

Astrocytes are an integral component of the neurovascular unit where they act as homeostatic regulators, especially after brain injuries, such as stroke. One process by which astrocytes modulate homeostasis is the release of functional mitochondria (Mt) that are taken up by other cells to improve their function. However, the mechanisms underlying the beneficial effect of Mt transfer are unclear and likely multifactorial.
Keywords: mitochondria, humanin, intracerebral hemorrhage, microglia, astrocyte, peroxisome proliferator-activated receptor gamma
Abstract
Astrocytes are an integral component of the neurovascular unit where they act as homeostatic regulators, especially after brain injuries, such as stroke. One process by which astrocytes modulate homeostasis is the release of functional mitochondria (Mt) that are taken up by other cells to improve their function. However, the mechanisms underlying the beneficial effect of Mt transfer are unclear and likely multifactorial. Using a cell culture system, we established that astrocytes release both intact Mt and humanin (HN), a small bioactive peptide normally transcribed from the Mt genome. Further experiments revealed that astrocyte-secreted Mt enter microglia, where they induce HN expression. Similar to the effect of HN alone, incorporation of Mt by microglia (1) upregulated expression of the transcription factor peroxisome proliferator-activated receptor gamma and its target genes (including mitochondrial superoxide dismutase), (2) enhanced phagocytic activity toward red blood cells (an in vitro model of hematoma clearance after intracerebral hemorrhage [ICH]), and (3) reduced proinflammatory responses. ICH induction in male mice caused profound HN loss in the affected hemisphere. Intravenously administered HN penetrated perihematoma brain tissue, reduced neurological deficits, and improved hematoma clearance, a function that normally requires microglia/macrophages. This study suggests that astrocytic Mt-derived HN could act as a beneficial secretory factor, including when transported within Mt to microglia, where it promotes a phagocytic/reparative phenotype. These findings also indicate that restoring HN levels in the injured brain could represent a translational target for ICH. These favorable biological responses to HN warrant studies on HN as therapeutic target for ICH.
SIGNIFICANCE STATEMENT Astrocytes are critical for maintaining brain homeostasis. Here, we demonstrate that astrocytes secrete mitochondria (Mt) and the Mt-genome-encoded, small bioactive peptide humanin (HN). Mt incorporate into microglia, and both Mt and HN promote a “reparative” microglia phenotype characterized by enhanced phagocytosis and reduced proinflammatory responses. Treatment with HN improved outcomes in an animal model of intracerebral hemorrhage, suggesting that this process could have biological relevance to stroke pathogenesis.
Introduction
Intracerebral hemorrhage (ICH) is a subtype of stroke caused by leaking or rupture of cerebral blood vessels within the brain parenchyma; it carries very high mortality rate (30%–67%) and poor prognosis for recovery, with limited therapeutic options (Qureshi et al., 2009; Adeoye and Broderick, 2010). After ICH, extravasated blood has direct contact with the brain parenchyma where it acts as a “reservoir” of toxicity for brain tissue, leading to progressive secondary “(bio)chemical” injury (Felberg et al., 2002; Wagner et al., 2003; Xi et al., 2004; X. Zhao et al., 2009a) through various mechanisms, including oxidative insult and complex proinflammatory responses (Hickenbottom et al., 1999; Wu et al., 2002; Aronowski and Hall, 2005; Keep et al., 2012). Ultimately, the hematoma is cleared by microglia/macrophages (MΦ) through phagocytosis, a process controlled by many factors, including peroxisome proliferator-activated receptor gamma (PPARγ) (Ricote et al., 1999; X. Zhao et al., 2007b, 2015c). This transcription factor induces the expression of key proteins controlling phagocytosis and associated cleanup processes after ICH, including antioxidative enzymes, such as superoxide dismutase (SOD) and catalase (X. Zhao et al., 2006, 2007b; Tureyen et al., 2007; Roszer et al., 2011). PPARγ also inhibits proinflammatory responses at multiple levels (Ricote et al., 1999; Sundararajan and Landreth, 2004; X. Zhao et al., 2007b).
Blood cell components (e.g., hemoglobin, heme, and iron) in hematomas are a major source of noxious free radicals that can cause oxidative damage to neighboring brain tissue (Regan and Panter, 1996; Felberg et al., 2002; Wagner et al., 2003; Nakamura et al., 2004; Aronowski and Hall, 2005). Impaired expression and dysfunction of antioxidative enzymes may amplify oxidative damage; for example, levels of the essential intramitochondrial antioxidant enzyme manganese SOD (Mn-SOD) are reduced in ICH-affected brain tissue (Wu et al., 2002). Since Mn-SOD shields mitochondria (Mt) and cells from oxidative damage, therapeutic approaches designed to upregulate Mn-SOD in the perihematoma zone could mitigate ICH-mediated damage.
Hayakawa et al. (2016) demonstrated that astrocytes transfer functional Mt to adjacent neurons, where they support prosurvival functions. However, the mechanism of how transferred Mt exert protective effects remains unclear. Mt are involved in myriad essential functions that can benefit recipient cells. Here we propose that one of the important tasks of transferred astrocytic Mt is supplying recipient cells with humanin (HN), a small peptide transcribed from MtDNA. HN is a 24-amino acid Mt-derived peptide discovered in the brain of an Alzheimer's patient (Hashimoto et al., 2001; Guo et al., 2003). The rat HN homolog rattin (HNr) is a 38 amino acid, and both HN and HNr are transcribed from an open reading frame within the 16S ribosomal RNA locus of MtDNA and are translated in the Mt and cytoplasm (Matsuoka and Hashimoto, 2010; Paharkova et al., 2015). HN can be stored in Mt or secreted into the extramitochondrial and extracellular spaces as an Mt autocrine, paracrine, and endocrine signal (Matsuoka and Hashimoto, 2010; Yen et al., 2013). Extracellularly secreted HN activates a cytoprotective signal by binding to a cytokine-like trimeric receptor composed of CNFR/WSX1/gp130 (Hashimoto et al., 2009a,b).
HN′s biological role has largely been studied in age-related contexts, including regulation of healthy aging, longevity, metabolism, and inflammation (Matsuoka, 2011; S. T. Zhao et al., 2013; Gong et al., 2014; Fuku et al., 2015; Kim et al., 2017). Less is known about HN in cerebrovascular diseases. It was shown to reduce brain infarction in a model of focal ischemia (Xu et al., 2006) and ameliorate neurological deficits in mice subjected to ICH (Wang et al., 2013). Here we demonstrate that astrocytes transfer Mt to microglia where, similar to HN alone, they promote “reparative” microglia function. This suggests that the beneficial effects of Mt transfer between astrocytes and microglia include generation of HN, and this process could be relevant to the pathogenesis of ICH.
Materials and Methods
Animals.
All animal studies were performed in accordance with the Guide for the care and use of laboratory animals from the National Institutes of Health guidelines and were approved by the Animal Welfare Committee of the University of Texas Health Science Center at Houston. All animal experiments and analyses used the approach of randomization, and investigators were blinded from treatments. Rodents were housed in standard cages under a 12 h inverted light-dark cycle. Three-month-old male C57BL/6J mice were purchased from The Jackson Laboratory, and female Sprague Dawley rats (embryonic 17 d timed pregnant) for primary cerebral cortical astrocyte and microglia cultures were purchased from Charles River Laboratories.
ICH.
Intrastriatal injection of autologous blood was used to simulate human ICH in mice as we described previously (X. Zhao et al., 2007b, 2017). Briefly, male C57BL/6J mice (25–30 g) were anesthetized with chloral hydrate (0.35 g/kg; i.p.) and placed in a stereotaxic apparatus before a 1-mm-diameter burr hole was drilled in the skull at the following coordinates: 0.0 mm anterior to bregma, 3.0 mm lateral to midline. A 30-gauge cannula was then inserted into the left striatum, 3.5 mm deep from the skull for blood (collected from femoral vein) infusion (18 μl at 1 μl/min). The body temperature was maintained at 37 ± 0.5°C during the procedure and for 2 h after surgery. Sham-operated mice underwent the same surgical procedures without blood infusion.
HN administration.
HN peptide was purchased from ANA SPEC (AS-60886). C57BL/6J male mice were randomly assigned to ICH surgery and treatments. Mice received HN (one injection per mouse per day for a total of 7 administrations) either intraperitoneally (1 μg/g in PBS) or intranasally (1 μg/g in PBS), starting 60 min (i.p.) or 3 h (intranasal) after the onset of ICH. PBS was used as vehicle control. To assess whether systemic administered HN enters ICH-injured brain, FITC-conjugated HN (1 μg/g in PBS) was injected into mice via femoral vein, 24 h after the onset of ICH. The HN-FITC peptide was customized/synthesized by WatsonBio Sciences.
Neurological deficit score (NDS) measurement.
All mouse behavioral tests were conducted under quiet, low-light conditions. The behavioral outcomes were measured by an investigator blinded to the treatment groups before and on day 3 for intranasal intervention and on days 3, 7, and 14 for intraperitoneal intervention after ICH. Foot-fault, postural flexing, wire, and corner tests were used to calculate the NDS as we reported previously (X. Zhao et al., 2007b, 2009c, 2017). Pretesting excluded animals with a preference during corner testing and/or >20% foot faults before being subjected to ICH.
Brain tissue harvesting.
Mice anesthetized with chloral hydrate (0.5 g/kg, i.p.) were transcardially perfused with ice-cold PBS. The brains were immediately frozen by submersion in 2-methylbutane (−80°C) and stored at −80°C until cryo-sectioning or RNA or protein extraction.
