

Abstract
Polymorphism in the apolipoprotein E (APOE) gene is a major genetic risk determinant of late-onset Alzheimer disease (AD), with the APOE*ε4 allele conferring an increased risk and the APOE*ε2 allele conferring a decreased risk, relative to the common APOE*ε3 allele. Strong evidence from clinical and basic research suggests that a major pathway by which APOE4 increases the risk of AD is by driving earlier and more abundant amyloid pathology in the brains of APOE*ε4 carriers. The list of amyloid-β (Aβ)-dependent and Aβ-independent pathways that are known to be differentially modulated by APOE isoforms is increasing. For example, evidence is accumulating that APOE influences tau pathology, tau-mediated neurodegeneration, and microglial responses to AD-related pathologies. In addition, APOE4 is either pathogenic or shows reduced efficiency in multiple brain homeostatic pathways, including lipid transport, synaptic integrity and plasticity, glucose metabolism, and cerebrovascular function. Here, we review the recent progress in clinical and basic research into the role of APOE in AD pathogenesis. We also discuss how APOE can be targeted for AD therapy using a precision medicine approach.
Introduction
Polymorphism in the apolipoprotein E (APOE) gene is a major risk determinant of late-onset Alzheimer disease (AD), the symptoms of which develop after the age of 65 years (Box 1)1. AD is the leading cause of dementia in elderly individuals2, and its pathological hallmarks include the deposition of extracellular amyloid-β (Aβ) aggregates as amyloid plaques and intracellular hyperphosphorylated tau aggregates as neurofibrillary tangles, along with neuronal loss and glial activation3. Given that individuals with late-onset AD account for more than 95% of the total AD population, efforts to elucidate the role of APOE, in particular its links to the pathological hallmarks of the disease, are relevant to the vast majority of patients with AD seeking new therapies.
Box 1: APOE polymorphism as a risk determinant for AD.
Numerous susceptibility genes (or loci) and coding variants associated with the risk of developing Alzheimer disease (AD) have been identified284,285. However, owing to its large effect size and high prevalence, the apolipoprotein E (APOE) polymorphism is considered to be the strongest genetic risk determinant for late-onset AD (Table 1). According to a meta-analysis, which included African American, white, Hispanic and Japanese individuals, the odds ratio (OR) for AD development in individuals with one allele of APOE2 is 0.621 (95% CI 0.456–0.85) compared with individuals homozygous for APOE*ε3. By contrast, the OR for AD development in individuals carrying one APOE*ε4 allele is 3.68 (95% CI 3.30–4.11) compared with individuals homozygous for APOE*ε3286. Importantly, APOE*ε4 both increases the risk of AD6,8,9 and lowers the age of disease onset4,10 in an allele number-dependent manner. A study that included >17,000 white individuals showed that the ORs for AD development were 2.64 and 3.63, respectively, in individuals with the APOE*ε2/ε4 and APOE*ε3/ε4 genotypes, in relation to those with the APOE*ε3/ε3 genotype9. The OR increased dramatically to 14.49 for individuals with the APOE*ε4/ε4 genotype (Table 2)9. In addition, compared with APOE*ε4 non-carriers, carrying one APOE*ε4 allele brings the onset of AD forward by 2–5 years4,10, and carrying two APOE*ε4 alleles brings onset forward by 5–10 years. The average allele frequency of APOE*ε4 in cognitively healthy individuals across African American, white, Hispanic and Japanese populations is 9–20%286; however, it is dramatically increased to ~40% among patients with AD (Table 1)6,10, further highlighting the strong association of APOE*ε4 with the risk of AD. A study published in 2018 showed that the prevalence of APOE*ε4 was 66% in individuals with biomarker-confirmed AD-type dementia287.
Ethnicity might influence the magnitude of the APOE*ε4-associated risk of developing AD. For example, when compared with white individuals6, the association of APOE*ε4 with AD risk is weaker among African American and Hispanic individuals, but is stronger in Japanese individuals. Understanding the mechanism by which traits such as ethnicity and sex (Box 3) influence the association of APOE*ε4 with AD risk might lead to the identification of modifiable factors that promote cognitive resilience in certain APOE*ε4 carriers.
Of the three major APOE allelic variants, ε2, ε3 and ε4, APOE*ε4 is associated with an increased risk4,5 and APOE*ε2 is associated with a decreased risk6,7 of AD relative to the common APOE*ε3 allele. Carrying one APOE*ε4 allele increases the risk of late-onset AD 3–4-fold, and carrying two alleles increases the risk 9–15-fold6,8,9. Among individuals with AD, APOE*ε4 is also associated with lower age of disease onset (Box 1)4,10. Mounting evidence suggests that the APOE4 protein isoform drives amyloid pathology and impairs multiple aspects of normal brain function, thereby increasing the risk of AD11 (Table 1). Studies have also shed light on the associations between APOE genotype and tau-mediated neurodegeneration12, the risk of dementia with Lewy bodies (DLB)13–15, Parkinson disease dementia (PDD)15–18 and the degree of TAR DNA-binding protein 43 (TDP43) pathology in the brains of individuals with AD19–21. In addition, advances in disease modelling and molecular profiling technology have provided insights into the role of APOE in AD pathogenesis at the molecular, cellular and organism level.
Table 1.
APOE variants and key AD-related clinical features
| APOE allelic variant | Allele frequency in cognitively healthy individuals (%)286 | Allele frequency in patients with AD (%)286 | Odds ratio for AD development286 | Key clinical features |
|---|---|---|---|---|
| APOE*ε2 | 7 | 4 | 0.621 |
|
| APOE*ε3 | 79 | 58 | 1.000 |
|
| APOE*ε4 | 14 | 38 | 3.680 |
|
AD, Alzheimer disease; APOE, apolipoprotein E.
In light of these findings, in this Review we describe the emerging link between APOE genotype and pathogenic proteins found in the brains of individuals with AD. We summarize recent progress in understanding the mechanisms by which different APOE isoforms contribute to or counteract AD pathogenesis via Aβ-dependent and Aβ-independent pathways. Finally, we discuss opportunities and strategies for developing APOE-targeted AD therapies in the future.
Biology of APOE
Human APOE is a 34 kDa glycoprotein22 that is composed of 299 amino acids after cleavage of the 18-amino-acid signal peptide. In the CNS, APOE is abundantly expressed in astrocytes, microglia, vascular mural cells and choroid plexus cells, and to a lesser extent in stressed neurons23,24. Once APOE has been secreted from the cells, the cell surface ATP-binding cassette transporters ABCA125 and ABCG126 transfer cholesterol and phospholipids to nascent APOE to form lipoprotein particles. The size and density of APOE lipoprotein particles in the cerebrospinal fluid (CSF) are similar to those of HDL27. APOE plays a critical role in redistributing cholesterol and other lipids to neurons through binding to cell-surface APOE receptors. The LDL receptor (LDLR) family members, including LDLR and LDLR-related protein 1 (LRP1), are major APOE receptors involved in APOE-mediated lipid metabolism. APOE has two main functional regions (FIG. 1): the receptor-binding region in the amino-terminal domain and the lipid-binding region in the carboxy-terminal domain28,29.
Figure 1: Structure of lipid-free and lipid-bound APOE3.
