar]
- George A.L., Jr 2005. Inherited disorders of voltage-gated sodium channels. J. Clin. Invest. 115:1990–1999. 10.1172/JCI25505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B., Rhodes K.J., Bekele-Arcuri Z., and Trimmer J.S.. 1999. Type I and type II Na(+) channel alpha-subunit polypeptides exhibit distinct spatial and temporal patterning, and association with auxiliary subunits in rat brain. J. Comp. Neurol. 412:342–352. [DOI] [PubMed] [Google Scholar]
- Guan D., Horton L.R., Armstrong W.E., and Foehring R.C.. 2011. Postnatal development of A-type and Kv1- and Kv2-mediated potassium channel currents in neocortical pyramidal neurons. J. Neurophysiol. 105:2976–2988. 10.1152/jn.00758.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallermann S., de Kock C.P., Stuart G.J., and Kole M.H.. 2012. State and location dependence of action potential metabolic cost in cortical pyramidal neurons. Nat. Neurosci. 15:1007–1014. 10.1038/nn.3132 [DOI] [PubMed] [Google Scholar]
- Herlenius E., Heron S.E., Grinton B.E., Keay D., Scheffer I.E., Mulley J.C., and Berkovic S.F.. 2007. SCN2A mutations and benign familial neonatal-infantile seizures: the phenotypic spectrum. Epilepsia. 48:1138–1142. 10.1111/j.1528-1167.2007.01049.x [DOI] [PubMed] [Google Scholar]
- Heron S.E., Crossland K.M., Andermann E., Phillips H.A., Hall A.J., Bleasel A., Shevell M., Mercho S., Seni M.H., Guiot M.C., et al. . 2002. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 360:851–852. 10.1016/S0140-6736(02)09968-3 [DOI] [PubMed] [Google Scholar]
- Howell K.B., McMahon J.M., Carvill G.L., Tambunan D., Mackay M.T., Rodriguez-Casero V., Webster R., Clark D., Freeman J.L., Calvert S., et al. . 2015. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology. 85:958–966. 10.1212/WNL.0000000000001926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., and Bean B.P.. 2018. Differential Control of Axonal and Somatic Resting Potential by Voltage-Dependent Conductances in Cortical Layer 5 Pyramidal Neurons. Neuron. 99:1355 10.1016/j.neuron.2018.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., Tian C., Li T., Yang M., Hou H., and Shu Y.. 2009. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat. Neurosci. 12:996–1002. 10.1038/nn.2359 [DOI] [PubMed] [Google Scholar]
- Isom L.L., De Jongh K.S., Patton D.E., Reber B.F.X., Offord J., Charbonneau H., Walsh K., Goldin A.L., and Catterall W.A.. 1992. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 256:839–842. 10.1126/science.1375395 [DOI] [PubMed] [Google Scholar]
- Isom L.L., Ragsdale D.S., De Jongh K.S., Westenbroek R.E., Reber B.F.X., Scheuer T., and Catterall W.A.. 1995. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 83:433–442. 10.1016/0092-8674(95)90121-3 [DOI] [PubMed] [Google Scholar]
- Kahlig K.M., Rhodes T.H., Pusch M., Freilinger T., Pereira-Monteiro J.M., Ferrari M.D., van den Maagdenberg A.M., Dichgans M., and George A.L. Jr. 2008. Divergent sodium channel defects in familial hemiplegic migraine. Proc. Natl. Acad. Sci. USA. 105:9799–9804. 10.1073/pnas.0711717105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai N., Fukushima K., Ueki Y., Prasad S., Nosakowski J., Sugata K., Sugata A., Nishizaki K., Meyer N.C., and Smith R.J.. 2001. Genomic structures of SCN2A and SCN3A - candidate genes for deafness at the DFNA16 locus. Gene. 264:113–122. 10.1016/S0378-1119(00)00594-1 [DOI] [PubMed] [Google Scholar]
- Le Bouter S., Demolombe S., Chambellan A., Bellocq C., Aimond F., Toumaniantz G., Lande G., Siavoshian S., Baró I., Pond A.L., et al. . 2003. Microarray analysis reveals complex remodeling of cardiac ion channel expression with altered thyroid status: relation to cellular and integrated electrophysiology. Circ. Res. 92:234–242. 10.1161/01.RES.0000053185.75505.8E [DOI] [PubMed] [Google Scholar]
- Liao Y., Deprez L., Maljevic S., Pitsch J., Claes L., Hristova D., Jordanova A., Ala-Mello S., Bellan-Koch A., Blazevic D., et al. . 2010. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 133:1403–1414. 10.1093/brain/awq057 [DOI] [PubMed]
