

Abstract
The apelinergic system comprises the apelin receptor and its cognate apelin and elabela peptide ligands of various lengths. This system has become an increasingly attractive target for pulmonary and cardiometabolic diseases. Small molecule regulators of this receptor with good drug-like properties are needed. Recently, we discovered a novel pyrazole based small molecule agonist 8 of APJ (EC50 = 21.5 μM, Ki = 5.2 μM) through focused screening which was further optimized to initial lead 9 (EC50 = 0.800 μM, Ki = 1.3 μM). In our efforts to synthesize more potent agonists and to explore the structural features important for apelin receptor agonism, we carried out structural modifications at N1 of the pyrazole core as well as the amino acid side-chain of 9. Systematic modifications at these two positions provided potent small molecule agonists exhibiting EC50 values of < 100 nM. Recruitment of β-arrestin as a measure of desensitization potential of select compounds was also investigated. Functional selectivity was a feature of several compounds with a bias towards calcium mobilization over β-arrestin recruitment. These compounds may be suitable as tools for in vivo studies of APJ function.
Keywords: Apelin, APJ, agonist, pyrazole, AGTRL1, APLNR
Graphical Abstract
Introduction
The apelinergic system comprises the endogenous apelin peptides and its cognate G-protein coupled receptor, APJ, encoded by the gene AGTRL1 (angiotensin receptor like 1).1 The apelin receptor is considered physiologically important due to its presence across many species including human, monkey, pig, rodents, frog and zebrafish.2 The apelin receptor is a 380 amino acid protein having ~33% sequence identity to the AT1 receptor.1 Apelin-36 was the first endogenous peptide identified along with other smaller peptides (apelin-17, apelin-13, apelin-12, pyr-apelin-13) that were derived from the cleavage of paired basic residues (Arg-Arg and Arg-Lys) of the 77 amino acid precursor protein.3–7 Known apelin peptides are readily hydrolyzed in vivo (half-life ~5 min).8 These proteins belong to an understudied component of the renin-angiotensin system with angiotensin-converting-enzyme II (ACE II) being involved in degradation of apelin in vivo.8,9 However, none of the known angiotensin receptor ligands or peptides activate apelin receptor.1 Recently, Elabela/Toddler was identified as the second endogenous ligand of this receptor.10–12 Identification of these two sets of endogenous ligands facilitated molecular characterization of apelin receptor. Apelin receptors are primarily coupled to inhibitory Gαi proteins and their activation is associated with inhibition of forskolin-stimulated cAMP production.3 Additionally, apelin receptor can also activate PLC mediated calcium flux through Gαq coupling13,14 and recruit β-arrestin that desensitizes the receptor utilizing clathrin mediated endocytosis.15 Apelin and apelin receptor are widely expressed across tissues in both human and rodents. In rat CNS, apelin mRNA is expressed within the hypothalamus where it is co-expressed with vasopressin mRNA in paraventricular (PVN) and supraoptic nuclei (SON).16,17 Peripherally, apelin receptor is expressed in many organs including kidney, lungs, adipose tissue, liver and heart.18–20 Activation of the apelinergic system has many beneficial effects including protection against hypertensive disorders, excitotoxic neuronal damage, pulmonary arterial hypertension, metabolic syndrome and heart failure.21,22 The receptor also plays an important role during embryonal development through regulation of vasculogenesis.23
Efforts are underway to produce small molecule agonists of apelin receptor with improved drug-like properties because the endogenous peptide ligands have short half-lives and limited utility as therapeutics. A number of small molecules have been reported. Iturrioz and colleagues reported a non-peptide functional apelin receptor agonist E339–3D6 (Figure 1) that exhibited an EC50 of 90 nM (FRET) and Ki value of ~400 nM in both rat and human apelin receptor.24
Figure 1.

Non-peptide apelin receptor agonists reported in the literature
The ML233 core (Figure 1) ((E)-2-cyclohexyl-5-methyl-4-(phenylsulfonyloxyimino)cyclohexa-2,5-dienone) was identified through a high throughput screen of ~330 600 compounds from the NIH small molecule library.25 ML233 is a full agonist and exhibits an EC50 of 3.7 μM (β-arrestin), but it displayed poor solubility and stability in microsomal fractions. In addition, ML233 had significant off-target binding at several other receptors including the 5-HT1A, α2C adrenergic, mu-opioid, benzylpiperazine receptors and norepinephrine transporter. A number of patent disclosures have also emerged. Sanofi Aventis,26 and Sanford-Burnham Medical Research Institute,27 have disclosed small molecule agonists based on the substituted benzimidazole (US20140094450 A1) and triazole core (WO 2015/184011 A2) respectively (Figure 1). In addition, Bristol-Myers Squibb and Amgen have disclosed small molecules based on dihydropyrimidine-4-one core (WO 2017/096130A1)28 and triazole core (WO 2017/192485A1)29 respectively. A small molecule benzimidazole derivative related to the Sanofi Aventis scaffold CMF-019 (Figure 1) was reported to be G protein biased small molecule apelin receptor agonist. CMF-019 was ~400 fold bias towards Gαi signaling over β-arrestin recruitment and ~6000 fold bias over receptor internalization. Studies using these biased agonists that preferentially stimulate G-protein activation over β-arrestin recruitment have demonstrated improved in vivo efficacy (vasodilation and inotropic actions) without β-arrestin dependent cardiac hypertrophy.30–32
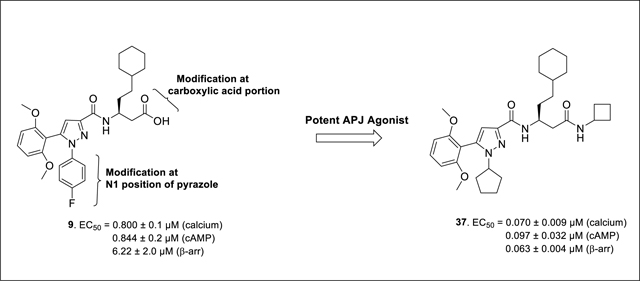
Our laboratory has previously reported a novel pyrazole scaffold 8 (EC50 = 21.5 μM (calcium), Ki = 5.2 μM) identified through focused screening, which was further optimized to an initial lead 9 (EC50 = 0.800 μM (calcium), Ki = 1.3 μM) (Figure 2).33 In our efforts to identify more potent agonists as tool compounds and to explore the structural features important for apelin receptor agonism, we carried out modifications at N1 position of the pyrazole core as well as the amino acid side-chain of the lead compound 9. Synthesized compounds were characterized using calcium mobilization, inhibition of forskolin-induced cAMP accumulation and β-arrestin recruitment assays. Considering the potential importance of selectivity over the β-arrestin pathway,30,31 bias factors for the most promising compounds were calculated.
Figure 2.

Chemical structure of initial hit 8, identified through focused screen and optimized to lead compound 9
Chemistry
Scheme 1 depicts the synthetic route employed to prepare target compounds 9 – 38. Synthesis of analogs 25 and 37 are shown as representative examples. 2, 6-Dimethoxy acetophenone 39 was condensed with diethyl oxalate to afford the sodium salt of diketone 40 in quantitative yield. Reaction of 40 with isobutylhydrazine or cyclopentylhydrazine trifluoroacetate in refluxing ethanol provided 1, 5-pyrazoles 41a, 42a and 1,3-pyrazoles 41b, 42b in a ratio of 4:1. Transformation of 41a and 42a to acids 43 and 44 was achieved using alkaline hydrolysis with lithium hydroxide monohydrate. Coupling of carboxylic acids 43 and 44 with (S)-tert-butyl 3amino-5-cyclohexylpentanoate33,34 in the presence of benzotriazole-1-yl-oxy-tris(dimethylamino)-phosphonium hexafluorophosphate (BOP) afforded 45 and 46. Tert-butyl esters 45 and 46 were deprotected using trifluoroacetic acid to provide acids 12 and 13. Finally, carboxylic acids 12 and 13 were coupled to cyclobutylamine using BOP to obtain 25 and 37 respectively.
Scheme 1:

Synthesis of compound 25 and 37
Reagents and conditions: a) Diethyl oxalate, EtONa, EtOH, reflux, 6 h, 97%; b) Isobutylhydrazine trifluoroacetate/cyclopentylhydrazine trifluoroacetate, EtOH, reflux, 16 h; 40/73% c) LiOH, MeOH/THF/H2O, rt, 18 h, 96/98%; d) (S)-tert-butyl 3-amino-5-cyclohexylpentanoate, BOP, Et3N, THF, rt, 1.5 h, 91/87%; e) TFA, DCM, rt, 1.5 h, 54/94%; f) cyclobutylamine, BOP, Et3N, THF, rt, 3 h, 61/75%.
As shown in Scheme 1 the synthesis of compounds 41 and 42 provides a mixture of isomers at the N1 and N2 pyrazole positions in ratio 4:1 that are readily separable using normal phase chromatography. The structures of hydrolyzed carboxylic acids 43 and 44 were not previously described but were confirmed using X-Ray crystallography (Figures 3 & 4). The NMR shift assignments for 41 and 42 were used as a guide to identify the additional N1 vs N2 alkyl substituted pyrazoles synthesized in this study.
Figure 3.

Crystal structure of compound 43
Figure 4.

Crystal structure of compound 44
Results and Discussion
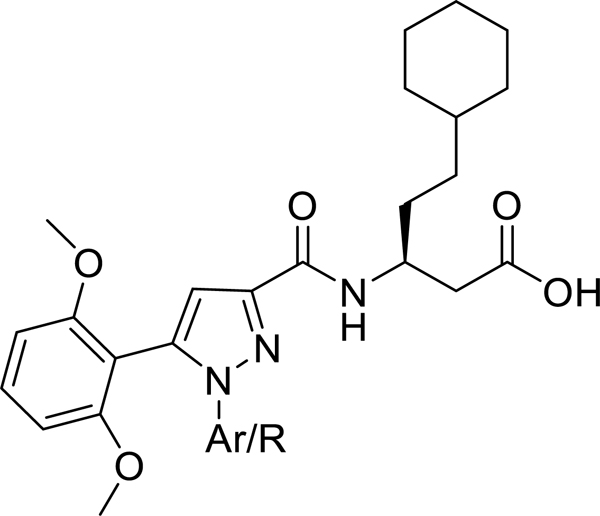
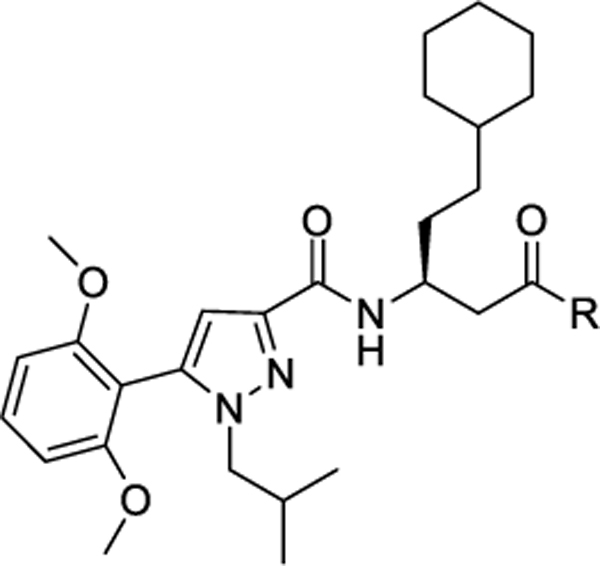
Our laboratory has previously reported the novel pyrazole based small molecule agonist hit 8 (Ca2+ EC50 = 21.5 μM, Ki = 5.2 μM) identified through focused screening that was further optimized to an initial lead 9 (Ca2+ EC50 = 0.800 μM, cAMP EC50 = 844 nM, Ki = 1.3 μM). In addition to improving potency and affinity, compound 9 exhibited significantly reduced off-target NTR2 activity and did not activate or inhibit the AT1 receptor (Table 1).33 Considering the structural similarity with the Sanofi scaffold 7, we hypothesized that modification of 9 at N1 position could potentially enhance apelin receptor potency. Substitution of 4-fluorophenyl in 9 with various alkyl or cycloalkyl groups were initially considered as these were envisioned to provide similar hydrophobicity and steric bulk (Table 1) as phenyl. In addition, these analogs would also help determine whether aromatic substitutions were optimal at the N1 position for apelin receptor potency with this scaffold. The agonist activities of synthesized analogs were evaluated using both calcium mobilization and cAMP generation functional assays. In addition to potency and efficacy, recruitment of β-arrestin was also evaluated.
Table 1.
Modification at N1 pyrazole core of compound with cyclohexyl ethyl amino acid side chain
 |
| Compd. | R | APJ Calcium EC50* μM±SEM (%Emax) |
APJ cAMP EC50* μM±SEM (%Emax) |
APJ β-arrestin EC50* μM±SEM (%Emax) |
APJ Binding Ki* (μM±SEM) |
AT1 Calcium EC50* (μM±SEM) |
|---|---|---|---|---|---|---|
| Pyr-Apelin-13 | - | 0.002 ± 0.0003 | 0.0003 ± 0.0001 | 0.001 ± 0.0005 | 0.001 ± 0.0005 | ND* |
| 9. | 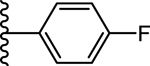 |
0.800 ± 0.1 (91) | 0.844 ± 0.2 (103) | 6.22 ± 2.0 (86) | 1.3 ± 0.3 | >10 |
| 10. | CH3 | 1.46 ± 0.4 (65) | 2.55 ± 1.6 (106) | 9.94 ± 1.4 (75) | ND | ND |
| 11. | CH2-CH2-CH3 | 0.416 ± 0.02 (89) | 0.281 ± 0.09 (109) | 3.28 ± 0.4 (67) | ND | ND |
| 12. | 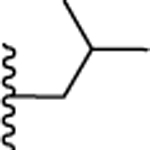 |
0.649 ± 0.1 (101) | 0.915 ± 0.5 (108) | 5.59 ± 0.06 (81) | ND | ND |
| 13. | 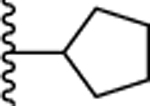 |
0.424 ± 0.07 (96) | 0.238 ± 0.05 (103) | 2.47 ± 0.4 (77) | ND | ND |
| 14. | 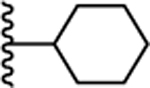 |
0.380 ± 0.03 (91) | 0.518 ± 0.08 (113) | 3.26 ± 0.08 (83) | ND | ND |
| 15. | 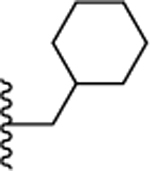 |
>10 | >10 | >10 | ND | ND |
| 16. | 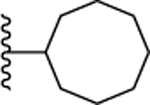 |
>10 | >10 | >10 | ND | ND |
EC50 and Ki values are averages of multiple experiments performed in duplicate unless otherwise stated ± standard error of the mean.
ND = Not Done
A. Modification at N1 pyrazole core of compound with cyclohexyl ethyl amino acid side chain:
The N1 methyl substituted analog 10 (Ca2+ EC50 = 1.46 μM, cAMP EC50 = 2.55 μM) exhibited approximately 2-fold reduced potency compared to the aromatic N1 phenyl substituent 9 (Ca2+ EC50 = 0.800 μM). Increasing the chain length to three carbon propyl 11, enhanced potency by 2-fold (Ca2+ EC50 = 0.416 μM) compared to 9. Adding bulk with addition of branched alkyl substitution (isobutyl) 12, however, did not enhance potency (Ca2+ EC50 = 0.649 μM). Further, increasing the hydrophobic volume to five carbon cyclopentyl 13 (Ca2+ EC50 = 0.424 μM) and six carbon cyclohexyl 14 (Ca2+ EC50 = 0.380 μM) retained the 2-fold enhanced potency. Extending the cyclohexyl ring by one methylene 15 (Ca2+ EC50 >10 μM) significantly reduced the potency suggesting tolerance for alkyl/cycloalkyl moiety with optimum hydrophobic volume of 3–6 carbons. This was further confirmed by replacing 4-fluorophenyl with larger hydrophobic cyclooctyl moiety 16 (Ca2+ EC50 >10 μM) that proved detrimental for activity. The agonist activities of these compounds were also evaluated using cAMP assay to monitor the influence of these modifications on functional selectivity. Compounds 11 (cAMP EC50 = 0.281 μM), 13 (cAMP EC50 = 0.238 μM) and 14 (cAMP EC50 = 0.518 μM) exhibited 2 to 3-fold enhanced potency compared to the N1 phenyl substituted lead compound 9 (cAMP EC50 = 0.844 μM). By contrast, 12 had similar potency to 9 in the cAMP assay. Compounds 15 and 16 with bulky cyclohexyl methyl and cyclooctyl group were completely inactive. Recruitment of β-arrestin was also investigated as a potential measure of desensitization. Compounds 11, 12, 13, 14 exhibited > 2 μM potencies in β-arrestin assay suggesting the possibility of reduced receptor internalization and desensitization with these analogs. These studies suggested that an aryl ring was not required at N1 position of the pyrazole core to retain APJ receptor activity. Compounds substituted with alkyl/cycloalkyl moieties and with optimum hydrophobic volumes (n-propyl, cyclopentyl, cyclohexyl) exhibited enhanced potency and preference towards calcium/cAMP signaling over β-arrestin recruitment.