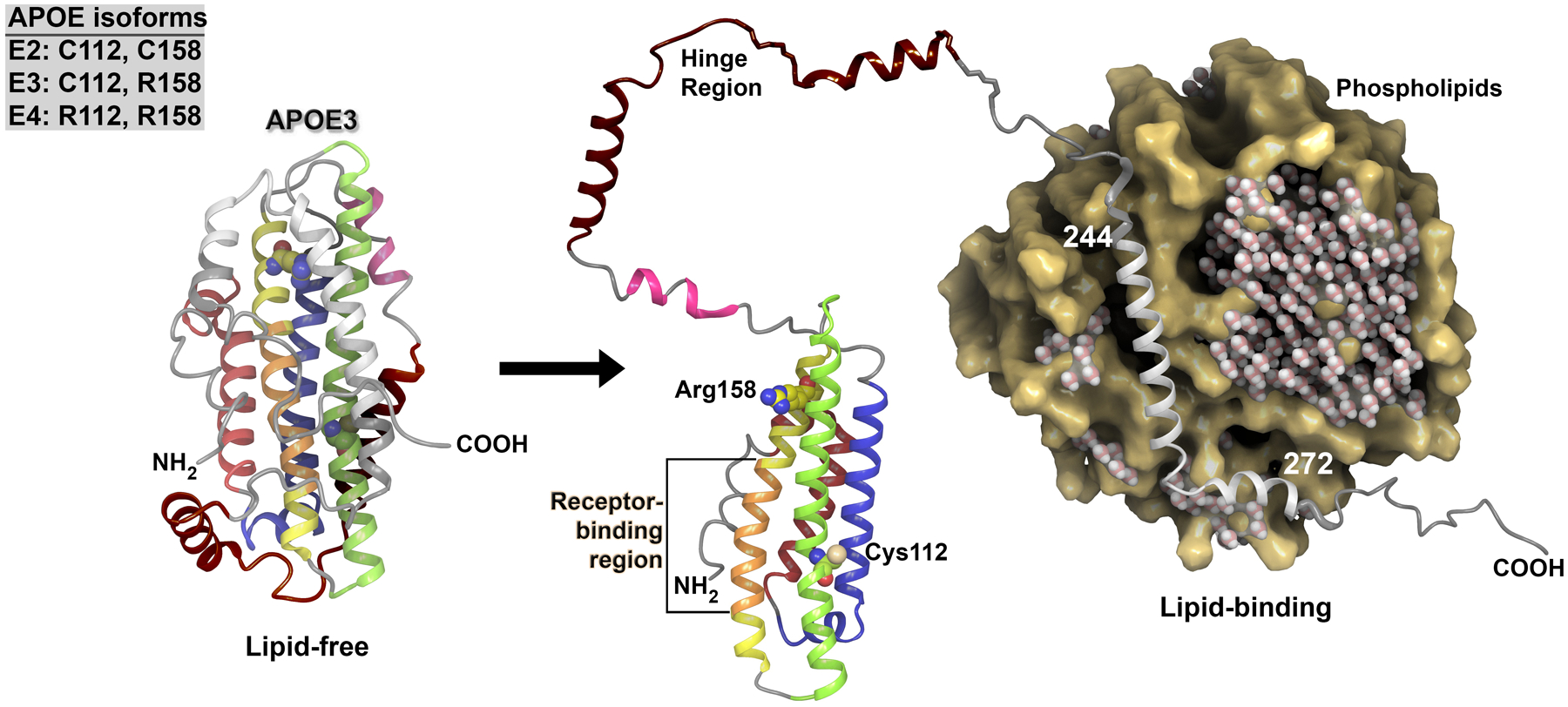
The structure of apolipoprotein E3 (APOE3) in the lipid-free state is shown on the left. APOE consists of multiple helices: a 4-helix bundle that consists of helix 2 (red), helix 3 (blue), helix 4 (green) and helix 5 (yellow and orange), hinge helices (pink and brown), and carboxyl-terminal domains that include lipid-binding residues and helices (translucent grey). The receptor-binding region (orange) is located on helix 5. The APOE isoforms differ at two residues only, and the residue composition for each isoform is given in the inset box. Isoform-specific residues (Arg158 and Cys112) are indicated on the structure of APOE3, with labels and carbons, shown as a van der Waals representation and coloured to match the helix with the residue. The lipid-bound structure of APOE3 is shown on the right. In its lipid-bound state, APOE3 demonstrates release of both the hinge region and the lipid-binding region, which causes the receptor binding region to be exposed. The crucial lipid-binding region includes residues 244–272. The lipid particle is shown in cross-section to allow better visualization of the APOE binding region. Water molecules depicted within the lipid particle illustrate the aqueous conditions and demonstrate the exclusion of water on binding of APOE3. The positively charged residues within the receptor-binding region in helix 5 (residues 136–150) interact with the negatively charged residues in the ligand-binding domains of LDL receptor family members; however, the precise binding structure of APOE with its receptors remains to be determined.
Computer-assisted modelling has been performed to examine the conformational changes in APOE that potentially occur on lipid binding. Structural modelling of human APOE3 was accomplished using data on APOE from the Protein Data Bank30–37. The modelling technique has been well documented38–44 and enables composite protein structures to be built from multiple structural templates. The resulting lipid-free and lipid-bound structures are shown in FIG. 145–48. The structures show that the lipid-binding region (residues 244–272) of APOE interacts directly with the lipid particle. Lipid binding to APOE increases the accessibility of the receptor-binding region (residues 136–150), thereby enabling cellular lipid delivery. The structural changes to APOE that are predicted to take place on lipid binding are consistent with those documented in the existing structural literature for this protein30,31,34,35,37,49–54.
The APOE isoforms encoded by the three corresponding gene alleles differ from one another only at positions 112 and 158 (APOE2: Cys112, Cys158; APOE3: Cys112, Arg158; APOE4: Arg112, Arg158). However, these single amino acid polymorphisms substantially alter the structure and function of APOE, thereby modulating its binding properties with regard to both lipids and receptors. For example, the binding of APOE2 to LDLR is more than 50 times weaker than the binding of APOE3 or APOE4 to this receptor55. In the periphery, the relatively low affinity of APOE2 for LDLR impairs the clearance of triglyceride-rich lipoprotein remnant particles and, as a consequence, APOE2 contributes to the onset of type III hyperlipoproteinaemia22,56. By contrast, enhanced binding of APOE4 to VLDL particles, as compared with the other APOE isoforms55, impairs lipolytic processing of VLDL in the periphery; thus, APOE4 is associated with pro-atherogenic changes in lipoprotein distribution56.
In addition to their role in lipid homeostasis in the periphery, APOE isoforms differentially modulate multiple pathways in the brain, including lipid transport, synaptic integrity and plasticity, glucose metabolism and cerebrovascular function. However, the correlation between the structure of individual APOE isoforms and their modulation of these pathways in the brain is less clear than for their actions on peripheral lipid metabolism. Possible mechanisms by which structural differences between APOE isoforms could modulate these brain pathways include differences in protein conformation, post-translational modification, lipoprotein preference and binding affinity for receptors29.
Studies have also shed light on the potential role of plasma APOE in brain homeostasis. Synaptic dysfunction in Apoe-knockout mice can be partially restored, and cognition improved, by genetic restoration of peripheral APOE57. These effects occur despite the presence of the blood–brain barrier (BBB), which blocks APOE influx from the periphery. The relative ratio of APOE4 to APOE3 in plasma positively correlates with the loss of regional grey matter volume and abnormal cerebral glucose metabolism in cognitively healthy APOE*ε3/ε4 carriers58, linking the isoform composition of plasma APOE to structural and metabolic changes of the brain. Therefore, plasma APOE is a potential determinant of brain structure and function.
In APOE*ε3/ε4 individuals, the APOE4:APOE3 ratio is >1 in the brain and CSF but <1 in the plasma59,60, suggesting that the metabolic pathways of APOE isoforms differ between the CNS and plasma. Thus, further studies exploring the similarities and differences in APOE metabolism between the CNS and periphery, and the functional crosstalk between brain and plasma APOE, would aid a better understanding of the impact of APOE isoforms on brain physiology.
APOE4 and Aβ pathology
Although evidence from clinical and pathological studies shows a link between APOE genotype and multiple proteinopathies, including those involving tau, α-synuclein and TDP43, the best-established association is between APOE genotype and Aβ. Pathological studies of post-mortem brain tissue from patients with AD have found that APOE*ε4 exacerbates the intra-neuronal accumulation of Aβ61, plaque deposition in the brain parenchyma62–65, formation of neurotoxic Aβ oligomers66 and the severity of cerebral amyloid angiopathy (CAA)67,68. By contrast, APOE*ε2 is associated with reduced numbers of neuritic plaques69 but somewhat increased risks of CAA and CAA-related intracranial haemorrhage70–73.
Imaging and CSF biomarker studies have shown that APOE*ε4 is consistently associated with greater Aβ deposition in the brains of cognitively healthy elderly individuals74–78, individuals with mild cognitive impairment (MCI)79 and individuals with AD80. A longitudinal study also showed that APOE*ε4 carriers exhibit increased Aβ deposition in the cortex compared with APOE*ε4 non-carriers81. Of note, APOE*ε4 is associated with an increased rate of longitudinal Aβ accumulation among cognitively healthy individuals who are amyloid-negative, whereas no differences in rates of Aβ deposition are observed among amyloid-positive individuals with different APOE genotypes82. Assuming that longitudinal Aβ accumulation represents an early phase of amyloid deposition in amyloid-negative individuals and a later phase in amyloid-positive individuals, these results suggest that the effect of APOE*ε4 on Aβ metabolism and aggregation is most pronounced during the initiation phase of Aβ deposition. By contrast, in cross-sectional studies, APOE*ε2 was found to be associated with reduced brain Aβ deposition in cognitively healthy elderly people77,83,84 and individuals with MCI85, particularly those without an APOE*ε4 allele. APOE*ε2 is also protective against longitudinal Aβ accumulation, especially among individuals without amyloid deposition82. Together, multiple layers of evidence indicate that the APOE genotype, in particular the presence of APOE*ε4, modulates amyloid pathology in the brain.
The effect of APOE4 in driving amyloid pathology is likely to outweigh the protective effect of APOE2 (FIG. 2, Table 2)85, as APOE*ε2/ε4 and APOE*ε3/ε4 individuals have comparable probabilities of exhibiting amyloid-β (Aβ) deposition on an amyloid-PET scan. In addition, among cognitively healthy individuals and people with MCI, the prevalence of amyloid pathology is higher in those with APOE*ε2/ε4 than in those with APOE*ε3/ε3. These findings indicate that APOE4 is a strong driver of Aβ deposition irrespective of the presence of APOE2 or APOE3. Importantly, the pattern of estimated probabilities of amyloid positivity associated with different APOE genotypes is similar to that of AD risk6,8,9 (Table 2), reinforcing the idea that APOE4 increases the risk of AD largely by causing earlier and more abundant amyloid pathology in the brain. Likewise, the presence of APOE*ε2 might reduce AD risk by decreasing amyloid pathology, especially in the absence of APOE*ε4. However, given that APOE*ε2 is associated not only with intact cognition but also, paradoxically, with increased AD pathology86 in the oldest old population, the protective effect of APOE*ε2 might also be mediated by Aβ-independent mechanisms. Supporting this notion, APOE*ε2 is associated with reduced cognitive decline during ageing even after adjustment for Aβ pathologies87.
