

Abstract
Background:
Imatinib mesylate (imatinib) is the first-line treatment for newly diagnosed chronic myeloid leukemia (CML) due to its remarkable hematologic and cytogenetic responses. We previously demonstrated that the imatinib-resistant CML cells (Myl-R) contained elevated Lyn activity and intracellular creatine pools compared to imatinib-sensitive Myl cells.
Methods:
Stable isotope metabolic labeling, media creatine depletion, and Na+/K+-ATPase inhibitor experiments were performed to investigate the origin of creatine pools in Myl-R cells. Inhibition and shRNA knockdown were performed to investigate the specific role of Lyn in regulating the Na+/K+-ATPase and creatine uptake.
Results:
Inhibition of the Na+/K+-ATPase pump (ouabain, digitoxin), depletion of extracellular creatine or inhibition of Lyn kinase (ponatinib, dasatinib), demonstrated that enhanced creatine accumulation in Myl-R cells was dependent on uptake from the growth media. Creatine uptake was independent of the Na+/creatine symporter (SLC6A8) expression or de novo synthesis. Western blot analyses showed that phosphorylation of the Na+/K+-ATPase on Tyr 10 (Y10), a known regulatory phosphorylation site, correlated with Lyn activity. Overexpression of Lyn in HEK293 cells increased Y10 phosphorylation (pY10) of the Na+/K+-ATPase, whereas Lyn inhibition or shRNA knockdown reduced Na+/K+-ATPase pY10 and decreased creatine accumulation in Myl-R cells. Consistent with enhanced uptake in Myl-R cells, cyclocreatine (Ccr), a cytotoxic creatine analog, caused significant loss of viability in Myl-R compared to Myl cells.
Conclusions:
These data suggest that Lyn can affect creatine uptake through Lyn-dependent phosphorylation and regulation of the Na+/K+-ATPase pump activity.
General Significance:
These studies identify kinase regulation of the Na+/K+-ATPase as pivotal in regulating creatine uptake and energy metabolism in cells.
Keywords: Lyn, kinase, metabolomics, metabolites, chronic myelogenous leukemia, 1H NMR spectroscopy, drug resistance, imatinib, creatine, phosphocreatine, sodium/potassium-ATPase, creatine transporter, ponatinib, cyclocreatine, cell viability, apoptosis
INTRODUCTION
The defining characteristic of Philadelphia chromosome-positive chronic myelogenous leukemia (Ph+ CML) is the presence of the Bcr-Abl fusion protein resulting from the reciprocal translocation of chromosomes 9 and 221,2. Bcr-Abl protein possesses constitutive kinase activity and is the principal cause of CML development and progression3,4. A major advance in CML cancer therapy was the approval of imatinib mesylate (Gleevec, Novartis), a 2-phenylaminopyrimidine compound that selectively inhibits the enzymatic activity of the BCR-ABL protein5,6. While imatinib has been extremely successful in the treatment and management of CML, the development of acquired imatinib-resistance in patients remains a clinical problem7.
Multiple Bcr-Abl-dependent mechanisms contribute to imatinib resistance including increased transcript and protein expression levels, gene amplification, extra chromosomal aberrations, and specific point (“gatekeeper”) mutations that sterically inhibit imatinib binding to the inactive configuration of Bcr-Abl8–14. In addition to alterations in molecular signaling events, Bcr-Abl is linked to cellular metabolic alterations that underlie enhanced cell proliferation and viability15. Hematopoietic cells transfected with Bcr-Abl show increased GLUT1 transporter expression and glucose uptake16. Metabolomics studies demonstrated that imatinib treatment alters glucose carbon flux involved in the de novo synthesis of nucleic and fatty acids thereby limiting Bcr-Abl transformed cells of important macromolecule substrates essential for proliferation17. In addition, imatinib treatment also results in decreased mitochondrial activity18,19, reduced glycolytic activity, and internalization of the GLUT1 transporter in Bcr-Abl-positive CML cells that consequently leads to reduced glucose uptake20–22. In fact, an important hallmark of imatinib-resistance in CML cell lines is up-regulated glucose uptake mediated by increased glycolytic activity and retention of GLUT1 transporters in the cell membrane. The increased glucose metabolism phenotype in these cell lines is further evidenced by high lactate synthesis and elevations in phosphocholine, which are believed to support enhanced cell proliferation23.
Bcr-Abl-independent mechanisms such as the overexpression of the Src-family kinase Lyn or Hck also contribute to imatinib resistance in CML3,4,12,24–26. Our lab previously showed that increased Lyn activity in imatinib-resistant CML cells (Myl-R) leads to upregulation of anti-apoptotic proteins such as Mcl-1 and BIRC6 resulting in increased imatinib resistance5,6,27,28. Furthermore, using high-resolution NMR spectroscopy to analyze water-soluble metabolites revealed that in addition to decreased glycolysis, there was a significant elevation of intracellular creatine in the imatinib-resistant Myl-R cells7,29 unlike in previously studied imatinib-resistant cells15,23. Over 50% of the creatine was in the form of phosphocreatine under these conditions, and considering its role as a high-energy phosphate donor, it was speculated that elevated phosphocreatine might confer a survival advantage on Myl-R cells. In the present study, we investigated the molecular mechanisms involved in the accumulation of creatine in Myl-R cells. The results of these studies demonstrate that Myl-R creatine levels were dependent both on Na+ -dependent creatine uptake from the media and the Na+/K+-ATPase activity. Moreover, we now provide evidence for the direct involvement of Lyn in creatine uptake through phosphorylation and regulation of the Na+/K+-ATPase pump. Thus, these studies demonstrate a pivotal role for the Na+/K+ ATPase pump in regulating creatine uptake and suggest that increased creatine uptake and metabolism may be an important cellular adaptation mechanism utilized by the imatinib-resistant CML sub-line (Myl-R cells).
MATERIALS AND METHODS
Cells, Cell Culture and Reagents
The human chronic myelogenous leukemia cell line, Myl, and its imatinib-resistant sub-line, Myl-R, were generous gifts from Dr. Hideo Tanaka (Department of Haematology and Oncology, Hiroshima University, Hiroshima, Japan)8–14. Cells were cultured in culture flasks suspended in RPMI 1640 medium (Gibco® by Life Technologies™, U.S.A.) supplemented with 10% fetal bovine serum (Atlanta Biologicals; Norcross, GA), and 1% antibiotic/antimycotic (Invitrogen; Carlsbad, Ca) as previously described15,29. Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere in concentrations of approximately 0.6×106 cells mL−1. Culture medium was replaced every 2 to 4 days. As originally reported, Myl-R cells can be maintained in imatinib-free culture medium for up to ~6 months without a change in their sensitivity to imatinib treatment12,16. For most experiments described here, cells were harvested by low-speed centrifugation and washed with cold 1X PBS prior to lysis.
For the labeled glycine experiments, an additional 10 mg/L 2-13C-glycine (Isotec Inc., Miamisburg, OH) was added to the growth medium that already contained 10 mg/L unlabeled glycine, thereby resulting in a 50% labeled glycine pool. As a control, more unlabeled glycine was added, resulting in a final concentration of 20 mg/L. Myl and Myl-R cells were maintained under these culture conditions for 5 days prior to extraction.
Reagents were obtained from the following sources: ponatinib and dasatinib were from LC Laboratories (Woburn, MA); imatinib was from Selleckchem (Houston, TX). Ouabain, DMSO, and 3-Guanidinopropionic acid were from Sigma-Aldrich (St. Louis, MO). Polyethylenimine (P.E.I.) was from Polysciences Inc. (Warrington, PA). The primary human antibodies used include: SLC6A8 and creatine kinase B (Abcam, Cambridge, MA), Phospho-Na+/K+-ATPase α1 (Tyr10), Na+/K+-ATPase, phospho-Src (Y416), PTMScan® Phospho-Tyrosine (Cell Signaling Technology, CST, Danvers, MA).), Lyn and β-actin (Santa Cruz Biotechnology, SCBT, Dallas, TX); with secondary antibodies, anti-mouse and anti-rabbit IgG-HRP conjugated (Promega {Madison, WI}). The primary antibodies were diluted following supplier recommendations: 1:200 to 1:1000 in 5% BSA in TBS-T. Secondary antibodies were diluted at 1:10,000 in 5% dry, non-fat milk in TBS-T.
The Lyn overexpression plasmids {pEGFP-Lyn wild type and -Lyn mutant (Y508F)} were kind gifts from Dr. Klaus Hahn’s Lab (Department of Pharmacology, UNC-Chapel Hill School of Medicine, Chapel Hill, NC, U.S.A.). pEGFP-Lyn kinase dead (K275R) was made from p-EGFP-Lyn-wild type using site-directed mutagenesis.
For experiments examining creatine depletion, cells were grown in RPMI medium containing 10% heat-inactivated dialyzed FBS (Sigma-Aldrich Company, St. Louis, MO) in place of normal FBS for 5 days prior to extraction.
