

Abstract
Here we present a novel protocol for the construction of saturation single-site—and massive multisite—mutant libraries of a bacteriophage. We segmented the ΦX174 genome into 14 nontoxic and nonreplicative fragments compatible with Golden Gate assembly. We next used nicking mutagenesis with oligonucleotides prepared from unamplified oligo pools with individual segments as templates to prepare near-comprehensive single-site mutagenesis libraries of genes encoding the F capsid protein (421 amino acids scanned) and G spike protein (172 amino acids scanned). Libraries possessed greater than 99% of all 11 860 programmed mutations. Golden Gate cloning was then used to assemble the complete ΦX174 mutant genome and generate libraries of infective viruses. This protocol will enable reverse genetics experiments for studying viral evolution and, with some modifications, can be applied for engineering therapeutically relevant bacteriophages with larger genomes.
Keywords: ΦX174 bacteriophage, genome engineering, nicking mutagenesis, Golden Gate cloning, oligo pool, deep mutational scanning
Graphical Abstract
Predicting the tempo and trajectory of evolutionary change in the complex environments encountered by viruses and bacteria remains a challenge.1–3 Understanding such changes is important for fundamental evolutionary studies as well as biotechnology applications like phage therapy for multidrug resistant bacteria.4 Two obstacles preventing better predictions are the oversimplification of the environment in experimental evolution studies compared to wild conditions and the vast number of experimentally unexplored mutational combinations that can occur, even in short adaptive walks and small genomes. Advances in DNA sequencing and synthesis have opened new ways to study microbial evolution and overcome some of these obstacles.5,6 Unfortunately, suitable methods do not exist for generating comprehensive bacteriophage mutant libraries. This technology gap hinders those seeking to engineer phages for biotechnological applications and for those seeking a deeper understanding of how viruses evolve.
Methods for genetically engineering phages have recently been reviewed7 and commonly involve homologous recombination and recombineering. While such approaches can be improved by clever incorporation of CRISPR-Cas systems,8–10 the overall modest efficiencies largely limits mutagenesis to the generation and single-site mutation of chimeric genomes,11,12 gene deletions,13 and limited multisite mutagenesis.14 A recent approach combines mutagenesis by PCR with degenerate (NNK) primers with in-cell recombination to create variation at specific residues in the tail fiber of T3 phage.15 By contrast, human viruses often have reverse genetics systems in place where comprehensive mutant libraries can be prepared for single genes16 or regions within a gene17,18 using replicative plasmids that encode whole viruses or viral components. Similarly, deep mutational scanning can be performed on plasmid-encoded phage proteins like the MS2 capsid protein.19 As described by Hartman et al. 2018,19 such experiments can identify mutations that confer useful characteristics, such as MS2 capsid stability mutations that could improve the targeted delivery of therapeutics to particular cellular compartments.
Advances in the technologies for generating mutant libraries, synthesizing DNA, and for the assembly of large DNA fragments20 allow for the construction and assembly of user-defined mutagenesis of long nucleic acids. Nicking mutagenesis (NM) can be used to construct comprehensive single-site or other user-defined mutant libraries21 using dsDNA as a template. In NM, oligos encode the desired mutations by mismatch with the parental template. On-chip inkjet printed oligo pools have been integrated into NM,22 so one can now construct heterogeneous libraries of oligos that contain tens to hundreds of thousands of high fidelity, unique sequences23,22 with a low per base pair cost.
We therefore suspected that large user-defined libraries of phage could be generated by combining the advantages of replicative plasmids with advances in DNA synthesis. The bacteriophage ΦX174 is an excellent system to test this approach. High resolution X-ray crystallography structures of the capsid and spike proteins24,25 are available for ΦX174, making it amenable to compare empirical mutagenesis results and molecular modeling predictions, it has a very small genome26 of 5386 nucleotides that is readily sequenced, and it is easily grown on a variety of lab E. coli strains. These attractive features of the ΦX174 experimental system have led to proof-of-principle demonstrations from other groups for synthetic genome assembly from oligonucleotides27 and complete refactoring of the phage.28,29
ΦX174 has also been extensively studied,30–33 especially as a model system for experimental evolution34 since populations grow rapidly to very large size, mutation rate is high, and the fitness differences between even single mutations can be very large. One of the focal areas of work on ΦX174 and related microvirid phage has been testing models of adaptive evolution and epistasis with empirical data.33,35,36 The inferential power of these studies has been limited by the modest sizes of the mutational libraries.