B. Modification of carboxylic acid moiety of 12:
Next, we focused on optimizing the carboxyl acid of 12. Simple conversion of carboxylic acid to primary amide 17 (Table 2) was detrimental for activity (Ca2+ EC50 > 5.75 μM). This suggested that either an acidic group or bulky amide substitution was necessary to retain activity. The first substituted amide compound 18 with glycine methyl ester displayed ~2-fold enhanced potency (Ca2+ EC50 = 0.288 μM) compared to the carboxylic acid derivative 12 (Ca2+ EC50 = 0.649 μM) suggesting substituted amides versus acids could also retain or enhance potency. The N-methyl analog 19 was synthesized to probe the importance of amide proton for receptor interaction. Compound 19 exhibited similar potency (Ca2+ EC50 = 0.237 μM) compared to the unsubstituted amide, suggesting the amide proton is not critical for activity through the calcium signaling pathway. In contrast, compound 19 exhibited 3-fold reduced potency (cAMP EC50 = 1.85 μM) in cAMP assay confirming the importance of the amide N-H to engage this pathway. Similarly, conversion of the glycine ester 18 (Ca2+ EC50 = 0.288 μM) to glycine acid 20 displayed 6-fold reduced potency (Ca2+ EC50 = 1.85 μM) signifying a preference for the hydrophobic ester over the polar carboxylic acid group. Since esters are metabolically labile and easily hydrolyzed in vivo, we synthesized relatively stable analogs 21 and 23 with 2-butanol and 2-butanone substitution respectively. Interestingly, compound 21 and 23 displayed 14-fold (Ca2+ EC50 = 0.046 μM) and 4-fold (Ca2+ EC50 = 0.144 μM) enhanced potency compared to the carboxylic acid derivative 12 (Ca2+ EC50 = 0.649 μM). In addition, compound 25 with cyclobutyl amide substitution also exhibited ~ 8-fold enhanced potency (Ca2+ EC50 = 81 nM). Taken together, these data suggest the hydrophobic amide substitution is important to enhance agonist potency. However, replacing glycine methyl ester with the more stable ethyl ether 24 exhibited reduced potency (Ca2+ EC50 = 1.70 μM). Compounds 17-25 were also evaluated for their ability to inhibit forskolin stimulated cAMP accumulation. Compounds 21 (cAMP EC50 = 0.118 μM), 22 (cAMP EC50 = 0.157 μM), 23 (cAMP EC50 = 0.246 μM) and 25 (cAMP EC50 = 0.283 μM) exhibited 2 to 3-fold enhanced potency compared to the carboxylic acid analog 12 (cAMP EC50 = 0.915 μM). Compounds 18, 21, 22, 23, 24, 25 were also potent in β-arrestin recruitment and exhibited an EC50 value < 0.5 μM. Thus, conversion of carboxylic acid to alkyl and cycloalkyl amides retained or enhanced potency for β-arrestin recruitment indicating a preference for hydrophobic amides is favorable for activating this signaling pathway. Binding affinities of select compounds were also evaluated using [125I]-apelin-13 in radioligand displacement assay. High binding affinities of compounds 21 (Ki = 0.036 μM), 22 (Ki = 0.038 μM), 23 (Ki = 0.172 μM), 25 (Ki = 0.054 μM) indicated that these compounds competitively displaced [125I]-apelin-13 and were bound to the orthosteric site of apelin receptor (Table 2). Compounds 21, 22, 23, 25 also did not activate AT1 receptor (EC50 > 10 μM), exhibiting selectivity of cyclohexyl ethyl series over the off-target AT1 receptor.
Table 2.
Modification of carboxylic acid moiety of 12
 |
| Compd. | R | APJ Calcium EC50* μM±SEM (%Emax) |
APJ cAMP EC50* μM±SEM (%Emax) |
APJ β-arrestin EC50* μM±SEM (%Emax) |
APJ Binding Ki* (μM±SEM) |
AT1 Calcium EC50* (μM±SEM) |
|---|---|---|---|---|---|---|
| 17. | -NH2 | 5.75 ± 1.1 (94) | >10 | 7.24 ± 0.9 (133) | ND* | ND |
| 18. | 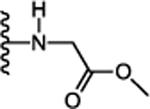 |
0.288 ± 0.03 (92) | 0.684 ± 0.1 (105) | 0.280 ± 0.001 (89) | ND | ND |
| 19. | 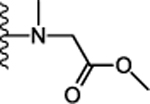 |
0.237 ± 0.04 (91) | 1.85 ± 0.2 (106) | 0.453 ± 0.014 (87) | ND | ND |
| 20. | 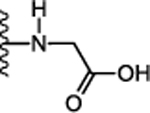 |
1.85 ± 0.5 (77) | 4.82 ± 1.1 (129) | 8.87 ± 2.7 (87) | ND | ND |
| 21. | 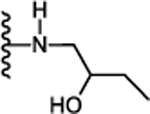 |
0.046 ± 0.008 (88) | 0.118 ± 0.02 (104) | 0.113 ± 0.02 (89) | 0.036 ± 0.006 | >10 |
| 22. | 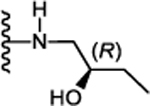 |
0.069 ± 0.02 (139) | 0.157 ± 0.08 (104) | 0.114 ± 0.005 (90) | 0.038 ± 0.01 | >10 |
| 23. | 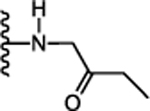 |
0.144 ± 0.03 (102) | 0.246 ± 0.02 (106) | 0.182 ± 0.045 (87) | 0.172 ± 0.1 | >10 |
| 24. | 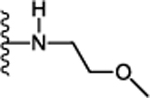 |
1.70 ± 0.04 (94) | 3.17 ± 0.8 (134) | 0.313 ± 0.04 (81) | ND | ND |
| 25. | 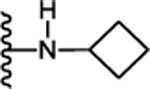 |
0.081 ± 0.02 (91) | 0.283 ± 0.08 (97) | 0.144 ± 0.008 (90) | 0.054 ± 0.01 | >10 |
EC50 and Ki values are averages of multiple experiments performed in duplicate unless otherwise stated ± standard error of the mean.
ND = Not Done
C. Modification of carboxylic acid moiety of 26:
Early on in our program we had evaluated if phenyl ethyl could replace the cyclohexyl ethyl sidechain because structural modifications of an aryl ring appeared more synthetically feasible. Since we had identified amide substituents that greatly enhanced agonist potency such as cyclobutyl amide (25), we evaluated if potency for (26, Ca2+ EC50 >10 μM) could be rescued with a similar set of modifications of the carboxylic acid moiety as shown in Table 2. All of the new amides with the phenylethyl sidechain (Table 3) had significantly enhanced potency over the parent acid 26, in keeping with the observations for the cyclohexyl ethyl analogs in Table 2. Compound 27 with glycine methyl ester exhibited > 400fold (Ca2+ EC50 = 0.021 μM) enhanced potency compared to the carboxylic acid derivative 26 (Ca2+ EC50 >10 μM). In addition, compound 27 displayed > 100-fold functional selectivity toward calcium signaling over cAMP (cAMP EC50 = 3.09 μM) and β-arrestin pathway (β-arr EC50 = 2.33 μM). However, functional selectivity was lost when the amide proton was replaced with methyl in analog 28 (Ca2+ EC50 = 0.027 μM, cAMP EC50 = 0.266 μM). Similarly, analogs 29 (Ca2+ EC50 = 0.411 μM, cAMP EC50 >10 μM, β-arr EC50 >10 μM), 30 (Ca2+ EC50 = 0.313 μM, cAMP EC50 >10 μM, β-arr EC50 >10 μM), 31 (Ca2+ EC50 = 0.162 μM, cAMP EC50 >10 μM, β-arr EC50 = 8.67 μM) with ethyl ether, 2-butanol and 2-butanone substitution were also selectively potent in the calcium assay. The cyclobutyl amide compound 32, however, had moderate potency (Ca2+ EC50 = 1.26 μM, cAMP EC50 > 3.48 μM) and did not demonstrate signaling bias. Interestingly, the cyclobutyl amide β-amino acid analog 34 displayed enhanced potency (Ca2+ EC50 = 0.083 μM) and was also selective towards calcium signaling over cAMP pathway. Thus, elongation of the carboxylic acid by one methylene in 34 demonstrated signaling bias for calcium over cAMP compared to 32. However, no signaling bias was observed in the cyclohexyl series with β-amino acid derivative 25. Binding affinities of select compounds were also evaluated. Compounds 27 (Ki = 0.187 μM), 28 (Ki = 0.134 μM), 31 (Ki = 0.571 μM), 34 (Ki = 0.449 μM) competitively displaced [125I]-apelin-13. Compounds 27, 28, 31, 34 were also selective over the off-target AT1 receptor (Table 3).
Table 3.
Modification of carboxylic acid moiety of 26.
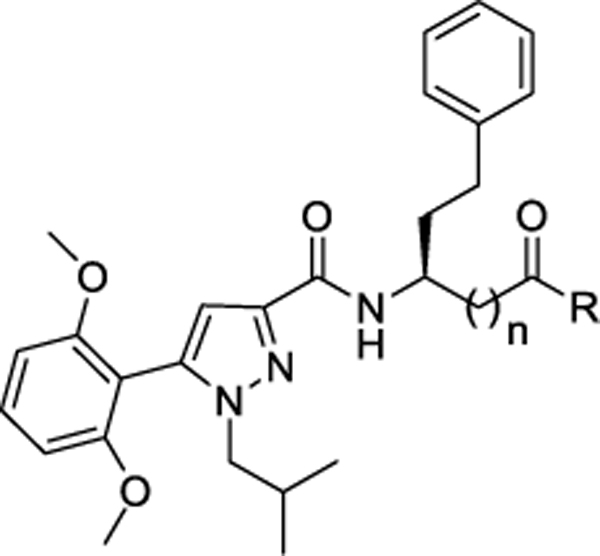 |
| Compd. | R | n | APJ Calcium EC50* μM±SEM (%Emax) |
APJ cAMP EC50* μM±SEM (%Emax) |
APJ β-arrestin EC50* μM±SEM (%Emax) |
APJ Binding Ki* (μM±SEM) (%Emax) |
AT1 Calcium EC50* (μM±SEM) |
|---|---|---|---|---|---|---|---|
| 26. | -OH | 0 | >10 | >10 | >10 | ND* | ND |
| 27. | 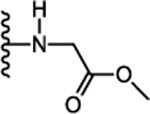 |
0 | 0.021± 0.007 (131) | 3.09 ± 3.0 (124) | 2.33 ± 0.9 (119) | 0.187± 0.06 | ND |
| 28. | 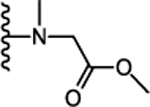 |
0 | 0.027 ± 0.02 (106) | 0.266 ± 0.057 (106) | 0.207 ± 0.002 (99) | 0.134± 0.09 | >10 |
| 29. | 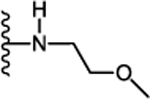 |
0 | 0.411± 0.09 (97) | >10 | >10 | ND | ND |
| 30. | 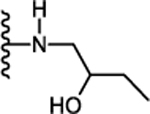 |
0 | 0.313± 0.01 (93) | >10 | >10 | ND | ND |
| 31. | 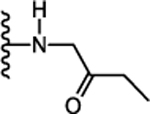 |
0 | 0.162± 0.001 (98) | >10 | 8.67± 0.1 (54) | 0.571± 0.3 | >10 |
| 32. | 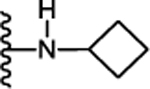 |
0 | 1.26 ± 0.8 (81) | 3.48± 0.1 (13) | >10 | ND | ND |
| 33. | -OH | 1 | >10 | >10 | >10 | ND | ND |
| 34. | 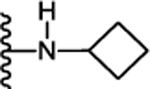 |
1 | 0.083± 0.01 (96) | >10 | 3.11 ± 0.2 (80) | 0.449± 0.08 | >10 |
EC50 and Ki values are averages of multiple experiments performed in duplicate ± standard error of the mean.
ND = Not Done
D. Modification at N1 position of pyrazole of the compound with cyclohexyl ethyl carboxyl amide side chain:
As noted earlier in Table 1, modification at the N1 position of pyrazole core had been critical in enhancing potency of carboxylic acid scaffold. To monitor the effects of similar N1 substituents on cyclobutylamide scaffold, compounds 35 – 38 (Table 4) were synthesized. The cyclobutyl carboxamide scaffold was chosen for modification since it showed better potency (Table 2) and was considered more stable than the ester or other alkyl moieties. At first, substitution at N1 with aromatic 4-fluorophenyl moiety 35 displayed moderate potency (Ca2+ EC50 = 0.425 μM). However, alkyl and cycloalkyl substituents 36, 25, 37, 38 significantly enhanced the potency of compounds compared to their carboxylic acid derivatives 11-14. Specifically, compound 36 (Ca2+ EC50 = 0.049 μM) and 37 (Ca2+ EC50 = 0.070 μM) with propyl and cyclopentyl substitution displayed 8-fold and 6-fold enhanced potency compared to the corresponding carboxylic acid derivative 11 (Ca2+ EC50 = 0.416 μM) and 13 (Ca2+ EC50 = 0.424 μM). Thus, modification at N1 position resulted in potent compounds 25, 36, 37, 38 that exhibited EC50 values ≤ 100 nM. However, these compounds were not functionally selective. Select potent compounds were also evaluated in radioligand binding assay. Compounds 25 (Ki = 0.054 μM), 36 (Ki = 0.060 μM), 37 (Ki = 0.059 μM), 38 (Ki = 0.147 μM) exhibited high binding affinities and competitively displaced [125I]-apelin-13.
Table 4.
Modification at N1 position of pyrazole core
 |
| Compd. | R | APJ Calcium EC50* μM±SEM (%Emax) |
APJ cAMP EC50* μM±SEM (%Emax) |
APJ β-arrestin EC50* μM±SEM (%Emax) |
APJ Binding Ki* μM±SEM), (%Emax) |
AT1 Calcium EC50* (μM) |
|---|---|---|---|---|---|---|
| 35. | 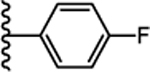 |
0.425 ± 0.06 (45) | 0.344 ± 0.004 (91) | 0.422 ± 0.008 (75) | ND* | ND |
| 36. |  |
0.049 ± 0.004 (91) | 0.107 ± 0.046 (110) | 0.078 ± 0.02 (77) | 0.060 ± 0.03 | >10 |
| 25. | 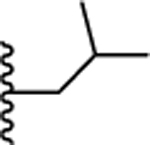 |
0.081 ± 0.025 (91) | 0.282 ± 0.08 (97) | 0.144 ± 0.008 (90) | 0.054 ± 0.01 | >10 |
| 37. | 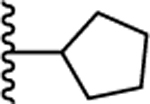 |
0.070 ± 0.009 (94) | 0.097 ± 0.032 (106) | 0.063 ± 0.004 (89) | 0.059 ± 0.02 | >10 |
| 38. | 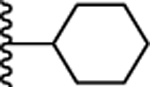 |
0.101 ± 0.001 (87) | 0.151 ± 0.04 (101) | 0.092 ± 0.007 (78) | 0.147 ± 0.06 | >10 |
EC50 and Ki values are averages of multiple experiments performed in duplicate unless otherwise stated ± standard error of the mean.