- Liu Y., Schubert J., Sonnenberg L., Helbig K.L., Hoei-Hansen C.E., Koko M., Rannap M., Lauxmann S., Huq M., Schneider M.C., et al. . 2019. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain. 142:376–390. 10.1093/brain/awy326 [DOI] [PubMed] [Google Scholar]
- Loewen J.L., Barker-Haliski M.L., Dahle E.J., White H.S., and Wilcox K.S.. 2016. Neuronal Injury, Gliosis, and Glial Proliferation in Two Models of Temporal Lobe Epilepsy. J. Neuropathol. Exp. Neurol. 75:366–378. 10.1093/jnen/nlw008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago L.F., Yuan Y., Wagnon J.L., Hull J.M., Frasier C.R., O’Malley H.A., Meisler M.H., and Isom L.L.. 2017. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc. Natl. Acad. Sci. USA. 114:2383–2388. 10.1073/pnas.1616821114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossin C., Wang D.W., Rhodes T.H., Vanoye C.G., and George A.L. Jr. 2002. Molecular basis of an inherited epilepsy. Neuron. 34:877–884. 10.1016/S0896-6273(02)00714-6 [DOI] [PubMed] [Google Scholar]
- Mason E.R., Wu F., Patel R.R., Xiao Y., Cannon S.C., and Cummins T.R.. 2019. Resurgent and Gating Pore Currents Induced by De Novo SCN2A Epilepsy Mutations. eNeuro. 6:ENEURO.0141-19.2019. [DOI] [PMC free article] [PubMed]
- Misra S.N., Kahlig K.M., and George A.L. Jr. 2008. Impaired NaV1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia. 49:1535–1545. 10.1111/j.1528-1167.2008.01619.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki H., Oyama F., Inoue R., Aosaki T., Abe T., Kiyonari H., Kino Y., Kurosawa M., Shimizu J., Ogiwara I., et al. . 2014. Singular localization of sodium channel β4 subunit in unmyelinated fibres and its role in the striatum. Nat. Commun. 5:5525 10.1038/ncomms6525 [DOI] [PubMed] [Google Scholar]
- Nakamura K., Kato M., Osaka H., Yamashita S., Nakagawa E., Haginoya K., Tohyama J., Okuda M., Wada T., Shimakawa S., et al. . 2013. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 81:992–998. 10.1212/WNL.0b013e3182a43e57 [DOI] [PubMed] [Google Scholar]
- O’Roak B.J., Deriziotis P., Lee C., Vives L., Schwartz J.J., Girirajan S., Karakoc E., Mackenzie A.P., Ng S.B., Baker C., et al. . 2011. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43:585–589. 10.1038/ng.835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I., Ito K., Sawaishi Y., Osaka H., Mazaki E., Inoue I., Montal M., Hashikawa T., Shike T., Fujiwara T., et al. . 2009. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 73:1046–1053. 10.1212/WNL.0b013e3181b9cebc [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I., Miyamoto H., Morita N., Atapour N., Mazaki E., Inoue I., Takeuchi T., Itohara S., Yanagawa Y., Obata K., et al. . 2007. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27:5903–5914. 10.1523/JNEUROSCI.5270-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva M., Berkovic S.F., and Petrou S.. 2012. Sodium channels and the neurobiology of epilepsy. Epilepsia. 53:1849–1859. 10.1111/j.1528-1167.2012.03631.x [DOI] [PubMed] [Google Scholar]
- Onkal R., Mattis J.H., Fraser S.P., Diss J.K., Shao D., Okuse K., and Djamgoz M.B.. 2008. Alternative splicing of Nav1.5: an electrophysiological comparison of ‘neonatal’ and ‘adult’ isoforms and critical involvement of a lysine residue. J. Cell. Physiol. 216:716–726. 10.1002/jcp.21451 [DOI] [PubMed] [Google Scholar]
- Rhodes T.H., Lossin C., Vanoye C.G., Wang D.W., and George A.L. Jr. 2004. Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc. Natl. Acad. Sci. USA. 101:11147–11152. 10.1073/pnas.0402482101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders S.J., Campbell A.J., Cottrell J.R., Moller R.S., Wagner F.F., Auldridge A.L., Bernier R.A., Catterall W.A., Chung W.K., Empfield J.R., et al. . 2018. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 41:442–456. 10.1016/j.tins.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L., et al. . 2012. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 485:237–241. 10.1038/nature10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Hieber C., and Bischofberger J.. 2010. Fast sodium channel gating supports localized and efficient axonal action potential initiation. J. Neurosci. 