Here we have combined Golden Gate assembly, nicking mutagenesis, and oligo pool technology to construct near comprehensive single-site and expansive multisite mutant libraries for the genes encoding the capsid F protein and spike G protein of the bacteriophage ΦX174. To our knowledge this is the first time near comprehensive single site mutant libraries of full capsid and spike proteins have been generated for an infective bacteriophage.
MATERIALS AND METHODS
Reagents.
All purchased enzymes and DNA purification kits were from New England Biolabs, antibiotics were purchased from GoldBio, other chemicals were purchased from Sigma-Aldrich. Individual primers were purchased from Integrated DNA Technologies.
Segmentation of the ΦX174 Genome.
A phage assembly platform for ΦX174 was devised following.37 The ΦX174 chromosome was divided into 14 genomic fragments designed to avoid host cell toxicity by separating genes from their promoters and breaking large genes into multiple segments (Supporting Information). Each segment is flanked by unique five nucleotide overlaps of WT ΦX174 sequence so that they can be amplified from the ancestral ΦX174 using PCR primers designed to incorporate terminal BsmB1 restriction sites. Amplicons were cloned into pCR2.1 using the Invitrogen TOPO TA cloning system (Life Technologies, Grand Island, NY) and verified by Sanger sequencing.
Introduction of Nicking Site.
The gene fragments—F1, F2, G1, G2—in the pCR2.1-TOPO plasmid (Supporting Information) had the BbvCI nicking site introduced via overhang PCR, type I restriction enzyme cutting, and ligation. First, PCR was performed with overhang primers (Supplemental Table S1) to introduce the BbvCI site. Standard Phusion Polymerase HF reaction conditions were used with 4 ng of template DNA, cycling conditions as follows: 98 °C, 1 min; 25× cycles of 98 °C, 10 s; 67 °C, 15 s; 72 °C, 2.5 min; followed by 72 °C for 10 min. PCR products were run on a 1% agarose gel stained with SYBR safe stain (Invitrogen) and the DNA bands at ∼4600 bp were extracted using a Monarch DNA Gel Extraction Kit. Next, 1 μg of the PCR product was digested with KpnI (20 U) in NEB Buffer 1.1 at 37 °C for 2 h. Digested DNA was cleaned and concentrated with a Monarch PCR and DNA Clean and Concentrate Kit and eluted into 20 μL nuclease free H2O. One microliter of the purified and digested DNA was ligated using T4 DNA ligase at ∼25 °C for 1 h in standard conditions in a 20 μL reaction. Five microliters of the ligation reaction were transformed into chemically competent XL1-Blue E. coli via standard protocols. Cells were plated on LB agar containing 100 μg/mL carbenicillin and 50 μg/mL kanamycin and grown at 37 °C for ∼16 h. Cells were picked from transformation plates and grown in 50 mL TB with 100 μg/mL carbenicillin and 50 μg/mL kanamycin for ∼12 h, cells were pelleted, and DNA purified using compact midi-preps.
Construction of Single Site Mutant Libraries.
Comprehensive mutant libraries were generated using nicking mutagenesis (NM) as in Wrenbeck et al. 201621 with modifications for using oligo pool mutagenic primers as noted in Medina-Cucurella et al. 2019.22 A single oligo pool encoding for all possible single missense and nonsense substitutions in F and G was designed using the custom python scripts from Medina-Cucurella et al. 201922 and synthesized by Agilent (full sequences of all oligos are given in the Supporting Information). Oligo pools were designed with 20–24 bases of gene overlap flanking the mutated codon. Codons were chosen based on E. coli codon usage frequency, where the highest frequency codon that encoded the mismatch mutation was selected. 14 853 unique oligos were ordered from Agilent, where 14 853 = 12 453 ΦX174 primers – 2400 other primers. These oligos encode WT (593 primers) and all 20 possible single point mutations (including stop) (11 860 primers). This oligo pool was used directly in NM without further amplification using 2 μg of the relevant Golden Gate plasmid as a template. Libraries were electroporated into high efficiency electrocompetent XL1-Blue E. coli (Agilent cat #: 200228) using 1 mm electroporation cuvettes at 1200 V using a Eppendorf Eporator. Cells were plated on large bioassay plates (245 mm × 245 mm × 25 mm, Sigma-Aldrich) containing LB agar + 100 μg/mL carbenicillin and 50 μg/mL kanamycin and grown at 37 °C for ∼16 h. Plates were scraped in 10 mL plain LB, broken into ∼1.2 mL aliquots, pelleted, and stored −80 °C. DNA was purified from 1× aliquot using a MonarchPlasmid Mini-Prep Kit.