Figure 2: APOE genotype and amyloid positivity.
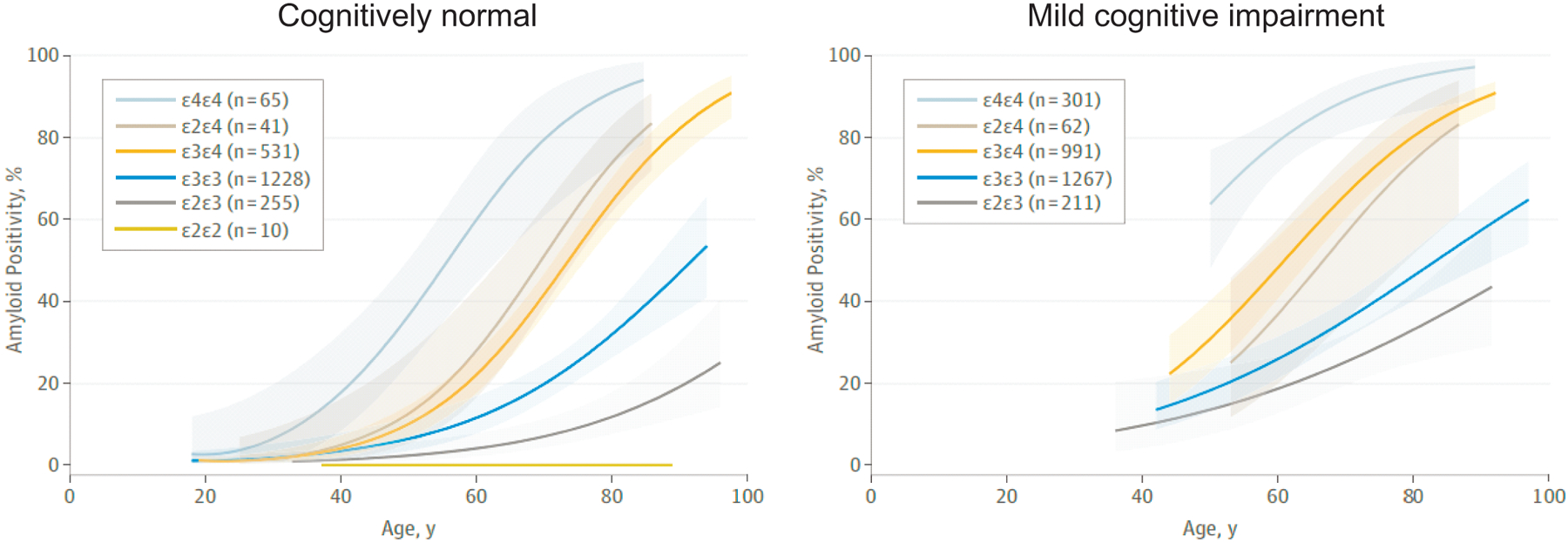
Estimated probabilities of amyloid positivity according to apolipoprotein E (APOE) genotype, plotted against age in cognitively healthy individuals and individuals with mild cognitive impairment (MCI). Shaded areas represent 95% CIs. In both groups, APOE*ε4/ε4 individuals are more likely to be positive for amyloid pathology than individuals with any other genotype, whereas APOE*ε2/ε2 individuals have the lowest probability of amyloid positivity. Also note that APOE*ε4 is a strong driver of Aβ positivity irrespective of the presence of APOE*ε2 or APOE*ε3. Data for APOE*ε2/ε2 individuals are not shown in the right panel owing to a small sample size. Adapted with permission from REF.85, American Medical Association.
Table 2.
APOE genotype, AD risk and amyloid-β deposition
| APOE genotype | Frequency in cognitively healthy individuals (%)286 | Frequency in patients with AD (%)286 | Odds ratio for AD development9 | Odds ratio for amyloid positivity at 70 years of age85 | |
|---|---|---|---|---|---|
| Cognitively healthy | Mild cognitive impairment | ||||
| ε2/ε2 | 0.7 | 0.3 | 0.56 | NA | NA |
| ε2/ε3 | 11.0 | 4.6 | 0.56 | 0.34 | 0.59 |
| ε3/ε3 | 62.3 | 34.3 | 1.00 | 1.00 | 1.00 |
| ε2/ε4 | 1.9 | 2.6 | 2.64 | 4.29 | 2.38 |
| ε3/ε4 | 22.2 | 43.4 | 3.63 | 2.94 | 3.52 |
| ε4/ε4 | 1.9 | 14.8 | 14.49 | 18.76 | 14.50 |
Carrying one or two apolipoprotein E ε4 (APOE*ε4) alleles increases the odds ratio for Alzheimer disease (AD) development 3–4-fold or 9–15-fold, respectively, regardless of the presence of APOE*ε2 or APOE*ε36,8,9. The pattern of estimated probabilities of amyloid positivity for the different APOE genotypes85 is similar to that of the risk of AD. NA, not available.
Aβ clearance.
As Aβ is continuously generated in the brain as a result of APP processing, efficient clearance is vital for preventing Aβ accumulation and subsequent aggregation88. The clearance of soluble Aβ from the brain interstitial fluid (ISF) occurs in an APOE isoform-dependent manner with APOE4 less efficient than APOE2 or APOE389. In addition, deficiency of ABCA1, which lipidates APOE, decreases Aβ clearance in APOE4-targeted replacement (TR) mice, in which murine Apoe is replaced by a specific allele of human APOE, whereas no such effect is observed in APOE3-TR mice90. Thus, the isoform and lipidation status of APOE is likely to influence the overall efficiency of Aβ clearance in the brain. Major pathways by which Aβ is removed from the brain include clearance through the BBB, cellular uptake and subsequent degradation, enzymatic degradation, clearance via ISF bulk flow, and CSF absorption into the circulatory and lymphatic systems88. The effects of individual APOE isoforms on each of these processes (FIG. 3) could contribute in a synergistic manner to variations in Aβ metabolism rates, in particular under pathogenic conditions.
Figure 3: APOE isoforms and Aβ aggregation and clearance.
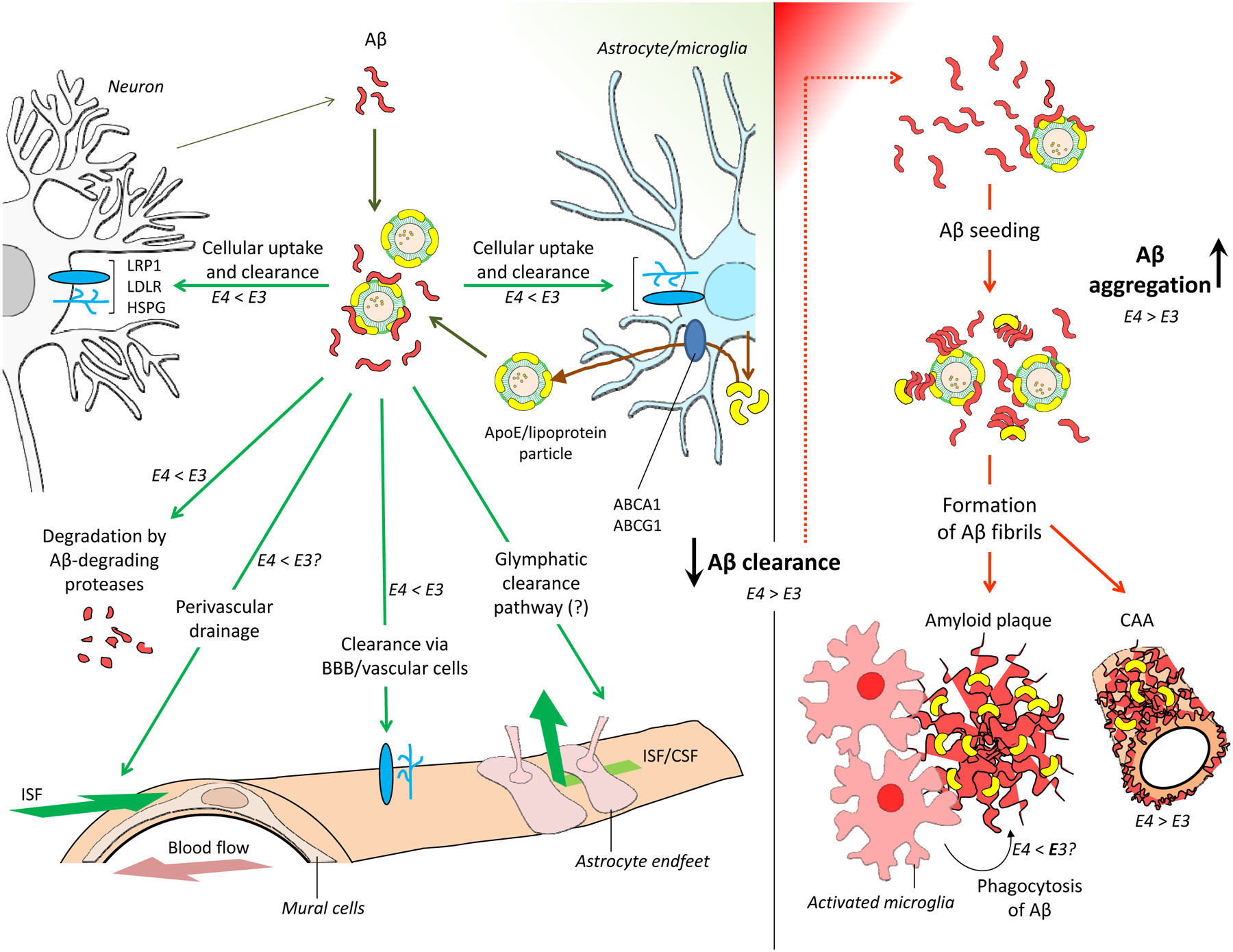
a | Amyloid-β (Aβ) production and clearance pathways. Aβ is produced primarily in neurons through proteolytic cleavage of amyloid precursor protein (grey arrow). Aβ is then removed from the brain by multiple Aβ clearance pathways (green boxes), including cellular uptake and subsequent degradation, enzymatic degradation, clearance through the blood–brain barrier (BBB), and clearance via interstitial fluid (ISF) bulk flow and, potentially, the glymphatic pathway. LDL receptor-related protein 1 (LRP1), LDL receptor (LDLR) and heparan sulfate proteoglycan (HSPG) are major APOE receptors that mediate cellular uptake of Aβ. Apolipoprotein E (APOE) is produced and lipidated primarily by astrocytes (brown arrow). A sub-pool of APOE lipoprotein particles interacts with soluble Aβ released into the brain interstitial fluid by neurons. b | Insufficient Aβ clearance from the brain leads to Aβ accumulation. This accumulation initiates Aβ oligomerization and accelerates subsequent aggregation and fibrillogenesis, leading to deposition of insoluble Aβ in the brain parenchyma (amyloid plaques) and in the vascular wall (cerebral amyloid angiopathy). APOE promotes the formation of Aβ fibrils by accelerating the initial seeding or nucleation of Aβ peptides. APOE can influence Aβ clearance and aggregation, either directly or indirectly, in an isoform-dependent manner. The relative abilities of APOE3 and APOE4 to promote each pathway are indicated alongside the arrows. ABCA1, ATP-binding cassette sub-family A member 1; ABCG1, ATP-binding cassette sub-family G member 1; CSF, cerebrospinal fluid.