Cell Treatments
Approximately 15 × 106 cells were grown and exposed to different drugs to examine the regulation of creatine uptake. Fewer cells, approximately 2 × 106, were used in experiments for immunoblot analyses. Drug dose-response experiments were initially performed with different compounds to determine the effective concentrations. For ouabain, cells were incubated in increasing concentrations, starting at 1.0 nM and increased by order of 10 until the concentration of 10 μM was achieved. Separate populations of the cells were also treated with dasatinib or ponatinib (Selleckchem), dual Bcr-Abl and Src family tyrosine kinase inhibitors. The creatine competitive inhibitor, 3-Guanidinopropionic Acid (Sigma-Aldrich, St. Louis, MO), was reconstituted in media (above) and a 30-mM concentration used to treat Myl-R cells. Cells were treated for 24 hours before metabolite extraction. The drugs/compounds were reconstituted in incubation media (3-Guanidinopropionic acid), hot, autoclaved water (ouabain), and DMSO (imatinib, ponatinib, dasatinib); likewise incubation media, diH2O, and DMSO were also used as the vehicle controls.
Cell Extraction and NMR Sample Preparation
Following cell treatments and incubations, cells were harvested by low-speed centrifugation and the metabolites were extracted using a cold methanol extraction method, as previously described17,29. Briefly, the cells were collected by centrifugation and washed three times with cold PBS. Following removal of the last wash, 500 μL of ice-cold 50% methanol was added to the cell pellet and vortexed. The cell extracts were then incubated for 30 minutes on dry ice and then allowed to thaw for 10 minutes on ice. The extracts were clarified by centrifugation at 16,000g for 10 minutes at 4° C. The methanol extract (supernatant) was collected and transferred to a new microcentrifuge tube, while an additional 500 μL of 50% methanol was added to the pellet for a second extraction. The second 50% methanol extract was collected and combined with the previous (first) extract. The total cell extract was evaporated to dryness using a SpeedVac lyophilizer.
Prior to NMR spectroscopy, the evaporated cell extract pellet was dissolved in 600 μL of D2O containing 0.5 mM (final concentration) trimethylsilyl-2,2,3,3-tetradeuteropropionic acid (TSP) and transferred to a 5 mm NMR tube for high resolution NMR analysis. For the Myl-R NMR studies on the regulation of creatine uptake, the pellet was suspended in 70 μL of D2O containing 0.1 mM TSP.
1D 1H and 2D 1H-{13C} HSQC NMR Spectroscopy
1D 1H NMR spectra characterizing the metabolic differences between Myl and Myl-R cells, and the Myl-R creatine regulation analysis were acquired at 16.4T using a Varian INOVA NMR spectrometer (700 MHz 1H, Varian Instruments) equipped with a 5 mm indirect cold probe. The FIDs were acquired using a one-pulse sequence with a total repetition time (TR) of 12.65s, number of transients (nt) of 64 and 1024, and a 90° flip angle. 2D 1H-{13C} heteronuclear single quantum coherence (HSQC) NMR spectra were acquired at 14.1 T NMR spectrometer equipped with 5 mm indirect HCN probe using 256 increments and zero filled in f1 and f2 to 4,096 points and with a shifted sine bell apodization.
Spectral Processing, Pattern Recognition and Metabolite determination
All NMR spectra were processed using ACD/1D NMR Manager, version 12.0 (Advanced Chemistry Development, Inc., Toronto, ON, Canada). Imported FIDs were zero filled to at least 32,000 points and an exponential line broadening of 0.1 to 1.0 Hz was applied prior to Fourier transformation. The 2D spectra were processed with Varian software and zero filled in both dimensions to 4,096 points and subjected to a shifted sine bell apodization. Spectra were phased, baseline corrected, and referenced to the TSP peak at 0.00 ppm. TSP (at 0.75 ppm and upfield), residual methanol, water, and formate were excluded from binning process. Metabolite identification and quantification were performed using Chenomx software (version 6.1; Chenomx Inc., Edmonton, Canada), as previously described (Dewar et al., 2010).
ShRNA Knockdown of Lyn
PLKO.1 lentiviral vectors containing shRNA directed against Lyn (TRCN0000010101, 04, 05, 06, and 07) or a non-targeting shRNA (shCtrl) were purchased from the UNC Lenti-shRNA Core Facility. Lentivirus transduction of MYL-R cells with shRNA was done per the protocol supplied by the RNAi Consortium (http://www.broadinstitute.org/rnai/public/resources/protocols), and as previously described20–22,27. Briefly, Myl-R cells were seeded at 5×105 cells/mL in 5 mL growth media containing 8 μg/mL polybrene, and incubated overnight with 1 mL of anti-Lyn viral shRNA (shLyn-01, −05, and −06) known to efficiently knock down Lyn. Non-targeting viral shRNA (shCtrl) was used as control. Stably transduced cells were selected for by exposure to 2 μg/mL puromycin in cell culture23,30. Aliquots of the cells were harvested one week after selection and immunoblot analyses performed to confirm Lyn knockdown. The rest of the cells were expanded in puromycin for another one week to obtain enough cells (~20 × 106) per condition for NMR analyses of total creatine pool.
Western Blotting Conditions
Cells were harvested by centrifugation, washed once in cold 1X PBS, and lysed in a modified RIPA (RIPA, no SDS) buffer (150 mM NaCl, 9.1 mM Na2HPO4, 1.7 mM NaH2PO4, 1% NP-40, and 0.5% deoxycholic acid; adjusted to pH 7.4) and supplemented with 2 mM sodium orthovanadate, 10 mM NaF, 0.0125 μM calyculin A, and Complete Protease Inhibitor Cocktail (Roche Diagnostics, U.S.A.). The lysates were clarified by centrifugation and the protein concentrations were normalized using a Bradford assay (BIO-RAD). Samples for gel electrophoresis were prepared by adding protein lysates to Laemmli sample buffer (final concentration: 0.25 M Tris pH 6.8, 10% glycerol, 5% β-mercaptoethanol, 0.001 μg/mL Bromophenol blue) and 30 μg of protein were loaded into each well of an SDS-polyacrylamide gel for protein separations. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (BIO-RAD) which were then blocked for 1 hr with 5% non-fat dry milk or 5 % BSA dissolved in Tris-buffered saline supplemented with Tween-20 (TBS-T). The membranes were then incubated in primary antibodies at 4° C overnight, washed 3 times with TBST, then incubated with anti-mouse / anti-rabbit IgG-HRP conjugated secondary antibodies for 1 hr at room temperature. The membranes were rinsed 3 times with TBS-T then developed using Clarity™ ECL Western Substrate (BIO-RAD), and imaged using a ChemiDoc™ Touch Imaging System (BIO-RAD).
Cell Transfection (Lyn overexpression and Na+/K+-ATPase activity)
HEK293 cells were passaged and split into four 60 mm plates a day before transfection. The following day, the cells were transfected with Lyn expression vectors: wild type or mutant (Y508F) or kinase dead (K275R), using polyethylenimine (PEI) transfection reagent (Polysciences Inc., Warrington, PA). A total of 5 μg DNA was used for each transfection. The fourth 60 mm plate was transfected with empty vector (mock). The cells were harvested 24 hours after transfection, washed in cold 1X PBS, and lysed in cold Western blot lysis buffer (above). Western blot analysis was performed as described above to examine the effect of Lyn overexpression on Na+/K+-ATPase (Tyr10) phosphorylation.
Rubidium Uptake Assay
Myl-R cells were treated with DMSO or dasatinib (1 nM) or ouabain (100 nM). After 24 hours, approximately 106 cells per condition were collected, pelleted and washed in rubidium-free uptake buffer. Cells were resuspended in uptake buffer supplemented with 5 mM RbCl. Cells from each condition were split into four aliquots and incubated at room temperature over four time-points: 0, 5, 10, and 15 minutes. Cells were then hydrolyzed in nitric acid at 70° for 24 hours. Rubidium (Rb+) uptake was measured using inductively coupled plasma optical emission spectroscopy (ICPOES; Model Optima 4300D, Perkin Elmer, Norwalk, CT) operated at wavelengths of 240.272w and 349.894 nm.
Cell Viability (MTS Assay)
Myl and Myl-R cells maintained in normal growth medium (10% FBS) or regular media containing various concentrations of cyclocreatine (CCr), a competitive inhibitor of creatine known to have high affinity for the Na+/creatine symporter31. Cyclocreatine was dissolved in regular cell growth medium at a stock concentration of 100 mM. In triplicate, CCr was added to 10 × 103/100 μL Myl or Myl-R cells in increasing concentrations in a 96-well plate. Regular cell culture medium was used as the vehicle control. After 48-hr incubation at 37°C with 5% CO2, cell viability was determined by MTS assay performed according to the manufacturer’s instructions (CellTiter 96® AQueous One Solution Reagent, Promega, Madison, WI). The absorbance was read at 490 nm using a SpectraMAX plate reader (Molecular Devices, Sunnyvale, CA). Trypan blue exclusion assay was performed using the Trypan blue reagent, and cells were counted using a hemacytometer (Hausser Scientific, Horsham, PA). Triplicate counts were obtained and used to determine cell proliferation. Data was analyzed for significant differences using a paired t-test.