Illumina Sequencing Prep and Analysis.
Purified library DNA was prepared for deep sequencing as in38 using the primers listed in Supplemental Table S2 and gene tiling as specified in Table 1. DNA was Illumina sequenced on a Mi-Seq platform with 250 BP paired end reads. The University of Colorado BioFrontiers Sequencing Core performed the Illumina sequencing. Data was processed using PACT39 to determine the library coverage with the following changes to the default options in the configuration file: fast_filter_translate: qaverage = 20, qlimit = 0, enrichment: ref_count_threshold = 5, sel_count_threshold = 0, strict_count_threshold = True. Library coverage statistics are shown in Supplemental Table S3 and S4.
Table 1.
Mutant Library NGS Statistics: Summary of the of the Libraries Prior to Viral Genome Assembly
| fragment | F1 | F2 | F3 | G1 | G2 |
|---|---|---|---|---|---|
| residues | 1–95 | 99–242 | 246–427 | 2–133 | 136–175 |
| sequencing reads post quality filter | 1 163 959 | 2 396 395 | 1 557 554 | 1 717 075 | 813 221 |
| fold oversampling of codon combinations | 441.1 | 792.5 | 412.1 | 619.4 | 821.9 |
| percent of reads with | |||||
| no nonsynonymous mutations | 23.0% | 33.1% | 29.7% | 33.3% | 25.2% |
| one nonsynonymous mutation | 52.6% | 37.4% | 35.5% | 38.9% | 59.7% |
| multiple nonsynonymous mutation | 24.4% | 29.4% | 34.7% | 27.7% | 15.1% |
| coverage of possible single nonsynonymous mutations | 100% | 99.9% | 100% | 99.8% | 100% |
Assembly of Mutant Genomes.
We pooled plasmid DNA containing all 14 of the phage DNA fragments (500 ng each) and digested them with BsmB1 (Fermentas Fast Digest, Life Technologies, Grand Island, NY) for 30 min to 1 h at 37 °C. The digested plasmids were subjected to agarose gel electrophoresis for 10 to 15 min using a 1.2% agarose gel to separate the vector from the insets. The inserts were excised from the gel, purified using the GeneJET gel extraction kit (Fermentas), ligated overnight at 14 °C with T4 DNA ligase (Promega Corporation, Madison, WI), and 4uL were transformed by electroporation into 50 μL of XL1-Blue electocompetent cells (Supplemental Table S7). The transformation mix was resuspended with 950 μL of SOC and either plated immediately or incubated for up to 120 min (Supplemental Table S5) at 37 °C in the presence of DNase1 (Supplemental Figure S18) to allow for cell lysis and the removal extracellular DNA, respectively. Aliquots of the cell lysates were added to 3 mL of ΦLB top agar containing 50 μL log-phage E. coli C cells and plated onto a ΦLB agar plate. After four to five hours of incubation at 37 °C, recombinant phage plaques were visible and plates were removed from the incubator. To verify that the recombinant phage encoded the intended sequence, we picked about 30 plaques for each intended mutational target and Sanger sequenced the entire targeted gene. We also sequenced the F and G genes for about 30 wildtype control plaques to ensure that no mutations were naturally accumulating. Briefly, individual plaques were picked with sterile toothpicks and placed in 200 μL ΦLB and gently swirled. One μL of this mix was used to PCR amplify approximately 1/2 of the ΦX174 genome using ΦX-0F (5′-GAGTTTTATCGCTTCCATG-3′) and ΦX-2953R (5′-CCGCCAGCAATAGCACC-3′) primers. Internal sequencing primers ΦX-979F (5′-CGGCCCCTTACTTGAGG-3′) and ΦX-1500R (5′-TTGAGATGGCAGCAACGG-3′) were used to sequence gene F. ΦX-2953R was used to sequence gene G. PCR cleanups and sequencing was done at Eurofins Genomics. PCR reaction conditions were: 5 uL10X Taq buffer, 2.5 μL 10 μM ΦX-0F primer, 10 μM ΦX-2953F primer, 0.8 μL 12.5 μM dNTPs, 0.5 μL Taq polymerase (NEB #M0273), 1 μL template, 37.7 μL H20. Thermocycling conditions were 1 cycle at 95 °C for 2 min, 30 cycles at 95 °C for 15 s, 52 °C for 30 s, 68 °C for 2 min, 1 cycle at 68 °C for 5 min.