Hematoma cleanup model (hematoma resolution).
To assess hematoma resolution, the remaining amount of hemoglobin present in hematoma-affected brain tissue was measured on day 7 after ICH, as we described previously (X. Zhao et al., 2007b, 2009c, 2017). Briefly, anesthetized mice were transcardially perfused with ice-cold PBS to remove intravascular blood. Brains were collected, and coronal hematoma-containing sections were prepared using a brain matrix and razor blade. Following homogenization and centrifugation, hemoglobin in the supernatant was calorimetrically measured with Drabkin's reagent (D5941-6VL; Sigma Millipore) (X. Zhao et al., 2007b, 2009c, 2017). For a calibration curve, various volumes of peripheral blood were added to naive brain homogenates. The data are expressed as blood volume per brain homogenate.
Immunohistochemistry.
Rodents were placed under chloral hydrate anesthesia and subjected to transcardial perfusion with ice-cold PBS followed by 4% PFA. Brain tissue was collected in 30% sucrose solution for 24 h before snap freezing and cryosectioning at 20 μm. Sections were incubated in 0.1% Triton X-100 for 15 min at room temperature and blocked with PBS containing 3% BSA for 1 h at room temperature before incubation with primary antibodies (1:50 dilution in PBS containing 0.1% Tween and 0.3% BSA). We used the following primary antibodies: anti-rattin (HNr; rabbit-polyclonal, PA1-46257; Thermo Fisher Scientific), anti-ionized calcium-binding adaptor molecular 1 (Iba1; rabbit-polyclonal, 019–19741; Wako Life Science), and anti-GFAP (mouse-monoclonal, G7171, Sigma Millipore). Secondary antibodies (1:100 dilution in PBS containing 0.1% Tween and 0.3% BSA) included the following: goat anti-rabbit IgG (AlexaFluor-488 or 546, Thermo Fisher Scientific) and goat anti-mouse IgG (AlexaFluor-488, Thermo Fisher Scientific). Brain images were captured using an LSM 800 confocal microscope with Airyscan high-resolution detectors (Carl Zeiss). For immunofluorescent analysis of primary rat astrocytes, we used anti-lysosomal-associated membrane protein 1 (LAMP1; 1:250 dilution, rabbit-polyclonal, ab24170; Abcam) and goat anti-rabbit IgG (1:500 dilution, AlexaFluor-488, Thermo Fisher Scientific). The procedure for immunofluorescence was published previously (Jung et al., 2015).
Primary cerebral cortical glial cell cultures.
Rat astrocyte and microglia cultures were generated using embryonic day 17 or 18 brains, as we previously described (X. Zhao et al., 2007b, 2017). Briefly, mixed cells from brain cortices were seeded in poly-d-lysine-coated 75 cm2 flasks and cultured in DMEM containing 10% FBS, penicillin (50 U/ml), and streptomycin (50 μg/ml) for 10–14 d. Once cells were confluent in the flask, adherent microglia were harvested using a slight shaking method (37°C at 220 rpm for 30–60 min). Harvested microglia were replated onto poly-d-lysine-coated plates with DMEM containing 10% FBS, penicillin (50 U/ml), and streptomycin (50 μg/ml). After microglia harvest, the cocultures were trypsinized with 0.25% Trypsin-EDTA to collect the mixed astroglial cells attached to the bottom of the flask. These were replated onto 75 cm2 flasks and cultured in DMEM containing 10% FBS, penicillin (50 U/ml), and streptomycin (50 μg/ml). Once the cells were slightly attached to the bottom (4–6 h after replating), the flask was transferred into a 37°C shaking incubator and continuously shaken (220 rpm) overnight to remove weakly attached cells. The medium containing floating cells was discarded the next day, and new fresh culture medium was added to the strongly attached astrocytes.
FACS analysis for Mt.
FACS analysis to detect secreted Mt from cultured astrocytes was performed on a Cytoflex S cytometer (Beckman Coulter). The data were analyzed with FloJo version 10. Rat cortical astrocytes were incubated with Mitotracker Red CMXRos (M7512, Thermo Fisher Scientific) for 30 min to label intracellular Mt. After extensive washing, the astrocytes received fresh culture medium and were incubated for 24 h to allow secreted Mt accumulation. The culture medium was collected and briefly spun down at 1000 rpm for 5 min to remove cellular debris. Half of the supernatant was saved as astrocyte-conditioned medium (ACM), and the other half was filtered through a column with 0.22 μm pores to remove Mt particles (Filtered-ACM) as previously described (Hayakawa et al., 2016). Unstained ACM (without Mitotracker treatment) was used as a negative control for proper gating.
Transmission electron microscope (TEM) analysis.
To probe ACM for extracellularly released Mt, collected ACM was subjected it to 4020 × g centrifugation for 30 min at 4°C to produce a pellet containing Mt. The pellet was resuspended, transferred to a 1.5 ml Eppendorf tube, and further spun down at 13,400 × g for 15 min at 4°C. The final Mt-containing pellet was washed with PBS and fixed with 2% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.4. Fixed samples were washed in 0.1 m sodium cacodylate buffer and treated with 0.1% cacodylate-buffered tannic acid, postfixed with 1% buffered osmium, and stained with 1% uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol and embedded in LX-112 medium. The samples were polymerized in a 60°C oven for ∼3 d. Ultrathin sections were cut using a microtome (Leica Microsystems), stained with uranyl acetate and lead citrate in a Leica Microsystems EM Stainer, and examined in a JEM 1010 transmission electron microscope (JEOL) at an accelerating voltage of 80 kV. Digital images were obtained using an AMT Imaging System (Advanced Microscopy Techniques). Sample processing and TEM evaluation were conducted at the High Resolution Electron Microscopy Facility of the University of Texas M.D. Anderson Cancer Center.
Red blood cell (RBC) isolation.
RBCs from rat peripheral blood were isolated using column density gradient centrifugation (BD Vacutainer CPT, BD Biosciences) as we described previously (X. Zhao et al., 2007b).
Phagocytosis in vitro.
To measure microglial phagocytic activity, in vitro phagocytosis assays were performed as described previously (X. Zhao et al., 2007b). Briefly, RBCs were added to the cultured microglia and incubated for 2 h. Then, the culture plate was briefly centrifuged to remove unphagocytosed RBCs. Microglia containing phagocytosed RBCs were fixed with 4% PFA and stained with anti-CD11b (mouse monoclonal, MCA275G; Bio-Rad) and anti-rat RBC (rabbit polyclonal, 20R-RR012; Fitzgerald Industries) antibodies to visualize internalized RBCs. The number of engulfed RBCs in microglia was used as a phagocytosis index.
Small interfering RNA (siRNA) transfection.
siRNA probes targeting HNr (GGTTCAACTGTCTCTTACT and GGGATAACAGCGCAATCCT) were obtained from Sigma Millipore. The nontargeting, negative control, scrambled siRNA (SIC001) was obtained from Sigma Millipore. Primary rat cortical astrocytes were seeded on 6-well plates (2 × 106 cell/well) and transfected with each siRNA (10 nmol/L) using HiPerFect siRNA transfection reagent (301704; QIAGEN) for 24 h following the manufacturer's instructions. The ACM for transfer onto microglia was collected as described above.
RT-PCR analysis.
Total RNA was extracted from rat primary microglia or the contralateral and ipsilateral ICH-affected mouse brain using the RNeasy Plus Mini Kit (74134, QIAGEN). The cDNAs were generated using amfiRivert Plantinum ONE cDNA Synthesis Master Mix (R6100, GenDEPOT). The amfiSure qGreen Q-PCR Master Mix without ROX (Q5600, GenDEPOT) was used for SYBR real-time RT-PCR analysis. The following sets of primers (5′-3′) were used for experiments: HNr, TTAGGGACTAGAATGAATGG and GGAGCTTCAATTTACTAGTT; rat PPARγ, ACCAACTTCGGAATCAGCTC and AGGCTCTACTTTGATCGCAC; rat lipoprotein lipase (LPL), CAGCTGGGCCTAACTTTGAG and TGCTGGGGTTTTCTTCATTC; rat catalase, ATGCAAAGGGAGCAGGTG and AATGGGAAGGTTTCTGCC; rat IL-10, TCCTTGGAAAACCTCGTTTG and GCTTTCGAGACTGGAAGTGG; rat IL-1β, CAGGAAGGCAGTGTCACTCA and GGGATTTTGTCGTTGCTTGT; rat CD68, AATGTGTCCTTCCCACAAGC and AGAGGGGCTGGTAGGTTGAT; rat GAPDH, AGACAGCCGCATCTTCTTGT and TACTCAGCACCAGCATCACC; and mouse GAPDH, TGTTCCTACCCCCAATGTGT and TGTGAGGGAGATGCTCAGTG.
Western blot.
Protein samples were prepared from whole-cell lysates as previously described (Jung et al., 2009, 2015). Briefly, protein samples were separated on 4%–20% gradient Tris-glycine SDS gels (EC6021, Thermo Fisher Scientific) followed by electrophoretic transfer onto nitrocellulose membranes using an iBlot2 Dry Blotting System (IB21001, Thermo Fisher Scientific). Antibodies against Mn-SOD (1:1000 dilution; rabbit polyclonal, ADI-SOD-110-D; Enzo Life Science), PPARγ (1:1000 dilution; rabbit polyclonal, sc-7196; Santa Cruz Biotechnology), and β-tubulin (1:5000 dilution; mouse monoclonal, T8328, Sigma Millipore) were used for immunoblotting. HRP-conjugated IgG and an enhanced chemiluminescent substrate (Pierce ECL kit 32106, Thermo Fisher Scientific) were used to visualize protein bands on x-ray films. The band intensity was analyzed by ImageJ program (U.S. National Institutes of Health) and normalized by intensity of control band (β-tubulin).