The removal of Aβ via transport through the BBB is mediated by APOE in an isoform-dependent manner. An in vivo study showed that APOE2–Aβ and APOE3–Aβ complexes were cleared by both VLDLR and LRP1 at the BBB, whereas APOE4 binding to Aβ switched the clearance pathway of Aβ at the BBB from LRP1 to the VLDL receptor (VLDLR)91. As VLDLR mediates the internalization of APOE–Aβ complex at a slower rate than LRP1, redirection of the clearance pathway for the APOE4–Aβ complex might contribute to the slower clearance of Aβ at the BBB, when compared with APOE2–Aβ and APOE3–Aβ complexes91. Consistent with the findings in vivo, a study utilizing a tissue engineering approach to generate a 3D model of human brain blood vessels showed that APOE4 was less efficient than APOE2 at promoting Aβ transport across vessel walls92. Furthermore, an in vitro study showed that clearance of Aβ aggregates by BBB-associated pericytes was impaired in the presence of astrocyte-derived APOE4 compared with APOE393. The precise mechanisms by which APOE isoforms interact with Aβ receptors to mediate Aβ efflux from or influx into the brain require further investigation.
Cellular uptake and subsequent degradation of Aβ by glial cells represents a crucial Aβ clearance pathway in the brain. Human iPSC-derived astrocytes that are homozygous for APOE*ε4 show impaired uptake of Aβ1–42 in vitro compared with APOE*ε3-homozygous iPSCs94. A study using mouse brain slices indicated that astrocyte-mediated degradation of Aβ occurs through a mechanism that is dependent on both APOE and LRP1, suggesting that APOE is essential for receptor-mediated Aβ uptake by astrocytes95. However, another study showed that downregulation of LDLR reduced Aβ uptake, whereas upregulation of LDLR enhanced both the uptake and clearance of Aβ in astrocytes, independently of APOE96. A more recent study showed that the interaction between APOE and Aβ under physiological conditions is minimal, and that APOE might not be required for the cellular clearance of Aβ in astrocytes97. Instead, APOE competes with Aβ for the LRP1-dependent cellular uptake pathway, thereby blocking ~50% of the total Aβ cellular clearance by astrocytes in vitro97. Therefore, receptor-mediated internalization of Aβ in astrocytes might at least be partially affected by the presence of different APOE isoforms and APOE receptors, with APOE4 impairing the uptake of Aβ by these cells.
Cellular uptake and degradation of Aβ by microglia is also differentially influenced by the various APOE isoforms. Degradation of Aβ peptides by neprilysin within microglia is markedly facilitated in the presence of APOE, with APOE4 being less efficient at promoting the degradation of soluble Aβ than APOE2 and APOE398. In addition, APOE-mediated cholesterol efflux facilitates the delivery of Aβ to lysosomes and increases the efficiency of intracellular Aβ degradation in microglia99. The clearance of extracellular Aβ has been found to be impaired in human iPSC-derived microglia-like cells expressing APOE*ε4 compared with those expressing APOE*ε394.
Studies have also investigated the effects of APOE isoforms on other clearance pathways, including perivascular drainage and proteolytic degradation of Aβ by Aβ-degrading proteases (Aβ-DPs). A wide array of Aβ-DPs, including neprilysin and insulin degrading enzyme (IDE), determine Aβ levels in the brain100. Expression of neprilysin in both brain parenchyma and vasculature and that of IDE in hippocampus are significantly lower in post-mortem brains from individuals with at least one copy of APOE*ε4 than in those from individuals without APOE*ε4101,102. The perivascular drainage pathway has a major role in ISF bulk-flow clearance of Aβ103. In a study that examined this pathway, after intracerebral injections of Aβ1–40, aggregation of Aβ1–40 in the peri-arterial drainage pathway was observed in APOE4-TR mice but not in APOE3-TR mice104. Thus, the clearance of Aβ mediated by proteolytic degradation and perivascular drainage pathways seems to be impaired by the presence of APOE4.
The influence of APOE isoforms on the glymphatic clearance of Aβ105 is less clear. In one study, APOE derived from the choroid plexus and CSF was shown to be delivered into the brain via the glymphatic fluid transporting system in an APOE isoform-dependent manner106, although the relevance of these findings to disease needs to be further investigated.
Formation of Aβ fibrils.
Aβ aggregation occurs when Aβ is over-produced and/or inefficiently cleared. The formation of Aβ fibrils, a major component of amyloid plaques, follows three kinetic stages: a lag phase, a growth phase and a plateau phase107. Although the effects of APOE on the formation of Aβ fibrils are inconsistent in vitro29,108, in vivo studies have shown a crucial role for APOE in the initial seeding stage of Aβ deposition. Through a novel approach involving conditional expression of different APOE isoforms in a mouse model of amyloid pathology, astrocytic overexpression of APOE4, but not APOE3, during the initial Aβ seeding stage was found to exacerbate amyloid pathology. However, expression of APOE4 or APOE3 during the period of rapid plaque growth had no effect on Aβ deposition109. In another study published in parallel, reduction of APOE expression by antisense oligonucleotide (ASO) treatment beginning at postnatal day 0 (Aβ seeding stage) led to a significant reduction in amyloid pathology in amyloid mouse models that were homozygous for APOE*ε3 or APOE*ε4110. By contrast, ASO treatment beginning at the onset of amyloid deposition did not change the overall plaque load110. These in vivo results align with the notion that APOE, and in particular APOE4, promotes the formation of Aβ fibrils by accelerating the initial seeding or nucleation of Aβ peptides (FIG. 3).
Aβ production.
Given that the generation of Aβ peptide from amyloid precursor protein (APP) represents a pivotal event in AD pathogenesis111, researchers have sought to identify the role of APOE in this process. A study published in 2017 found that HEK293 cell-derived APOE induced APP transcription and Aβ production in human embryonic stem cell-derived and induced pluripotent stem cell (iPSC)-derived neurons. The effects were isoform-dependent, with APOE4 stimulating Aβ production more potently than other isoforms112. Aβ secretion is significantly higher in human iPSC-derived neurons carrying APOE*ε4 than in those with APOE*ε3, probably as a result of increased APP transcription or processing94,113. However, transcriptomic analysis of cortical tissue from APOE-TR mice did not reveal the transcriptional regulation of App associated with APOE4114. Furthermore, in mouse models of amyloid pathology that expressed human APOE, no APOE isoform-dependent differences in the amount of APP fragment C99 or β-secretase activity were observed, suggesting that amyloidogenic processing of APP does not vary in the presence of different APOE isoforms in vivo89. Species-dependent (human versus mouse) differences in APOE isoform-regulated Aβ production113 may have contributed to these conflicting results.
APOE in AD pathogenesis: beyond amyloid
APOE allele effects have also been implicated in proteinopathies characterized by tau, α-synuclein, and TDP43 pathology in the brains of individuals with AD. In addition, emerging evidence suggests that APOE can modulate tau-mediated neurodegeneration as well as microglial homeostasis, synaptic integrity, lipid transport, glucose metabolism and cerebrovascular function (FIG. 4). In this section, we discuss the disease relevance of these findings within the context of APOE genotype and its association with the risk of AD.