RESULTS
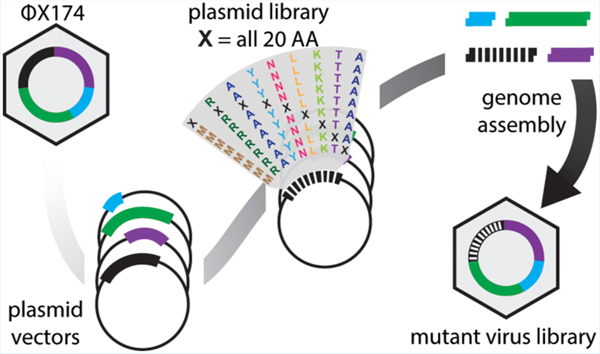
Our strategy for generating large user-defined mutagenesis libraries of bacteriophage ΦX174 is shown in Figure 1. The circular 5386 nucleotide genome compactly encodes 11 genes where the F and G genes encode the capsid and spike proteins, respectively (Figure 1A). The genome was segmented onto 14 separate plasmids, including 3 plasmids for the F gene and 2 for the G gene (Figure 1B). Fragment boundaries were first engineered to divide large genes, separate regulatory regions from protein coding genes, and result in fragments of similar size. Initial attempts to clone some fragments resulted in low plasmid yield, presumably due to gene toxicity. These genes were further divided and tested empirically, resulting in the final set of 14 fragments. User-defined comprehensive mutations on F and G were encoded using nicking mutagenesis with a single unamplified oligo pool containing all 11 860 mutagenic oligonucleotides (Figure 1C). ΦX174 mutant genomes were reconstituted from individual plasmids using Golden Gate cloning and transformation into a host E. coli strain (Figure 1D). Harvested phages contained all possible nonsynonymous mutations in the F and G genes depending on the mutagenized segments used for Golden Gate cloning (Figure 1E).
Figure 1.

ΦX174 mutant library assembly. (A) Cartoon structure of the ΦX174 virion. F and G proteins are organized into pentamers and are the only exposed proteins in the mature virion. (B) A schematic of the circular ΦX174 genome showing the size and location of the synthetic genome fragments. The locations of mutated residues in genes F and G are indicated. (C) An oligo pool containing all mutagenic oligos for genes F and G was generated and used in Nicking Scanning Mutagenesis for the generation of saturation mutant libraries for genes F and G. (D) Golden Gate cloning was used to assemble the mutant genomes, which were transformed into XL1-Blue competent cells (Agilent) and plated on susceptible E. coli C cells. The resulting plaques were sequenced. (E) Linear schematics depicting some of the possible mutant libraries that can be generated using the workflow and existing libraries.
We sought a reverse genetics system for ΦX174 wherein the virus chromosome was segmented and encoded on individual vector plasmids. Our method of construction closely followed that for the assembly of human coronavirus NL63,37 where each plasmid contained unique BsmB1 type IIS restriction endonuclease sites flanking unique five nucleotide overlaps of wild-type (WT) ΦX174. This architecture allows for faithful and easy assembly of the complete genome via Golden Gate cloning. The ΦX174 genome was separated into 14 separate nontoxic fragments by PCR and cloned into vector plasmids where genes were separated from their promoters and larger genes were segmented (full sequences for all plasmid inserts are given in Supporting Information). The segments ranged from 129 to 634 base pairs in length, with necessary additional truncations of genes D and A. ΦX174 phage could be reconstituted by digesting pooled plasmids with BsmB1, ligating inserts overnight, and transforming into electrocompetent E. coli C cells.
The F and G genes were targeted for saturation mutagenesis. We introduced mutations by nicking mutagenesis, which requires the presence of a BbvCI nicking site on the dsDNA plasmid. The F gene plasmid F3 encoding residues 246–427 of the F gene product contained a unique BbvCI sequence, while the remaining four plasmids (F1, F2, G1, G2) required introduction of BbvCI nicking sites in the vector backbone. The presence in each plasmid of the unique BbvCI nicking site was verified with the successful generation of circular ssDNA from the modified plasmids by BbvCI.Nt and exonuclease digestion (Supplemental Figure S1).