Dot blot.
To detect HNr that is released into media, we collected ACM in 75 cm2 tissue culture flasks. The astrocytes were incubated with 5–6 ml of serum-free DMEM for 24 h before concentrating ACM to 100 μl to reach a concentration of 30 μg of proteins in 10 μl of loading volume for dot blotting. To concentrate released HNr, collected ACM were centrifuged with Vivaspin 20 Centrifuge Concentrators (VS2091, 3000 Dalton Max Molecular Weight Cut Off, PES membrane; Sartorius). Dot blot analysis using a Bio-Dot SF Microfiltration Apparatus (1706545, Bio-Rad) was used to assess HNr concentration. Briefly, ACM protein was deposited onto nitrocellulose membranes in the dot blot apparatus. The membranes were blocked with 5% BSA in Tris-buffered saline with Tween and then incubated with anti-rattin (HNr) antibody (1:1000 dilution; rabbit-polyclonal, PA1-46257, Thermo Fisher Scientific). Secondary HRP-conjugated IgG and an enhanced chemiluminescent substrate were used to visualize HNr as described above. In addition, an Odyssey CLx imaging system (LI-COR) using IRDye 800CW goat anti-rabbit IgG secondary antibody (1:5000 dilution; 925-32211, LI-COR) or IRDye 800CW Goat anti-Mouse IgG secondary antibody (1:5000 dilution; 925-32210, LI-COR) was used to visualize HNr or β-tubulin (see Figure 9B). The immunointensity of each of the dots was analyzed by ImageJ program and normalized to intensity of β-tubulin, control.
Figure 9.
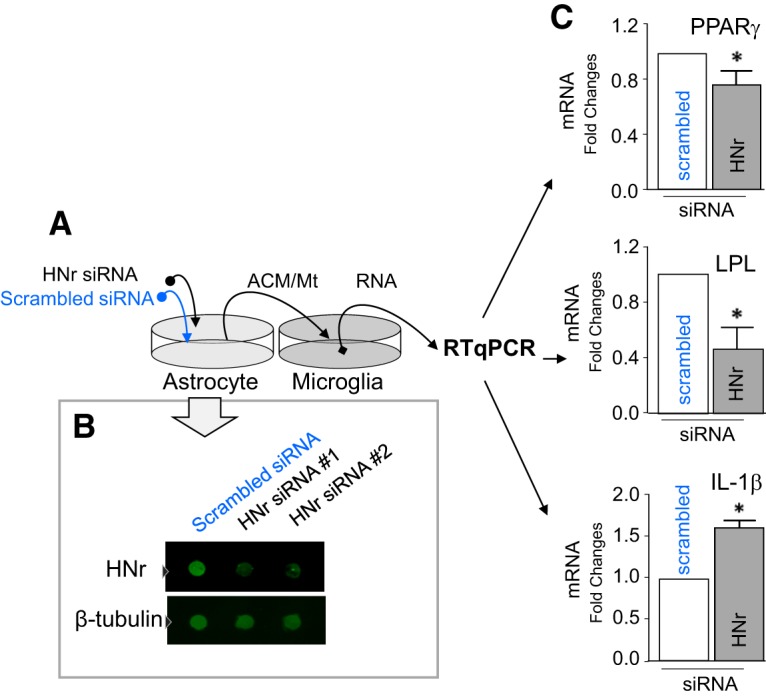
ACM from HN-deficient astrocytes has reduced capacity to modulate microglia. Rat astrocytes were transfected with HNr-specific siRNAs or scrambled RNA for 24 h. After 24 h, siRNA-containing media was replaced with fresh culture medium. Forty-eight hours later, ACM was collected and transferred onto cultured microglia (see schematic; A). The microglia were incubated with ACM for 24 h. The knockdown efficiencies of two different siRNAs in astrocytes were assessed by confirming reduced protein level using immune dot blot analysis for HNr (B). C, Gene expression profiles of PPARγ, LPL, and IL-1β by SYBR real-time RT-PCR in microglia treated for 24 h with ACM collected from astrocytes treated with siRNA targeting HNr or scrambled siRNA. Data are mean ± SEM and assessed for difference between scrambled siRNA and HNr. For scrambled versus HNr: scrambled ACM (n = 3), HNr knockdown ACM (n = 5), two-tailed unpaired t test, PPARγ; *p = 0.0357, LPL; *p = 0.0430, t values (t = 2.558), IL-1β; *p = 0.0024, t values (t = 5.007).
Experimental design and statistical analyses.
For all the in vivo animal studies, animals were randomly divided for treatments and ICH surgery. The sample size calculation was based on our past experience with mouse ICH model (X. Zhao et al., 2007a,b, 2009b, 2017), regarding reproducibility in the amount of damage, and power analysis using STATMATE 3 software (a significant difference at a power of 90%, estimated SD of mean at ∼0.1–0.2, and expected difference between groups at 10%–20%) to obtain the minimal number of animals for statistical significance. For animal behavior tests, two-way ANOVA followed by the Fisher LSD method was used to compare NDS between two groups at different time points. Data are presented as mean ± SEM. Most in vitro studies were analyzed by one-way ANOVA followed by the Fisher's Least Significant Difference (LSD) method, or followed by Kruskal–Wallis test or Bonferroni test between experimental groups. Paired or unpaired t tests were used to compare two groups. Statistically significant differences between mean values were considered only at p < 0.05. Prism 7.04 software (GraphPad Software) was used for statistical analysis. The detailed results of statistical analyses, including the number or size of each experiment, are listed in each figure legend.
Results
ICH reduces HN level in the affected brain hemisphere
Reduced HN levels adversely affect many pathological processes, longevity, and aging (Muzumdar et al., 2009; Voigt and Jelinek, 2016). It is therefore relevant to understand how cerebrovascular events, such as ICH, affect HN and influence disease progression. Hence, we evaluated mRNA and protein levels of HN in the ICH-affected brain. Using SYBR real-time RT-PCR and brains harvested at 24 h after ICH, we found a robust ∼50% reduction in HNr mRNA levels in the ICH-affected forebrain compared with the homologous contralateral hemisphere (mRNA ratio of contralateral vs ipsilateral is 1:0.574, n = 13, two-tailed paired t test, p = 0.000198, t = 5.27). We also observed a profound loss of HNr immunoreactivity throughout the ICH-affected hemisphere compared with the contralateral brain (Fig. 1). Considering the localized nature of the ICH lesion (5%–10% of the hemisphere), the extent of HNr immunoreactivity loss was widespread in the ipsilateral brain.
Figure 1.
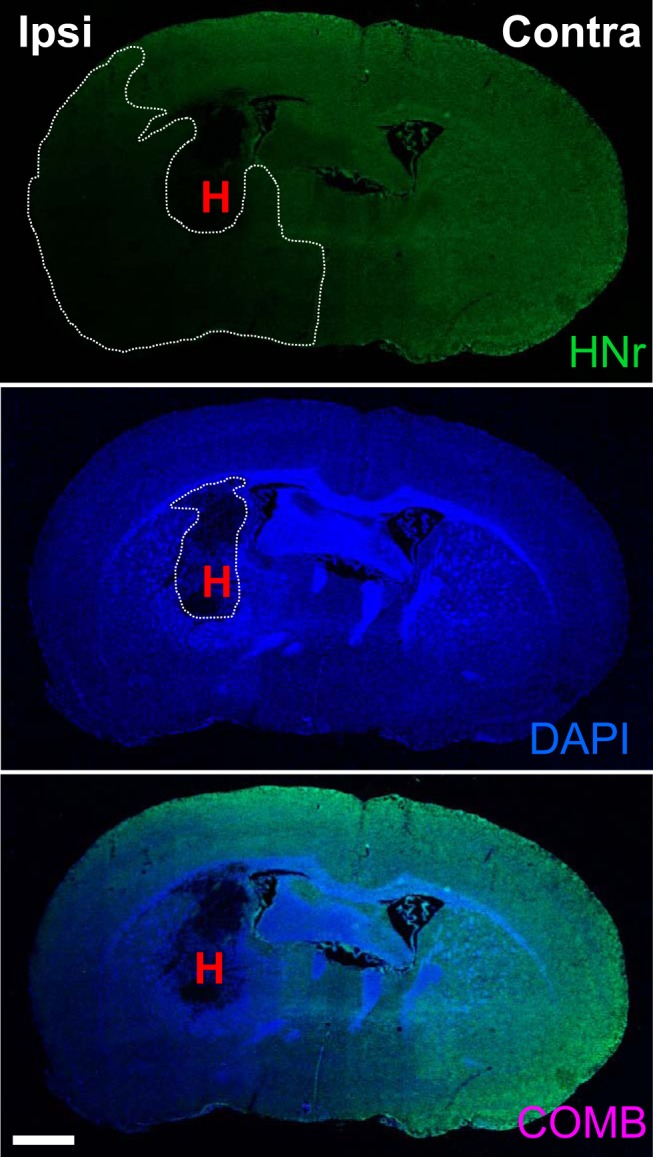
ICH reduces HN expression in the affected hemisphere. HNr immunoreactivity is reduced in the ICH-affected forebrain compared with contralateral hemisphere at 24 h after ICH. Representative image reconstructing a coronal section of the ICH mouse brain under fluorescence microscope (10×) showing the HN signal detected using anti-HNr (Rattin; a rat homolog of HN) antibody. Scale bar, 1000 μm. DAPI was used to visualize nuclei. H, Hematoma; Contra, contralateral; Ipsi, ipsilateral; COMB, combined.