ND = Not Done
Human liver microsomal stability studies
Our long-term goal is to develop potent, metabolically stable APJ biased agonists for chronic in vivo studies to determine the importance of individual signaling cascades. Early in vitro assessment of metabolic stability of compounds is necessary to determine possible in vivo metabolic liabilities. Liver microsomal assays that includes cytochrome P450 system are the primary assays used to evaluate the stability of compounds. A small set of potent compounds (Table 5) were therefore evaluated for their stability in human liver microsomes. Our lead compound 9 demonstrated excellent stability (t1/2 = 229 min, CLINT = 5 mL/min/kg). All the other analogs tested except 31 (t1/2 = 34.71 min, CLINT = 36 mL/min/kg) with 2-butanone substitution are having very poor stability as indicated by high clearance values. This may be because all the other analogs contain alkyl/cycloalkyl substitution at N1 that possibly undergo oxidative metabolism at α-carbon attached to the N1 nitrogen. Excellent stability of compound 9 is possibly due to the presence of stable aromatic 4-fluorophenyl moiety at N1 pyrazole.
Table 5:
Microsomal stability of synthesized compounds
| Compd. | Half-life, t1/2 (min) |
Clearance, CLINT (mL/min/kg) |
|---|---|---|
| 7-ethoxy-4-methylcoumarin | 3.84 | 325 |
| 9. | 229.59 | 5 |
| 21. | 3.21 | 389 |
| 22. | 3.10 | 402 |
| 23. | 3.55 | 351 |
| 25. | 3.26 | 383 |
| 28. | 8.18 | 153 |
| 31. | 34.71 | 36 |
| 34. | 3.59 | 347 |
| 36. | 2.90 | 431 |
| 37. | 6.25 | 199 |
| 38. | 6.28 | 199 |
Effects of Modifications on Ligand Bias
As noted earlier, small molecule apelin receptor agonists have been generated that are G protein biased and result in increased cardiac contractility in vivo.30 We tested the effect of these site modifications on bias relative to the reference endogenous agonist Pyr-Apelin-13 by calculating bias factors35,36 between assays for G protein signaling (calcium and cAMP) and β-arrestin recruitment (Table 5). In general, two patterns were observed (Figure 5): compounds that displayed bias towards calcium signaling compared to cAMP inhibition and β-arrestin recruitment (Figure 5A) and compounds that did not display significant bias (< 10-fold) between pathways (Figure 5B). For those compounds that displayed significant bias between calcium signaling and cAMP or β-arrestin, bias factors generally ranged between 2 and 3 on a log scale (corresponding to 100- to 1000-fold bias towards calcium signaling).
Figure 5.

Radar plots of bias factors for (A) compounds that display bias towards calcium signaling and (B) compounds that have minimal (< 10-fold) bias towards any signaling pathways compared to the reference agonist Pyr-Apelin-13. Bias factors are shown on a logarithmic scale.
Conclusions
Systematic modifications at N1 position of the pyrazole core as well as the amino acid side chain resulted in potent small molecule agonists exhibiting EC50 values of ≤ 100 nM. Functional selectivity was a feature of several compounds with a bias towards calcium mobilization over β-arrestin recruitment. Modification at N1 position of the pyrazole core resulted in compounds 11, 13, 14 (n-propyl, cyclopentyl, cyclohexyl substitutions) that exhibited enhanced potency and functional selectivity towards calcium/cAMP over β-arrestin signaling. Conversion of the carboxylic acid to alkyl and cycloalkyl amides were well tolerated and enhanced potency. Compounds 21, 22, 25 with cyclohexyl ethyl side chain exhibited potency ≤ 100 nM in calcium assay were equally potent towards other signaling cascades (cAMP and β-arrestin). Modification at carboxylic acid portion of compounds having a phenylethyl side chain resulted in compounds 29, 30, 31 that displayed significant bias toward calcium signaling over β-arrestin cascade (bias factors between 2 (100-fold) and 3 (1000-fold) on a log scale). Similarly, compounds 27–31, 34 exhibited significant bias toward calcium signaling over cAMP cascade. Among the set of biased compounds, compound 31 displayed better metabolic stability (t1/2 = 34.71 min, CLINT = 36 mL/min/kg) and also did not activate the off-target AT1 receptor. Interestingly, elongation of the carboxylic acid by one methylene (β-amino acid) in 34 demonstrated signaling bias for calcium over cAMP compared to the one methylene short carboxylic acid derivative 32. However, no signaling bias was observed in cyclohexyl series with β-amino acid 25. Modification at N1 position of the pyrazole resulted in compounds 25, 36, 37, 38 that exhibited high potency across all the three signaling cascade. Specifically, compound 37 with N1 cyclopentyl substitution exhibited the highest potency (cAMP EC50 = 0.097 μM) inhibiting forskolin-induced cAMP accumulation. Compound 37 (cAMP EC50 = 0.097 μM) also exhibited ~10-fold enhanced potency compared to the starting lead 9 (cAMP EC50 = 0.844 μM). Lipinski’s “Rule of 5” have been useful guides for predicting oral absorption of compounds. The calculated properties of 37 (clogP 5.5, 2 donors, 5 acceptors, TPSA of 94.5 and a MW of 550.7) suggested that this compound might potentially be orally bioavailable. In conclusion, we have identified potent small molecule apelin receptor agonist 37 including several others that can be suitable as tool compounds for in vivo studies of apelin receptor upon additional characterization.
Experimental Section:
Reagents and starting materials were obtained from commercial suppliers and were used without purification. Reactions were conducted under N2 atmosphere using oven-dried glassware. All solvents and chemicals used were reagent grade. Anhydrous tetrahydrofuran (THF), dichloromethane (DCM), and N, N-dimethylformamide (DMF) were purchased from Fisher Scientific and used as such. Flash column chromatography was carried out using a Teledyne ISCO Combiflash Rf system and Redisep Rf gold pre-packed HP silica columns. Purity and characterization of compounds were established by combination of HPLC, TLC, LC-MS, and NMR analytical technique described below. 1H NMR spectra were recorded on a Bruker Avance DPX300 (300 MHz) spectrometer using CHCl3-d, MeOH-d4, DMSO-d6 with tetramethylsilane (TMS) (0.00 ppm) as the internal reference. Chemical shifts are reported in ppm relative to the solvent signal and coupling constants (J) values are reported in Hertz (Hz). Thin-layer chromatography (TLC) was performed on precoated silica gel GF Uniplates from Analtech and spots were visualized with UV light or I2 detection or phosphomolybdic acid stain. Low-resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). Optical rotations were measured on an Auto Pol III polarimeter at the sodium D line. Analytical and preparative HPLC was performed on an automated Varian ProStar HPLC system equipped with a Xterra® C18 RP18 (4.6 × 100 mm i. d., 3.5 μm) column, with a flow rate of 0.1 mL/min.; λmax = 254 nm; mobile phase A: H2O and mobile phase B: CH3CN. Purity of the target compounds was determined to be ≥ 95% by HPLC. Chemical names were generated using ChemDraw Ultra (CambridgeSoft, version 10.0).
Procedure: Scheme 1
Ethyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (40):
To a solution of sodium ethoxide (21% in EtOH) (5.4 mL, 14.37 mmol) was added dropwise a mixture of diethyl oxalate (1.85 mL, 13.690 mmol) and 2,6-dimethoxy acetophenone (2.45 g, 13.69 mmol) in anhydrous ethanol (15 mL). The resultant mixture was stirred at room temperature for 30 minutes, upon which yellow suspension formed. The reaction mixture was heated to reflux for 4 h. The reaction was cooled to room temperature. Ethanol was evaporated in vacuo. The resultant residue was triturated with diethyl ether (30 mL) and filtered to obtain sodium salt of ethyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate as yellow solid (4.0 g, 97 %). MS (ESI) m/z: Calcd. for C14H16O6 280.09 [M]+, found 279.3 [M-H]−.
Ethyl 5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylate (41a)
Step 1: Preparation of Isobutylhydrazine trifluoroacetate:
Isobutyraldehyde (1g, 13.87 mmol) and tert-butyl carbazate (1.8 g, 13.87 mmol) in methanol (25 mL) was stirred at room temperature for 1 h. The solvent was evaporated and the resulting solid was dried under in vacuo to give white solid of (E)-tert-butyl 2-(2-methylpropylidene)hydrazinecarboxylate in quantitative yield. Sodium cyanoborohydride (1.2 g, 20.13 mmol) was added portionwise to a mixture of the (E)-tert-butyl 2-(2-methylpropylidene)hydrazinecarboxylate (2.5 g, 13.42 mmol) in 75 % of aqueous acetic acid (25 mL) at room temperature. The resultant solution was stirred for 3 h at room temperature. The reaction mixture was neutralized with 1N NaOH, extracted with CH2Cl2 (3 × 25 mL), washed with saturated NaHCO3, dried with Na2SO4, filtered, and evaporated to give tert-butyl 2-isobutylhydrazinecarboxylate as oil (2.40 g, 95 %). 1H NMR (CDCl3, 300 MHz) δ 0.93 (d, J=6.78 Hz, 6 H), 1.46 (s, 9 H), 1.64 – 1.82 (m, 1 H), 2.43 (br. s., 1 H), 2.67 (d, J=6.78 Hz, 2 H). MS (ESI) m/z: Calcd. for C9H20N2O2 188.15 [M]+, found 189.3 [M+H]+.
Trifluoroacetic acid (12 mL) was added dropwise to a solution of the tert-butyl 2-isobutylhydrazinecarboxylate (2.4 g, 12.75 mmol) in CH2Cl2 (12 mL). The reaction mixture was stirred at room temperature for 1.5 h. The solvent was evaporated to give the trifluoroacetate salt of the title compound as colorless oil in quantitative yield. 1H NMR (CDCl3, 300 MHz) δ 1.04 (dd, J=9.04, 6.78 Hz, 6 H), 2.04 – 2.25 (m, 1 H), 3.02 (dd, J=6.97, 3.96 Hz, 2 H). MS (ESI) m/z: Calcd. for C4H12N2 88.10 [M]+, found 89.4 [M+H]+.
Step 2: Preparation of ethyl 5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylate (41a):
Sodium salt of ethyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (40) (1.2 g, 3.96 mmol) and isobutylhydrazine trifluoroacetate (0.962 g, 4.76 mmol) was mixed with glacial acetic acid (25 mL) and conc. HCl (0.6 mL). The reaction mixture was heated to reflux for 3.5 h. After cooling, reaction mixture was poured into water (25 mL). The aqueous layer was extracted with CH2Cl2 (3 × 30 mL) and the combined CH2Cl2 layer was washed with saturated aqueous NaHCO3. The organic layer was then washed with saturated brine, dried over Na2SO4, followed by filtration. The solvent was evaporated in vacuo. The residue was purified by silica gel flash chromatography (0–40% EtOAc:Hex) to give major N1-substituted title compound as colorless oil (535 mg, 40 %) and less polar N2-substituted, 2-isobutyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate as colorless oil (190 mg, 14 %).
Ethyl 5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylate (41a)
1H NMR (CDCl3, 300 MHz) δ 0.72 (d, J=6.78 Hz, 6 H), 1.39 (t, J=7.15 Hz, 3 H), 2.10–2.24 (m, 1 H), 3.72 (d, J=6.0 Hz, 2 H), 3.74 (s, 6 H), 4.41 (q, J=7.16 Hz, 2 H), 6.62 (d, J=8.67 Hz, 2 H), 6.73 (s, 1 H), 7.38 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C18H24N2O4 332.17 [M]+, found 333.4 [M+H]+.
Ethyl 5-(2,6-dimethoxyphenyl)-2-isobutyl-1H-pyrazole-3-carboxylate (41b)
1H NMR (CDCl3, 300 MHz) δ 0.94 (d, J=6.78 Hz, 6 H), 1.37 (t, J=7.16 Hz, 3 H), 2.26 (m, 1 H), 3.76 (s, 6 H), 4.33 (q, J=7.16 Hz, 2 H), 4.44 (d, J=7.16 Hz, 2 H), 6.61 (d, J=8.67 Hz, 2 H), 6.92 (s, 1 H), 7.28 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C18H24N2O4 332.17 [M]+, found 333.3 [M+H]+.
Ethyl 1-cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate (42a)
Step 1: Preparation of Cyclopentylhydrazine trifluoroacetate:
Cyclopentanone (2.0 g, 23.77 mmol) and tert-butyl carbazate (3.1 g, 23.77 mmol) in methanol (20 mL) was stirred at room temperature for 3 h. The solvent was evaporated and the resulting solid was dried under in vacuo to give white solid of Tert-butyl 2-cyclopentylidenehydrazinecarboxylate (4.6 g, 98 %). Sodium cyanoborohydride (2.1 g, 34.06 mmol) was added portionwise to a mixture of tert-butyl 2-cyclopentylidenehydrazinecarboxylate (4.5 g, 22.697 mmol) in 75 % of aqueous acetic acid (25 mL) at room temperature. The resultant solution was stirred for 3 h at room temperature. The reaction mixture was neutralized with 1N NaOH, extracted with CH2Cl2 (3 × 30 mL), washed with saturated NaHCO3, dried with Na2SO4, filtered, and evaporated to give tert-butyl 2-cyclopentylhydrazinecarboxylate as oil (4.4 g, 97 %). 1H NMR (CDCl3, 300 MHz) δ 1.40–1.55 (m, 4 H), 1.46 (s, 9 H), 1.63–1.75 (m, 4 H), 3.45–3.58 (m, 1 H). MS (ESI) m/z: Calcd. for C10H20N2O2 200.15 [M]+, found 223.3 [M+Na]+.
Trifluoroacetic acid (24 mL) was added dropwise to a solution of the tert-butyl 2-cyclopentylhydrazinecarboxylate (4.4 g, 21.97 mmol) in CH2Cl2 (24 mL). The reaction mixture was stirred at room temperature for 2.5 h. The solvent was evaporated to give the trifluoroacetate salt of the title compound as colorless oil in quantitative yield. 1H NMR (CDCl3, 300 MHz) δ 1.58 – 1.70 (m, 2 H), 1.72– 1.92 (m, 4 H), 1.95–2.09 (m, 2 H), 3.60–3.78 (m, 1 H). MS (ESI) m/z: Calcd. for C5H12N2 100.10 [M]+, found 101.2 [M+H]+.
Step 2: Preparation of ethyl 1-cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate (42a):
Sodium salt of ethyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (40) (1.2 g, 3.96 mmol) and cyclopentylhydrazine trifluoroacetate (1.3 g, 5.95 mmol) was mixed with glacial acetic acid (25 mL) and conc. HCl (0.6 mL). The reaction mixture was heated to reflux for 3 h. After cooling, reaction mixture was poured into water (50 mL). The aqueous layer was extracted with DCM (3 × 30 mL). The combined DCM layer was washed with saturated aqueous NaHCO3. The organic layer was then washed with saturated brine, and then dried over Na2SO4, followed by filteration. The solvent was evaporated in vacuo. The residue was purified by silica gel flash chromatography (0–40% EtOAc:Hex) to give N1-substituted title compound (1.0 g, 73 %, major product) as light yellow solid and less polar N2-substituted, 2-cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate as colorless oil (50 mg, 5 %).
1-cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate (42a):
1H NMR (CDCl3, 300 MHz) δ 1.38 (t, J=7.2 Hz, 3 H), 1.44 – 1.57 (m, 2 H), 1.84 – 1.98 (m, 4 H), 2.08 – 2.26 (m, 2 H), 3.74 (s, 6 H), 4.21–4.31 (m, 1 H), 4.39 (q, J=6.9 Hz, 2 H), 6.63 (d, J=8.3 Hz, 2 H), 6.69 (s, 1 H), 7.38 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C19H24N2O4 344.41 [M]+, found 345.0 [M+H]+.