Statistical Analyses
Data are reported as the mean ± standard error of the mean (S.E.M); S.E.M. was performed on all datasets to determine positive and negative error. Two-tailed Student’s t-test was used to make comparisons between groups, and p values below 0.05 at the 95% confidence level were considered to be statistically significant. Calculations were performed using GraphPad Prism and Microsoft Excel. Analysis of variance (ANOVA) was used to test whether differences in intracellular creatine between Myl and Myl-R cells were significant.
RESULTS
Increased steady state levels of creatine in Myl-R cells detected by 1H NMR
1H NMR spectroscopy was used to examine the metabolic differences between the CML cell line Myl and its imatinib-resistant counterpart, Myl-R12,29. Consistent with our previous study, the 1D 1H NMR spectra obtained from Myl and Myl-R cell extracts demonstrated increased levels of intracellular creatine in the Myl-R cells (Supplementary Fig. 1B) compared to Myl cells (Supplementary Fig. 1A). We observed the 1H and 13C chemical shifts of the creatine methyl group (*) at 3.023 ppm and 39.56 ppm respectively, and the two protons of the methylene group (**) at chemical shifts of 3.93 ppm and 56.57 ppm respectively (Supplementary Fig. 1B). To verify the identity of creatine, 2D 1H-{13C} HSQC NMR was performed on the Myl-R cell extracts and this confirmed the creatine signal by correlating the 1H and 13C resonances (Supplementary Fig. 1, inset).
De novo synthesis does not account for elevated creatine levels in Myl-R cells
To determine if de novo synthesis was contributing to the elevated levels of creatine observed in Myl-R cells, Myl and Myl-R cells were maintained in total growth medium (RPMI + 10%FBS) with or without the addition of 2-13C-glycine for 5 days. Glycine and arginine are required for the de novo biosynthesis of creatine, therefore, incubation with 2-13C-glycine, results in creatine labeled at the 2-position through this two-part reaction (Fig.1A)32.
Figure 1. Increased intracellular creatine in Myl-R cells is due to uptake from cell media, and not de novo synthesis.
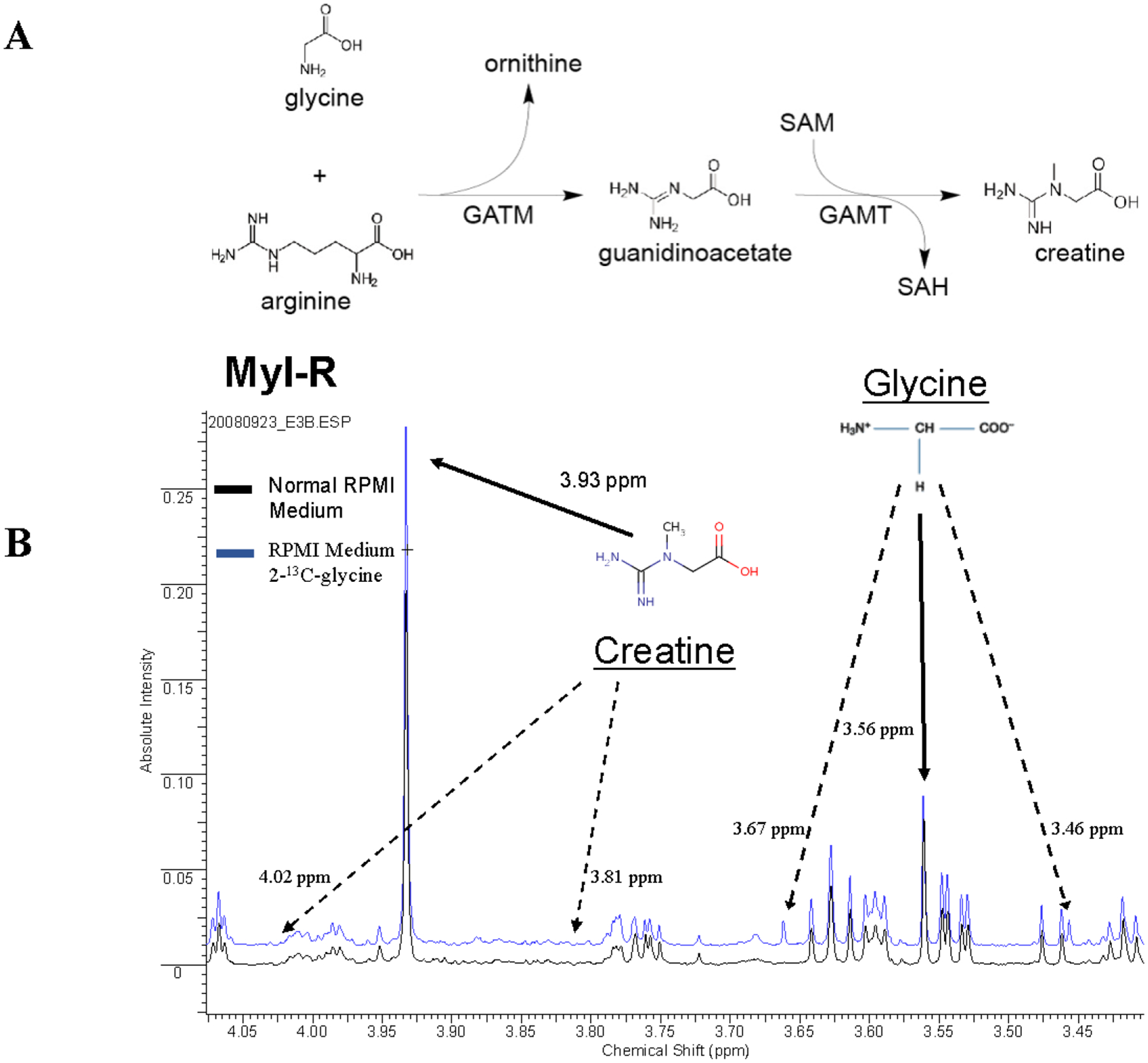
(A) Glycine and arginine are required for the biosynthesis of creatine in a two-part reaction. Myl and Myl-R cells were maintained in total growth medium with or without the addition of 2-13C-glycine for 5 days. (B) Analysis of 1H NMR spectra of each cell extract revealed that there was no statistically significant difference in percent incorporation of labeled glycine in the cell extracts from Myl and Myl-R cells. Whereas satellite peaks from 2-13C-glycine were only observed in Myl-R cells incubated with heavy glycine, satellite peaks associated with creatine were not observed under these conditions. The data shown are representative of three independent experiments.
After 5 days, the cells were harvested, metabolites extracted, and 1H NMR spectra of each cell extract were obtained. Following incubation in medium containing 50% 2-13C-glycine, the percent incorporation of labeled glycine in the cell extracts was found to be 31.3 ± 4.1% and 31.0 ± 2.6% for Myl and Myl-R cells, respectively. These data indicate that there were no statistically significant differences in the ability of each cell type to synthesize labeled creatine de novo. Shown in Fig. 1B are the stacked 1H NMR spectra from Myl-R cells incubated with (blue) or without (black) labeled 2-13C-glycine. The proton peaks representing the unlabeled (12C) glycine and creatine were observed at 3.56 and 3.93 ppm, respectively. As expected, satellite peaks from 2-13C-glycine were only observed in Myl-R cells incubated with labeled glycine (blue line). However, no satellite peaks were observed associated with creatine under these conditions indicating that de novo synthesis does not account for the elevated creatine levels observed in Myl-R cells.
Media creatine is a major source of intracellular creatine in both Myl and Myl-R cells
Since de novo creatine synthesis was not higher in Myl-R than Myl cells, we examined whether uptake from the cell culture medium was a potential source of elevated creatine in Myl-R cells. 1H NMR analysis of RPMI and RPMI + 10% FBS media indicated that FBS is a significant source of creatine with an average concentration of 37.4 ± 3.04 μM (data not shown). Dialyzed FBS (dFBS) is depleted of creatine and other small molecules, including growth factors, cytokines, and metabolites. Incubating Myl and Myl-R cells in RPMI medium supplemented with 10% dFBS resulted in a significant decline in creatine in both cell types (Fig. 2). Addition of 100 μM creatine to dFBS containing culture medium significantly restored the total creatine pool (creatine and phosphocreatine) in the Myl cells to basal levels, and significantly increased (p<0.1) the total creatine pool in Myl-R cells, nearly 2-fold greater compared to Myl-R cells incubated in RPMI + 10% FBS (Fig. 2).
Figure 2. Media creatine is responsible for observed differences in intracellular creatine levels between Myl and Myl-R cells.