HN reduces neurological deficits and improves hematoma clearance in ICH mice
Since ICH dramatically decreased HNr in the brain, we examined whether HN administration could affect the severity of ICH-induced injury. First, we intravenously injected mice with HN-FITC 24 h after ICH. Confocal microscopy was performed to inspect brain tissue for the presence of green fluorescence. We established that Iba1-positive cells (e.g., microglia and macrophages) are one of the prominent cell targets for HN-FITC, based on colocalization of fluorescence signals (Fig. 2A). By probing cells in randomly selected locations around hematoma location and in homolog locations of the contralateral hemisphere, we found that ∼67% of all Iba1+ cells were also positive for FITC (double-positive) (Fig. 2B), indicating an overall abundant internalization of HN to the brain and microglia.
Figure 2.
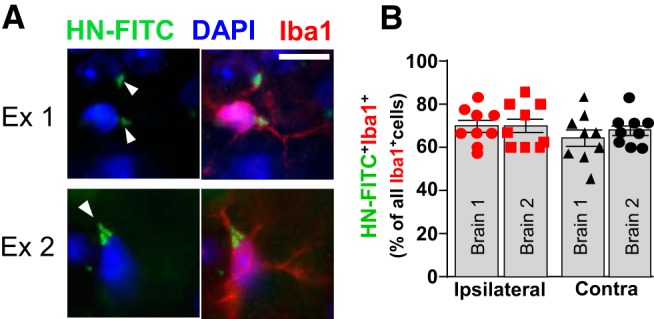
Intravenously administered HN is found in microglia in the mouse brain. A, HN-FITC (1 μg/g in PBS) was injected via the femoral vein 24 h after ICH onset. After another 24 h, confocal microscopy was used to detect HN-FITC (green, white arrowheads) in the ICH-affected brain (ipsilateral) and their colocalization with Iba1+ cells (red). Representative examples of two randomly selected HN-FITC-positive cells are shown. Nuclei were stained with DAPI (blue). Scale bar, 10 μm. B, Quantitative bar graph (mean ± SEM) showing the percentage of HN-FITC/Iba1 double-positive cells among all the Iba1+ cells, as assessed in nine or 10 randomly selected perihematoma (ipsilateral) and homologous contralateral locations (Contra) in two independently analyzed mouse brains.
Next, we used a mouse ICH model to determine whether HN has a therapeutic effect in ICH-induced brain injury. Specifically, we tested whether HN could reduce neurological deficits (NDS; measured with corner, postural flexing, or foot-fault testing) resulting from ICH injury. HN was injected once a day (1 μg/g in PBS, i.p., starting 1 h after ICH) for a total of 7 administrations. This significantly reduced NDS in HN-treated mice (Fig. 3A,B). In addition to the functional benefit, HN accelerated hematoma clearance, a process that is mediated by phagocytic cells, including microglia/macrophages. This was established by reduced hemoglobin content (a major hematoma component) 7 d after ICH in HN-treated mice compared with vehicle-treated mice (Fig. 3C). In a parallel experiment, we assessed whether the therapeutic effect of HN could also be achieved with intranasal HN. We extended the therapeutic window for HN to 3 h after ICH, which is a more clinically relevant protocol. Mice were treated with HN at 1 μg/g in PBS for a total of 7 administrations. Similar to the intraperitoneal route, intranasal HN reduced NDS (Fig. 3D).
Figure 3.

HN treatment reduces neurological deficits and improves hematoma clearance in an ICH mouse model, suggesting a role of phagocytic cells. NDSs were calculated from corner, postural flexing, or foot-fault testing in mice treated either with HN given intraperitoneally (1 μg/g) (A,B) or intranasally (1 μg/g) (D) starting 1 h (A,B) or 3 h (D) after ICH onset. PBS was used as a vehicle control. C, Hemoglobin content as an index of hematoma clearance in ICH-affected brains on day 7 after ICH in mice treated with intraperitoneal HN or PBS. Data are mean ± SEM. A, Two-way ANOVA/Fisher's LSD test, n = 8 per group, *p < 0.05 (p = 0.0271), t values (t = 2.275). B, Two-way ANOVA/Fisher's LSD test, n = 8 per group, *p < 0.05 (p = 0.0299), t values (t = 2.232) at day 7; *p < 0.05 (p = 0.0455), t values (t = 2.049) at day 14. C, Two-tailed unpaired t test, n = 7 per group, *p < 0.05 (p = 0.0322), t values (t = 2.423). D, Two-way ANOVA/Fisher's LSD test, n = 8 per group, *p < 0.05 (p = 0.0195), t values (t = 2.479) at day 3.
Astrocytes in culture secrete HN, and HN is present in astrocytes in the brain
HN is a peptide encoded by Mt DNA (Yen et al., 2013). Once translated, HN is primarily confined to Mt, but it can also be released to the cytoplasm (Yen et al., 2013). Mt themselves are the major storage site for HN. A recent study demonstrated that astrocytes release Mt, and other cell types can uptake these Mt (Hayakawa et al., 2016). This suggests that the biological effects of HN could be mediated through its release to the extracellular space and/or cell-to-cell transfer via astrocyte-secreted Mt.
To test this hypothesis, we examined whether HN is produced and secreted by primary rat astrocytes in culture. ACM was collected in an established culture system. Using dot blots and an antibody against HNr, we demonstrated that cultured astrocytes indeed produce and secrete HNr (Fig. 4A). Specifically, we collected ACMs from five rat astrocyte preps, and performed dot immunoblots to quantitate secreted HNr. Compared with DMEM, we found ∼14-fold increase in HNr level in ACM (DMEM vs ACM = 1:14.506, n = 5, two-tailed unpaired t test, p = 0.0075, t = 3.547). In addition, immunohistochemistry experiments confirmed strong presence of HNr immunoreactivity in GFAP+ cells in the inspected cerebral cortex of the normal rat brain (Fig. 4B). We quantified the numbers of GFAP+/HNr+ double-positive cells in the randomly selected areas of cerebral cortex of 3 naive rats. Approximately 61% of HNr+ cells showed GFAP-positive (Fig. 4C), confirming that astrocytes could be an important source of HN.
Figure 4.

Astrocytes in culture secrete HN, and HN is present in astrocytes in the brain. A, Rat ACM was concentrated using Vivaspin and spotted onto nitrocellulose membranes. HN (2 μl from 10 mm HN stock) and DMEM were used as positive and negative controls, respectively. Image represents two different ACM samples from each culture that were spotted. Extracellularly released HN was detected using anti-HNr (a rat homolog of HN) antibody. B, A strong HNr immunofluorescence signal (red) was observed in GFAP+ (green) rat astrocytes. Confocal image. Scale bar, 6.25 μm. C, Quantitative bar graph (mean ± SEM) showing the percentage of HNr/GFAP double-positive cells among all the HNr+ cells, as assessed in four randomly selected locations in three independently analyzed mouse brains.
Astrocyte-secreted Mt incorporate into microglia and subsequently increase HNr levels
To confirm that astrocytes release Mt, primary astrocyte cultures were incubated with or without Mitotracker Red CMXRos to fluorescently label Mt for FACS analysis of ACM (Fig. 5A,B). Mt were abundant in the ACM, suggesting that astrocytes indeed secrete large quantities of Mt (Fig. 5B). Hayakawa et al. (2016) used qNano analysis to show that astrocyte-released Mt range in size from 300 to 1100 nm and, as such, could be removed from conditioned media with a 0.22 μm filter. Quantification of MitoTracker-positive particles revealed that ACM filtration through a 0.22 μm filter removed 78% of Mt (Fig. 5C). TEM confirmed the presence of Mt in ACM (Fig. 5D).
Figure 5.

Mt are released by astrocytes in vitro. Rat cortical astrocytes were incubated for 30 min with Mitotracker/Red/CMXRos to label Mt. After extensive washing, astrocytes were incubated with fresh culture medium for 24 h to accumulate Mt in the media. ACM was collected and briefly spun down to remove cellular debris and assessed for the presence of Mitotracker-positive Mt using FACS analysis. A, Negative control, ACM from astrocytes that were not stained with Mitotracker/Red/CMXRos. B, ACM collected from astrocytes stained with Mitotracker/Red/CMXRos. C, ACM collected as in B, but filtered through a 0.22 μm pore-size column to remove Mt (Filtered-ACM). FACS analysis data for the above samples. D, An example of Mt found in ACM as detected using TEM. Scale bar, 100 nm.