2-cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate (42b):
1H NMR (CDCl3, 300 MHz) δ 1.36 (t, J=7.2 Hz, 3 H), 1.62 – 1.74 (m, 2 H), 1.85 – 1.98 (m, 2 H), 2.08 – 2.26 (m, 4 H) 3.77 (s, 6 H), 4.33 (q, J=7.16 Hz, 2 H), 5.68 (quin, J=7.49 Hz, 1 H), 6.62 (d, J=8.48 Hz, 2 H), 6.91 (s, 1 H), 7.27 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C19H24N2O4 344.41 [M]+, found 345.3 [M+H]+.
5-(2,6-Dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylic acid (43):
Lithium hydroxide monohydrate (189 mg, 4.51 mmol) in 1 mL of water was added to a solution of ethyl 5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylate (500 mg, 1.50 mmol) in MeOH (11 mL) and THF (2 mL). The mixture was stirred at room temperature for 18 h. The reaction mixture was concentrated to about half the volume and then extracted with ether (2 × 15 mL). The aqueous layer was acidified with 1 N HCl and extracted with CH2Cl2 (3 × 25 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4. The solvent was evaporated in vacuo to give the title compound as white solid (440 mg, 96 %). 1H NMR (CDCl3, 300 MHz) δ 0.74 (d, J=6.40 Hz, 6 H), 2.10–2.24 (m, 1 H), 3.72 (d, J=7.54 Hz, 2 H), 3.75 (s, 6 H), 6.63 (d, J=9.0 Hz, 2 H), 6.79 (s, 1 H), 7.40 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C16H20N2O4 304.14 [M]+, found 303.3 [M-H]−.
1-Cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylic acid (44):
Following the procedure described in 43, compound 44 was obtained from ethyl 1-cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate. 98 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 1.48 – 1.64 (m, 2 H), 1.84 – 2.02 (m, 4 H), 2.03 – 2.20 (m, 2 H), 3.75 (s, 6 H), 4.30 (quin, J=7.4 Hz, 1 H), 6.64 (d, J=8.3 Hz, 2 H), 6.76 (s, 1 H), 7.40 (t, J=8.3 Hz, 1 H). MS (ESI) m/z: Calcd. for C17H20N2O4 316.14 [M]+, found 317.2 [M+H]+, 315.4 [M-H]−.
(S)-Tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoate (45):
5-(2,6-Dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylic acid (43) (40 mg, 0.13 mmol) was dissolved in THF (2 mL). To the solution was added BOP (58 mg, 0.13 mmol) and triethylamine (0.055 mL, 0.39 mmol). The resulting mixture was stirred at room temperature for 15 minutes. (S)-Tert-butyl 3-amino-5-cyclohexylpentanoate (37 mg, 0.14 mmol) was added and stirred at room temperature for 3 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4. The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (0–50 % EtOAc:Hex) to give (S)-tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoate (45) as foam (65 mg, 91%). 1H NMR (CDCl3, 300 MHz) δ 0.75 (d, J=5.65 Hz, 6 H), 0.81–0.96 (m, 2 H), 1.10 – 1.36 (m, 6 H), 1.47 (s, 8 H), 1.61 – 1.73 (m, 8 H), 2.12 (dt, J=13.75, 7.06 Hz, 1 H), 2.54 (d, J=5.65 Hz, 2 H), 3.58 – 3.67 (m, 2 H), 3.72 (s, 3 H), 3.74 (s, 3 H), 4.31 – 4.42 (m, 1 H), 6.61 (d, J=8.67 Hz, 2 H), 6.69 (s, 1 H), 7.23 (s, 1 H), 7.37 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C31H47N3O5 541.35 [M]+, found 542.6 [M+H]+.
(S)-Tert-butyl 5-cyclohexyl-3-(1-cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3carboxamido)pentanoate (46):
Following the procedure described in 45, compound 46 was obtained using 1-cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylic acid (44) and (S)-tert-butyl 3-amino-5-cyclohexylpentanoate. 87% yield; yellow solid; 1H NMR (CDCl3, 300 MHz) δ 0.81 – 0.96 (m, 2 H), 1.10 – 1.38 (m, 8 H), 1.48 (s, 9 H), 1.59 – 1.77 (m, 8 H), 1.81 – 1.96 (m, 4 H), 2.00 – 2.16 (m, 2 H), 2.55 (d, J=5.46 Hz, 2 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 4.21 – 4.30 (m, 1 H), 4.30 – 4.43 (m, 1 H), 6.62 (d, J=8.48 Hz, 2 H), 6.67 (s, 1 H), 7.25 (br. s., 1 H), 7.37 (t, J=8.38 Hz, 1 H). MS (ESI) m/z: Calcd. for C32H47N3O5 553.35 [M]+, found 555.0 [M+H]+
(S)-5-Cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoic acid (12):
Trifluoroacetic acid (0.6 mL) was added dropwise to a solution of the (S)-tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoate (45) (60 mg, 0.11 mmol) in CH2Cl2 (1.5 mL). The reaction mixture was stirred at room temperature for 2.5 h. The solvent was evaporated to give the trifluoroacetate salt of the title compound. 54 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (d, J=6.78 Hz, 6 H), 0.81–0.98 (m, 3 H), 1.11–1.40 (m, 6 H), 1.60 – 1.78 (m, 8 H), 2.02–2.17 (m, 1 H), 3.66 (d, J=7.54 Hz, 2 H), 3.74 (s, 6 H), 4.29–4.44 (m, 1 H), 6.62 (d, J=8.67 Hz, 2 H), 6.72 (s, 1 H), 7.38 (t, J=8.48 Hz, 1 H), 7.37–7.51(m, 1 H). MS (ESI) m/z: Calcd. for C27H39N3O5 485.29 [M]+, found 486.5 [M+H]+.
(S)-5-Cyclohexyl-3-(1-cyclopentyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (13):
Following the procedure described in 12, compound 13 was obtained using (S)-tert-butyl 5-cyclohexyl-3-(1-cyclopentyl-5-(2,6-dimethoxyphenyl)-1Hpyrazole-3-carboxamido)pentanoate (46). 94% yield; white solid; 1H NMR (CDCl3, 300 MHz) δ ppm 0.78 – 1.00 (m, 3 H), 1.09 – 1.40 (m, 6 H), 1.49 – 1.77 (m, 4 H), 1.71 (t, J=7.44 Hz, 4 H), 1.821.98 (m, 4 H), 1.99 – 2.13 (m, 2 H), 2.65 – 2.80 (m, 2 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 4.21–4.33 (m, 2 H), 6.63 (d, J=8.48 Hz, 2 H), 6.68 (s, 1 H), 7.29 (s, 1 H), 7.38 (t, J=8.38 Hz, 1 H). MS (ESI) m/z: Calcd. for C28H39N3O5 497.29 [M]+, found 496.7[M-H]−
(S)-N-(1-(Cyclobutylamino)-5-cyclohexyl-1-oxopentan-3-yl)-5-(2,6-dimethoxyphenyl)-1isobutyl-1H-pyrazole-3-carboxamide (25):
Following the procedure described in 45, compound 25 was synthesized using (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole3-carboxamido)pentanoic acid (12) and cyclobutylamine. 61 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (d, J=6.40 Hz, 6 H), 0.80 – 0.98 (m, 3 H), 1.10 – 1.40 (m, 8 H), 1.61–1.72 (m, 5 H), 1.77 – 1.93 (m, 2 H), 2.05 – 2.17 (m, 1 H), 2.20–2.34 (m, 2 H), 2.52 (d, J=6.03 Hz, 2 H), 3.63 (d, J=7.16 Hz, 2 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 4.19 – 4.29 (m, 1 H), 4.32 – 4.43 (m, 1 H), 6.62 (d, J=8.67 Hz, 2 H), 6.67–6.72 (s, 1 H), 6.69 (s, 1 H), 7.09 (d, J=8.67 Hz, 1 H), 7.37 (t, J=8.67 Hz, 1 H). MS (ESI) m/z: Calcd. for C31H46N4O4 538.35 [M]+, found 539.6 [M+H]+.
(S)-N-(1-(Cyclobutylamino)-5-cyclohexyl-1-oxopentan-3-yl)-1-cyclopentyl-5-(2,6dimethoxyphenyl)-1H-pyrazole-3-carboxamide (37):
Following the procedure described in 45, compound 37 was synthesized using (S)-5-cyclohexyl-3-(1-cyclopentyl-5-(2,6dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (13) and cyclobutylamine. 75 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.88 (d, J=11.30 Hz, 2 H), 1.12 – 1.39 (m, 6 H), 1.60 – 1.77 (m, 9 H), 1.80 – 1.97 (m, 6 H), 2.00–2.13 (m, 2 H), 2.18–2.32 (m, 2 H), 2.24 (d, J=7.16 Hz, 2 H), 2.53 (d, J=6.40 Hz, 2 H), 3.74 (s, 3 H), 3.73 (s, 3 H), 4.15 – 4.46 (m, 3 H), 6.63 (d, J=8.29 Hz, 2 H), 6.67 (s, 1 H), 6.75 (d, J=8.29 Hz, 1 H), 7.06 (d, J=9.04 Hz, 1 H), 7.38 (t, J=8.38 Hz, 1 H). MS (ESI) m/z: Calcd. for C32H46N4O4 550.35 [M]+, found 551.6 [M+H] +.
Preparation of Analogs:
(S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxamido)pentanoic acid (10)
Step 1: Preparation of ethyl 5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxylate
Sodium salt of ethyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (40) (1.5 g, 4.96 mmol) and commercially available, methylhydrazine (0.26 ml, 4.96 mmol), was mixed with glacial acetic acid (30 mL) and conc. HCl (0.6 mL). The reaction mixture was heated to reflux for 4 h. After cooling, reaction mixture was poured into water (30 mL). The aqueous layer was extracted with CH2Cl2 (3 × 30 mL) and the combined CH2Cl2 layer was washed with saturated aqueous NaHCO3. The organic layer was then washed with saturated brine, dried over Na2SO4, followed by filtration. The solvent was evaporated in vacuo and the residue was purified by silica gel flash chromatography (0–40 % EtOAc:Hex) to give the title compound as oil (160 mg, 11 %). 1H NMR (CDCl3, 300 MHz) δ 1.40 (t, J=7.0 Hz, 3 H), 3.72 (s, 3 H), 3.76 (s, 6 H), 4.42 (q, J=7.2 Hz, 2 H), 6.64 (d, J=8.3 Hz, 2 H), 6.79 (s, 1 H), 7.39 (t, J=8.3 Hz, 1 H). MS (ESI) m/z: Calcd. for C15H18N2O4 290.13 [M]+, found 291.2 [M+H]+.
Step 2: Preparation of 5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxylic acid
Following the procedure as described in 45, compound 5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxylic acid was obtained using ethyl 5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxylate. 87 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 3.73 (s, 3 H), 3.78 (s, 6 H), 6.65 (d, J=8.5 Hz, 2 H), 6.84 (s, 1 H), 7.40 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C13H14N2O4 262.10 [M]+, found 263.3 [M+H]+, 261.3 [M-H]−.
Step 3: Preparation of (S)-tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxamido)pentanoate:
Following the procedure described in 45, compound (S)-tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxamido)pentanoate was obtained using 5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxylic acid and (S)-tert-butyl 3-amino-5-cyclohexylpentanoate. 87% yield; yellow solid; 1H NMR (CDCl3, 300 MHz) δ 0.78–0.95 (m, 2 H), 1.08–1.32 (m, 7 H), 1.48 (s, 9 H), 1.52 – 1.78 (m, 8 H), 2.53 (dd, J=5.46, 3.01 Hz, 1 H), 3.65 (s, 3 H), 3.76 (s, 6 H), 6.63 (d, J=8.48 Hz, 2 H), 6.76 (s, 1 H), 7.31–7.33 (br. s., 1 H), 7.37 (t, J=8.38 Hz, 1 H). MS (ESI) m/z: Calcd. for C28H41N3O5 499.30 [M]+, found 501.0 [M+H]+.
Step 4: Preparation of (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole3-carboxamido)pentanoic acid (10):
Using the procedure described in 12, compound (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxamido)pentanoic acid was obtained from (S)-tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-methyl-1Hpyrazole-3-carboxamido)pentanoate. 51 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.80 – 0.97 (m, 2 H), 1.11 – 1.37 (m, 6 H), 1.59–1.78 (m, 7 H), 2.71 (d, J=5.09 Hz, 2 H), 3.66 (s, 3 H), 3.76 (s, 6 H), 4.29–4.42 (m, 1 H), 6.63 (d, J=8.48 Hz, 2 H), 6.78 (s, 1 H), 7.38 (t, J=8.48 Hz, 1 H), 7.39–7.43 (m, 1 H). MS (ESI) m/z: Calcd. for C24H33N3O5 443.24 [M]+, found 442.6 [M-H]−.
Preparation of (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxamido)pentanoic acid (11)
Step 1: Preparation of ethyl 5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxylate
Step 1.1: Preparation of propylhydrazine trifluoroacetate :
Propionaldehyde (1g, 17.22 mmol) and tert-butyl carbazate (2.3 g, 17.22 mmol) in methanol (20 mL) was stirred at room temperature for 2 h. The solvent was evaporated and the resulting solid was dried under in vacuo to give white solid of (E)-tert-butyl 2-propyldiazenecarboxylate in quantitative yield. Sodium cyanoborohydride (1.5 g, 24.39 mmol) was added portionwise to a mixture of the (E)-tert-butyl 2-propyldiazenecarboxylate (2.8 g, 16.26 mmol) in 75 % of aqueous acetic acid (20 mL) at room temperature. The resultant solution was stirred for 3 h at room temperature. The reaction mixture was neutralized with 1N NaOH, extracted with CH2Cl2 (3 × 25 mL), washed with saturated NaHCO3, dried with Na2SO4, filtered, and evaporated to give title compound as oil in quantitative yield. 1H NMR (CDCl3, 300 MHz) δ 0.92 (t, J=9.0 Hz, 3 H),1.46 (s, 9 H), 1.44–1.55 (m, 2 H), 2.62 (bs, 2 H).
Trifluoroacetic acid (11 mL) was added dropwise to a solution of the tert-butyl 2-propylhydrazinecarboxylate (2.3 g, 13.20 mmol) in CH2Cl2 (11 mL). The reaction mixture was stirred at room temperature for 2.5 h. The solvent was evaporated to give the trifluoroacetate salt of the title compound as colorless oil in quantitative yield. 1H NMR (CDCl3, 300 MHz) δ 1.01 (t, J=9.0 Hz, 3 H), 1.71–1.87 (m, 2 H), 3.10 (t, J=7.0 Hz, 2 H). MS (ESI) m/z: Calcd. for C3H10N2 74.08 [M]+, found 75.2 [M+H]+.
Step 1.2: Preparation of ethyl 5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxylate
Sodium salt of ethyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (1.2 g, 3.96 mmol) and propylhydrazine trifluoroacetate (1.12 g, 5.95 mmol) was mixed with glacial acetic acid (30 mL) and conc. HCl (0.6 mL). The reaction mixture was heated to reflux for 3 h. After cooling, reaction mixture was poured into water (30 mL). The aqueous layer was extracted with CH2Cl2 (3 × 30 mL) and the combined CH2Cl2 layer was washed with saturated aqueous NaHCO3. The organic layer was then washed with saturated brine, dried over Na2SO4, followed by filtration. The solvent was evaporated in vacuo and the residue was purified by silica gel flash chromatography (0–40% EtOAc:Hex) to give the title light yellow foam (158 mg, 15 %). 1H NMR (CDCl3, 300 MHz) δ 0.76 (t, J=7.3 Hz, 3 H), 1.39 (t, J=7.2 Hz, 3 H), 1.69–1.82 (m, 2 H), 3.74 (s, 6 H), 3.88 (t, J=7.5 Hz, 2 H), 4.41 (q, J=7.2 Hz, 2 H), 6.63 (d, J=8.3 Hz, 2 H), 6.73 (s, 1 H), 7.39 (t, J=8.5 Hz, 1 H). MS (ESI) m/z: Calcd. for C17H22N2O4 318.16 [M]+, found 319.3 [M+H]+.