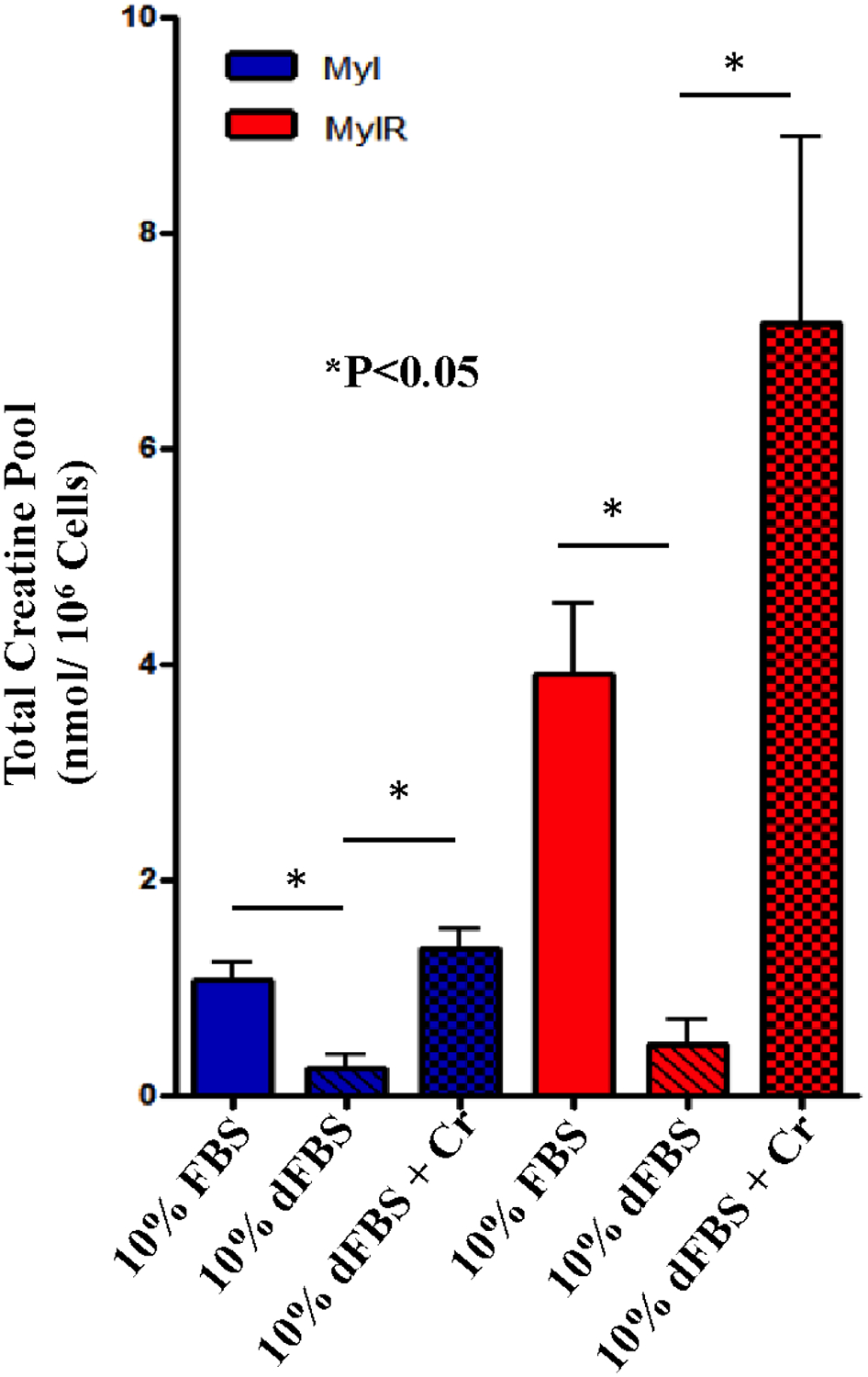
Myl and Myl-R cells were cultured in RPMI medium supplemented with 10% dialyzed FBS (dFBS) and intracellular creatine concentrations analyzed as described in Materials and Methods. Compared to cells cultured in RPMI supplemented with 10% FBS, there was significant reduction in creatine observed in both cell lines. Addition of 100 μM creatine to the dFBS-containing media restored creatine in Myl cells to normal levels and increased creatine levels in Myl-R cells by ~2-fold over that observed with 10% FBS-containing media. Approximately 50 × 106 Myl or Myl-R cells were cultured in RPMI media supplemented with 10% dFBS five days prior to extraction. Creatine was added to 10% dFBS to a final concentration of 100 μM two days prior to extraction. Cells were harvested and metabolites extracted for 1H NMR analysis of total intracellular creatine. Metabolites for Myl and Myl-R cells cultured under normal conditions (10% FBS) were similarly analyzed for comparison. Error was determined as deviation from the mean of all data from three independent experiments. The error bars reflect the SEM. (See Statistical Analysis in Methods.)
The creatine transporter and creatine kinase B protein levels are comparable in Myl and Myl-R cells
Creatine is imported into the cell through the SLC6A8 transporter. Brain type creatine kinase (creatine kinase B, CKB) phosphorylates creatine to generate phosphocreatine, a source of high-energy phosphate for ATP generation in low-energy states33. CKB is expressed mainly in the brain, but is also found expressed in other tissues. Overexpression of CKB has been observed in many tumor types and has been linked to adverse prognostic outcomes34. Hence, we compared the protein levels of both the creatine transporter (SLC6A8) and CKB by Western blotting to determine if differences in these proteins contributed to the increased total creatine pool observed in Myl-R cells (Figure 2, Figure 3A, and Supplementary Fig. 1B). These analyses showed that SLC6A8 and CKB protein levels were comparable in Myl and Myl-R cells (Fig. 3A), suggesting that the observed increase in total creatine pool was independent of the expression levels of these two proteins.
Figure 3. The Na+/K+-ATPase pump is required for creatine uptake by Myl-R cells.
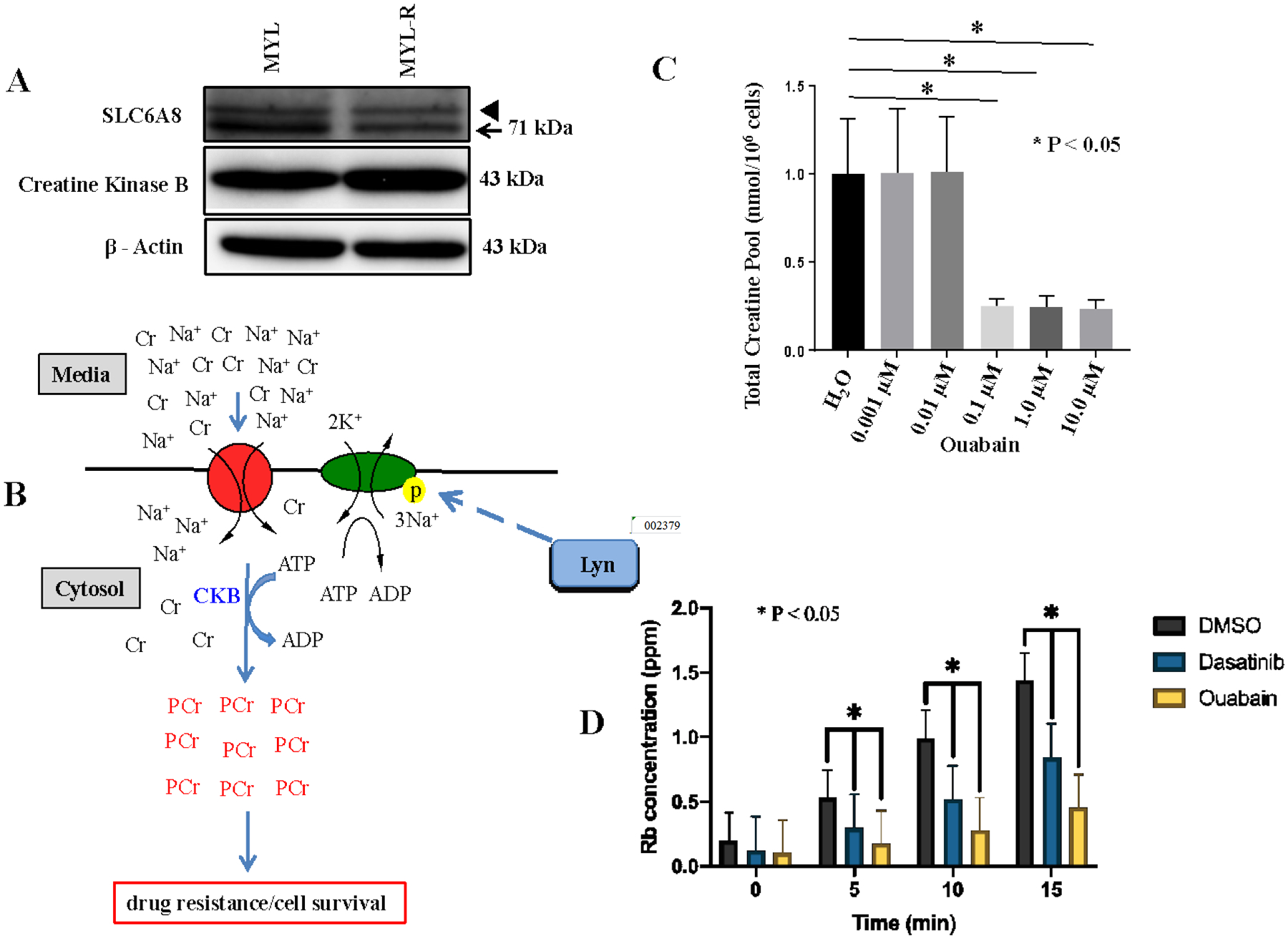
(A) Creatine kinase B (CKB) and SLC6A8 protein levels were compared in Myl and Myl-R cells by Western blotting. Arrowhead represents cross reactivity with an off-target protein of unknown origin. (B) A model diagram depicting creatine uptake and subsequent phosphorylation by CKB to generate phosphocreatine. (C) Treatment of Myl-R cells with increasing concentrations of ouabain significantly reduced total intracellular creatine in a dose-dependent manner as determined by 1H NMR analysis. Myl-R cells were treated for 24 hours with increasing concentrations of ouabain and 1H NMR used to measure total creatine. (D) Treatment of Myl-R cells with dasatinib (1 nM) or ouabain (100 nM) significantly reduced rubidium (Rb) uptake as determined using the Rb uptake assay. Rb+ uptake measurements were performed courtesy of Dr. Andrew Ghio’s lab (EPA, UNC Chapel Hill, NC). The data presented in this figure are averages of three independent experiments, and * represents p < 0.05.