Next, we tested whether Mt released by astrocytes can enter microglia. We labeled Mt in rat astrocytes with MitoTracker/CMX/Ros (Fig. 6A,B, red, white arrowheads) and collected ACM-containing MitoTracker-positive Mt for transfer into rat primary microglia cultures (Fig. 6A). This approach revealed that astrocytic Mt effectively integrated into microglia (Fig. 6C,D). Time-lapse imaging confirmed Mt transfer into microglia. The images of two representative microglial cells (Fig. 6C, color-coded blue and red) taken before Mt transfer (0 min, top), at the indicated times after ACM addition between 15 min and 11 h 11 min (Fig. 6C, middle), and at 14 h at the end of the recording (Fig. 6C, bottom) show the dynamics of microglial Mt accumulation. All the microglial cells showed the presence of MitoTracker red-positive Mt (Fig. 6-1). Upon adding ACM, Mt incorporate into the cultured microglia almost immediately and continue to accumulate for hours. It is essential to note that Mt that entered microglia remained intact for at least 72 h (Fig. 6D), suggesting that these Mt are not just a target of phagocytosis and consequent degradation. To strengthen this claim, we quantified the numbers of LAMP-1+/MitoTracker+ (double-positive) particles within 14 randomly selected microglia (Fig. 6E). We established that 31.24% of MitoTracker+ speckles were also LAMP-1+. This indicates that most of the internalized Mt (∼69%) did not colocalize with the phagosome marker LAMP-1 at 72 h.
Figure 6.

Astrocyte-secreted Mt incorporate into microglia and elevate intracellular HN levels. A, Astrocytes in culture were treated with MitoTracker/CMX/Ros to red-fluorescently label Mt. After extensive washing to remove MitoTracker and additional 24 h incubation to accumulate Mt in the media, the media containing red-labeled astrocytic Mt were transferred to rat primary microglia in culture. B, Representative confocal image of rat astrocytes in culture treated with MitoTracker/CMX/Ros to label Mt (red puncta, white arrowheads). Scale bar, 20 μm. C, Microglia taking up astrocytic Mt. Time-lapse images were collected to document Mt (red) transfer into microglia over a 14 h period. Images of two representative microglial cells (color-coded as blue and red) before Mt transfer (0 min, top) and at the indicated times (15 min to 11 h, middle; and at 14 h, bottom). Scale bar, 20 μm. At the end of the recording, all the microglial cells showed the presence of MitoTracker (Figure 6-1). D, Confocal image of microglial cells containing astrocytic Mt (MitoTracker red+ puncta). These astrocyte-derived Mt (red, white arrowheads) are retained in microglia for at least 72 h after internalization. Most of the Mt (Mito+ red puncta) did not colocalize with the LAMP-1 (green puncta), suggesting that Mt are not merely undergoing lysosomal degradation. Scale bar, 10 μm. E, Quantitative data showing the percent of LAMP-1/MitoTracker double-positive puncta among all the MitoTracker red+ puncta, as determined for 14 randomly selected microglial cells.
Microglia taking up astrocytic Mt. Astrocytes in culture were treated with MitoTracker/CMX/Ros to label Mt. After extensive washing to remove MitoTracker and 24 h incubation to accumulate Mt in the media, the media containing labeled astrocytic Mt were transferred to rat primary microglia in culture. Time-lapse images (0 min, top panel and 14 h, bottom panel) were collected to document Mt transfer into microglia over a 14 h period. Images of three representative microglial cells (color-coded as blue, red and yellow) prior to Mt transfer (0 min, top panel) and at the end time point (14 h, bottom panel) are shown. Vast majority of microglia in a culture dish took up astrocytic Mt. Download Figure 6-1, EPS file (11.5MB, eps)
Since Mt are the main source of HN, astrocyte-released Mt could transport HN in the form of DNA, mRNA, and/or protein. We therefore tested whether astrocytic Mt accumulated in microglia could elevate HN content in these cells. ACM or filtered ACM (mdACM; Mt-depleted ACM) (Fig. 7A,B) was added to rat microglia and incubated for 24 h. Indeed, incorporation of astrocytic Mt into microglia significantly increased levels of HNr mRNA (Fig. 7A) and protein (Fig. 7B). When Mt were removed by passage through a 0.22 μm filter, there was no increase in microglial HNr (Fig. 7A,B). These data suggest that astrocytic Mt transfer HNr to microglia, which increases HNr levels.
Figure 7.
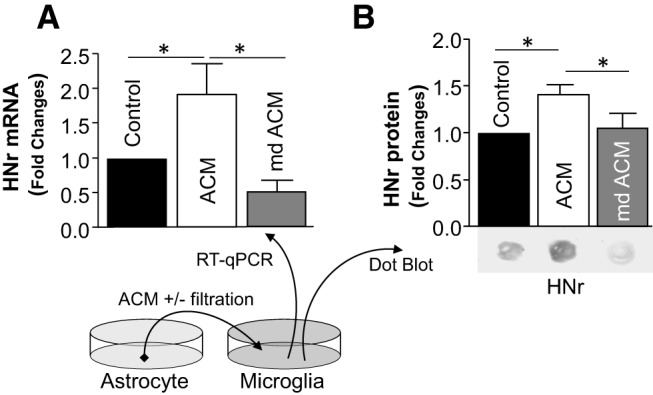
ACM containing Mt, but not Mt-depleted ACM, induced microglial HN synthesis. ACM or Mt-depleted ACM (mdACM, filtered ACM through a 0.22 μm pore-size column to remove Mt) was transferred onto cultured microglia. After 24 h, HNr mRNA levels (A) were assessed by SYBR real-time RT-PCR. Data are mean ± SEM; n = 5. One-way ANOVA/Fisher's LSD test: *p < 0.05 (p = 0.0380, Control vs ACM), t values (t = 2.358); *p < 0.01 (p = 0.0062, ACM vs mdACM), t values (t = 3.373). B, HNr protein levels were assessed by immune dot blot. Data are mean ± SEM; n = 3. One-way ANOVA/Fisher's LSD test: *p < 0.05 (p = 0.0366, Control vs ACM), t values (t = 2.678); *p < 0.05 (p = 0.0479, ACM vs mdACM), t values (t = 2.478).
HN or Mt upregulate PPARγ/Mn-SOD and other prototypic PPARγ target genes in microglia
Following ICH, the transcription factor PPARγ and its target genes play central roles in promoting a “reparative” microglia phenotype that is protrophic and phagocytic but anti-inflammatory phenotype (Ricote et al., 1999; X. Zhao et al., 2007b; Yi et al., 2008). Mn-SOD (a transcriptional target of PPARγ), an enzyme exclusively confined to Mt, plays an essential role by maintaining the oxidative balance necessary to maintain this “reparative” microglial phenotype. Hence, we tested whether microglial PPARγ and Mn-SOD are upregulated in response to HN or after astrocytic Mt transfer (another source of intra-Mt HNr). ACM (Fig. 8A,B) or mdACM (Fig. 8B) was incubated with rat microglia for 24 h. In a parallel experiment, HN alone (Fig. 8A,B) or in the presence of HN-neutralizing antibody (Fig. 8B) was added to microglia and incubated for 24 h. Western blotting revealed that PPARγ and Mn-SOD were significantly upregulated by HN or ACM (Fig. 8A). However, removal of Mt from ACM (mdACM) by filtration before ACM transfer significantly reduced these responses in microglia (Fig. 8B, column 2), suggesting that PPARγ and Mn-SOD induction are achieved through Mt transfer rather than small particles or soluble factor(s) present in ACM. Interestingly, addition of HN to mdACM significantly restored Mn-SOD and PPARγ induction (Fig. 8B, columns 1, 2, and 4). This effect was partially ameliorated with an antibody targeting HN (Fig. 8B, columns 4 and 5).
Figure 8.

HN or ACM upregulates PPARγ/Mn-SOD and other prototypic PPARγ target genes in microglia, and enhances microglial phagocytic activity in an in vitro hematoma clearance model. A, Western blots determine microglial induction of PPARγ and Mn-SOD at 24 h after exposure to ACM or HN (20 μm). The significance of induction was as follows: CON versus ACM in PPARγ, n = 3, two-tailed unpaired t test, *p = 0.0310, t values (t = 5.548). CON versus HN in PPARγ, n = 4, two-tailed unpaired t test, *p = 0.0337, t values (t = 2.741). CON versus ACM in Mn-SOD, n = 4, two-tailed unpaired t test, *p = 0.0289, t values (t = 2.857). CON versus HN in Mn-SOD, n = 4, two-tailed unpaired t test, *p = 0.0209, t values (t = 3.109). B, Mn-SOD and PPARγ protein levels in the microglia. ACM after removing Mt (Mt depleted ACM; mdACM, by filtration with 0.22 μm filter alone) or in combination with HN neutralizing antibody (Ab-HNr, 100 ng) was added to cultured microglia and incubated for 24 h, as above. The significant changes in PPARγ; n = 3 or 4. One-way ANOVA/Kruskal–Wallis test: ACM versus mdACM treatment, *p = 0.0486, mean rank difference (7.0) and for mdACM versus mdACM + HN, *p = 0.0011, mean rank difference (−12.5). The significant changes in Mn-SOD; n = 3–5. One-way ANOVA/Kruskal–Wallis test: ACM versus mdACM, *p = 0.0173, mean rank difference (8.4); mdACM versus HN, *p = 0.0210, mean rank difference (−9.4); and mdACM versus mdACM + HN, *p = 0.0084, mean rank difference (−10.73). C, Gene expression profile by RT-PCR (SYBR real-time analysis) in microglia treated with ACM or HN (20 μm) for 24 h. Data are mean ± SEM; n = 3. One-way ANOVA/Fisher's LSD test: PPARγ, *p = 0.0131, t values (t = 3.484) at control versus HN; LPL, *p = 0.0386, t values (t = 2.638) at control versus ACM, *p = 0.0007, t values (t = 6.355) at control versus HN; catalase, *p = 0.0353, t values (t = 2.705) at control versus ACM, *p = 0.0063, t values (t = 4.102) at control versus HN; IL-1β, *p = 0.0018, t values (t = 6.057) at control versus ACM, *p = 0.0131, t values (t = 3.484) at control versus HN. D, E, Phagocytosis assay. Rat primary microglia were treated with ACM, HN (20 μm), or vehicle (control) for 24 h. Rat RBCs (1 × 106 RBCs/well) were added as a phagocytic target. After 2 h, the cells were fixed and processed for immunostaining to determine the number of engulfed RBCs. D, Representative confocal image of microglia/CD11b+ (green) containing engulfed RBCs (red). Scale bar, 10 μm. E, Index of phagocytosis illustrating increased levels with ACM or HN treatment. Data are mean ± SEM; n = 3 or 4 per group. One-way ANOVA/Bonferroni's test: *p = 0.0013, CON versus ACM, t values (t = 6.227); *p = 0.0002, CON versus HN, t values (t = 8.228). CON, Control.