Figure 4: Effects of APOE4 on AD pathogenesis pathways.
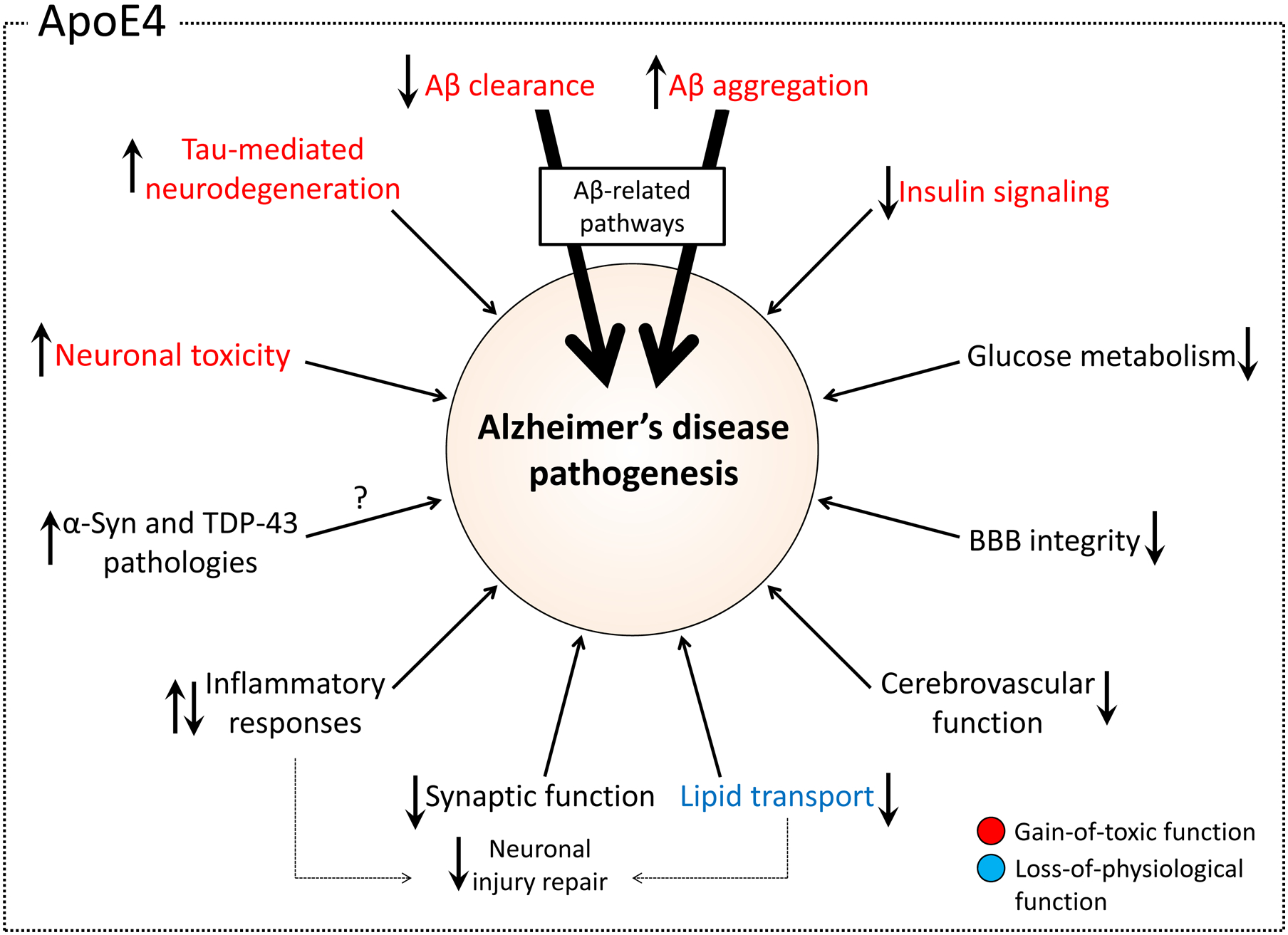
Apolipoprotein E4 (APOE4) affects multiple different pathways in Alzheimer disease (AD) pathogenesis. Key functional pathways are shown, and the arrows within the boxes depict the effects of APOE4 compared with APOE3. Pathways are shown in red boxes if evidence suggests that APOE4 increases risk of AD via a gain of toxic function. The effects on the lipid transport pathway, shown in blue, represent a potential loss of physiological function of APOE4 relative to APOE3. Pathways shown in grey are unclassified either owing to insufficient evidence or the potential for both gain of toxic and loss of physiological function. The two thicker arrows indicate the importance of the Aβ-related pathway as the key mechanism by which APOE influences AD. Aβ, amyloid-β; α-syn, α-synuclein; BBB, blood–brain barrier; TDP43, TAR DNA-binding protein 43.
Tau, α-synuclein and TDP43 proteinopathies.
Findings suggest that APOE alleles exert an effect on neurodegenerative diseases marked by the aggregation of tau, a microtubule-associated protein encoded by the MAPT gene115. Individuals with primary tauopathies, such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD)115,116, exhibit tau but not amyloid pathology. AD, though characterized by extensive tau pathology, is considered a secondary tauopathy as the tau pathology is accompanied by earlier development of amyloid pathology. Of note, postmortem brains from individuals carrying two APOE*ε4 alleles have more tau aggregates than those carrying either one or no APOE*ε4 alleles when Aβ pathology is present65, but no such association is observed in brains without Aβ117,118. Moreover, among patients with PSP, the presence of concomitant amyloid pathology is associated with an increased likelihood of carrying APOE*ε4119. Thus, APOE*ε4 is likely to be a risk factor for amyloid pathology but not necessarily for tau burden in primary tauopathies119,120. Interestingly, although APOE*ε2 is protective in the setting of AD, in the absence of amyloid pathology it has been implicated as a risk factor for tau-related neurodegeneration121. A study showed that APOE*ε2 was associated with increased risk and greater severity of tau pathology in patients with PSP122. Taken together, these results indicate that the effects of APOE alleles on tauopathies are influenced by the presence of amyloid pathology.
The pathological aggregation of tau strongly correlates with patterns of neurodegeneration and clinical manifestations in AD123–126. Studies have shown that neuron-specific overexpression of APOE4 increases tau phosphorylation in mice127. Subsequently, Shi et al. crossed the TauP301S transgenic mouse model, which overexpresses human tau containing the frontotemporal dementia-linked Pro301Ser mutation, with human APOE-TR mice. TauP301S–APOE4-TR mice developed markedly more brain atrophy and neuroinflammation than TauP301S–APOE2-TR and TauP301S–APOE3-TR mice at 9 months of age12. By contrast, Zhao et al. introduced tau with another frontotemporal dementia-linked mutation, Pro301Leu, to APOE-TR mice via viral delivery, and found more severe tau pathology and behavioural deficits at 6 months of age in TauP301L–APOE2-TR mice than in TauP301L–APOE3-TR and TauP301L–APOE4-TR mice122. These seemingly contradictory results are likely to be a result of differences in the experimental model systems that the two groups used. Furthermore, Shi et al. measured tau-mediated neurodegeneration, whereas Zhao et al. focused on tau pathology.
In addition to the findings from mouse models, human iPSC-derived APOE*ε4-expressing neurons have higher levels of tau phosphorylation than neurons expressing APOE*ε3113. Taken together, these results suggest that APOE influences tau pathology and tau-mediated neuronal toxicity in an isoform-dependent manner. Understanding the relevance of these findings to different neurodegenerative diseases with tauopathy as either a primary or concurrent pathology will require further studies using models that are more relevant to the specific disease conditions.
APOE*ε4 is also a genetic risk factor for DLB13–15 and PDD15–18. These conditions are both classified as synucleinopathies, a spectrum of neurodegenerative disorders that also includes PD128,129 and is characterized by the presence of hallmark accumulations of α-synuclein, termed Lewy bodies, within neuronal cell bodies130,131. Patients with DLB or PDD often have some degree of concomitant AD-type pathology132,133, and 30–40% of individuals with AD also present with Lewy bodies134. Therefore, determining whether APOE*ε4 contributes to α-synuclein pathology through an Aβ-dependent mechanism, similar to that proposed for tau pathology in the amyloid cascade hypothesis135, or through an Aβ-independent mechanism136 remains challenging. However, a study published in 2018 demonstrated that in individuals with Lewy body disease, APOE*ε4 was associated with increased α-synuclein pathology irrespective of the degree of AD pathology137, suggesting that APOE*ε4 impacts the severity of α-synuclein pathology independently of tau and Aβ. Of note, the gene encoding LDLR-related protein 10 (LRP10), which is thought to play a role in the metabolism of APOE lipoproteins138 and the trafficking of APP139, is associated with the development of inherited forms of DLB and PD140.
APOE*ε4 might also directly increase the risk of TDP43 pathology in the brains of people with AD. TDP43 proteinopathy is a core pathological hallmark of amyotrophic lateral sclerosis and of frontotemporal lobar degeneration with TDP43 pathology141, and often coexists with AD pathology. Of note, APOE*ε4 is associated with the presence of comorbid TDP43 proteinopathy in the brains of individuals with AD19, as well as the severity of TDP43 proteinopathy, even after adjusting for the presence of Aβ, tau and Lewy body pathologies21. These observations suggest that a direct association between APOE*ε4 and TDP43 proteinopathy exists in AD. A study published in 2018 confirmed that APOE*ε4 is associated with TDP43 pathology independently of Aβ in the brains of individuals with AD20.