In nicking mutagenesis, desired mutations are introduced by libraries of mutagenic oligonucleotides, which can be sourced from unamplified oligo pools.22 We used a custom pool of 11 860 oligos that encoded nearly every missense and nonsense mutation in the F and G genes. Each missense or nonsense mutation was encoded by a single oligonucleotide. Application of nicking mutagenesis using this oligo pool for different plasmids (F1, F2, F3, G1, G2) resulted in at least 14-fold excess transformants required for 99.9% theoretical coverage of the desired library (Supplemental Table S3). The diversity of the mutant plasmid libraries was validated using deep sequencing on an Illumina MiSeq platform. All plasmid libraries have >99.7% coverage of all possible single mutations (n = 11 821/11 860, minimum counts for a mutation >5, full library statistics are given in Table 1 and Supplemental Table S4). Heatmaps showing mutation-specific frequencies for all segments are shown in Supplemental Figures S2–S17.
Mutant dsDNA libraries were transformed into nonsusceptible hosts (Agilent XL1-Blue), then plated on susceptible hosts (E. coli C) after a 30 min recovery in SOC. Viral titers increase rapidly after 30 min, suggesting that the XL1-Blue cells do not burst until after this time point (Supplemental Table S5). Accordingly, we reasoned that the number of plaques at 30 min represents the number of XL1-Blue cells that were transformed with viable genomes. The number of transformants varied considerably between constructs (Table 2, Supplemental Figure S19). 19 200 plaques were recovered from the cotransformed G1/G2 libraries, where only 3440 total variants were expected in the pool. On the contrary, 460 plaques were observed for the F2 library, when 2880 total variants were in the starting plasmid pool. The under-representation of the F2 library at this scale could be remedied by transforming more competent cells. We expect that differences between fragments in the number of recovered viruses is due to in part to the tolerability of fragments to mutation, as inviable mutants would not form plaques. Estimates from deep mutational scanning of model proteins show that approximately 18–38% of all mutations are nonviable,40,41 and this percentage may be larger for structural proteins like F and G.
Table 2.
Characterization of Viable Virus Genotypes in Mutant Phage Librarya
| fragment | F1 | F2 | F3 | G1/G2 | WT |
|---|---|---|---|---|---|
| possible mutants | 1900 | 2880 | 3640 | 3440 | NA |
| viable transformants | 2300 | 460 | 5.840 | 19 200 | 6400 |
| plaques sequenced | 34 | 25 | 30 | 60 | 30 |
| wildtype plaques | 17 (50%) | 11 (44%) | 15 (50%) | 30 (50%) | 30 (100%) |
Summary table of number of possible mutant viruses, number of viable viruses recovered from one transformation, and the number of mutant and wildtype genotypes recovered. No mutant genotypes were observed more than once.
To test the validity of the mutagenesis and phage reconstitution method, we picked about 30 plaques generated from each library to Sanger sequence. Of these, 40–50% were wildtype and the remaining contained exactly one mutation (Table 2). No variants were observed more than once, supporting the deep sequencing results showing that the libraries consist of thousands of variants. Interestingly, none of the exact substitutions in these sequences have been previously observed (Supplemental Table S6), although mutations at the same residues (G106, F29, F45, F84, F185, F204, F205, and F393) have been seen in previous experiments.34 In all cases the percentage of WT plaques is slightly higher than in the starting plasmid pools (p-values F1: 0.001, F2: 0.088, F3: 0.015, G: 0.001, 1-tailed binomial distribution), consistent with purifying selection against mutations (Table 1). In contrast, for the control where all plasmids in the Golden Gate reaction were WT, 0% (0/30) plaques contained mutations in the encoded regions.
DISCUSSION
Here we present a novel method for generating comprehensive single-site and multisite saturation mutant libraries of the spike and capsid proteins of the bacteriophage ΦX174. This method uses unamplified oligo pools, nicking scanning mutagenesis, and Golden Gate cloning of the fragmented genome.
The fragment-based system provides an ideal way to generate libraries for the study of epistasis and multimutational step regions of the neighborhood around wild type. When a gene is in multiple fragments, the number of mutational variants we expect from the assembled mutant fragments is the product of their individual variant number. Hence, since G1 contains 2640 variants and G2 contains 800 variants, they can be combined to generate 2.1 million double mutation variants. Further, nothing requires the fragments to be from the same gene. For example, since the F and G proteins bind to each other to form the capsid of ΦX174 (Figure 2), libraries combining mutations in F with mutations in G will capture mutations that can readily interact in the protein and have the potential to generate interesting instances of epistasis (Figure 2). The central challenge then becomes how to survey the vast size of these combinatorial plasmid libraries. Right now, our transformational efficiencies vary, but even at the upper end of hundreds of thousands of transformants per reaction, only a fraction of the total diversity in the library can be surveyed.