Gene expression changes were analyzed to further clarify microglial responses to HN or ACM. The expression of PPARγ and its prototypic target genes, LPL and catalase (a potent antioxidant), was increased by HN and ACM. At the tested doses, HN was more potent (Fig. 8C). Overall, these data suggest that HN and Mt affect PPARγ signaling in microglia. Interestingly, HN and ACM significantly reduced IL-1β expression, the outcome that could occur upon increased PPARγ signaling that normally leads to anti-inflammatory responses (Sundararajan and Landreth, 2004). There was no significant effect on IL-10 (p = 0.8572 in Control vs ACM, p = 0.5483 in Control vs HN). Considering the unaltered expression of CD68 and microglial cell counting (data not included), HN or Mt did not affect the number of microglia, suggesting no obvious effect on proliferation. Overall, these data suggest that HN and astrocyte-derived Mt are capable of inducing a “reparative” microglial phenotype.
HN or Mt enhance microglial phagocytic activity toward RBCs: in vitro hematoma cleanup model
Enhanced phagocytic function is one of the microglial responses to PPARγ activation (X. Zhao et al., 2007b). Together with the in vivo hematoma clearance data (Fig. 3C), the results suggest that HN and Mt could regulate the phagocytic function of microglia. To test this question, we used primary microglia treated with ACM or HN for 24 h before adding RBCs as a phagocytic target. After 2 h of incubation, the numbers of internalized RBCs per CD11b+ microglia were counted under a confocal microscope. An example of microglia containing engulfed RBCs is shown in Figure 8D. HN and ACM effectively increased the numbers of engulfed RBCs by microglia compared with vehicle control (Fig. 8E). This result suggests that both HN and astrocytic Mt can enhance microglial phagocytic activity.
Mt from HN-deficient astrocytes fail to induce a healing microglial phenotype
As discussed above, astrocytic Mt likely modulate microglia by transporting HN to microglia. Thus, we tested whether HN deficiency in astrocytes inhibited the ability of ACM to modulate microglial expression of PPARγ and a prototypic target gene, LPL. Rat astrocytes were transfected with HNr-specific siRNAs (Fig. 9A), which unlike scrambled siRNA, reduced HNr expression in these cells (Fig. 9B). Next, ACM from HNr-deficient and -proficient astrocytes were collected and transferred to cultured microglia (Fig. 9A). In contrast to ACM from astrocytes treated with scrambled siRNA, ACM from HNr-knockdown astrocytes was significantly less effective in inducing PPARγ and LPL expression (Fig. 9C). ACM from HNr knockdown was also less effective in suppressing IL-1β expression by microglia (Fig. 9C).
Discussion
A limited number of studies addressed the existence of Mt transfer between cells, with most assuming that the main effect is to enhance the bioenergetic capacity of the recipient cell (Spees et al., 2006; Plotnikov et al., 2010; Islam et al., 2012; Pasquier et al., 2013; Liu et al., 2014; Wang and Gerdes, 2015; Hayakawa et al., 2016, 2018). Even fewer investigated the role of Mt transfer in the CNS (Hayakawa et al., 2016, 2018). It was recently proposed that, following brain ischemia, astrocytes increase the ATP supply by transferring functional Mt to neurons (Hayakawa et al., 2016). However, while the beneficial effect of Mt transfer is fairly well documented, the specific factor(s) provided by transferred Mt remain unclear. This benefit can extend beyond the energetic aspect of Mt transfer. Here, we propose that astrocytic Mt-derived HN acts as a transcellular signaling molecule that can modulate “reparative” microglial functions after being secreted by astrocytes as a peptide or while being transferred within Mt. We also propose that HN may help reduce damage caused by ICH.
Our study demonstrates that astrocyte-secreted Mt are taken up by microglia, where they are detected for at least several days at the intracellular locations that do not colocalize with phagosomes, suggesting that these “adopted” Mt may not be just a target for phagocytosis. Mt transfer promoted a “reparative” microglial phenotype. Importantly, we showed that HN, a small secretory peptide normally encoded by the Mt genome, may be an essential component of biological effect following Mt transfer. Astrocytic Mt or HN stimulated the expression of microglial PPARγ and several PPARγ-regulated target genes with well-defined roles in controlling antioxidative, anti-inflammatory (Yi et al., 2008; Aronowski and Zhao, 2011) and phagocytic functions (Ricote et al., 1999; X. Zhao et al., 2007b, 2015c; Roszer et al., 2011; Natrajan et al., 2015). Notably, both astrocytic Mt and HN increased the phagocytic activity of microglia toward RBCs, a principal constituent of hematomas and an object of phagocytosis-mediated cleanup after ICH (X. Zhao et al., 2007b, 2009a).
Through multiple mechanisms, including iron-catalyzed Fenton's reaction, ICH leads to excessive generation of free radicals and simultaneous loss of antioxidative defenses, which leads to irreversible mitochondrial injury and oxidative brain tissue damage in the ICH-affected hemisphere (Peeling et al., 1998; Aronowski and Hall, 2005; X. Zhao et al., 2007a, 2015a,b,c). Several experimental and human studies have documented that ICH affects Mt stability and causes Mt dysfunction, including excessive superoxide production at the perihematoma zone (Wu et al., 2002; Kim-Han et al., 2006; Swanson, 2006; Lu et al., 2015; Hu et al., 2016; Qu et al., 2016). Since Mn-SOD acts as a shield preventing Mt and cells from harmful superoxides (Wu et al., 2002), any approach that upregulates Mn-SOD could mitigate ICH-mediated damage. Our present data demonstrate that both astrocytic Mt and HN significantly increase the microglial expression of both Mn-SOD and the potent antioxidative enzyme catalase. Interestingly, the expression of both proteins is under transcriptional control of PPARγ (X. Zhao et al., 2006, 2007b, 2009c), expression of which was effectively upregulated by both HN and Mt. This suggests that PPARγ activation could play important roles in beneficial HN- and Mt-mediated effects after ICH. In support of this hypothesis, we further showed that microglia receiving Mt from HN knockdown astrocytes failed to upregulate the expression of PPARγ and its prototypic target gene LPL. This suggests that HN in astrocytic Mt are indeed important for the Mt transfer-induced change in microglial phenotype. In addition to acting as a carrier of presynthesized HN, Mt entering microglia may also deliver MtDNA to allow the recipient cell to increase HN transcription. To test this model, we showed that HN mRNA and protein were significantly increased in cultured microglia following astrocytic Mt incorporation. This suggests that astrocytic Mt adopted by microglia may indeed stimulate additional HN synthesis. Although unlikely, we cannot fully exclude the possibility that Mt transfer could induce local production of HN mRNA from its own MtDNA.
Results from our mouse ICH model indicate that mRNA and protein levels of HN are robustly reduced in the ICH-affected hemisphere. Loss extended far beyond the perihematoma tissue, suggesting that ICH may affect mitochondrial gene expression throughout most of the hemisphere. Presuming the previously described beneficial role of HN (Guo et al., 2003; Matsuoka, 2011; Gong et al., 2014; Cui et al., 2017; Kim et al., 2017), our results showing decreased HN following ICH suggest that HN supplementation could exert beneficial effects. Experiments with fluorescently tagged HN showed that systemically administered HN entered the brain and localized in Iba1+ microglia and neurons (data not included), suggesting that this cell type could be an important target for HN′s biological effects. Encouraged by these results, we delivered HN in an ICH mouse model via two routes and found that it reduced neurological deficits when given intraperitoneally or intranasally. In an in vitro model of hematoma clearance, HN effectively increased RBC phagocytosis. This was expected based on our finding that HN induces expression of PPARγ, a transcription factor that we and others demonstrated to promote the phagocytic function of microglia/macrophages, including during hematoma clearance after ICH (Ricote et al., 1999; X. Zhao et al., 2007b, 2015c; Roszer et al., 2011; Natrajan et al., 2015). We believe that the present study warrants additional translational work with HN, or synthetic HN analogs (Tajima et al., 2005; Xu et al., 2006; Wang et al., 2013), to determine the efficacy, appropriate doses, and therapeutic window during pleiotropic therapy for ICH, especially assuming that HN could be used via a noninvasive intranasal route of delivery. Effects of age and sex also need to be examined. Collectively, these results suggest that astrocyte-derived Mt are transported to microglia where they stimulate the production and release of HN to prevent damage caused by ICH.