Role of the Na+/ K+-ATPase in regulating creatine uptake in Myl-R cells
Since our data indicated that the total creatine pool in Myl-R cells was dependent on uptake from the media, we examined possible mechanisms for this regulation. Creatine uptake is mediated by a Na+ /creatine symporter, SLC6A832,35–37, and is dependent on gradients of both Na+ and Cl− ions38. Thus we tested whether creatine uptake was dependent on the activity of the Na+-K+-ATPase pump in Myl-R cells (Fig 3B). Myl-R cells were incubated with increasing concentrations of ouabain, a glycoside that has been shown to bind and inhibit the Na+-K+-ATPase39. As expected, oubain significantly (p<0.05) depleted the total creatine pool in Myl-R cells at 100 nM or greater concentrations (Fig 3C). Similar results were obtained with digitoxin, another well-established Na+/K+-ATPase inhibitor (data not shown). A rubidium uptake assay further confirmed that these drug treatments directly inhibited the Na+/K+-ATPase activity (Fig 3D). Compared to DMSO treatment, 1 nM dasatinib and 100 nM ouabain significantly lowered Rb uptake in Myl-R cells (Fig 3D). These studies demonstrate an important role of the Na+/K+-ATPase activity in affecting creatine levels in Myl-R cells.
Role of Lyn in mediating creatine uptake in Myl-R cells
Lyn has been shown to be an important tyrosine kinase for the survival of Myl-R cells12,30,27,28. Immunoprecipitation data showed that Lyn is able to bind in a complex with the Na+/K+-ATPase pump resulting in phosphorylation and activation of the pump40–42. Specifically, phosphorylation of the α-subunit of the Na+/K+-ATPase on tyrosine 10 (pY10) is critical for the enzyme’s pumping activity41,42. To test the potential involvement of Lyn in the regulation of the Na+/K+-ATPase, Myl-R cells were incubated with dasatinib, a broad tyrosine kinase inhibitor known to inhibit Lyn, and the intracellular creatine pool measured. Compared to DMSO, dasatinib substantially reduced the total intracellular creatine pool in Myl-R cells (Supplementary Fig. 2A). Western blot analyses were used to compare both Lyn activation (as measured by pY416) and Na+/K+-ATPase α−1 activation (as measured by pY10) in Myl and Myl-R cells. As reported earlier12,30, both total Lyn and phospho-SFK (pY416) were higher in the Myl-R compared to the Myl cells (Fig 4A). Phospho-Na+/K+-ATPase α−1 (pY10) was also substantially higher in Myl-R compared to Myl cells whereas total Na+/K+-ATPase protein was comparable in the two cell lines (Fig 4A). Ponatinib, a more selective Bcr-Abl and Lyn inhibitor27,43, inhibited Lyn activity (pY416) and significantly reduced the amount of creatine in Myl-R cells (Fig 4C & Fig 4B). While phospho-Na+/K+-ATPase α−1 (pY10) was substantially reduced by this treatment, neither the total amount of the Na+/K+-ATPase nor the SLC6A8 protein was substantially affected (Fig 4C). Incubation of Myl-R cells with dasatinib or ouabain resulted in ~65% and ~95% inhibition of creatine uptake respectively (Supplementary Fig. 2B).
Figure 4. Lyn regulates the Na+/K+-ATPase phosphorylation and creatine uptake in Myl-R cells.
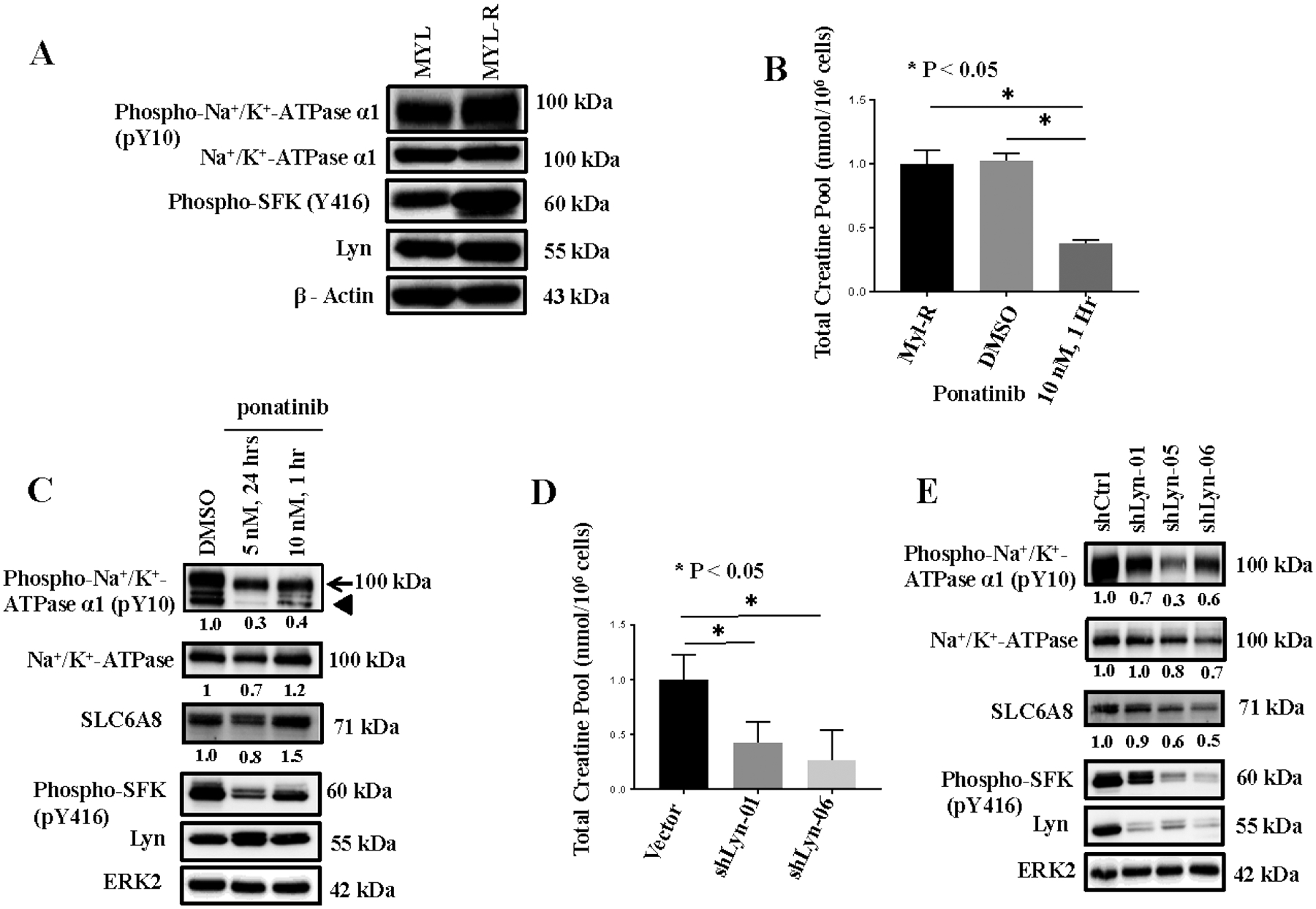
(A) Western blotting showed that tyrosine phosphorylation of Lyn and the Na+/K+-ATPase was higher in Myl-R compared to Myl cells. (B) Lyn inhibition significantly reduced the total creatine pool in Myl-R cells. Approximately 15 × 106 Myl-R cells were treated for 1 hour with DMSO or ponatinib (10 nM) and total intracellular creatine determined using 1H NMR as outlined in Materials and Methods. Control (DMSO) Myl-R cells were similarly analyzed for comparison. (C) Western blotting confirmed that Lyn inhibition reduced the phosphorylation of Lyn (pY416) and the Na+/K+-ATPase (pY10). Arrowhead represents cross reactivity with an induced 75–80 kDa off-target protein of unknown origin. (D and E) Lyn knockdown significantly reduced Na+/K+-ATPase phosphorylation and the total intracellular creatine pool in Myl-R cells. Approximately 15 × 106 Myl-R cells were infected with lentiviral particles containing shRNA directed against Lyn (shLyn-01, shLyn-06) and intracellular creatine levels measured. Lyn shRNA constructs (shLyn-01, shLyn-05, and shLyn-06) reduced Na+/K+-ATPase and Lyn phosphorylation. The data are the averages of three independent experiments, and * represents p < 0.05.