Step 2: Preparation of 5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxylic acid
Following the procedure as described in 45, compound 5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxylic acid was obtained using ethyl 5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxylate. 88 % yield; light yellow solid; 1H NMR (CDCl3, 300 MHz) δ 0.78 (t, J=7.5 Hz, 3 H), 1.72 – 1.84 (m, 2 H), 3.76 (s, 6 H), 3.87 (t, J=7.5 Hz, 2 H), 6.64 (d, J=8.3 Hz, 2 H), 6.78 (s, 1 H), 7.40 (t, J=8.3 Hz, 1 H). MS (ESI) m/z: Calcd. for C15H18N2O4 290.13 [M]+, found 289.2 [M-H]−.
Step 3: Preparation of (S)-tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxamido)pentanoate:
Following the procedure described in 46, compound (S)-tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxamido)pentanoate was obtained using 5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxylic acid and (S)-tert-butyl 3-amino-5-cyclohexylpentanoate. 84% yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.77 (t, J=7.44 Hz, 3 H), 0.82 – 0.96 (m, 2 H), 1.10 – 1.36 (m, 7 H), 1.47 (s, 9 H), 1.61 – 1.80 (m, 7 H), 2.54 (d, J=5.65 Hz, 2 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 3.79 (t, J=9.0 Hz, 2 H), 4.30 – 4.47 (m, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.70 (s, 1 H), 7.25 (br. s., 1 H), 7.37 (t, J=8.38 Hz, 1 H). MS (ESI) m/z: Calcd. for C30H45N3O5 527.34 [M]+, found 529.1 [M+H]+.
Step 4: Preparation of (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole3-carboxamido)pentanoic acid (11):
Following the procedure described in 12, compound 11 was obtained from deprotection of (S)-tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxamido)pentanoate. 84 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ ppm 0.78 (t, J=7.44 Hz, 3 H), 0.84 – 0.99 (m, 2 H), 1.09 – 1.37 (m, 6 H), 1.60 – 1.80 (m, 9 H), 2.65 – 2.78 (m, 2 H), 3.74 (s, 3 H), 3.75 (s, 3 H), 3.81 (t, J=6.0 Hz, 3 H), 4.28 – 4.39 (m, 1 H), 6.62 (d, J=8.48 Hz, 2 H), 6.72 (s, 1 H), 7.32 – 7.41 (m, 2 H). MS (ESI) m/z: Calcd. for C29H37N3O5 471.27 [M]+, found 470.6 [M-H]−.
Preparation of (S)-5-Cyclohexyl-3-(1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (14)
Step 1: Ethyl 1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate:
Sodium salt of methyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (40) (1.5 g, 3.31 mmol) and commercially available cyclohexylhydrazine hydrochloride (0.75 g, 3.31 mmol) was mixed with glacial acetic acid (30 mL) and conc. HCl (0.6 mL). The reaction mixture was heated to reflux for 4 h. After cooling, reaction mixture was poured into water (50 mL). The aqueous layer was extracted with CH2Cl2 (3 × 30 mL) and the combined CH2Cl2 layer was washed with saturated aqueous NaHCO3. The organic layer was then washed with saturated brine, dried over Na2SO4, followed by filteration. The solvent was evaporated in vacuo to give crude residue. The residue was purified by silica gel flash chromatography (0–40 % EtOAc:Hex) to give the title compound as white solid (820 mg, 46 %). 1H NMR (CDCl3, 300 MHz) δ 1.09 – 1.31 (m, 5 H), 1.37 (t, J=6.97 Hz, 3 H), 1.74 – 1.92 (m, 5 H), 1.98 – 2.11 (m, 3 H), 3.63 – 3.72 (m, 1 H), 3.74 (s, 3 H), 4.40 (q, J=7.16 Hz, 2 H), 6.64 (d, J=8.29 Hz, 2 H), 6.69 (s, 1 H), 7.39 (t, J=9.0 Hz, 1 H). MS (ESI) m/z: Calcd. for C20H26N2O4 358.19 [M]+, found 359.5 [M-H]−.
Step 2: 1-Cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylic acid:
Following the procedure as described in 45, compound 5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxylic acid was obtained using ethyl 1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate. 96 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 1.11 – 1.29 (m, 4 H), 1.57 – 1.67 (m, 1 H), 1.76– 2.06 (m, 6 H), 3.66 – 3.73 (m, 1 H), 3.75 (s, 6H), 6.65 (d, J=8.29 Hz, 2 H), 6.75 (s, 1 H), 7.40 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C18H22N2O4 330.38 [M]+, found 331.4 [M+H]+, 329.4 [M-H]−.
Step 3: (S)-Tert-butyl 5-cyclohexyl-3-(1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoate:
Following the procedure described in 45, compound (S)-tert-butyl 5-cyclohexyl-3-(1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoate was obtained using 1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylic acid and (S)-tert-butyl 3-amino-5-cyclohexylpentanoate. 93% yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.80 – 0.96 (m, 3 H), 1.10 – 1.35 (m, 10 H), 1.49 (s, 9 H), 1.61– 1.74 (m, 7 H), 1.77–1.95 (m, 5 H), 2.55 (d, J=5.27 Hz, 2 H), 3.58 – 3.70 (m, 1 H), 3.72 (s, 3 H), 3.74 (s, 3 H), 4.28 – 4.46 (m, 1 H), 6.63 (d, J=8.29 Hz, 2 H), 6.66 (s, 1 H), 7.29 (s, 1 H), 7.37 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C33H49N3O5 567.37 [M]+, found 568.7 [M+H]+.
Step 4: (S)-5-Cyclohexyl-3-(1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (14):
Following the procedure described in 12, compound 14 was obtained from deprotection of (S)-tert-butyl 5-cyclohexyl-3-(1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoate. 94 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ ppm 0.79 – 0.99 (m, 2 H), 1.10 – 1.37 (m, 10 H), 1.51 – 1.77 (m, 11 H), 1.78–1.94 (m, 2 H), 2.65 – 2.80 (m, 2 H), 3.60 – 3.71 (m, 1 H), 3.74 (s, 6 H), 4.20 – 4.33 (m, 1 H), 6.64 (d, J=8.48 Hz, 2 H), 6.67 (s, 1 H), 7.24 (br. s., 1 H), 7.38 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C29H41N3O5 511.30 [M]+, found 510.6 [M-H]−
(S)-5-Cyclohexyl-3-(1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (15):
Step 1: Preparation of ethyl 1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate
Step 1.1: Preparation of (Cyclohexylmethyl)hydrazine trifluoroacetate :
Commercially available cyclohexanecarbaldehyde (1g, 8.91 mmol) and tert-butyl carbazate (1.2 g, 8.91 mmol) in methanol (25 mL) was stirred at room temperature for 1 h. The solvent was evaporated and the resulting solid was dried under in vacuo to give white solid of (E)-tert-butyl 2-(cyclohexylmethylene)hydrazinecarboxylate in quantitative yield. Sodium cyanoborohydride (0.82 g, 13.12 mmol) was added portionwise to a mixture of the (E)-tert-butyl 2-(cyclohexylmethylene)hydrazinecarboxylate (2.0 g, 8.75 mmol) in 50 % of aqueous acetic acid (25 mL) at room temperature. The resultant solution was stirred for 2.5 h at room temperature. The reaction mixture was neutralized with 1N NaOH, extracted with CH2Cl2 (3 × 30 mL), washed with saturated NaHCO3, dried with Na2SO4, filtered, and evaporated to give title compound as oil (1.85 g, 92 %). 1H NMR (CDCl3, 300 MHz) δ 0.82 – 1.05 (m, 2 H), 1.13 – 1.33 (m, 4 H), 1.46 (s, 9 H), 1.59 – 1.84 (m, 5 H), 2.69 (d, J=6.78 Hz, 2 H). MS (ESI) m/z: Calcd. for C12H24N2O2 228.18 [M]+, found 229.4 [M+H]+.
Trifluoroacetic acid (8 mL) was added dropwise to a solution of the tert-butyl 2-isobutylhydrazinecarboxylate (2.2 g, 9.63 mmol) in CH2Cl2 (8 mL). The reaction mixture was stirred at room temperature for 1.5 h.The solvent was evaporated to give the trifluoroacetate salt of the title compound as colorless oil in quantitative yield. 1H NMR (CDCl3, 300 MHz) δ 0.20 – 0.85 (m, 4 H), 0.86 – 1.34 (m, 6 H), 1.62–1.69 (m, 1 H), 1. 70 – 1.82 (m, 2 H). MS (ESI) m/z: Calcd. for C7H16N2 128.13 [M]+, found 129.3 [M+H]+.
Step 1.2: Preparation of ethyl 1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole3-carboxylate:
Sodium salt of ethyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (1.2 g, 3.96 mmol) and (cyclohexylmethyl)hydrazine trifluoroacetate (0.960 g, 3.965 mmol) was mixed with glacial acetic acid (25 mL) and conc. HCl (0.6 mL). The reaction mixture was heated to reflux for 4 h. After cooling, reaction mixture was poured into water (25 mL). The aqueous layer was extracted with DCM (3 × 30 mL). The combined DCM layer was washed with saturated aqueous NaHCO3. The organic layer was then washed with saturated brine, and then dried over Na2SO4, followed by filteration. The solvent was evaporated in vacuo. The residue was purified by silica gel flash chromatography (0–50% EtOAc:Hex) to give the title compound as oil (0.510 g, 35 %) and 2-methyl cyclohexyl substituted isomer as oil (0.215 g, 15 %). 1H NMR (CDCl3, 300 MHz) δ 0.61 – 0.78 (m, 2 H), 0.97 – 1.19 (m, 2 H), 1.39 (t, J=8.48 Hz, 3 H), 1.45 – 1.66 (m, 6 H), 1.78–1.93 (m, 1 H), 3.74 (s, 6 H), 3. 74 (d, J=6.78 Hz, 2 H), 4.41 (q, J=7.16 Hz, 2 H), 6.63 (d, J=8.67 Hz, 2 H), 6.72 (s, 1 H), 7.39 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C21H28N2O4 372.20 [M]+, found 373.5 [M+H]+.
Step 2: Preparation of 1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylic acid:
Following the procedure as described in 45, compound 1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylic acid was obtained using ethyl 1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate. 95 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.62 – 0.81 (m, 2 H), 0.98 – 1.22 (m, 3 H), 1.43 – 1.66 (m, 5 H), 1.76–1.93 (m, 1 H), 3.73 (d, J=6.78 Hz, 2 H), 3.75 (s, 6 H), 6.63 (d, J=8.29 Hz, 2 H), 6.78 (s, 1 H), 7.40 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C19H24N2O4 344.17 [M]+, found 343.4 [M-H]−
Step 3: (S)-Tert-butyl 5-cyclohexyl-3-(1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoate:
Following the procedure described in 45, compound (S)-tert-butyl 5-cyclohexyl-3-(1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoate was obtained using 1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylic acid and (S)-tert-butyl 3-amino-5-cyclohexylpentanoate. 66% yield; clear foam; 1H NMR (CDCl3, 300 MHz) δ 0.63 – 0.78 (m, 2 H), 0.80 – 0.98 (m, 3 H), 1.02 – 1.36 (m, 9 H), 1.48 (s, 9 H), 1.53 – 1.85 (m, 12 H), 2.54 (d, J=5.46 Hz, 2 H), 3.62 – 3.69 (m, 2 H), 3.72 (s, 3 H), 3.74 (s, 3 H), 4.30 – 4.42 (m, 1 H), 6.62 (d, J=8.48 Hz, 2 H), 6.68 (s, 1 H), 7.22 (s, 1 H), 7.37 (t, J=8.29 Hz, 1 H). MS (ESI) m/z: Calcd. for C34H51N3O5 581.38 [M]+, found 582.8 [M+H]+.
Step 4: Preparation of (S)-5-cyclohexyl-3-(1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)1H-pyrazole-3-carboxamido)pentanoic acid (15):
Following the procedure described in 12, compound 15 was obtained from deprotection of (S)-tert-butyl 5-cyclohexyl-3-(1-(cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoate. 84 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.62–0.98 (m, 3 H), 1.02–1.87(m, 10 H), 1.421.84 (m, 13 H), 2.64–2.81 (m, 2 H), 3.62–3.72 (m, 2 H), 3.74 (s, 6 H), 4.24–4.39 (m, 1 H), 6.62 (d, J=8.10 Hz, 2 H), 6.71 (s, 1 H), 7.20–7.26 (m, 1 H), 7.28 – 7.41 (m, 1 H). MS (ESI) m/z: Calcd. for C30H43N3O5 525.32 [M]+, found 524.8 [M-H]−.
(S)-5-Cyclohexyl-3-(1-cyclooctyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (16)
Step 1: Ethyl 1-cyclooctyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate
Step 1.1: Preparation of tert-butyl 2-cyclooctylhydrazine carboxylate:
Octanone (1.43 g, 11.34 mmol) and tert-butyl carbazate (1.5 g, 11.34 mmol) in methanol (25 mL) was stirred at room temperature for 2 h. The solvent was evaporated and the resulting solid was dried under in vacuo to give white solid of (E)-tert-butyl 2-cyclooctyldiazenecarboxylate in quantitative yield. Sodium cyanoborohydride (0.980 g, 15.60 mmol) was added portionwise to a mixture of the (E)-tert-butyl 2-cyclooctyldiazenecarboxylate (2.50 g, 10.40 mmol) in 75 % of aqueous acetic acid (35 mL) at room temperature. The resultant solution was stirred for 3 h at room temperature. The reaction mixture was neutralized with 1N NaOH, extracted with CH2Cl2 (3 × 30 mL), washed with saturated NaHCO3, dried with Na2SO4, filtered, and evaporated to give title compound as oil (2.40 g, 95 %). 1H NMR (CDCl3, 300 MHz) δ 1.33–1.59 (m, 4 H), 1.46 (s, 9 H), 1.63–1.79 (m, 11 H). MS (ESI) m/z: Calcd. for C13H26N2O2 242.20 [M]+, found 243.4 [M+H]+.
Trifluoroacetic acid (10 mL) was added dropwise to a solution of the tert-butyl 2(naphthalen-2-ylmethyl)hydrazinecarboxylate (3.0 g, 12.38 mmol) in CH2Cl2 (10 mL). The reaction mixture was stirred at room temperature for 1.5 h. The solvent was evaporated to give the trifluoroacetate salt of the title compound as colorless oil in quantitative yield. 1H NMR (CDCl3, 300 MHz) δ 1.36 – 2.04 (m, 14 H), 3.22– 3.47 (m, 1 H). MS (ESI) m/z: Calcd. for C8H18N2 142.15 [M]+, found 143.3 [M+H]+.
Step 1.2: Preparation of ethyl 1-cyclooctyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate:
Sodium salt of ethyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (1.2 g, 3.97 mmol) and cyclooctylhydrazine trifluoroacetate (1.2 g, 4.76 mmol) was mixed with glacial acetic acid (25 mL) and conc. HCl (0.6 mL). The reaction mixture was heated to reflux for 3.5 h. After cooling, reaction mixture was poured into water (30 mL). The aqueous layer was extracted with DCM (3 × 30 mL). The combined DCM layer was washed with saturated aqueous NaHCO3. The organic layer was then washed with saturated brine, and then dried over Na2SO4, followed by filteration. The solvent was evaporated in vacuo. The residue was purified by silica gel flash chromatography (0–30% EtOAc:Hex) to give the title compound as gum (600 mg, 39 %). 1H NMR (CDCl3, 300 MHz) δ 1.18 – 1.29 (m, 2 H), 1.32 – 1.43 (m, 6 H), 1.37 (t, J=7.15 Hz, 3 H), 1.67 – 1.86 (m, 4 H), 2.15 – 2.29 (m, 2 H), 3.74 (s, 6 H), 3.98 – 4.09 (m, 1 H), 4.39 (q, J=7.03 Hz, 2 H), 6.64 (d, J=8.29 Hz, 2 H), 6.69 (s, 1 H), 7.39 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C22H30N2O4 386.22 [M]+, found 387.4 [M+H]+.