Figure 2.

Structural view of potential epistatic interactions between F and G libraries. (A) Pentameric complex between F and G shown in surface view. For both F and G, one monomer is detailed as a ribbon color-coded according to the saturation mutagenesis library prepared in this work. (B) Close-up of interaction surface between the F3 sublibrary of F protein (green ribbons) and G2 sublibrary of G protein (purple spheres). (C) Distance mapping between alpha carbons in F and G crystal structure. Residues within 10 Å are colors red, 10–20 are black, and >20 are blue. Euclidian distances calculated from F–G crystal structure. Heatmap shows that many opportunities exist for close proximity (interactions) both within and between fragments.
We are fortunate that our model virus has a small genome, under 6000 nts, which enabled us to assemble the 14 nontoxic fragments with sufficient transformants for near-complete coverage of the user-defined mutations. This method can be applied with little modification to other phages with small genomes like the microviridae. However, most biotech-relevant phages have larger genomes, and extension of this method to other viruses would encounter the following technical challenges. First, increasing genome sizes decrease transformational efficiencies, and larger genomes have increased susceptibility to DNA shearing.42 Both size-dependent effects can disrupt the integrity and coverage of the phage libraries.43 Second, our method includes restrictions on the DNA sequences that can be used. Golden Gate cloning requires a genome without unique Type IIS restriction sequences, while nicking mutagenesis requires that there be only one orientation of the BbvCI nicking site within the template DNA fragment. Third, ligating 14 fragments is close to the upper limit of Golden Gate cloning. Additionally, increasing the size of individual fragments is difficult as the presented method depends on replicating plasmids in E. coli, and larger fragments are more likely to be toxic in vivo.
Based on the above considerations, adapting this method to larger viruses will require modifications for the genome assembly steps. There are a variety of methods available for attempting to improve genome library assembly and amplification. For improving genome assembly, we speculate that a combination of hierarchical assembly44 with other yeast-based assembly methods45 will allow for large viral genomes to be efficiently assembled. Yehl et al. 201915 built large libraries of targeted epitope regions by combining methods; plasmid-based mutagenesis and cloning followed by recombination with wild-type phage during infection. Finally, we speculate that the TX-TL cell free expression system46 will be able to produce the viral libraries with less bias than in vivo amplification in E. coli. In summary, we have presented a method for efficient deep mutational scanning of the bacteriophage ΦX174. We anticipate that this method will find utility in fundamental molecular evolution studies as well as translate to potential medicinal applications.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation BEACON Centre for the Study of Evolution in Action and EPSCoR T2 under award numbers DBI-0939454 and OIA-1736253 and the National Institute of General Medical Sciences of the National Institutes of Health under award numbers P20GM104420 and R01GM076040. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.9b00411.
Figures S1, S18–19, Tables S1, S2, S5, S7: Experimental details that may assist the reader in replicating the experiment or applying the method to a new system; Figures S2–S17 and Tables S3, S4, S6: Characterization of the plasmid library by deep sequencing and the mutant virus library by plaque growth and isolate sequencing (PDF)
phix174_MutOligos.fna: oligo pool sequences; phix174_primer_generation_inputs: input files for generating the oligo pool and an example of the command line input to execute the script from Medina-Cucurella et al. 2019;22 phix174_fragments.fna: nucleotide sequences for all inserts in carrier plasmid; phix174_fragments.gff: annotation of fragments including restriction sites and coding regions; phix174_fragments.faa: amino acid sequences of mutated genes (TXT)
The authors declare no competing financial interest.
Raw sequencing reads for the mutant libraries have been deposited in the SRA database: SAMN12660301-SAMN12660308.