In summary, APOE allele-specific effects on tau and α-synuclein proteinopathies, and on TDP43 pathology in the brains of individuals with AD, have been identified; however, the relevance of these findings to AD risk and disease progression is currently unclear. Further studies addressing the associations between APOE isoforms and these pathogenic proteins, using a model that reflects disease-specific conditions, should increase our understanding of APOE pathobiology within the context of AD.
Neuroinflammation.
Evidence suggests that APOE has an important role in regulating the innate immune response to amyloid pathology and neurodegeneration. Microglia play a central role in the immune response in the brain, highlighted by the abundant reactive microgliosis surrounding Aβ plaques in post-mortem brain tissue from individuals with AD142. These disease-associated microglia (DAM)143–145, also referred to as a microglial neurodegenerative (MGnD) phenotype146, have a conserved transcriptional signature across mouse models of AD. APOE is among these DAM-associated genes, as APOE expression by microglia is upregulated in association with ageing and amyloid and tau pathology14. By contrast, APOE deficiency diminishes the DAM signature in AD mouse models146,147, highlighting the crucial role of APOE in regulating DAM phenotypes.
The various APOE isoforms seem to modulate microglial functions differently in AD pathogenesis148. Human iPSC-derived microglia-like cells carrying APOE*ε4 exhibit altered morphologies and reduced levels of Aβ phagocytosis when compared with those carrying APOE*ε394. APOE4 also increases microglial reactivity against Aβ plaques in a mouse model of amyloid pathology149. Furthermore, APOE4 boosts microglial proinflammatory activation and neurodegeneration in a tau transgenic mouse model12. One suggestion is that the specific lipid composition of the APOE4 lipoprotein affects lipid raft structures on microglial cell membranes to induce a stronger DAM phenotype, which exacerbates Aβ or tau pathogenesis and neurodegeneration150.
APOE is an endogenous ligand of triggering receptor expressed on myeloid cells 2 (TREM2)151–153, a cell surface receptor expressed exclusively by microglia in the brain. A rare coding variant of TREM2 is associated with increased risk of AD, with an effect size comparable to that of APOE*ε4154,155. Furthermore, TREM2 is likely to mediate the APOE-dependent molecular signatures in microglia146,150,156. As such, the APOE–TREM2 pathway might contribute to AD pathogenesis through the modulation of microglial homeostasis157,158.
A study published in 2019 demonstrated that amyloid plaque seeding was increased in the absence of functional TREM2, both in animal models and in individuals with AD159. Lack of TREM2 was also associated with a reduction in plaque-associated APOE. Of note, levels of plaque-associated APOE were substantially reduced on microglial depletion in a mouse model of amyloid pathology, indicating that microglia represent a prominent source of amyloid plaque-associated APOE. Thus, microglial APOE seems to be induced in a TREM2-dependent manner, suggesting an important association of APOE with TREM2 in the context of AD pathogenesis.
Synaptic function.
Increasing evidence indicates that the presence of APOE*ε4 is associated with increased synaptic degeneration160 and synaptic accumulation of Aβ oligomers66, along with reductions in dendritic density161, plasticity160 and numbers of glutamate receptors162, in the brains of individuals with AD. In addition, the levels of the synaptic markers synaptophysin, syntaxin 1 and postsynaptic density protein 95 (PSD95) are reduced in the brains of APOE*ε4 carriers, and levels of PSD95 are increased in the brains of APOE*ε2 carriers163. Consistent with findings from post-mortem human brains, reduced expression of synaptic proteins164,165, reduced dendritic spine density and length166, and impaired synaptic transmission167,168 have all been observed in APOE4-TR mice when compared with APOE3-TR mice. In addition, APOE4 reduces neuronal surface expression of the LDLR family member APOER2 (also known as LRP8), along with NMDA and AMPA receptors, by sequestering them in the intracellular endosomal compartments169, leading to suppression of reelin-mediated synaptic transmission. Therefore, the isoform-dependent effect of APOE on the intracellular trafficking of lipoprotein and glutamate receptors represents a potential mechanism by which APOE4 can impair synaptic plasticity170. Furthermore, hippocampal neurogenesis and hilar GABAergic neurotransmission are impaired in mice carrying APOE*ε4171–173.
Collectively, these results suggest that APOE4 is either less efficient at maintaining synaptic integrity and neurogenesis than other AOPE isoforms or has a toxic effect113 on these spine-related and synapse-related pathways. Given that synaptic loss strongly correlates with cognitive decline in AD, the impact of APOE4 on synaptic function and neurogenesis might combine with other APOE4-mediated pathways and AD pathology to contribute to the earlier onset of symptoms.
Lipid transport.
Several lipid transport-related properties of APOE have been shown to be isoform-dependent. Although in vitro studies using cell systems with limited relevance to human brain physiology such as HEK293 cells and immortalized mouse astrocytes generated inconsistent results174,175, a more recent study using iPSC-derived astrocytes showed that APOE4 is less lipidated than APOE3, which could potentially diminish the neurotrophic function of APOE4176. In addition, in the CSF, APOE particles are larger in APOE*ε2/ε3 individuals and smaller in both APOE*ε3/ε4 and APOE*ε4/ε4 individuals than in APOE*ε3/ε3 individuals177. Furthermore, cholesterol efflux capacity is impaired in the CSF of individuals who are homozygous for APOE*ε4178. The amount of poorly lipidated APOE in the CSF is higher in APOE*ε4 carriers than in APOE*ε4 non-carriers179. Overall, the APOE isoform effects on the APOE–lipid interaction support a loss-of-function effect of APOE*ε4, with APOE4 being less efficient than APOE2 or APOE3 at transporting lipids to neurons.
The overall lipid-transporting capacity of APOE could also be affected by the amount of this molecule within the brain180. In mice, the quantity of APOE in CSF181,182, ISF183 and brain parenchyma182 shows an isoform-dependent gradient, with APOE2 being the most abundant and APOE4 the least abundant of the three isoforms. By contrast, studies utilizing a mass spectrometry-based approach in young control individuals (aged 22–60 years, average 34.5 ± 10.3 years)60, cognitively healthy people (aged 43–80 years, average 61 years) and patients with AD (aged 60–94 years, average 78 years)59 found no isoform-dependent differences in APOE levels in the CSF. However, one of the studies found that in a cohort of Aβ-positive individuals composed of both cognitively healthy individuals and patients with MCI, APOE levels in the CSF were significantly lower in APOE*ε4 carriers than in non-carriers60. Thus, the presence of Aβ in APOE*ε4 carriers might further exacerbate APOE4-mediated loss of neuroprotective function by decreasing the amount of APOE4 available to transport lipids to neurons.
Glucose metabolism and insulin signalling.
Several studies have highlighted the need to consider the APOE*ε4 genotype when assessing the role of glucose metabolism and insulin signalling in AD. Cerebral glucose hypometabolism is an early biomarker of AD and exists in pre-symptomatic individuals long before the clinical onset of AD184–187. Altered levels of insulin, insulin receptors and insulin signalling are observed in the brain of individuals with AD compared with cognitively healthy individuals188–190, and epidemiological studies have confirmed that diabetes and midlife insulin resistance are risk factors for AD191,192. Interestingly, the APOE*ε4 genotype, independently of Aβ, contributes to age-related reductions in cerebral glucose metabolism and insulin signalling193–197. In the brain areas frequently affected in AD, APOE*ε4 carriers, both with and without dementia, have lower cerebral glucose metabolism than non-carriers195,197–203. Phase II clinical trials of an insulin nasal spray in individuals with AD have shown that the preventative effects of insulin on cognitive decline are modulated by APOE genotype status204–206. In the CSF, increased levels of insulin are associated with increased levels of total tau and phosphorylated tau among APOE*ε4 non-carriers207. Fasting plasma insulin levels and free fatty acid levels are also increased in APOE*ε4 non-carriers208.
Consistent with the clinical findings, preclinical studies using APOE-TR mice have also revealed that APOE isoforms differentially influence brain glucose metabolism and insulin signalling. Compared with APOE3-TR mice, APOE4-TR mice display downregulation of brain peroxisome proliferator-activated receptor γ (PPARγ) and PPARγ coactivator 1 (PGC1α) signalling, which is involved in the regulation of glucose uptake and metabolism209. Following treatment with a high-fat diet, APOE4-TR mice show more profound cognitive impairment, reduced cerebral blood volume, decreased glucose uptake, and impaired insulin signalling compared with APOE3-TR mice194,210, providing additional evidence that human APOE isoforms differentially modulate brain bioenergetic metabolism. Furthermore, APOE4-TR mice exhibit impaired brain insulin signalling and insulin resistance compared with APOE3-TR mice in an age-dependent manner194. Importantly, APOE4 interacts with insulin receptors and traps them in the endosomes, thus impairing insulin receptor trafficking and related signalling194. APOE receptors are also involved in brain glucose metabolism and insulin signalling. Conditional deletion of the gene encoding a major APOE receptor, LRP1, in mouse forebrain neurons leads to impaired brain insulin signalling and a reduced capacity to metabolize glucose211.