Step 2: Preparation of 1-cyclooctyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylic acid
Following the procedure as described in 46, compound 1-cyclooctyl-5-(2,6-dimethoxyphenyl)1H-pyrazole-3-carboxylic acid was obtained from hydrolysis of ethyl 1-cyclooctyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate. 77 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 1.21 – 1.65 (m, 8 H), 1.68 – 1.89 (m, 4 H), 2.10 – 2.33 (m, 2 H), 3.75 (s, 6 H), 3.98 – 4.09 (m, 1 H), 6.65 (d, J=8.29 Hz, 2 H), 6.74 (s, 1 H), 7.41 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C20H26N2O4 358.19 [M]+, found 357.5 [M-H]−.
Step 3: Preparation of (S)-tert-butyl 5-cyclohexyl-3-(1-cyclooctyl-5-(2,6-dimethoxyphenyl)1H-pyrazole-3-carboxamido)pentanoate:
Following the procedure described in 46, compound (S)-tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxamido)pentanoate was obtained using 5-(2,6-dimethoxyphenyl)-1-methyl-1H-pyrazole-3-carboxylic acid and (S)-tert-butyl 3-amino-5-cyclohexylpentanoate. 73% yield; clear foam; 1H NMR (CDCl3, 300 MHz) δ 0.80 – 0.97 (m, 2 H), 1.13 – 1.45 (m, 13 H), 1.48 (s, 9 H), 1.62 – 1.86 (m, 13 H), 2.02 – 2.18 (m, 2 H), 2.55 (d, J=5.46 Hz, 2 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 3.92–4.04 s, 1 H), 4.31 – 4.42 (m, 1 H), 6.62 (s, 1 H), 6.65 (d, J=4.33 Hz, 1 H), 7.25 – 7.28 (m, 1 H), 7.38 (t, J=8.38 Hz, 1 H). MS (ESI) m/z: Calcd. for C35H53N3O5 595.40 [M]+, found 596.9 [M+H]+.
Step 4: Preparation of (S)-5-cyclohexyl-3-(1-cyclooctyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (16):
Following the procedure described in 12, compound 16 was obtained from deprotection of (S)-tert-butyl 5-cyclohexyl-3-(1-cyclooctyl-5(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoate. 86 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.81 – 0.99 (m, 2 H), 1.11 – 1.47 (m, 13 H), 1.49–1.88 (m, 14 H), 2.03–2.18 (m, 2 H), 2.66 – 2.81 (m, 2 H), 3.74 (s, 5 H), 3.94 – 4.05 (m, 1 H), 4.21 – 4.32 (m, 1 H), 6.62 (s, 1 H), 6.66 (d, J=6.59 Hz, 2 H), 7.28 (br. s., 1 H), 7.39 (t, J=8.38 Hz, 1 H). MS (ESI) m/z: Calcd. for C31H45N3O5 539.34 [M]+, found 538.7 [M-H]−.
(S)-N-(1-amino-5-cyclohexyl-1-oxopentan-3-yl)-5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamide (17):
Following the procedure described in 45, compound 17 was synthesized using (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoic acid (12) and 2 M NH3. 72 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.75 (dd, J=6.59, 2.83 Hz, 6 H), 0.81 – 1.01 (m, 3 H), 1.11 – 1.36 (m, 6 H), 1.61 – 1.78 (m, 6 H), 2.06 – 2.17 (m, 1 H), 2.23 – 2.36 (m, 1 H), 2.58 (d, J=6.03 Hz, 2 H), 3.64 (d, J=7.16 Hz, 2 H), 3.74 (d, J=2.26 Hz, 6 H), 4.26 – 4.39 (m, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.68 (s, 1 H), 6.81 (br. s., 1 H), 7.09 (d, J=9.42 Hz, 1 H), 7.38 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C27H40N4O4 484.30 [M]+, found 485.6 [M+H]+.
(S)-Methyl 2-(5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanamido)acetate (18):
Following the procedure described in 45, compound 18 was synthesized using (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoic acid (12) and glycine methyl ester hydrochloride. 21 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (dd, J=6.78, 1.51 Hz, 6 H), 0.82 – 0.99 (m, 2 H), 1.10 – 1.36 (m, 7 H), 1.63 – 1.77 (m, 6 H), 2.11 (td, J=14.22, 7.35 Hz, 1 H), 2.62 (d, J=5.65 Hz, 2 H), 3.63 (d, J=7.16 Hz, 2 H), 3.68 (s, 3 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 4.04 (d, J=5.65 Hz, 2 H), 4.27 – 4.41 (m, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.68 (s, 1 H), 6.91 – 7.00 (m, 1 H), 7.18 (d, J=9.04 Hz, 1 H), 7.37 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C30H44N4O6 556.33 [M]+, found 557.8 [M+H]+.
(S)-Methyl 2-(5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)-N-methylpentanamido)acetate (19):
Following the procedure described in 45, compound 19 was synthesized using (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoic acid (12) and sarcosine methyl ester hydrochloride. 38 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (d, J=6.78 Hz, 6 H), 0.81 – 1.01 (m, 3 H), 1.10 – 1.40 (m, 6 H), 1.54 – 1.83 (m, 5 H), 2.06 – 2.18 (m, 1 H), 2.62 (dd, J=15.45, 6.78 Hz, 1 H), 2.84 – 2.96 (m, 2 H), 3.14 (s, 3 H), 3.63 (d, J=7.16 Hz, 2 H), 3.71 (s, 3 H), 3.72 (s, 3 H), 3.73 (s, 3 H), 4.04 – 4.21 (m, 2 H), 4.27–4.39 (m, 1 H), 6.61 (d, J=8.67 Hz, 2 H), 6.67 (s, 1 H), 7.30 (br. s., 1 H), 7.37 (t, J=8.29 Hz, 1 H). MS (ESI) m/z: Calcd. for C31H46N4O6 570.34 [M]+, found 572.0 [M+H]+.
(S)-2-(5-Cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanamido)acetic acid (20):
Following the procedure as described in 45, compound (S)-2-(5-Cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanamido)acetic acid (20) was obtained using (S)-methyl 2-(5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanamido)acetate (18). 52 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (d, J=6.40 Hz, 6 H), 0.85–0.95 (m, 2 H), 1.10 – 1.39 (m, 5 H), 1.55–1.87 (m, 8 H), 2.06 – 2.15 (m, 2 H), 2.50–2.82 (m, 2 H), 3.64 (d, J=7.54 Hz, 2 H), 3.74 (s, 6 H), 4.01 – 4.17 (m, 2 H), 4.26–4.39 (m, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.67 (s, 1 H), 7.16 (d, J=8.67 Hz, 1 H), 7.38 (t, J=8.48 Hz, 1 H), 7.42 (br. s., 1 H). MS (ESI) m/z: Calcd. for C29H42N4O6 542.31 [M]+, found 543.7 [M+H]+.
N-((3S)-5-Cyclohexyl-1-((2-hydroxybutyl)amino)-1-oxopentan-3-yl)-5-(2,6dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamide (21):
Following the procedure described in 45, compound 21 was synthesized using (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoic acid (12) and 1-amino-2-butanol. 65 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (d, J=6.00 Hz, 6 H), 0.90 (t, J=7.35 Hz, 3 H), 0.81–0.96 (m, 2 H), 1.10 – 1.46 (m, 7 H), 1.62 – 1.76 (m, 5 H), 2.04–2.19 (m, 1 H), 2.45 – 2.69 (m, 2 H), 2.88 – 3.03 (m, 1 H), 3.18 – 3.29 (m, 1 H), 3.35 – 3.52 (m, 3 H), 3.57 (br. s., 2 H), 3.64 (d, J=7.16 Hz, 2 H), 3.74 (m, 6 H), 4.25 – 4.46 (m, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.68 (s, 1 H), 6.88–6.98 (d, J=5.65 Hz, 1 H), 7.04 (t, J=9.61 Hz, 1 H), 7.38 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C31H48N4O5 556.36 [M]+, found 558.1 [M+H]+.
N-((S)-5-Cyclohexyl-1-(((R)-2-hydroxybutyl)amino)-1-oxopentan-3-yl)-5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamide (22):
Following the procedure described in 45, compound 22 was synthesized using (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoic acid (12) and (2R)-1-amino2-butanol. 63 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (dd, J=6.69, 1.41 Hz, 6 H), 0.79 – 0.98 (m, 2 H), 0.90 (t, J=6.0 Hz, 3 H), 1.08 – 1.47 (m, 8 H), 1.58 – 1.75 (m, 6 H), 1.89 (br. s., 1 H), 2.11 (m, 1 H), 2.50 – 2.64 (m, 2 H), 3.17 – 3.26 (m, 1 H), 3.37 (dd, J=5.65, 2.64 Hz, 1 H), 3.45–3.51 (m, 1 H), 3.54–3.61 (m, 1 H), 3.64 (d, J=8.48 Hz, 2 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 4.25 – 4.37 (m, 1 H), 6.62 (d, J=8.48 Hz, 2 H), 6.68 (s, 1 H), 6.97 (t, J=5.46 Hz, 1 H), 7.09 (d, J=9.23 Hz, 1 H), 7.38 (t, J=9.23 Hz, 1 H). MS (ESI) m/z: Calcd. for C31H48N4O5 556.36 [M]+, found 557.8 [M+H]+.
(S)-N-(5-Cyclohexyl-1-oxo-1-((2-oxobutyl)amino)pentan-3-yl)-5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamide (23):
To a solution of N-((S)-5-cyclohexyl-1-(((R)-2-hydroxybutyl)amino)-1-oxopentan-3-yl)-5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamide (21) (22 mg, 0.39 mmol) in CH2Cl2 (5 mL) was added PCC (15 mg, 0.06 mmol), Celite 545 (10 mg) and stirred at room temperature for 16 h. The reaction mixture was diluted with ethyl ether (1 mL), stirred at rt for 1 h, before it was filtered through celite and silica gel (1:1) pad. The filtrate was concentrated to give crude residue. Crude product was purified by silica gel flash chromatography (0–50% EtOAc/hexanes) to give the title compound. 27 % yield; light brown solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (d, J=6.03 Hz, 6 H), 0.81 – 1.01 (m, 2 H), 1.08 (t, J=7.16 Hz, 4 H), 1.13 – 1.39 (m, 7 H), 1.63–1.78 (m, 7 H), 2.46 (q, J=7.16 Hz, 2 H), 2.60 (br. s., 2 H), 3.63 (d, J=6.40 Hz, 2 H), 3.74 (s, 6 H), 4.12 (m, 2 H), 6.62 (d, J=8.29 Hz, 2 H), 6.68 (br. s., 1 H), 6.85 (br. s., 1 H), 7.24–7.31 (m, 1 H), 7.37 (t, J=9.00 Hz, 1 H). MS (ESI) m/z: Calcd. for C31H46N4O5 554.35 [M]+, found 556.2 [M+H]+.
(S)-N-(5-Cyclohexyl-1-((2-methoxyethyl)amino)-1-oxopentan-3-yl)-5-(2,6dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamide (24):
Following the procedure described in 45, compound 24 was synthesized using (S)-5-cyclohexyl-3-(5-(2,6dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)pentanoic acid (12) and 2-methoxy ethylamine. 91 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (d, J=6.78 Hz, 6 H), 0.82 – 1.01 (m, 2 H), 1.12 – 1.40 (m, 7 H), 1.63 – 1.77 (m, 7 H), 2.55 (d, J=6.03 Hz, 2 H), 3.27 – 3.31 (m, 3 H), 3.39 – 3.48 (m, 4 H), 3.63 (d, J=7.16 Hz, 2 H), 3.72 (s, 3 H), 3.74 (s, 3 H), 4.23–4.36 (m, 1 H), 6.51 (br. s., 1 H), 6.62 (d, J=8.67 Hz, 2 H), 6.68 (s, 1 H), 7.21 (d, J=8.29 Hz, 1 H), 7.37 (t, J=8.29 Hz, 1 H). MS (ESI) m/z: Calcd. for C30H46N4O5 542.35 [M]+, found 544.1 [M+H]+.
(S)-2-(5-(2,6-Dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)-4-phenylbutanoic acid (26):
Following the procedure described in 12, compound 26 was obtained from deprotection of (S)-tert-butyl 2-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)-4-phenylbutanoate. 80 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.75 (dd, J=6.59, 2.45 Hz, 6 H), 0.81–0.95 (m, 2 H), 2.08 – 2.26 (m, 2 H), 2.31 – 2.46 (m, 1 H), 2.82 (t, J=8.10 Hz, 2 H), 3.67 (d, J=7.35 Hz, 2 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 4.63 – 4.74 (m, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.73 (s, 1 H), 7.12 – 7.25 (m, 3 H), 7.27 – 7.32 (m, 1 H), 7.29 – 7.44 (m, 2 H). MS (ESI) m/z: Calcd. for C26H31N3O5 465.23 [M]+, found 464.8 [M-H]−.
(S)-Methyl 2-(2-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)-4-phenylbutanamido)acetate (27):
Following the procedure described in 45, compound 27 was synthesized using 5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylic acid (43) and (S)-methyl 2-(2-amino-4-phenylbutanamido)acetate trifluoroacetate. 50 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.76 (d, J=6.78 Hz, 3 H), 0.74 (d, J=6.78 Hz, 3 H), 2.07–2.21 (m, 2 H), 2.32 – 2.44 (m, 1 H), 2.79 (t, J=7.91 Hz, 2 H), 3.64 (d, J=7.16 Hz, 2 H), 3.73 (s, 6 H), 3.75 (s, 3 H), 4.05 (dd, J=5.65, 2.26 Hz, 2 H), 4.64 (td, J=8.10, 6.03 Hz, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.71 (s, 1 H), 6.85 – 6.95 (m, 1 H), 7.14 – 7.27 (m, 4 H), 7.28 – 7.33 (m, 2 H), 7.38 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C29H36N4O6 536.26 [M]+, found 537.5 [M+H]+.
(S)-Methyl 2-(2-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)-Nmethyl-4-phenylbutanamido)acetate (28):
Following the procedure described in 45, compound 28 was synthesized using 5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylic acid (43) and (S)-methyl 2-(2-amino-N-methyl-4-phenylbutanamido)acetate trifluoroacetate. 50 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.76 (d, J=4.52 Hz, 3 H), 0.73 (d, J=4.52 Hz, 3 H), 2.03 – 2.23 (m, 4 H), 2.70 – 2.84 (m, 2 H), 3.05 (s, 3 H), 3.63 (d, J=9.00 Hz, 3 H), 3.72 (s, 3 H), 3.74 (s, 6 H), 4.44–4.52 (m, 1 H), 5.20–5.28 (m, 1 H), 6.62 (dd, J=8.67, 1.51 Hz, 2 H), 6.69 (s, 1 H), 7.14 – 7.23 (m, 1 H), 7.29 (m, 3 H), 7.37 (t, J=8.29 Hz, 1 H), 7.65 (d, J=8.67 Hz, 1 H). MS (ESI) m/z: Calcd. for C30H38N4O6 550.28 [M]+, found 551.7 [M+H]+.
(S)-5-(2,6-Dimethoxyphenyl)-1-isobutyl-N-(1-((2-methoxyethyl)amino)-1-oxo-4phenylbutan-2-yl)-1H-pyrazole-3-carboxamide (29):
Following the procedure described in 45, compound 29 was synthesized using 5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylic acid (43) and (S)-2-amino-N-(2-methoxyethyl)-4-phenylbutanamide trifluoroacetate. 73 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.76 (d, J=6.78 Hz, 3 H), 0.74 (d, J=6.78 Hz, 3 H), 2.02 – 2.20 (m, 2 H), 2.25 – 2.39 (m, 1 H), 2.76 (t, J=7.91 Hz, 2 H), 3.34 (s, 3 H), 3.39 – 3.53 (m, 4 H), 3.64 (d, J=7.16 Hz, 2 H), 3.74 (s, 3 H), 3.75 (s, 3 H), 4.56–4.63 (m, 1 H), 6.53 (br. s., 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.70 (s, 1 H), 7.15 – 7.25 (m, 3 H), 7.28 – 7.33 (m, 3 H), 7.38 (t, J=9.00 Hz, 1 H). MS (ESI) m/z: Calcd. for C29H38N4O5 522.28 [M]+, found 523.9 [M+H]+.