REFERENCES
- (1).Holmes EC. (2013) What can we predict about viral evolution and emergence? Curr. Opin. Virol 3, 180–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Neher RA. (2013) Genetic draft, selective interference, and population genetics of rapid adaptation. Annu. Rev. Ecol. Evol. Syst 44, 195–215. [Google Scholar]
- (3).Lieberman TD, Flett KB, Yelin I, Martin TR, McAdam AJ, Priebe GP, and Kishony R. (2014) Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat. Genet 46, 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, Gilmour KC, Soothill J, Jacobs-Sera D, Schooley RT, Hatfull GF, and Spencer H. (2019) Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med 25, 730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Barrick JE, and Lenski RE. (2013) Genome dynamics during experimental evolution. Nat. Rev. Genet 14, 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fowler DM, and Fields S. (2014) Deep mutational scanning: a new stype of protein science. Nat. Methods 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pires DP, Cleto S, Sillankorva S, Azeredo J, and Lu TK. (2016) Genetically engineered phages: a review of advances over the last decade. Microbiol. Mol. Biol. Rev 80, 523–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kiro R, Shitrit D, and Qimron U. (2014) Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol 11, 42–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lemay ML, Tremblay DM, and Moineau S. (2017) Genome engineering of virulent lactococcal phages using CRIPSR-Cas9. ACS Synth. Biol 6, 1351–1358. [DOI] [PubMed] [Google Scholar]
- (10).Schilling T, Dietrich S, Hoppert M, and Hertel R. (2018) A CRISPR-Cas9-based toolkit for fast and precise in vivo genetic engineering of Bacillus subtilis phages. Viruses 10, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Doore SM, and Fane BA. (2015) The kinetic and thermodynamic aftermath of horizontal gene transfer governs evolutionary recovery. Mol. Biol. Evol 32, 2571–2584. [DOI] [PubMed] [Google Scholar]
- (12).Doore SM, Schweers NJ, and Fane BA. (2017) Elevating fitness after a horizontal gene exchange in bacteriophage Φ X174. Virology 501, 25–34. [DOI] [PubMed] [Google Scholar]
- (13).Uchiyama A, Heiman P, and Fane BA. (2009) N-terminal deletions of the Φ X174 external scaffolding protein affect the timing and fidelity of assembly. Virology 386, 303–309. [DOI] [PubMed] [Google Scholar]
- (14).Stockdale SR, Collins B, Spinelli S, Douillard FP, Mahony J, Cambillau C, and van Sinderen D. (2015) Structure and assembly of TP901–1 Virion unveiled by mutagenesis. PLoS One 10, No. e0131676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yehl K, Lemire S, Yang AC, Ando H, Mimee M, Torres MDT, de la Fuente-Nunez C, and Lu TK. (2019) Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 179, 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lee ME, DeLoache WC, Cervantes B, and Dueber JE. (2015) A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth. Biol 4, 975–986. [DOI] [PubMed] [Google Scholar]
- (17).Doud MB, Lee JM, and Bloom JD. (2018) How single mutations affect viral escape from broad and narrow antibodies to H1 influenza hemagglutinin. Nat. Commun 9, 1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wu NC, and Qi H. (2019) Application of deep mutational scanning in hepatitis C virus. Methods Mol. Biol 1911, 183–190. [DOI] [PubMed] [Google Scholar]
- (19).Hartman EC, Jakobson CM, Favor AH, Lobba MJ, Álvarez-Benedicto E, Francis MB, and Tullman-Ercek D. (2018) Quantitative characterization of all single amino acid variants of a viral capsid based drug delivery vehicle. Nat. Commun 9, 1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).van Dolleweerd CJ, Kessans SA, Van de Bittner KC, Bustamante LY, Bundela R, Scott B, Nicholson MJ, and Parker EJ. (2018) MIDAS: a modular DNA assembly system for synthetic biology. ACS Synth. Biol 7, 1018–1029. [DOI] [PubMed] [Google Scholar]
- (21).Wrenbeck EE, Klesmith JR, Stapleton JA, Adeniran A, Tyo KEJ, and Whitehead TA. (2016) Plasmid-based one-pot saturation mutagenesis. Nat. Methods 13, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Medina-Cucurella A, Steiner PJ, Faber MS, Beltrán J, Borelli A, Kirby MB, Cutler SR, and Whitehead TA. (2019) User-defined single pot mutagenesis using unpurified oligo pools. Protein Eng., Des. Sel 32 (1), 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kosuri S, and Church GM. (2014) Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 11, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).McKenna R, Xia D, Willingmann R, Ilag LL, Krishnaswamy S, Rossmann MG, Olson NH, Baker TS, and Incardona NL. (1992) Atomic structure of single-stranded DNA bacteriophage Φ X174 and its functional implications. Nature 355, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dokland T, McKenna R, Ilag LL, Bowman BR, Incardona NL, Fane BA, and Rossmann MG. (1997) Structure of a viral procapsid with molecular scaffolding. Nature 389, 308–313. [DOI] [PubMed] [Google Scholar]