Since this study was designed to study astrocyte–microglia interactions, we did not focus on other types of brain cells as potential donors or recipients of HN and Mt. We have shown that astrocytes release Mt; however, neurons and microglia may also release Mt that have distinct functions, be dysfunctional, or lack internalization capability. When comparing Mt from astrocytes, neurons, and microglia (ongoing studies), we observed that astrocyte-released Mt have the highest red/green fluorescence ratio by using JC-1 staining (a probe for Mt membrane potential), suggesting that these Mt are highly functional. Davis et al. (2014) showed that Mt secreted by retinal ganglion axons are internalized by the astrocytes and then degraded, suggestive of “transmitophage.” Thus, neurons may also secrete Mt, but these Mt could be less healthy, have lower HN levels, and possibly be cellular waste. Preliminary experiments suggest that oxidative stress-injured neurons secrete more Mt than uninjured neurons (data not included), but these Mt also exhibited a lower JC-1 red/green ratio, suggesting that they could be dysfunctional. Platelets and mast cells also release Mt (Zhang et al., 2012; Boudreau et al., 2014). Although outside the focus of this study, we have already initiated probing for the presence and release of HN in cultured astrocytes, neurons, oligodendrocytes, and microglia using immune dot blot and RT-PCR. Our early results suggest that astrocytes have the highest HN expression level (data not included).
HN was previously shown to interact with Bax (Bcl2-associated X protein) by suppressing Bax translocation to Mt, which influences mitochondrial cytochrome C release and triggers caspase-dependent apoptosis (Guo et al., 2003). In the context of the present study, we cannot exclude the possibility that HN-mediated protection in ICH could also involve interference with the Bax-dependent apoptotic pathway, adding to the overall benefit through involvement of apoptotic pathways. On the other hand, it appears that apoptosis is not a key death pathway after ICH or ICH-like injury (Wang et al., 2002; Aronowski and Hall, 2005; X. Zhao et al., 2006; Zille et al., 2017), suggesting that the Bax pathway may not be the sole reason for the therapeutic benefit of HN.
In conclusion, we demonstrated that Mt-derived HN, whether secreted, transferred within intact Mt, or injected as synthetic HN, can promote a “reparative” microglial phenotype. Our findings suggest that HN and its biological properties could represent a potential therapeutic target for ICH (Fig. 10).
Figure 10.
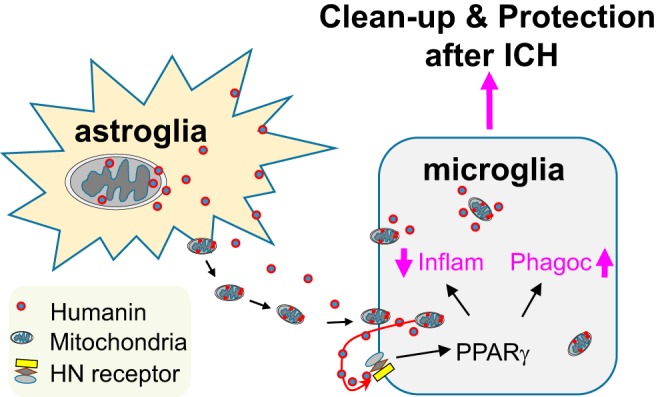
Astrocytes release Mt and HN that are taken up by microglia. The new microglia-adopted Mt and astrocyte-secreted HN may amplify microglial PPARγ production and PPARγ-dependent gene target induction, known to promote a “healing” microglial phenotype characterized by improved phagocytic efficiency and anti-inflammatory and trophic effects.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS111590-01.
The authors declare no competing financial interests.
References
- Adeoye O, Broderick JP (2010) Advances in the management of intracerebral hemorrhage. Nat Rev Neurol 6:593–601. 10.1038/nrneurol.2010.146 [DOI] [PubMed] [Google Scholar]
- Aronowski J, Hall CE (2005) New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res 27:268–279. 10.1179/016164105X25225 [DOI] [PubMed] [Google Scholar]
- Aronowski J, Zhao X (2011) Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 42:1781–1786. 10.1161/STROKEAHA.110.596718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Paré A, Rousseau M, Naika GS, Lévesque T, Laflamme C, Marcoux G, Lambeau G, Farndale RW, Pouliot M, Hamzeh-Cognasse H, Cognasse F, Garraud O, Nigrovic PA, Guderley H, et al. (2014) Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 124:2173–2183. 10.1182/blood-2014-05-573543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui AL, Zhang YH, Li JZ, Song T, Liu XM, Wang H, Zhang C, Ma GL, Zhang H, Li K (2017) Humanin rescues cultured rat cortical neurons from NMDA-induced toxicity through the alleviation of mitochondrial dysfunction. Drug Des Devel Ther 11:1243–1253. 10.2147/DDDT.S133042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N (2014) Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A 111:9633–9638. 10.1073/pnas.1404651111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felberg RA, Grotta JC, Shirzadi AL, Strong R, Narayana P, Hill-Felberg SJ, Aronowski J (2002) Cell death in experimental intracerebral hemorrhage: the “black hole” model of hemorrhagic damage. Ann Neurol 51:517–524. 10.1002/ana.10160 [DOI] [PubMed] [Google Scholar]
- Fuku N, Pareja-Galeano H, Zempo H, Alis R, Arai Y, Lucia A, Hirose N (2015) The mitochondrial-derived peptide MOTS-c: a player in exceptional longevity? Aging Cell 14:921–923. 10.1111/acel.12389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Tas E, Muzumdar R (2014) Humanin and age-related diseases: a new link? Front Endocrinol 5:210. 10.3389/fendo.2014.00210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC (2003) Humanin peptide suppresses apoptosis by interfering with bax activation. Nature 423:456–461. 10.1038/nature01627 [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K, Nishimoto I (2001) A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Abeta. Proc Natl Acad Sci U S A 98:6336–6341. 10.1073/pnas.101133498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Kurita M, Matsuoka M (2009a) Identification of soluble WSX-1 not as a dominant-negative but as an alternative functional subunit of a receptor for an anti-Alzheimer's disease rescue factor humanin. Biochem Biophys Res Commun 389:95–99. 10.1016/j.bbrc.2009.08.095 [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Kurita M, Aiso S, Nishimoto I, Matsuoka M (2009b) Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Mol Biol Cell 20:2864–2873. 10.1091/mbc.e09-02-0168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535:551–555. 10.1038/nature18928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Chan SJ, Mandeville ET, Park JH, Bruzzese M, Montaner J, Arai K, Rosell A, Lo EH (2018) Protective effects of endothelial progenitor cell-derived extracellular mitochondria in brain endothelium. Stem Cells 36:1404–1410. 10.1002/stem.2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickenbottom SL, Grotta JC, Strong R, Denner LA, Aronowski J (1999) Nuclear factor-kappaB and cell death after experimental intracerebral hemorrhage in rats. Stroke 30:2472–2477; discussion 2477–2478. 10.1161/01.STR.30.11.2472 [DOI] [PubMed] [Google Scholar]
- Hu X, Tao C, Gan Q, Zheng J, Li H, You C (2016) Oxidative stress in intracerebral hemorrhage: sources, mechanisms, and therapeutic targets. Oxid Med Cell Longev 2016:3215391. 10.1155/2016/3215391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J (2012) Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18:759–765. 10.1038/nm.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH (2009) Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci 29:7003–7014. 10.1523/JNEUROSCI.1110-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JE, Karatas H, Liu Y, Yalcin A, Montaner J, Lo EH, van Leyen K (2015) STAT-dependent upregulation of 12/15-lipoxygenase contributes to neuronal injury after stroke. J Cereb Blood Flow Metab 35:2043–2051. 10.1038/jcbfm.2015.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keep RF, Hua Y, Xi G (2012) Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol 11:720–731. 10.1016/S1474-4422(12)70104-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Xiao J, Wan J, Cohen P, Yen K (2017) Mitochondrially derived peptides as novel regulators of metabolism. J Physiol 595:6613–6621. 10.1113/JP274472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Han JS, Kopp SJ, Dugan LL, Diringer MN (2006) Perihematomal mitochondrial dysfunction after intracerebral hemorrhage. Stroke 37:2457–2462. 10.1161/01.STR.0000240674.99945.4e [DOI] [PubMed] [Google Scholar]
- Liu K, Ji K, Guo L, Wu W, Lu H, Shan P, Yan C (2014) Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 92:10–18. 10.1016/j.mvr.2014.01.008 [DOI] [PubMed] [Google Scholar]
- Lu H, Jiang M, Lu L, Zheng G, Dong Q (2015) Ultrastructural mitochondria changes in perihematomal brain and neuroprotective effects of huperzine A after acute intracerebral hemorrhage. Neuropsychiatr Dis Treat 11:2649–2657. 10.2147/NDT.S92158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M. (2011) Humanin signal for Alzheimer's disease. J Alzheimers Dis 24 [Suppl 2]:27–32. [DOI] [PubMed] [Google Scholar]
- Matsuoka M, Hashimoto Y (2010) Humanin and the receptors for humanin. Mol Neurobiol 41:22–28. 10.1007/s12035-009-8090-z [DOI] [PubMed] [Google Scholar]
- Muzumdar RH, Huffman DM, Atzmon G, Buettner C, Cobb LJ, Fishman S, Budagov T, Cui L, Einstein FH, Poduval A, Hwang D, Barzilai N, Cohen P (2009) Humanin: a novel central regulator of peripheral insulin action. PLoS One 4:e6334. 10.1371/journal.pone.0006334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G (2004) Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg 100:672–678. 10.3171/jns.2004.100.4.0672 [DOI] [PubMed] [Google Scholar]