To further study the role of Lyn in creatine uptake, we developed a lentiviral shRNA system to investigate the effects of Lyn knockdown. While three knockdown constructs (shLyn-01, −05, and −06) were equally effective at reducing Lyn as determined by Western blot analyses (Fig 4E), two constructs (shLyn-01 and −06) were selected for further studies. Next, the total intracellular creatine pool was measured as described above. As observed with Lyn inhibition (Fig 4B), Lyn knockdown substantially reduced the amount of intracellular creatine observed in these cells compared to the vector control (Fig 4D). ShLyn-06, the shRNA construct most effective at reducing Lyn protein, was also the most effective at reducing intracellular creatine levels (Fig 4D). These data further support the requirement of Lyn for mediating creatine uptake in Myl-R cells.
Since tyrosine 10 has been identified as a key phosphorylation site involved in regulating the Na+/K+-ATPase pump activity42,44, we examined the effects of Lyn knockdown on the phosphorylation of this site using a commercially available antibody (anti-phospho-Na+/K+-ATPase α1 (pY10)), Cell Signaling Technology). Lyn knockdown substantially reduced phosphorylation of the Na+/K+-ATPase pump on tyrosine 10 (pY10), consistent with inhibition of Lyn activity (Fig 4E).
Overexpression of Lyn in HEK293 cells increased Na+/K+-ATPase α1 tyrosine phosphorylation (pY10)
Since our data suggested that Lyn was involved in phosphorylating and regulating the Na+/K+-ATPase in Myl-R cells, we further examined if expression of active Lyn was capable of phosphorylating the Na+/K+-ATPase in HEK293 cells. Overexpression of wild type (WT) Lyn or a constitutively active (Y508F) Lyn in HEK293 cells resulted in substantial increase in phospho- Na+/K+-ATPase α1 (pY10). Phospho-STAT5A (pY694), a known marker of Lyn activity, was strongly increased in HEK293 cells expressing constitutively active (Y508F) Lyn. By comparison, the kinase dead (K275R) (KD) Lyn did not increase the phosphorylation of this protein over control (pEGFP) (Supplementary Fig. 3). Taken together, these data are consistent with Lyn catalyzing the phosphorylation and activation of the Na+/K+-ATPase pump.
Inhibitors of creatine uptake reduce cell viability
We next investigated the effect of intracellular creatine depletion on cell viability. Cyclocreatine (CCr) is a creatine analog with high affinity for the Na+/creatine symporter31 that is rapidly transported and phosphorylated like creatine, thereby, competing with phosphocreatine function. Myl and Myl-R cells were treated for 48 hours with increasing concentrations of CCr and cell viability was calculated as a percentage of the cells maintained in normal media only (0 mM CCr), as described in Materials and Methods. Under normal culture conditions (0 mM CCr), the percent of viable Myl and Myl-R cells remained unchanged after 48 hours. By contrast, CCr caused a dose-dependent loss in cell viability over the 48-hr period for both the Myl and Myl-R cells as shown in Figure 5. The loss in viability was significantly higher for the Myl-R compared to the Myl cells in response to 12.5, 25, and 50 mM CCr treatment for 48 hours (Fig 5). In addition to suggesting that Myl-R cells have a higher creatine uptake than Myl cells, these studies show that creatine may be necessary for maintaining cell viability. Similarly, we tested the effect of 3-Guanidinopropionic Acid (3-GPA), a creatine analog45, on creatine uptake by Myl-R cells. Treatment of Myl-R cells with 3-GPA (30 mM, 24 hrs.) reduced total intracellular creatine pool ten-fold, comparable to untreated Myl cells (Supplementary Fig. 4). MTS assay studies following treatment of Myl-R cells with increasing concentrations of 3-GPA revealed loss in cell viability at ≥30 mM concentrations (data not shown), confirming the importance of creatine uptake and metabolism for the viability of these cells.
Figure 5. Cyclocreatine treatment significantly reduced Myl-R cell viability.
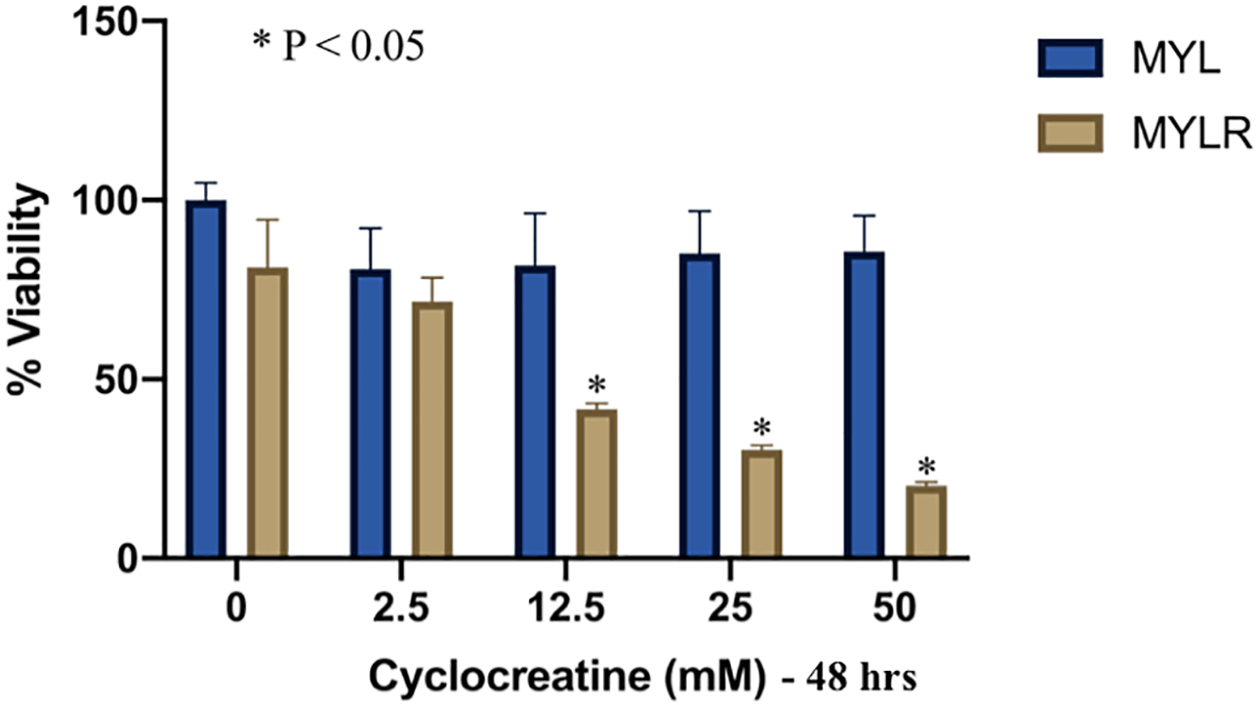
Treatment of Myl-R cells with cyclocreatine (CCr) significantly reduced cell viability in a dose-dependent manner compared to Myl cells. Myl and Myl-R cells were treated for 48 hours with increasing concentrations of CCr. Cell viability was determined in triplicate using the MTS Assay as described in Materials and Methods. The data is representative of three independent experiments.
Discussion
Similar to that originally reported by Ito et al., studies from our lab have confirmed that Lyn kinase is a major driver of drug resistance and cell survival in Myl-R cells12,27,28. Since these studies also demonstrated a substantial accumulation of creatine (and phosphocreatine) in Myl-R cells, the objective of the current study was to investigate the molecular basis for this difference. A second goal was to determine if increased creatine accumulation was important for Myl-R viability. Lastly, we sought to determine if hyperactivation of Lyn in Myl-R cells contributed to the increased accumulation of creatine in these cells. The results of these studies demonstrate three important findings. First, we established that the accumulation of creatine in Myl-R cells was dependent on uptake from FBS added to extracellular media, and was not derived from de novo synthesis. Second, our data demonstrated an essential role for Na+/K+-ATPase activity in creatine uptake in Myl-R cells and provided evidence for Lyn-dependent tyrosine phosphorylation of this protein. Our results also suggest that the increased accumulation of creatine (and phosphocreatine) may be crucial for maintaining Myl-R cell viability.
Multiple mechanisms are known to contribute to the development of imatinib-resistance in CML. While mutations in Bcr-Abl are frequent, additional mechanisms of resistance are now established, including hyperactivation of Lyn or Hck kinases and overexpression of anti-apoptotic proteins12,24–28,30,46. The development of second and third generation inhibitors targeting these kinases, in addition to Bcr-Abl, has been promising. Despite these clinical advances, the exact mechanisms by which Lyn or other Src family kinases contribute to acquired drug resistance are poorly understood. The data presented in this study suggest that Lyn contributes to altered cell metabolism through the regulation of creatine uptake.