- (26).Sanger F, Coulson AR, Friedmann T, Air GM, Barrell BG, Brown NL, Fiddes JC, Hutchinson CA, Slocombe PM, and Smith M. (1978) The nucleotide sequence of bacteriophage phiX174. J. Mol. Biol 125, 225–246. [DOI] [PubMed] [Google Scholar]
- (27).Smith HO, Hutchison CA, Pfannkoch C, and Venter JC. (2003) Generating a synthetic genome by whole genome assembly: Φ X174 bacteriophage from synthetic oligonucleotides. Proc. Natl. Acad. Sci. U. S. A 100, 15440–15445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Jaschke PR, Lieberman EK, Rodriguez J, Sierra A, and Endy D. (2012) A fully decompressed synthetic bacteriophage Φ X174 genome assembled and archived in yeast. Virology 434, 278–284. [DOI] [PubMed] [Google Scholar]
- (29).Jaschke PR, Dotson GA, Hung K, Liu D, and Endy D. (2018) Definitive demonstration by synthesis of genome annotation completeness. bioRxiv, 455428. [DOI] [PMC free article] [PubMed]
- (30).Aoyama A, Hamatake RK, and Hayashi M. (1981) Morphogenesis of Φ X174: in vitro synthesis of infectious phage from purified viral components. Proc. Natl. Acad. Sci. U. S. A 12, 7285–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hafenstein S, and Fane BA. (2002) Φ X174 genome-capsid interactions influence the biophysical properties of the virion: evidence for a scaffolding-like function for the genome during the final stages of morphogenesis. J. of Viro 76, 5350–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bernal RA, Hafenstein S, Esmeralda R, Fane BA, and Rossmann MG (2004) The Φ X174 protein J mediates DNA packaging and viral attachment to host cells. J. Mol. Biol 337, 1109–1122. [DOI] [PubMed] [Google Scholar]
- (33).Rokyta DR, Joyce P, Caudle SB, and Wichman HA. (2005) An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus. Nat. Genet 37, 441–444. [DOI] [PubMed] [Google Scholar]
- (34).Wichman HA, and Brown CJ. (2010) Experimental evolution of viruses: Microviridae as a model system. Philos. Trans. R. Soc., B 365, 2495–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Caudle SB, Miller CR, and Rokyta DR. (2014) Environment Determines Epistatic Patterns for a ssDNA Virus. Genetics 196, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sackman AM, and Rokyta DR. (2018) Additive Phenotypes Underlie Epistasis of Fitness Effects. Genetics 208, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Donaldson EF, Yount B, Sims AC, Burkett S, Pickles RJ, and Baric RS. (2008) Systematic Assembly of a Full-Length Infectious Clone of Human Coronavirus NL63. J. Virol 82, 11948–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kowalsky CA, Klesmith JR, Stapleton JA, Kelly V, Reichkitzer N, and Whitehead TA. (2015) High-Resolution Sequence-Function Mapping of Full-Length Proteins. PLoS One 10, No. e0118193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Klesmith JR, and Hackel BJ. (2019) Improved mutation function prediction via PACT: Protein Analysis and Classifier Toolkit. Bioinformatics 35 (6), 2707–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Firnberg E, Labonte JW, Gray JJ, and Ostermeier M. (2014) A comprehensive, high resolution map of a gene’s fitness landscape. Mol. Biol. Evol 31, 1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Faber MS, Wrenbeck EW, Azouz LR, Steiner PJ, and Whitehead TA. (2019) Impact of in vivo protein folding probability on local fitness landscapes. Mol. Biol. Evol 36, 2764. [DOI] [PubMed] [Google Scholar]
- (42).Rogers SO, and Bendich AJ. (1988) Extraction of DNA from plant tissues. Plant Mol. Biol. Man A6, 1–10. [DOI] [PubMed] [Google Scholar]
- (43).Edwards RA, and Rohwer F. (2005) Viral metagenomics. Nat. Rev. Microbiol 3, 504–509. [DOI] [PubMed] [Google Scholar]
- (44).Lee JM, Huddleston J, Doud MB, Hooper KA, Wu NC, Bedford T, and Bloom JD. (2018) Deep mutational scanning of hemagglutinin helps predict evolutionary fates of human H3N2 influenza variants. Proc. Natl. Acad. Sci. U. S. A 115, E8276–E8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ando H, Lemire S, Pires DP, and Lu TK. (2015) Engineering modular viral scaffolds for targeted bacterial population editing. Cell Systems 1, 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Garamella J, Marshall R, Rustad M, and Noireaux V. (2016) The all E. coli Toolbox 2.0: a platform for cell-free synthetic biology. ACS Synth. Biol 5, 344–355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.