30:10233–10242. 10.1523/JNEUROSCI.6335-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt P.W.E., Ben-Shalom R., Keeshen C.M., Burke K.J. Jr., Clarkson R.L., Sanders S.J., and Bender K.J.. 2019. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron. 103:673–685.e5. 10.1016/j.neuron.2019.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C.H., Kahlig K.M., and George A.L. Jr. 2011. SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia. 52:1000–1009. 10.1111/j.1528-1167.2011.03040.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C.H., Porter J.C., Kahlig K.M., Daniels M.A., and George A.L. Jr. 2012. Nontruncating SCN1A mutations associated with severe myoclonic epilepsy of infancy impair cell surface expression. J. Biol. Chem. 287:42001–42008. 10.1074/jbc.M112.421883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C., Wang K., Ke W., Guo H., and Shu Y.. 2014. Molecular identity of axonal sodium channels in human cortical pyramidal cells. Front. Cell. Neurosci. 8:297 10.3389/fncel.2014.00297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touma M., Joshi M., Connolly M.C., Grant P.E., Hansen A.R., Khwaja O., Berry G.T., Kinney H.C., Poduri A., and Agrawal P.B.. 2013. Whole genome sequencing identifies SCN2A mutation in monozygotic twins with Ohtahara syndrome and unique neuropathologic findings. Epilepsia. 54:e81–e85. 10.1111/epi.12137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uebachs M., Opitz T., Royeck M., Dickhof G., Horstmann M.T., Isom L.L., and Beck H.. 2010. Efficacy loss of the anticonvulsant carbamazepine in mice lacking sodium channel beta subunits via paradoxical effects on persistent sodium currents. J. Neurosci. 30:8489–8501. 10.1523/JNEUROSCI.1534-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon J.L., Barker B.S., Hounshell J.A., Haaxma C.A., Shealy A., Moss T., Parikh S., Messer R.D., Patel M.K., and Meisler M.H.. 2015. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann. Clin. Transl. Neurol. 3:114–123. 10.1002/acn3.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek R.E., Merrick D.K., and Catterall W.A.. 1989. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 3:695–704. 10.1016/0896-6273(89)90238-9 [DOI] [PubMed] [Google Scholar]
- Wolff M., Johannesen K.M., Hedrich U.B.S., Masnada S., Rubboli G., Gardella E., Lesca G., Ville D., Milh M., Villard L., et al. . 2017. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 140:1316–1336. 10.1093/brain/awx054 [DOI] [PubMed] [Google Scholar]
- Xu R., Thomas E.A., Jenkins M., Gazina E.V., Chiu C., Heron S.E., Mulley J.C., Scheffer I.E., Berkovic S.F., and Petrou S.. 2007. A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Mol. Cell. Neurosci. 35:292–301. 10.1016/j.mcn.2007.03.003 [DOI] [PubMed] [Google Scholar]
- Yamagata T., Ogiwara I., Mazaki E., Yanagawa Y., and Yamakawa K.. 2017. Nav1.2 is expressed in caudal ganglionic eminence-derived disinhibitory interneurons: Mutually exclusive distributions of Nav1.1 and Nav1.2. Biochem. Biophys. Res. Commun. 491:1070–1076. 10.1016/j.bbrc.2017.08.013 [DOI] [PubMed] [Google Scholar]
- Ye M., Yang J., Tian C., Zhu Q., Yin L., Jiang S., Yang M., and Shu Y.. 2018. Differential roles of NaV1.2 and NaV1.6 in regulating neuronal excitability at febrile temperature and distinct contributions to febrile seizures. Sci. Rep. 8:753 10.1038/s41598-017-17344-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.H., Mantegazza M., Westenbroek R.E., Robbins C.A., Kalume F., Burton K.A., Spain W.J., McKnight G.S., Scheuer T., and Catterall W.A.. 2006. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9:1142–1149. 10.1038/nn1754 [DOI] [PubMed] [Google Scholar]
- Zerem A., Lev D., Blumkin L., Goldberg-Stern H., Michaeli-Yossef Y., Halevy A., Kivity S., Nakamura K., Matsumoto N., Leshinsky-Silver E., et al. . 2014. Paternal germline mosaicism of a SCN2A mutation results in Ohtahara syndrome in half siblings. Eur. J. Paediatr. Neurol. 18:567–571. 10.1016/j.ejpn.2014.04.008 [DOI] [PubMed] [Google Scholar]