The Myl-R cells are a well characterized model system to investigate the role of Lyn kinase in imatinib resistance12. Our phosphoproteomics analyses also revealed changes in tyrosine phosphorylation of the Na+/K+-ATPase in a manner consistent with Lyn activation and inhibition (upon ponatinib treatment)47. Western blot analyses for phospho-Na+/K+-ATPase (pY10) confirmed Lyn-dependent changes in the phosphorylation of this protein (Fig 4 and Supplementary Fig. 3), and consistent with studies demonstrating the importance of pY10, inhibition or knockdown of Lyn reduced the uptake of creatine in Myl-R cells (Fig 4B and Fig 4D). While our studies focused on Lyn, the dominant Src family kinase in Myl-R cells, it is possible that other Src family kinases can also phosphorylate and regulate the Na+/K+-ATPase in other cell types.
Metabolic reprogramming is a hallmark of a variety of human cancers29,48–50. While high levels of creatine and phosphocreatine have been reported in human breast51,52 and colorectal33 cancers, the roles of these high-energy metabolites on drug resistance have not been extensively explored. Tokarska-Schlattner et al. showed that phosphocreatine interacts with plasma membrane phospholipids thereby altering membrane structure in ways that not only protect against various insults, but also influence the physiological role(s) of the membrane53. While our previous studies demonstrated that Myl-R cells contained elevated creatine29, the current study suggests that this may exert a protective effect that contributes to enhanced cell viability or anti-apoptosis. Even though the specific role of creatine depletion on mitochondrial function was not investigated, we previously showed that Lyn knockdown in Myl-R cells reduced mitochondrial membrane potential and increased caspase-3/7 activity27. Though outside the scope of this study, our data is consistent with other studies that have shown the creatine/phosphocreatine system is protective against apoptosis by regulating mitochondrial oxidative phosphorylation54. Additionally, the creatine/phosphocreatine system acts as an ATP buffering system where excess ATP in the cell is abstracted by creatine (Cr) to produce pools of phosphocreatine (PCr), a high-energy reservoir used by the cell when needed. Thus, the system functions to ensure energy/ATP homeostasis in the cell55,56. We did not, however, observe a significant shift in imatinib response when Myl-R cells were treated with combinations of imatinib and 3-GPA, suggesting that creatine depletion in the presence of inhibitor of apoptosis proteins (IAPs) was not sufficient to drive the Myl-R cells into apoptosis (data not shown). This may possibly be due to the fact that Myl-R cells express a high amount of BIRC6, which inhibits caspase-dependent cell death27.
To determine their effects on creatine uptake, Myl-R cells were also treated with dasatinib and bryostatin 1, therapeutic agents also used in the treatment of CML57,58. Bryostatin is a regulator of protein kinase C (PKC), and both PKC and protein kinase A (PKA) have been reported to phosphorylate the α-subunit of the Na+/K+-ATPase on serine and threonine residues. However, phosphorylation on these residues probably does not account for the basal phosphorylation level of this protein in Myl-R cells42. Treatment of Myl-R cells with bryostatin increased the total intracellular creatine levels compared to the DMSO control (Supplementary Fig. 2A). Conversely, data from dasatinib and ouabain treatments indicate that both agents inhibited creatine uptake by ~65% and ~95% respectively (Supplementary Fig. 2B). Given that dasatinib is a Lyn inhibitor and ouabain inhibits the Na+/K+-ATPase pump, our data suggest that Lyn can mediate creatine regulation in Myl-R cells through regulation of the Na+/K+-ATPase pump. Taken together, these data suggest that targeting Lyn or other kinases that regulate the Na+/K+-ATPase may have therapeutic potential in treating drug-resistant CML or other cancers.
Supplementary Material
Supplementary Figure 2. Lyn and Na+/K+-ATPase inhibitors suppress creatine uptake in Myl-R cells. (A) Lyn and Na+/K+-ATPase inhibitors reduced total creatine pool in Myl-R cells. My-R cells were treated overnight with 1 nM dasatinib or 0.1% DMSO or 10 nM bryostatin 1 or 100 nM ouabain and total intracellular creatine examined by 1H NMR. Creatine quantification was performed as described in Materials and Methods. Inhibition of Lyn or Na+/K+-ATPase with dasatinib or ouabain substantially reduced total intracellular creatine. Conversely, treatment of Myl-R cells with bryostatin 1 increased total intracellular creatine levels above those recorded for DMSO. (B) Percent inhibition of intracellular creatine was calculated from the values obtained in (A), with ouabain registering ~95% inhibition.
Supplementary Figure 3. Lyn mediates the phosphorylation and activation of the Na+/K+-ATPase pump. Transfection of constitutively active Lyn (Y508F) into HEK293 cells increased phospho-Na+/K+-ATPase α1 (pY10) levels. Phospho-STAT5A (pY694) levels were measured as an indication of Lyn activity and were similarly increased. No change in Na+/K+-ATPase, SLC6A8 or STAT5A protein levels were observed under these conditions. Arrowhead represents cross reactivity with an off-target protein of unknown origin. HEK293 cells were transiently transfected with wild type or constitutively active (Y508F) or kinase dead (K275R) Lyn DNA as described in Materials and Methods. Na+/K+-ATPase, STAT5A and Lyn activities, together with SLC6A8, STAT5A and Na+/K+-ATPase protein levels were measured by immunoblotting.
Supplementary Figure 4. Competitive inhibitors of creatine transport reduce creatine levels in Myl-R cells. Treatment of Myl-R cells with 3-Guanidinopropionic acid (3-GPA) reduced total intracellular creatine pool ten-fold, comparable to untreated Myl cells. Myl-R cells were treated for 24 hours with 3-GPA (30 mM), and total intracellular creatine pool determined using 1H NMR as outlined in Materials and Methods. Untreated Myl and Myl-R cells were similarly analyzed for comparison.
Supplementary Figure 1. Quantification of intracellular creatine in Myl (A) and Myl-R (B) cells. 1H NMR analysis showed that intracellular creatine was significantly higher in Myl-R compared to Myl cells 29. Creatine concentrations from the 1H NMR processed spectra were determined using Chenomx software and calculated as nmol/106 cells. Two-tailed Student’s t-test was used to test for significance (p < 0.05) in the difference in total intracellular creatine between Myl and Myl-R cells 29.