Cerebrovascular integrity and function.
APOE*ε4 is a risk factor for vascular cognitive impairment212,213, suggesting a possible link between this allele and neurovascular unit dysfunction. Imaging studies have shown that the presence of APOE*ε4 is associated with increased severity of white matter hyperintensities independently of AD diagnosis214,215. In cognitively healthy individuals, APOE*ε4 carriers show a higher CSF:plasma albumin quotient than APOE*ε4 non-carriers216, suggesting that BBB integrity is disrupted in the carriers. APOE*ε4 carrier status is also associated with accelerated pericyte degeneration and loss of BBB integrity in AD, as observed in post-mortem brain tissue217. Thus, multiple layers of clinical evidence suggest a link between APOE*ε4 and impaired cerebrovascular integrity and function, both in the presence and absence of AD pathology.
Consistent with these findings, reduced cerebral blood flow, vascular density and neurovascular coupling are observed in APOE4-TR mice compared with APOE2-TR and APOE3-TR mice218–220. Loss of BBB integrity has also been observed in APOE4-TR mice218,219, although conflicting results have been obtained221. Another study showed that cerebral hypoperfusion, white matter damage and cognitive impairment induced by bilateral carotid artery stenosis are more severe in APOE4-TR mice than in APOE3-TR mice220. Overall, these APOE isoform effects on cerebrovascular integrity and function suggest that APOE4 is less efficient than APOE2 or APOE3 at maintaining cerebrovascular homeostasis. Of note, analysis using data sets from the Alzheimer’s Disease Neuroimaging Initiative (ADNI)222, and a CSF biomarker study223 both indicated a role for vascular dysregulation in the early stages of the AD cascade. Thus, determining how the cerebrovascular effects of APOE4 drive AD pathogenesis could represent an important area for future studies.
The cerebrovascular contribution to cognitive decline and synaptic dysfunction probably combines with the accumulation of pathogenic proteins, such as Aβ224,225, in both a synergistic and an additive manner. Therefore, the role of cerebrovascular effects of APOE4 in AD pathogenesis needs to be investigated in the context of both Aβ-dependent and Aβ-independent mechanisms. Peripheral APOE4 might modulate cerebrovascular function by exerting direct actions on the endothelial cells226 that form the BBB, which exists as an interface between the CNS and the peripheral circulation. In addition, peripheral APOE4 could indirectly modulate the functions of brain endothelial cells and neurons through its effects on lipid metabolism, atherosclerosis and peripheral inflammation, perhaps acting synergistically with other vascular risk factors227.
Perhaps surprisingly, APOE*ε2 is associated with increased risk of CAA and CAA-related intracranial haemorrhage70–73 and severity of cerebral small vessel disease215,228 despite its protective effect against AD risk. However, the cerebrovascular effects of APOE2 remain relatively unexplored.
APOE-targeted therapies for AD
The current strategies for targeting of APOE to treat AD fall into three main categories: modulating APOE quantity and lipidation, targeting APOE structural properties and interactions with Aβ, and targeting APOE receptors.
Modulating APOE quantity and lipidation.
As discussed above, APOE has crucial roles in lipid transport and maintenance of synaptic homeostasis. Therefore, AD-related pathways might be modulated by increasing the quantity and/or degree of lipidation of APOE. In particular, administration of agonists of the liver X receptor (LXR) and the retinoid X receptor (RXR) has been shown to positively regulate transcription and secretion of APOE229, reduce Aβ deposition98,230–233 and restore cognitive function98,231,233,234 in mouse models of amyloid pathology. Oral administration of the RXR agonist bexarotene reduced Aβ deposition and improved cognitive function in amyloid mouse models235, including those that also expressed human APOE*ε3 and APOE*ε4236, and in APOE4-TR mice237. Bexarotene also seems to combat ageing-related loss of synaptic proteins in mice238. However, conflicting results with respect to the effects of bexarotene on amyloid pathology in mouse models have also been reported236,239–241. In addition, systemic adverse effects including liver toxicity are observed on bexarotene treatment in mice238,242.
Bexarotene treatment does not reduce amyloid burden in patients with AD243. Oral administration of bexarotene to healthy individuals induced a 25% increase in CSF APOE levels but had no effect on CSF Aβ metabolism244 and showed poor CNS penetration. The clinical utility of bexarotene as a drug for AD might also be hampered by systemic adverse effects, such as hypertriglyceridaemia243, which are mediated by the permissive action of the RXR–LXR heterodimer on target genes, leading to activation of specific metabolic pathways in the liver245. Therefore, the potential clinical application of bexarotene has major limitations. From the perspective of gaining insight into APOE-targeted therapeutic strategies, the extent to which these RXR and LXR treatment-related phenotypes are mediated by an increase in APOE lipidation remains unclear. High-throughput screening for APOE agonists using human astrocytes may help to identify APOE-inducing compounds with pharmacological actions that do not depend on the nuclear receptor–APOE axis246.
Studies utilizing virus-mediated gene transfer approaches have also provided insights into whether increasing the expression of APOE isoforms that are considered protective halts AD pathogenesis. Virus-mediated human APOE gene expression in mice has been shown to have APOE isoform-dependent effects on brain Aβ pathology247–249 and APOE lipidation250. Adeno-associated virus (AAV)-mediated expression of APOE*ε4 in a mouse model of amyloid pathology exacerbated synaptic loss and Aβ deposition, whereas expression of APOE*ε2 in the same model caused a reduction in brain Aβ levels249. Consistent with this finding, AAV-mediated delivery of APOE*ε2 reduced Aβ deposition in two different amyloid mouse models, crossed with APOE*ε4 knock-in mice248. Furthermore, overexpression of APOE*ε4 in APOE4-TR mice increased the levels of poorly lipidated APOE in the brain, whereas overexpression of APOE*ε2 in these mice enhanced APOE lipidation250. Therefore, increasing the expression of APOE*ε2, but not APOE*ε4, might be beneficial with regard to increasing APOE lipidation and reducing Aβ pathology.
The therapeutic potential of decreasing the expression levels of specific APOE isoforms in order to reduce Aβ deposition has also been investigated in mouse models. Decreasing APOE expression by APOE haploinsufficiency in an amyloid mouse model carrying APOE*ε3 or APOE*ε4 led to a reduction in Aβ deposition, which was independent of APOE isoform251,252. Furthermore, a study published in 2017 showed that reduction of APOE expression by ASOs significantly decreased Aβ pathology in an amyloid mouse model that was homozygous for APOE*ε3 or APOE*ε4110. Thus, decreasing the levels of APOE3 and APOE4, particularly during the initial Aβ seeding stage, may be beneficial.
Given that APOE4 is associated with reduced levels of lipid in the CNS179,253, which could contribute to the APOE4-mediated loss of neuroprotective function, the question of whether an increase in APOE lipidation can reduce Aβ pathology has been investigated. Deletion of the gene encoding ABCA1, which lipidates APOE in the CNS, resulted in a decrease in APOE lipidation and an increase in Aβ deposition in amyloid mouse models254,255. Conversely, overexpression of ABCA1 in an amyloid mouse model decreased Aβ deposition — an effect that was probably mediated by an increase in APOE lipidation256. In addition, an Abca1 hemizygous knockout exacerbated memory deficits and Aβ deposition in an amyloid mouse model carrying human APOE*ε4, but not in mice carrying APOE*ε390. The exacerbation of Aβ pathology in the mice carrying APOE*ε4 could have been attributable to an increase in the amount of poorly lipidated APOE4 in the absence of one allele of Abca1. Whether, and to what extent, increasing APOE4 lipidation is beneficial in the presence of Aβ pathology remains to be fully determined.
APOE-mediated plaque formation might also be targeted via APOE immunotherapy. Treatment with the anti-mouse APOE antibody HJ6.3 induced a significant reduction in Aβ deposition in a mouse model of amyloid pathology that expressed murine APOE257,258. In another study, Aβ deposition was reduced in an APOE*ε4-positive amyloid mouse model following treatment with an anti-human APOE antibody, HAE-4, which preferentially binds the non-lipidated, aggregated form of APOE259. By binding to aggregated APOE in Aβ plaques, HAE-4 is likely to induce activation of plaque phagocytosis by microglia, thereby reducing Aβ deposition. Importantly, peripheral administration of HJ6.3 or HAE-4 does not change the total amount of APOE in plasma and brain parenchyma257–259. Thus, the targeting of APOE that is specifically associated with amyloid plaques is an attractive approach that would have minimal impact on the physiological function of APOE.
Treatment with peptides that mimic the structural and biological properties of native APOE reduces Aβ deposition260,261, tau hyperphosphorylation262 and glial activation260–262 in mouse models of amyloid pathology. However, the effects of these peptides on Aβ deposition and other AD-related pathologies in the context of human APOE isoforms have not been fully determined.
Targeting APOE structural properties and Aβ interaction.
The pathological conformation of APOE4 is proposed to result from an interaction between its amino-terminal and carboxy-terminal domains263,264,265, and the use of small molecules to block this interaction has been explored in vitro264,266,267. The APOE4 structure corrector PH002, at a final concentration of 100 nM in the culture medium, was shown to decrease APOE4 fragmentation and reduce the effects of APOE4 on Aβ production, tau phosphorylation and GABAergic neuron degeneration in human iPSC-derived neurons113. Targeting the abnormal biophysical properties of APOE4 represents a potential therapeutic approach; however, the efficacy of this approach in vivo, in the context of human APOE isoforms and amyloid pathology, remains to be investigated.