- Natrajan MS, de la Fuente AG, Crawford AH, Linehan E, Nuñez V, Johnson KR, Wu T, Fitzgerald DC, Ricote M, Bielekova B, Franklin RJ (2015) Retinoid X receptor activation reverses age-related deficiencies in myelin debris phagocytosis and remyelination. Brain 138:3581–3597. 10.1093/brain/awv289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paharkova V, Alvarez G, Nakamura H, Cohen P, Lee KW (2015) Rat humanin is encoded and translated in mitochondria and is localized to the mitochondrial compartment where it regulates ROS production. Mol Cell Endocrinol 413:96–100. 10.1016/j.mce.2015.06.015 [DOI] [PubMed] [Google Scholar]
- Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, Le Foll F, Rafii A (2013) Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11:94. 10.1186/1479-5876-11-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeling J, Yan HJ, Chen SG, Campbell M, Del Bigio MR (1998) Protective effects of free radical inhibitors in intracerebral hemorrhage in rat. Brain Res 795:63–70. 10.1016/S0006-8993(98)00253-4 [DOI] [PubMed] [Google Scholar]
- Plotnikov EY, Khryapenkova TG, Galkina SI, Sukhikh GT, Zorov DB (2010) Cytoplasm and organelle transfer between mesenchymal multipotent stromal cells and renal tubular cells in co-culture. Exp Cell Res 316:2447–2455. 10.1016/j.yexcr.2010.06.009 [DOI] [PubMed] [Google Scholar]
- Qu J, Chen W, Hu R, Feng H (2016) The injury and therapy of reactive oxygen species in intracerebral hemorrhage looking at mitochondria. Oxid Med Cell Longev 2016:2592935. 10.1155/2016/2592935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi AI, Mendelow AD, Hanley DF (2009) Intracerebral haemorrhage. Lancet 373:1632–1644. 10.1016/S0140-6736(09)60371-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan RF, Panter SS (1996) Hemoglobin potentiates excitotoxic injury in cortical cell culture. J Neurotrauma 13:223–231. 10.1089/neu.1996.13.223 [DOI] [PubMed] [Google Scholar]
- Ricote M, Huang JT, Welch JS, Glass CK (1999) The peroxisome proliferator-activated receptor (PPARgamma) as a regulator of monocyte/macrophage function. J Leukoc Biol 66:733–739. 10.1002/jlb.66.5.733 [DOI] [PubMed] [Google Scholar]
- Roszer T, Menéndez-Gutierrez MP, Lefterova MI, Alameda D, Núñez V, Lazar MA, Fischer T, Ricote M (2011) Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol 186:621–631. 10.4049/jimmunol.1002230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spees JL, Olson SD, Whitney MJ, Prockop DJ (2006) Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A 103:1283–1288. 10.1073/pnas.0510511103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan S, Landreth GE (2004) Antiinflammatory properties of PPARgamma agonists following ischemia. Drug News Perspect 17:229–236. 10.1358/dnp.2004.17.4.829049 [DOI] [PubMed] [Google Scholar]
- Swanson RA. (2006) Intracerebral hematoma: beyond the mass lesion. Stroke 37:2445. 10.1161/01.STR.0000244060.83388.76 [DOI] [PubMed] [Google Scholar]
- Tajima H, Kawasumi M, Chiba T, Yamada M, Yamashita K, Nawa M, Kita Y, Kouyama K, Aiso S, Matsuoka M, Niikura T, Nishimoto I (2005) A humanin derivative, S14G-HN, prevents amyloid-beta-induced memory impairment in mice. J Neurosci Res 79:714–723. 10.1002/jnr.20391 [DOI] [PubMed] [Google Scholar]
- Tureyen K, Kapadia R, Bowen KK, Satriotomo I, Liang J, Feinstein DL, Vemuganti R (2007) Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J Neurochem 101:41–56. 10.1111/j.1471-4159.2006.04376.x [DOI] [PubMed] [Google Scholar]
- Voigt A, Jelinek HF (2016) Humanin: a mitochondrial signaling peptide as a biomarker for impaired fasting glucose-related oxidative stress. Physiol Rep 4:e12796. 10.14814/phy2.12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF (2003) Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab 23:629–652. 10.1097/01.WCB.0000073905.87928.6D [DOI] [PubMed] [Google Scholar]
- Wang T, Huang Y, Zhang M, Wang L, Wang Y, Zhang L, Dong W, Chang P, Wang Z, Chen X, Tao L (2013) [Gly14]-humanin offers neuroprotection through glycogen synthase kinase-3beta inhibition in a mouse model of intracerebral hemorrhage. Behav Brain Res 247:132–139. 10.1016/j.bbr.2013.03.023 [DOI] [PubMed] [Google Scholar]
- Wang X, Gerdes HH (2015) Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ 22:1181–1191. 10.1038/cdd.2014.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Mori T, Sumii T, Lo EH (2002) Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke 33:1882–1888. 10.1161/01.STR.0000020121.41527.5D [DOI] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, Xi G (2002) Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res 953:45–52. 10.1016/S0006-8993(02)03268-7 [DOI] [PubMed] [Google Scholar]
- Xi G, Fewel ME, Hua Y, Thompson BG Jr, Hoff JT, Keep RF (2004) Intracerebral hemorrhage: pathophysiology and therapy. Neurocrit Care 1:5–18. 10.1385/NCC:1:1:5 [DOI] [PubMed] [Google Scholar]
- Xu X, Chua CC, Gao J, Hamdy RC, Chua BH (2006) Humanin is a novel neuroprotective agent against stroke. Stroke 37:2613–2619. 10.1161/01.STR.0000242772.94277.1f [DOI] [PubMed] [Google Scholar]
- Yen K, Lee C, Mehta H, Cohen P (2013) The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J Mol Endocrinol 50:R11–R19. 10.1530/JME-12-0203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JH, Park SW, Brooks N, Lang BT, Vemuganti R (2008) PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res 1244:164–172. 10.1016/j.brainres.2008.09.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Asadi S, Weng Z, Sismanopoulos N, Theoharides TC (2012) Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PLoS One 7:e49767. 10.1371/journal.pone.0049767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ST, Zhao L, Li JH (2013) Neuroprotective peptide humanin inhibits inflammatory response in astrocytes induced by lipopolysaccharide. Neurochem Res 38:581–588. 10.1007/s11064-012-0951-6 [DOI] [PubMed] [Google Scholar]
- Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J (2006) 15d-prostaglandin J2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab 26:811–820. 10.1038/sj.jcbfm.9600233 [DOI] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, Grotta JC, Aronowski J (2007a) Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke 38:3280–3286. 10.1161/STROKEAHA.107.486506 [DOI] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J (2007b) Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol 61:352–362. 10.1002/ana.21097 [DOI] [PubMed] [Google Scholar]
- Zhao X, Grotta J, Gonzales N, Aronowski J (2009a) Hematoma resolution as a therapeutic target: the role of microglia/macrophages. Stroke 40:S92–S94. 10.1161/STROKEAHA.108.533158 [DOI] [PubMed] [Google Scholar]
- Zhao X, Song S, Sun G, Strong R, Zhang J, Grotta JC, Aronowski J (2009b) Neuroprotective role of haptoglobin after intracerebral hemorrhage. J Neurosci 29:15819–15827. 10.1523/JNEUROSCI.3776-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Strong R, Zhang J, Sun G, Tsien JZ, Cui Z, Grotta JC, Aronowski J (2009c) Neuronal PPARgamma deficiency increases susceptibility to brain damage after cerebral ischemia. J Neurosci 29:6186–6195. 10.1523/JNEUROSCI.5857-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Ting SM, Gonzales N, Aronowski J (2015a) Dimethyl fumarate protects brain from damage produced by intracerebral hemorrhage by mechanism involving Nrf2. Stroke 46:1923–1928. 10.1161/STROKEAHA.115.009398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sun G, Ting SM, Song S, Zhang J, Edwards NJ, Aronowski J (2015b) Cleaning up after ICH: the role of Nrf2 in modulating microglia function and hematoma clearance. J Neurochem 133:144–152. 10.1111/jnc.12974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Gonzales N, Aronowski J (2015c) Pleiotropic role of PPARgamma in intracerebral hemorrhage: an intricate system involving Nrf2, RXR, and NF-kappaB. CNS Neurosci Ther 21:357–366. 10.1111/cns.12350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Ting SM, Liu CH, Sun G, Kruzel M, Roy-O'Reilly M, Aronowski J (2017) Neutrophil polarization by IL-27 as a therapeutic target for intracerebral hemorrhage. Nat Commun 8:602. 10.1038/s41467-017-00770-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, Milner TA, Jonas EA, Ratan RR (2017) Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 48:1033–1043. 10.1161/STROKEAHA.116.015609 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Microglia taking up astrocytic Mt. Astrocytes in culture were treated with MitoTracker/CMX/Ros to label Mt. After extensive washing to remove MitoTracker and 24 h incubation to accumulate Mt in the media, the media containing labeled astrocytic Mt were transferred to rat primary microglia in culture. Time-lapse images (0 min, top panel and 14 h, bottom panel) were collected to document Mt transfer into microglia over a 14 h period. Images of three representative microglial cells (color-coded as blue, red and yellow) prior to Mt transfer (0 min, top panel) and at the end time point (14 h, bottom panel) are shown. Vast majority of microglia in a culture dish took up astrocytic Mt. Download Figure 6-1, EPS file (11.5MB, eps)