5-(2,6-Dimethoxyphenyl)-N-((2S)-1-((2-hydroxybutyl)amino)-1-oxo-4-phenylbutan-2-yl)-1isobutyl-1H-pyrazole-3-carboxamide (30):
Following the procedure described in 45, compound 30 was synthesized using 5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylic acid (43) and (2S)-2-amino-N-(2-hydroxybutyl)-4-phenylbutanamide trifluoroacetate. 90 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.76 (d, J=3.39 Hz, 3 H), 0.74 (d, J=3.77 Hz, 3 H), 0.95 (t, J=6.00 Hz, 3 H), 1.38–1.52 (m, 2 H), 2.06 – 2.18 (m, 2 H), 2.30 – 2.44 (m, 1 H), 2.75 – 2.85 (m, 2 H), 3.09 – 3.19 (m, 1 H), 3.40 – 3.54 (m, 1 H), 3.64 (d, J=7.54 Hz, 3 H), 3.74 (m, 6 H), 4.48 – 4.58 (m, 1 H), 6.63 (d, J=9.00 Hz, 2 H), 6.69 (s, 1 H), 6.81 (br. s., 1 H), 7.15 – 7.24 (m, 2 H), 7.28 – 7.32 (m, 4 H), 7.38 (t, J=8.29 Hz, 1 H). MS (ESI) m/z: Calcd. for C30H40N4O5 536.30 [M]+, found 535.7 [M-H]−.
(S)-5-(2,6-Dimethoxyphenyl)-1-isobutyl-N-(1-oxo-1-((2-oxobutyl)amino)-4-phenylbutan-2yl)-1H-pyrazole-3-carboxamide (31):
Following the procedure described in 23, compound 31 was synthesized using 5-(2,6-dimethoxyphenyl)-N-((2S)-1-((2-hydroxybutyl)amino)-1-oxo-4-phenylbutan-2-yl)-1-isobutyl-1H-pyrazole-3-carboxamide (31). 45 % yield; light brown solid; 1H NMR (CDCl3, 300 MHz) δ 0.75 (dd, J=6.78, 3.39 Hz, 6 H), 1.10 (t, J=7.16 Hz, 3 H), 2.04 – 2.20 (m, 2 H), 2.28–2.41 (m, 1 H), 2.47 (q, J=7.16 Hz, 2 H), 2.78 (t, J=7.72 Hz, 2 H), 3.64 (d, J=7.54 Hz, 2 H), 3.74 (s, 6 H), 4.09–4.18 (m, 2 H), 4.60–4.71 (m, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.70 (s, 1 H), 6.94 (br. s., 1 H), 7.13 – 7.25 (m, 3 H), 7.27 – 7.34 (m, 3 H), 7.38 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C30H38N4O5 534.28 [M]+, found 535.6 [M+H]+.
(S)-N-(1-(Cyclobutylamino)-1-oxo-4-phenylbutan-2-yl)-5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamide (32):
Following the procedure described in 45, compound 32 was synthesized using 5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxylic acid (43) and (S)-2-amino-N-cyclobutyl-4-phenylbutanamide trifluoroacetate. 85 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.75 (dd, J=6.78, 2.26 Hz, 6 H), 1.66 – 1.74 (m, 2 H), 1.81 – 1.96 (m, 2 H), 2.01 – 2.18 (m, 2 H), 2.26 – 2.38 (m, 3 H), 2.75 (t, J=7.91 Hz, 2 H), 3.64 (d, J=7.16 Hz, 2 H), 3.75 (s, 6 H), 4.29 – 4.41 (m, 1 H), 4.47 – 4.56 (m, 1 H), 6.50 (d, J=7.91 Hz, 1 H), 6.63 (d, J=8.29 Hz, 2 H), 6.71 (m, 1 H), 7.14 – 7.24 (m, 3 H), 7.28 – 7.33 (m, 3 H), 7.38 (t, J=8.48 Hz, 1 H). MS (ESI) m/z: Calcd. for C30H38N4O4 518.29 [M]+, found 519.9 [M+H]+.
(S)-3-(5-(2,6-Dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)-5-phenylpentanoic acid (33):
Following the procedure described in 12, compound 33 was obtained from deprotection of (S)-tert-butyl 3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamido)-5-phenylpentanoate. 69 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (dd, J=6.59, 1.88 Hz, 6 H), 2.00–2.17 (m, 4 H), 2.72–2.81 (m, 2 H), 2.76 (d, J=6.00 Hz, 2 H), 3.66 (d, J=7.54 Hz, 2 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 4.38 – 4.49 (m, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.72 (s, 1 H), 7.15 – 7.23 (m, 3 H), 7.26 – 7.34 (m, 2 H), 7.38 (t, J=8.48 Hz, 1 H), 7.44 (m, 1 H). MS (ESI) m/z: Calcd. for C27H33N3O5 479.24 [M]+, found 478.7 [M-H]−.
(S)-N-(1-(Cyclobutylamino)-1-oxo-5-phenylpentan-3-yl)-5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3-carboxamide (34):
Following the procedure described in 45, compound 34 was synthesized using (S)-3-(5-(2,6-dimethoxyphenyl)-1-isobutyl-1H-pyrazole-3carboxamido)-5-phenylpentanoic acid (33) and cyclobutylamine. 93 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.74 (d, J=6.78 Hz, 6 H), 1.61 – 1.72 (m, 2 H), 1.73 – 1.87 (m, 2 H), 1.92 – 2.18 (m, 3 H), 2.19 – 2.32 (m, 2 H), 2.55 (d, J=6.22 Hz, 2 H), 2.68 – 2.79 (m, 2 H), 3.64 (d, J=7.35 Hz, 2 H), 3.73 (m, 3 H), 3.74 (m, 3 H), 4.28 – 4.41 (m, 2 H), 6.62 (d, J=8.29 Hz, 2 H), 6.70 (s, 1 H), 7.15 – 7.26 (m, 5 H), 7.28 – 7.41 (m, 3 H). MS (ESI) m/z: Calcd. for C31H40N4O4 532.30 [M]+, found 533.7 [M+H]+.
(S)-N-(1-(Cyclobutylamino)-5-cyclohexyl-1-oxopentan-3-yl)-5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamide (35):
Following the procedure described in 45, compound 35 was synthesized using (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (9) and cyclobutylamine. 91 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 0.78 – 0.97 (m, 2 H), 1.10 – 1.40 (m, 7 H), 1.57 – 1.75 (m, 8 H), 1.79 – 1.91 (m, 2 H), 2.21 – 2.35 (m, 2 H), 2.49 – 2.54 (m, 2 H), 3.55 (s, 3 H), 3.60 (s, 3 H), 4.27 – 4.42 (m, 2 H), 6.47 – 6.60 (m, 3 H), 6.94 (s, 1 H), 6.97 (d, J=8.29 Hz, 2 H), 7.19 – 7.25 (m, 2 H), 7.27 – 7.34 (m, 2 H). MS (ESI) m/z: Calcd. for C33H41FN4O4 576.31 [M]+, found 577.5 [M+H]+.
(S)-N-(1-(Cyclobutylamino)-5-cyclohexyl-1-oxopentan-3-yl)-5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxamide (36):
Following the procedure described in 45, compound 36 was synthesized using (S)-5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-propyl-1H-pyrazole-3-carboxamido)pentanoic acid (11) and cyclobutylamine. 75 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ ppm 0.78 (t, J=7.44 Hz, 3 H), 0.82 – 0.98 (m, 2 H), 1.12 – 1.35 (m, 7 H), 1.62 – 1.95 (m, 12 H), 2.18–2.35 (m, 2 H), 2.52 (d, J=9.0 Hz, 2 H), 3.73 (s, 3 H), 3.74 (s, 3 H), 3.79 (t, J= 9.0 Hz, 2 H), 4.20 – 4.30 (m, 1 H), 4.31 – 4.44 (m, 1 H), 6.64 (d, J=9.0 Hz, 2 H), 6.65–6.68 (m, 1 H), 6.69 (s, 1 H), 7.10 (d, J=8.67 Hz, 1 H), 7.38 (t, J=8.38 Hz, 1 H). MS (ESI) m/z: Calcd. for C30H44N4O4 524.34 [M]+, found 526.0 [M+H]+
(S)-N-(1-(Cyclobutylamino)-5-cyclohexyl-1-oxopentan-3-yl)-1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamide (38):
Following the procedure described in 45, compound 38 was synthesized using (S)-5-cyclohexyl-3-(1-cyclohexyl-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (14) and cyclobutylamine. 61 % yield; white solid; 1H NMR (CDCl3, 300 MHz) δ ppm 0.80 – 0.97 (m, 2 H), 1.09 – 1.38 (m, 9 H), 1.58 – 1.76 (m, 10 H), 1.77 – 2.00 (m, 8 H), 2.18 – 2.33 (m, 2 H), 2.53 (d, J=6.40 Hz, 2 H), 3.58 – 3.69 (m, 1 H), 3.74 (s, 3 H), 3.73 (s, 3 H), 4.19 – 4.30 (m, 1 H), 4.31 – 4.44 (m, 1 H), 6.63 (d, J=9.0 Hz, 2 H), 6.66 (s, 1 H), 6.74 (d, J=7.54 Hz, 1 H), 7.08 (d, J=9.23 Hz, 1 H), 7.38 (t, J=8.38 Hz, 1 H). MS (ESI) m/z: Calcd. for C33H48N4O4 564.37 [M]+, found 565.7 [M+H]+.
Bioassays
Lance™ cAMP accumulation assay
Stimulation buffer containing 1X Hank’s Balanced Salt Solution (HBSS), 5 mM HEPES, 0.1% BSA stabilizer, and 0.5 mM final IBMX was prepared and titrated to pH 7.4 at room temperature. Serial dilutions of the test compounds and 1 μM forskolin, both prepared at 4x the desired final concentration in stimulation buffer, were added to a 96-well white ½ area microplate (PerkinElmer). A cAMP standard curve prepared at 4x the desired final concentration in stimulation buffer was added to the assay plate. The total assay volume for 96-well plate was 40uL. The final DMSO concentration in all wells was 1.5%. Stable hAPJ-CHO cells were lifted with a non-enzymatic solution (Cell-stripper, Mediatech Inc., Orlando, FL) and spun at 270xg for 10 min. The cell pellet was resuspended in stimulation buffer and 4000 cells were added to each well except wells containing the cAMP standard curve. After incubating for 30 min at RT, Eu-cAMP tracer and uLIGHT-anti-cAMP working solutions were added per the manufacturer’s instructions. After incubation at RT for 1 h, the TR-FRET signal (ex 337 nm, em 620 and 650 nm) was read on a CLARIOstar multimode plate reader (BMG Biotech, Cary, NC). For the screen, the same procedure was followed except that instead of adding serial dilutions of the test compounds, a single concentration (1 μM, prepared at 4× final) of each test compound was added to the assay plates. Data were analyzed using Prism software (GraphPad, La Jolla, CA). Nonlinear regression analysis was performed to fit data and obtain maximum response (Emax), EC50, correlation coefficient (r2), and other parameters. All experiments were performed in duplicate at least two times to ensure reproducibility and data are reported as mean ± SEM, unless noted otherwise.
Calcium mobilization assay
CHO cells stably expressing either the Gαq16 protein (CHO-RD-HGA16; Molecular Devices) plus the human apelin receptor (CHO hAPJ) or human angiotensin II receptor, type I (CHO hAGTR1; DiscoverX) were removed from flasks using Cell-stripper and quenched with DMEM/F12, 10 % FBS and 1X Penicillin/Streptomycin, centrifuged, and re-suspended in the serum-containing media. Cells were counted with a hemocytometer and 30,000 cells of CHO hAPJ and 20,000 cells of CHO hAGTR1 were transferred to each well of a black Costar 96-well optical bottom plate (Corning Corporation, Corning, NY). Each plate was incubated at 37°C for 24 h. The culture media was removed from the plates, cells were washed with DPBS and were subsequently loaded with a fluorescent calcium probe (Calcium 5 dye; Molecular Devices, Sunnyvale, CA) in an HBSS-based buffer containing 20 mM HEPES, 1% BSA and 10 μM probenecid (Sigma) in a total volume of 225 μL. Cells were incubated at 37°C for 1h and then stimulated with test compounds or pyrapelin-13 (Anaspec, Freemont, CA) at various concentrations using a FLIPR Tetra plate-reader. Agonist-mediated change in fluorescence (ex 488 nm, em 525 nm) was monitored in each well at 1 sec intervals for 90 sec. Data were collected using Softmax version 4.8 (MDS Analytical Technologies) and analyzed using Prism software. Nonlinear regression analysis was performed to fit data and obtain maximum response (Emax), EC50, correlation coefficient (r2), and other parameters. All experiments were performed in duplicate at least two times to ensure reproducibility and data are reported as mean ± SEM, unless noted otherwise.
PathHunter β-arrestin recruitment assay
The stable CHO-K1 human AGTRL1 β-arrestin-2 cell line (DiscoverX) was maintained in TC-treated flasks with growth media containing DMEM/F12 (Corning Cellgro, Manassas, VA), 10% FBS, 1XPenicillin-streptomycin L-glutamine (Gibco, Carlsbad, CA). The selection reagents used were geneticin (800 μg/mL) and hygromycin (300 μg/mL). The cells were removed from the flasks using Cell stripper. Cells were resuspended in complete media and spun at 300xg for 5 min. The supernatant was discarded and cells were resuspended in Assay Complete Cell plating 2 Reagent (DiscoverX). Cells were counted using hemocytometer and seeded in white clear bottom TC-treated 96-well plate (DiscoverX) at 10,000 cells per well. The plates were incubated at 37°C and 5% CO2 for 24 hours. Compound dilutions were prepared at 10X final in DPBS (1X) buffer with 10% DMSO and 1% BSA. Compound dilutions were added to the 80–90% confluent assay plate and the plates were incubated at 37°C and 5% CO2 for 90 min, followed by the addition of PathHunter Detection Reagent (DiscoverX), prepared as per the manufacturer’s instructions. The assays were run at 1% DMSO and 0.1% BSA final. The culture plates were incubated another 60 min at room temperature. The luminescent signal was detected on an Enspire multimode plate reader (PerkinElmer CT, USA). Data were collected using EnSpire Software 4.1 and analyzed using GraphPad Prism. Nonlinear regression analysis was performed to fit data and obtain maximum response (Emax), EC50, correlation coefficient (r2), and other parameters. All experiments were performed in duplicate at least two times to ensure reproducibility and data reported as mean ± SEM, unless noted otherwise.
Radioligand displacement assay
Competitive inhibition binding was performed using [125I]-apelin-13 (Perkin Elmer, Akron, OH). Eleven concentrations (ranging from 10−11 M to 3.16X10−5 M) of compound were incubated in assay buffer (0.025 M HEPES, (pH=7.4), 0.01 M MgCl2, 0.001 M CaCl2, 0.5% fatty acid-free BSA) with 7.5X10−11 M [125I]-apelin-13 (Perkin-Elmer) in the presence of 0.125 μg of CHO-K1 cell membrane homogenates expressing the human apelin receptor (Perkin Elmer). Non-specific binding (NSB) was determined in the presence of 10−6 M unlabeled apelin-13 (Anaspec). The assays were run in 1.4 mL polypropylene tubes (Thermo Scientific, Waltham, MA) in a 96-well format. All assays were incubated at room temperature on a shaking platform for two hours. Incubation was terminated and bound radioligand was separated from free radioligand by rapid vacuum filtration over 0.5% PEI treated GF/C filter plates using ice-cold 50 mM Tris-HCl (pH=7.4) in a Brandel Scientific (Gaithersburg, MD) 96-well harvester. Filter plates were allowed to dry and Microscint 20 scintillation cocktail (Perkin-Elmer) was added to each well. Bound radioactivity was determined with a TopCount 12-detector instrument (Packard Instruments) using standard scintillation counting techniques. The binding data from each assay plate were normalized to NSB and total counts per minute (CPM) bound values prior to analysis. The Ki values were calculated from a three-parameter logistic curve fit to the data with Prism (version 6.0, GraphPad Software, Inc., San Diego, CA) using 2.1X10−10 M as the Kd for [125I]-apelin-13. All experiments were performed in duplicate at least two times to ensure reproducibility and data reported as mean ± SEM, unless noted otherwise.