ACKNOWLEDGMENTS
We gratefully acknowledge R01 GM075941 and NIEHS T32 EX007126 to J.M.M. and L.M.G. D.O.O., L.J.A.-C., and I.M.M were supported by NIH 5 T32GM007040. E.H. was supported by a grant from the Russian Science Foundation (19-75-20145). We thank the laboratory of Dr. Andrew Ghio (US EPA, Chapel Hill, NC) for assistance with the rubidium uptake assays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Rowley JD Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 243, 290–293 (1973). [DOI] [PubMed] [Google Scholar]
- 2.Melo JV, Gordon DE, Cross NC & Goldman JM The ABL-BCR fusion gene is expressed in chronic myeloid leukemia. Blood 81, 158–165 (1993). [PubMed] [Google Scholar]
- 3.Daley GQ, Van Etten RA & Baltimore D Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 247, 824–830 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Evans CA, Owen-Lynch PJ, Whetton AD & Dive C Activation of the Abelson tyrosine kinase activity is associated with suppression of apoptosis in hemopoietic cells. Cancer Res. 53, 1735–1738 (1993). [PubMed] [Google Scholar]
- 5.Deininger MW, Goldman JM, Lydon N & Melo JV The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood 90, 3691–3698 (1997). [PubMed] [Google Scholar]
- 6.Druker BJ et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med 2, 561–566 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Apperley JF Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 8, 1018–1029 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Mahon FX et al. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood 96, 1070–1079 (2000). [PubMed] [Google Scholar]
- 9.Campbell LJ et al. BCR/ABL amplification in chronic myelocytic leukemia blast crisis following imatinib mesylate administration. Cancer Genet. Cytogenet 139, 30–33 (2002). [DOI] [PubMed] [Google Scholar]
- 10.le Coutre P et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood 95, 1758–1766 (2000). [PubMed] [Google Scholar]
- 11.Hochhaus A et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 16, 2190–2196 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Ito T, Tanaka H & Kimura A Establishment and characterization of a novel imatinib-sensitive chronic myeloid leukemia cell line MYL, and an imatinib- resistant subline MYL- R showing overexpression of Lyn. European journal of haematology 78, 417–431 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Gorre ME et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Shah NP et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Klawitter J et al. Metabolic characteristics of imatinib resistance in chronic myeloid leukaemia cells. Br. J. Pharmacol 158, 588–600 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bentley J et al. Glucose transport regulation by p210 Bcr-Abl in a chronic myeloid leukaemia model. Br J Haematol 112, 212–215 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Boren J et al. Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J. Biol. Chem 276, 37747–37753 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Vitiello GA et al. Mitochondrial Inhibition Augments the Efficacy of Imatinib by Resetting the Metabolic Phenotype of Gastrointestinal Stromal Tumor. Clin. Cancer Res 24, 972–984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kluza J et al. Exploiting mitochondrial dysfunction for effective elimination of imatinib-resistant leukemic cells. PLoS ONE 6, e21924 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klawitter J et al. Time-dependent effects of imatinib in human leukaemia cells: a kinetic NMR-profiling study. Br J Cancer 100, 923–931 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottschalk S, Anderson N, Hainz C, Eckhardt SG & Serkova NJ Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin. Cancer Res 10, 6661–6668 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Barnes K et al. Chronic myeloid leukaemia: an investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene 24, 3257–3267 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Kominsky DJ et al. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin. Cancer Res 15, 3442–3450 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Donato NJ et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood 101, 690–698 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Dai Y, Rahmani M, Corey SJ, Dent P & Grant S A Bcr/Abl-independent, Lyn-dependent form of imatinib mesylate (STI-571) resistance is associated with altered expression of Bcl-2. J. Biol. Chem 279, 34227–34239 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Ptasznik A, Nakata Y, Kalota A, Emerson SG & Gewirtz AM Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat. Med 10, 1187–1189 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Okumu DO et al. BIRC6 mediates imatinib resistance independently of Mcl-1. PLoS ONE 12, e0177871 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmerman EI et al. Lyn kinase-dependent regulation of miR181 and myeloid cell leukemia-1 expression: implications for drug resistance in myelogenous leukemia. Molecular Pharmacology 78, 811–817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dewar BJ et al. Metabolic assessment of a novel chronic myelogenous leukemic cell line and an imatinib resistant subline by H NMR spectroscopy. Metabolomics 6, 439–450 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper MJ et al. Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS ONE 8, e66755 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maril N, Degani H, Rushkin E, Sherry AD & Cohn M Kinetics of cyclocreatine and Na(+) cotransport in human breast cancer cells: mechanism of activity. Am. J. Physiol 277, C708–16 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Wyss M & Kaddurah-Daouk R Creatine and creatinine metabolism. Physiol. Rev 80, 1107–1213 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Loo JM et al. Extracellular metabolic energetics can promote cancer progression. Cell 160, 393–406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lillie JW et al. Cyclocreatine (1-carboxymethyl-2-iminoimidazolidine) inhibits growth of a broad spectrum of cancer cells derived from solid tumors. Cancer Res. 53, 3172–3178 (1993). [PubMed] [Google Scholar]
- 35.Loike JD, Somes M & Silverstein SC Creatine uptake, metabolism, and efflux in human monocytes and macrophages. Am. J. Physiol 251, C128–35 (1986). [DOI] [PubMed] [Google Scholar]
- 36.Gregor P, Nash SR, Caron MG, Seldin MF & Warren ST Assignment of the creatine transporter gene (SLC6A8) to human chromosome Xq28 telomeric to G6PD. Genomics 25, 332–333 (1995). [DOI] [PubMed] [Google Scholar]
- 37.Sora I et al. The cloning and expression of a human creatine transporter. Biochem. Biophys. Res. Commun 204, 419–427 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Bröer S & Gether U The solute carrier 6 family of transporters. Br. J. Pharmacol 167, 256–278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandtner W et al. Ouabain binding site in a functioning Na+/K+ ATPase. Journal of Biological Chemistry 286, 38177–38183 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y et al. Oxidized LDL-bound CD36 recruits an Na+ /K+ -ATPase-Lyn complex in macrophages that promotes atherosclerosis. Sci Signal 8, ra91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bozulic LD, Dean WL & Delamere NA The influence of SRC-family tyrosine kinases on Na,K-ATPase activity in lens epithelium. Investigative Ophthalmology & Visual Science 46, 618–622 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Wang XQ & Yu SP Novel regulation of Na, K-ATPase by Src tyrosine kinases in cortical neurons. J. Neurochem 93, 1515–1523 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Wu P, Nielsen TE & Clausen MH Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov. Today 21, 5–10 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Féraille E et al. Insulin-induced stimulation of Na+,K(+)-ATPase activity in kidney proximal tubule cells depends on phosphorylation of the alpha-subunit at Tyr-10. Mol. Biol. Cell 10, 2847–2859 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oudman I, Clark JF & Brewster LM The effect of the creatine analogue beta-guanidinopropionic acid on energy metabolism: a systematic review. PLoS ONE 8, e52879 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donato NJ et al. Imatinib mesylate resistance through BCR-ABL independence in chronic myelogenous leukemia. Cancer Res. 64, 672–677 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Krulikas LJ et al. Application of Integrated Drug Screening/Kinome Analysis to Identify Inhibitors of Gemcitabine-Resistant Pancreatic Cancer Cell Growth. SLAS Discov 23, 850–861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beloueche-Babari M et al. Acquired resistance to EGFR tyrosine kinase inhibitors alters the metabolism of human head and neck squamous carcinoma cells and xenograft tumours. Br J Cancer 112, 1206–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jain M et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang WC et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 148, 259–272 (2012). [DOI] [PubMed] [Google Scholar]
- 51.More TH et al. Metabolomic alterations in invasive ductal carcinoma of breast: A comprehensive metabolomic study using tissue and serum samples. Oncotarget 9, 2678–2696 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan KWY et al. CEST-MRI detects metabolite levels altered by breast cancer cell aggressiveness and chemotherapy response. NMR Biomed 29, 806–816 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tokarska-Schlattner M et al. Phosphocreatine interacts with phospholipids, affects membrane properties and exerts membrane-protective effects. PLoS ONE 7, e43178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun Z et al. Phosphocreatine protects against LPS-induced human umbilical vein endothelial cell apoptosis by regulating mitochondrial oxidative phosphorylation. Apoptosis 21, 283–297 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Yan Y-B Creatine kinase in cell cycle regulation and cancer. Amino Acids 48, 1775–1784 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Wallimann T The extended, dynamic mitochondrial reticulum in skeletal muscle and the creatine kinase (CK)/phosphocreatine (PCr) shuttle are working hand in hand for optimal energy provision. J. Muscle Res. Cell. Motil 36, 297–300 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Bradner JE A pulse at the heart of targeted therapy. Nature chemical biology 5, 144–145 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Jørgensen HG et al. Enhanced CML stem cell elimination in vitro by bryostatin priming with imatinib mesylate. Experimental Hematology 33, 1140–1146 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 2. Lyn and Na+/K+-ATPase inhibitors suppress creatine uptake in Myl-R cells. (A) Lyn and Na+/K+-ATPase inhibitors reduced total creatine pool in Myl-R cells. My-R cells were treated overnight with 1 nM dasatinib or 0.1% DMSO or 10 nM bryostatin 1 or 100 nM ouabain and total intracellular creatine examined by 1H NMR. Creatine quantification was performed as described in Materials and Methods. Inhibition of Lyn or Na+/K+-ATPase with dasatinib or ouabain substantially reduced total intracellular creatine. Conversely, treatment of Myl-R cells with bryostatin 1 increased total intracellular creatine levels above those recorded for DMSO. (B) Percent inhibition of intracellular creatine was calculated from the values obtained in (A), with ouabain registering ~95% inhibition.
Supplementary Figure 3. Lyn mediates the phosphorylation and activation of the Na+/K+-ATPase pump. Transfection of constitutively active Lyn (Y508F) into HEK293 cells increased phospho-Na+/K+-ATPase α1 (pY10) levels. Phospho-STAT5A (pY694) levels were measured as an indication of Lyn activity and were similarly increased. No change in Na+/K+-ATPase, SLC6A8 or STAT5A protein levels were observed under these conditions. Arrowhead represents cross reactivity with an off-target protein of unknown origin. HEK293 cells were transiently transfected with wild type or constitutively active (Y508F) or kinase dead (K275R) Lyn DNA as described in Materials and Methods. Na+/K+-ATPase, STAT5A and Lyn activities, together with SLC6A8, STAT5A and Na+/K+-ATPase protein levels were measured by immunoblotting.
Supplementary Figure 4. Competitive inhibitors of creatine transport reduce creatine levels in Myl-R cells. Treatment of Myl-R cells with 3-Guanidinopropionic acid (3-GPA) reduced total intracellular creatine pool ten-fold, comparable to untreated Myl cells. Myl-R cells were treated for 24 hours with 3-GPA (30 mM), and total intracellular creatine pool determined using 1H NMR as outlined in Materials and Methods. Untreated Myl and Myl-R cells were similarly analyzed for comparison.
Supplementary Figure 1. Quantification of intracellular creatine in Myl (A) and Myl-R (B) cells. 1H NMR analysis showed that intracellular creatine was significantly higher in Myl-R compared to Myl cells 29. Creatine concentrations from the 1H NMR processed spectra were determined using Chenomx software and calculated as nmol/106 cells. Two-tailed Student’s t-test was used to test for significance (p < 0.05) in the difference in total intracellular creatine between Myl and Myl-R cells 29.