Hepatic Liver Microsome Stability Studies
Human liver microsomal (HLM) stability assays were performed as described previously.37 Briefly, test compounds were incubated at a 1 μM final concentration with 0.5 mg/ml pooled human liver microsomes from 200 unidentified donors (Xenotech, LLC, Lenexa, KS) in a 100 mM phosphate buffer (pH 7.4) containing 3 mM MgCl2, 1 mM nicotinamide adenine dinucleotide phosphate (NADPH), 5 mM uridine diphosphate glucuronic acid (UDPGA), and 50 μg/ml alamethicin. Triplicate samples were incubated for up to 120 min. Samples were removed at regular intervals. Reactions were terminated by addition of 3 volumes of methanol and processed for LC-MS by centrifugation. Standard curves were prepared in blank matrix for each compound for quantitative assessment. Intrinsic clearance rate was calculated for each compound using the formula: Clint (μl/min/mg) = 0.693/(t1/2 X microsomal protein concentration). Data reported are average values from 3 measurements.
Bias Factor Calculation
Data from technical replicates on the same day were baseline-corrected and normalized relative to the reference agonist Pyr-Apelin-13. Data from multiple days were then combined and fit to logistic functions with Hill coefficient equal to 1 to determine Emax, EC50 and their standard errors. Bias factors, which quantify the degree of signaling through pathway 1 compared to pathway 2 for a ligand relative to a reference agonist on a logarithmic scale, were calculated as previously described. 35,36
Supplementary Material
Table 6:
Bias factors for a series of APJ agonists (shown in dimensionless units on a logarithmic scale ± standard error of the mean). Positive values denote bias towards the first pathway listed and negative values denote bias towards the second pathway listed (pathway 1: pathway 2).
| Compd. | Bias Ca:cAMP |
Bias Ca:βarr | Bias cAMP:βarr |
|---|---|---|---|
| Pyr-Apelin-13 | 0 ± 0.136 | 0 ± 0.194 | 0 ± 0.180 |
| 9. | 0.793 ± 0.172 | 0.89 ± 0.239 | 0.097 ± 0.243 |
| 10. | 0.743 ± 0.229 | 0.952 ± 0.189 | 0.209 ± 0.23 |
| 11. | 0.549 ± 0.133 | 1.18 ± 0.153 | 0.634 ± 0.157 |
| 12. | 0.947 ± 0.173 | 1.13 ± 0.17 | 0.182 ± 0.178 |
| 13. | 0.606 ± 0.163 | 1.06 ± 0.157 | 0.454 ± 0.189 |
| 14. | 0.895 ± 0.132 | 1.14 ± 0.164 | 0.241 ± 0.167 |
| 19. | 1.75 ± 0.169 | 0.509 ± 0.185 | −1.24 ± 0.199 |
| 27. | 2.45 ± 0.149 | 1.95 ± 0.215 | −0.498 ± 0.231 |
| 28. | 2.27 ± 0.217 | 1.56 ± 0.392 | −0.709 ± 0.344 |
| 29. | 2.73 ± 0.39 | 2.82 ± 0.805 | 0.095 ± 0.882 |
| 30. | 2.75 ± 0.376 | 2.46 ± 0.32 | −0.288 ± 0.479 |
| 31. | 2.72 ± 0.161 | 2.14 ± 0.219 | −0.584 ± 0.243 |
| 34. | 2.45 ± 0.17 | 1.8 ± 0.193 | −0.65 ± 0.186 |
ACKNOWLEDGEMENTS
Authors wish to thank Dr. Elaine Gay, Dr. Ann Decker for help with various assays and Dr. Thomas Prisinzano, Victor Day, and the University of Kansas X-Ray Crystallography Laboratory for the crystal structures.
Funding Sources
This work was supported by NIH grants: 1R01HD079547-01A1 and 1R01DK103625-01A1
ABBREVIATIONS
- AT1
Angiotensin 1
- NPS
Neuropeptide S receptor
- NTR-1
Neurotensin receptor 1
- NTR-2
Neurotensin receptor 2
- BOP
Benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate
- LiOH.H2O
Lithium hydroxide monohydrate
- TFA
Trifluroacetic acid
- HCl
Hydrochloric acid
- Et3N
Triethylamine
- DCM
Dichloromethane
- THF
Tetrahydrofuran
- MeOH
Methanol
- SAR
Structure Activity Relationship
- TLC
thin layer chromatography
Footnotes
X-RAY CRYSTALLOGRAPHIC DATA
Atomic coordinates for 43 and 44 have been deposited with the Cambridge Crystallographic Data Centre (deposition numbers 1946460, 1946461). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK [fax: +44(0)-1223–336033 or e-mail: deposit@ccdc.cam.ac.uk.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.O’Dowd BF; Heiber M; Chan A; Heng HH; Tsui LC; Kennedy JL; Shi X; Petronis A; George SR; Nguyen T A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 1993; 136, 355–360. [DOI] [PubMed] [Google Scholar]
- 2.Tatemoto K; Hosoya M; Habata Y; Fujii R; Kakegawa T; Zou MX; Kawamata Y; Fukusumi S; Hinuma S; Kitada C; Kurokawa T; Onda H; Fujino M Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun 1998; 251, 471–476. [DOI] [PubMed] [Google Scholar]
- 3.Hosoya M; Kawamata Y; Fukusumi S; Fujii R; Habata Y; Hinuma S; Kitada C; Honda S; Kurokawa T; Onda H; Nishimura O; Fujino M Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J Biol Chem 2000; 275, 21061–21067. [DOI] [PubMed] [Google Scholar]
- 4.Maguire JJ; Kleinz MJ; Pitkin SL; Davenport AP [Pyr(1)]Apelin-13 Identified as the Predominant Apelin Isoform in the Human Heart Vasoactive Mechanisms and Inotropic Action in Disease. Hypertension 2009; 54, 598–U296. [DOI] [PubMed] [Google Scholar]
- 5.Langelaan DN; Bebbington EM; Reddy T; Rainey JK Structural insight into G-protein coupled receptor binding by apelin. Biochemistry 2009; 48, 537–548. [DOI] [PubMed] [Google Scholar]
- 6.De Mota N; Goazigo ARL; El Messari S; Chartrel N; Roesch D; Dujardin C; Kordon C; Vaudry H; Moos FO; Llorens-Cortes C Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. P Natl Acad Sci USA 2004; 101, 10464–10469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azizi M; Iturrioz X; Blanchard A; Peyrard S; De Mota N; Chartrel N; Vaudry H; Corvol P; Llorens-Cortes C Reciprocal regulation of plasma apelin and vasopressin by osmotic stimuli. J Am Soc Nephrol 2008; 19, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murza A; Parent A; Besserer-Offroy E; Tremblay H; Karadereye F; Beaudet N; Leduc R; Sarret P; Marsault E Elucidation of the structure-activity relationships of apelin: influence of unnatural amino acids on binding, signaling, and plasma stability. Chemmedchem 2012; 7, 318–325. [DOI] [PubMed] [Google Scholar]
- 9.Murza A; Belleville K; Longpre JM; Sarret P; Marsault E Stability and Degradation Patterns of Chemically Modified Analogs of Apelin-13 in Plasma and Cerebrospinal Fluid. Biopolymers 2014; 102, 297–303. [DOI] [PubMed] [Google Scholar]
- 10.Pauli A; Norris ML; Valen E; Chew GL; Gagnon JA; Zimmerman S; Mitchell A; Ma J; Dubrulle J; Reyon D; Tsai SQ; Joung JK; Saghatelian A; Schier AF Toddler: An Embryonic Signal That Promotes Cell Movement via Apelin Receptors. Science 2014; 343, 746-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chng SC; Ho LN; Tian J; Reversade B ELABELA: A Hormone Essential for Heart Development Signals via the Apelin Receptor. Dev Cell 2013; 27, 672–680. [DOI] [PubMed] [Google Scholar]
- 12.Laflamme B ELABELA, a peptide hormone for heart development. Nat Genet 2014; 46, 7. [Google Scholar]
- 13.Szokodi I; Tavi P; Foldes G; Voutilainen-Myllya S; Ilves M; Tokola H; Pikkarainen S; Piuhola J; Rysa J; Toth M; Ruskoaho H Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res 2002; 91, 434–440. [DOI] [PubMed] [Google Scholar]
- 14.Lee DK; Jeong JH; Oh S; Jo YH Apelin-13 Enhances Arcuate POMC Neuron Activity via Inhibiting M-Current. PLoS One 2015; 10, e0119457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El Messari S; Iturrioz X; Fassot C; De Mota N; Roesch D; Llorens-Cortes C Functional dissociation of apelin receptor signaling and endocytosis: implications for the effects of apelin on arterial blood pressure. J Neurochem 2004; 90, 1290–1301. [DOI] [PubMed] [Google Scholar]
- 16.De Mota N; Lenkei Z; Llorens-Cortes C Cloning, pharmacological characterization and brain distribution of the rat apelin receptor. Neuroendocrinology 2000; 72, 400–407. [DOI] [PubMed] [Google Scholar]
- 17.Reaux-Le Goazigo A; Morinville A; Burlet A; Llorens-Cortes C; Beaudet A Dehydration-induced cross-regulation of apelin and vasopressin immunoreactivity levels in magnocellular hypothalamic neurons. Endocrinology 2004; 145, 4392–4400. [DOI] [PubMed] [Google Scholar]
- 18.Medhurst AD; Jennings CA; Robbins MJ; Davis RP; Ellis C; Winborn KY;Lawrie KW; Hervieu G; Riley G; Bolaky JE; Herrity NC; Murdock P; Darker JG Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J Neurochem 2003; 84, 1162–1172. [DOI] [PubMed] [Google Scholar]
- 19.Reaux-Le Goazigo A; Alvear-Perez R; Zizzari P; Epelbaum J; Bluet-Pajot MT; Llorens-Cortes C Cellular localization of apelin and its receptor in the anterior pituitary: evidence for a direct stimulatory action of apelin on ACTH release. Am J Physiol Endocrinol Metab 2007; 292, E7–15. [DOI] [PubMed] [Google Scholar]
- 20.Yokomori H; Oda M; Yoshimura K; Machida S; Kaneko F; Hibi T Overexpression of apelin receptor (APJ/AGTRL1) on hepatic stellate cells and sinusoidal angiogenesis in human cirrhotic liver. J Gastroenterol 2011; 46, 222–231. [DOI] [PubMed] [Google Scholar]
- 21.Narayanan S; Harris DL; Maitra R; Runyon SP Regulation of the Apelinergic System and Its Potential in Cardiovascular Disease: Peptides and Small Molecules as Tools for Discovery. J Med Chem 2015; 58, 7913–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wysocka MB; Pietraszek-Gremplewicz K; Nowak D The Role of Apelin in Cardiovascular Diseases, Obesity and Cancer. Front Physiol 2018; 9, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papangeli I; Kim J; Maier I; Park S; Lee A; Kang Y; Tanaka K; Khan OF; Ju H; Kojima Y; Red-Horse K; Anderson DG; Siekmann AF; Chun HJ MicroRNA 139–5p coordinates APLNR-CXCR4 crosstalk during vascular maturation. Nature communications 2016; 7, 11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iturrioz X; Alvear-Perez R; De Mota N; Franchet C; Guillier F; Leroux V; Dabire H; Le Jouan M; Chabane H; Gerbier R; Bonnet D; Berdeaux A; Maigret B; Galzi JL; Hibert M; Llorens-Cortes C Identification and pharmacological properties of E339–3D6, the first nonpeptidic apelin receptor agonist Faseb J 2010; 24, 1506–1517. [DOI] [PubMed] [Google Scholar]
- 25.Khan P; Maloney PR; Hedrick M; Gosalia P; Milewski M; Li L; Roth GP; Sergienko E; Suyama E; Sugarman E; Nguyen K; Mehta A; Vasile S; Su Y; Shi S; Stonich D; Nguyen H; Zeng FY; Novo AM; Vicchiarelli M; Diwan J; Chung TDY; Pinkerton AB; Smith LH In Probe Reports from the NIH Molecular Libraries Program Bethesda (MD), 2010. [Google Scholar]
- 26.Hachtel SW, Wolfart P; Weston J; Mueller M; Defossa E; Mertsch K; Weng JH;Binnie RA; Abdul-Latif F; Bock WJ; Walser A; ; Benzoimidazole-carboxylic acid amide derivatives as APJ Receptor Modulators.; US20140094450 A1; Sanofi[Fr]: 2014. [Google Scholar]
- 27.Pinkerton ABS, L. H.; Agonists of the Apelin Receptor and Methods of Use Thereof.; WO2015/184011 A2; Sanford-Burnham Medical Research Institute: 2015.
- 28.Meng WC, H. J.; Finlay H; Lawrence RM; Myers MC Apelin Receptor Agonists and Methods of Use. WO2017096130A1 2017, Bristo-Myers Squibb Company.
- 29.Chen XC, Y.; Cheng AC; Connors RV; Debenedetti MV; Dransfield PJ; Fu Z; Harvey JS; Heath JA; Hedley SJ; Houze J; Judd TC; Khakoo AY; Kopecky DJ; Lai SJ; Ma Z; Nishimura N; Olson SH; Pattaropong V; Swaminath G; Wang X; Yeh WC; Heterocyclic Triazole Compounds as Agonists of the APJ Receptor. WO2017192485A1 2017, Amgen Inc.
- 30.Read C; Fitzpatrick CM; Yang P; Kuc RE; Maguire JJ; Glen RC; Foster RE; Davenport AP Cardiac action of the first G protein biased small molecule apelin agonist. Biochemical pharmacology 2016; 116, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brame AL; Maguire JJ; Yang P; Dyson A; Torella R; Cheriyan J; Singer M; Glen RC; Wilkinson IB; Davenport AP Design, characterization, and first-in-human study of the vascular actions of a novel biased apelin receptor agonist. Hypertension 2015; 65, 834–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trifonov L; Afri M; Palczewski K; Korshin EE; Gruzman A An Expedient Synthesis of CMF-019: (S)-5-Methyl-3-{1-(pentan-3-yl)-2-(thiophen-2-ylmethyl)-1H-benzo[d]imidazole5-carboxamido}hexanoic Acid, a Potent Apelin Receptor (APJ) Agonist. Med Chem 2018; 14, 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Narayanan S; Maitra R; Deschamps JR; Bortoff K; Thomas JB; Zhang Y; Warner K; Vasukuttan V; Decker A; Runyon SP Discovery of a novel small molecule agonist scaffold for the APJ receptor. Bioorg Med Chem 2016; 24, 3758–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davies SG; Mulvaney AW; Russell AJ; Smith AD Parallel synthesis of homochiralb-amino acids. Tetrahedron: Asymmetry 2007; 18, 1554–1566. [Google Scholar]
- 35.Rajagopal S; Ahn S; Rominger DH; Gowen-MacDonald W; Lam CM; DeWire SM; Violin JD; Lefkowitz RJ Quantifying Ligand Bias at Seven-Transmembrane Receptors. Molecular Pharmacology 2011; 80, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onaran HO; Ambrosio C; Ugur O; Koncz EM; Gro MC; Vezzi V; Rajagopal S; Costa T Systematic errors in detecting biased agonism: Analysis of current methods and development of a new model-free approach. Sci Rep-Uk 2017; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fulp A; Bortoff K; Seltzman H; Zhang YA; Mathews J; Snyder R; Fennell T; Maitra R Design and Synthesis of Cannabinoid Receptor 1 Antagonists for Peripheral Selectivity. J Med Chem 2012; 55, 2820–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.