Conversion of the APOE4 amino acid sequence into that of APOE3 or APOE2 seems to be a more straightforward approach to modulate the pathobiology of APOE4. Conversion of APOE*ε4 to APOE*ε3 by gene editing considerably alters cellular phenotypes94,113. Reductions in APOE fragmentation, Aβ production, tau phosphorylation and GABAergic neuron degeneration are observed in iPSC-derived neurons when APOE*ε4 is converted to APOE*ε3, suggesting that the detrimental effect of APOE4 could be abolished by gene editing113. Similarly, converting APOE*ε4 to APOE*ε3 attenuates several AD-related phenotypes in glial cells and organoids: this intervention enhances the ability of glial cells to endocytose extracellular Aβ and significantly reduces the amount of Aβ deposition in organoids after 6 months of culture94. Despite these promising in vitro findings, the in vivo feasibility and clinical translatability of this therapeutic concept remain to be determined.
Inhibition of the APOE–Aβ interaction by the synthetic peptide Aβ12–28P, which is homologous to the APOE binding site on the full-length Aβ molecule, reduced Aβ deposition268–270 and insoluble tau accumulation270 in AD mouse models, and intra-neuronal Aβ accumulation271 in primary hippocampal neurons. In addition, treatment with Aβ12–28P decreased brain Aβ accumulation, co-deposition of APOE within Aβ plaques and neuritic degeneration in amyloid mouse models with APOE2-TR and APOE4-TR backgrounds272. Therefore, blocking APOE-mediated facilitation of Aβ assembly and/or deposition with a synthetic peptide seems to be beneficial in reducing Aβ pathology, irrespective of APOE isoforms.
Targeting APOE receptors.
Given that clearance of Aβ in the brain is partially mediated by APOE receptors, including LDLR and LRP128,29,273, increasing the expression of these receptors is a potential therapeutic strategy to reduce Aβ pathology. In mouse models of amyloid pathology, Ldlr deficiency is associated with increased Aβ deposition274,275, whereas overexpression of Ldlr leads to enhanced Aβ clearance and decreased Aβ deposition276. Conditional knockout of Lrp1 in neurons277, astrocytes278 and vascular smooth muscle cells279 led to increased Aβ deposition in amyloid mouse models. Furthermore, treatment of animal models with compounds280 that have been clinically explored for AD therapy, including fluvastatin281, decreases Aβ deposition and/or enhances Aβ clearance, probably by increasing Lrp1 expression. Whether these APOE receptor-related effects are mediated by APOE remains to be elucidated.
Challenges and considerations.
As with any attempts to target proteins that have essential physiological functions, a major challenge for APOE-targeted therapy is to minimize potential adverse effects on APOE-dependent brain homeostasis and systemic physiology. One individual with an ablative homozygous APOE frameshift mutation did not exhibit substantial neurocognitive deficits at the age of 40 years282. However, the impact of APOE deficiency on brain physiology in the context of ageing and AD development has not been documented. Thus, it will be crucial to monitor the effects of modulating APOE levels not only on AD pathogenesis, but also on brain physiology at different stages of AD development. The potential impact of APOE-targeted therapy on peripheral lipid metabolism and related physiology should also be considered. For example, modulating the amount of APOE or the expression of APOE receptors in the periphery could increase the risk of hyperlipidaemia, atherosclerosis and cardiovascular events, owing to impaired lipoprotein metabolism. Although increasing APOE2 levels in the brain might be beneficial, long-term expression of APOE2 could increase the risk of CAA, CAA-related intracerebral haemorrhage70–73 and perhaps primary tauopathy122.
These adverse effects could be managed by optimizing the treatment strategy for each APOE genotype, taking into consideration the treatment duration, the therapeutic window, and the specific biochemical properties and in vivo distribution of the pathogenic forms of APOE. Furthermore, the identification of APOE downstream effectors219 that modulate AD pathogenesis could provide therapeutic options with a limited impact on APOE-related physiology. Finally, the development and integration of novel technologies, such as targeted in vivo gene editing and more efficient drug delivery methods, could accelerate the clinical translation of APOE-targeted therapeutic concepts that have been uncovered in basic studies.
Conclusions
Strong evidence suggests that human APOE isoforms modulate AD pathogenesis primarily through their differential effects on Aβ clearance and aggregation. However, APOE isoforms also differentially affect multiple pathways that are not necessarily dependent on Aβ, with APOE4 exhibiting either a gain of toxic function or a loss of physiological function. Associations between APOE isoforms and tau-mediated neurodegeneration, and the risk of multiple neurodegenerative proteinopathies found in the brains of individuals with AD, are also recognized. Furthermore, a growing body of evidence links APOE with TREM2, highlighting the crucial role of APOE in the innate immune response in the brain.
To elucidate the precise pathological mechanism by which the APOE polymorphism determines the risk of AD (Table 1), several important yet underappreciated aspects of APOE pathobiology need to be determined. These aspects include the structure–function relationships for the different APOE isoforms, the impact of peripheral APOE on brain neurobiology and pathobiology, Aβ-dependent and independent protective pathways activated by APOE2, similarities and differences in the role of APOE in late-onset and early-onset AD (Box 2), sex–APOE genotype interactions (Box 3), and the role of APOE in immune responses and cerebrovascular pathways. As APOE is expressed by multiple cell types in the brain and periphery, the specific contributions of cell-autonomous versus cell-non-autonomous effects of APOE at different stages of AD development will also need to be investigated, perhaps using conditional mouse models and human cerebral organoids283, combined with human studies. Finally, a precision medicine approach based on APOE genotype status could facilitate the development of different AD treatment strategies (Box 4, FIG. 5). Establishment of APOE-based therapeutics is a considerable challenge; however, the targeting of APOE and its pathogenic interconnections offers great promise for the prevention and/or treatment of AD.
Box 2: APOE polymorphism and early-onset AD.
The apolipoprotein E ε4 allele (APOE*ε4) is associated with an increased risk of early-onset Alzheimer disease (EOAD)9,288, the symptoms of which develop at ≤65 years of age. The presence of two APOE*ε4 alleles is sufficient to increase the risk of EOAD regardless of family history of dementia289. By contrast, carrying one allele of APOE*ε4 increases the risk of EOAD only in individuals with a positive family history of the disease289. The allele frequency of APOE*ε4 is higher in individuals with EOAD than in healthy controls290,291, further supporting an association of APOE*ε4 with the risk of EOAD. Furthermore, APOE*ε4 decreases the age of onset of EOAD in families carrying amyloid precursor protein (APP)292, presenilin 1 (PSEN1)293 or PSEN2294 mutations, whereas APOE*ε2 delays age of onset in those carrying PSEN1 mutations295. Overall, the impact of APOE polymorphism on the risk and age of onset of EOAD seems to be similar to that of APOE polymorphism on late-onset AD, at least with respect to the directionality of effect. However, the available evidence is insufficient to draw conclusions about the relative dose effects of APOE*ε4 and APOE*ε2 alleles in individuals with EOAD who have different genetic backgrounds. Of note, in individuals with autosomal dominant AD, family history and mutation type explain substantial portions of the observed variance in the age of symptom onset296, suggesting that the impact of APOE polymorphism on age of onset differs among subtypes of individuals with EOAD.
Box 3: Sex-specific association of APOE*ε4 with AD.
The apolipoprotein E ε4 allele (APOE*ε4) seems to interact with sex to modify the risk of Alzheimer disease (AD). Women carrying one or two copies of APOE*ε4 have a greater risk of developing AD than men with the same APOE genotype6. In addition, cognitively healthy women carrying APOE*ε4 are more likely to progress to mild cognitive impairment and Alzheimer disease (AD) than are men carrying APOE*ε4297. One study showed that among APOE*ε4 carriers aged 65–75 years, women had a greater risk of AD than did men8. A post hoc analysis of imaging data sets from cognitively healthy individuals who carried APOE*ε4 and were positive for amyloid-β (Aβ) deposition on an amyloid-PET scan suggested that age-related cognitive decline was faster in the women than in the men298. A stronger association between APOE*ε4 and elevated cerebrospinal fluid tau levels297,299,300 was observed in women than in men, particularly among Aβ-positive individuals299,300. Thus, the increased risk of AD in women carrying APOE*ε4 might be attributed to increased susceptibility to Aβ pathology, leading to accelerated neuronal damage. By contrast, no sex difference was observed in the association of APOE*ε4 with regional tau deposition in clinically healthy individuals301. Comprehensive studies investigating sex differences in the association of APOE*ε4 with AD-related biomarkers at different stages of AD development302 would help us to better understand the mechanisms by which APOE*ε4 confers an increased risk of AD in women.
Box 4: APOE genotype as a guide for precision medicine.
The aim of precision medicine is to tailor therapeutic interventions to an individual’s predisposition to disease, disease progression and response to therapy. A strategy in which medical interventions for Alzheimer disease (AD) are guided by apolipoprotein E (APOE) polymorphisms, within the framework of precision medicine, might hold promise. In cognitively healthy individuals, the presence of APOE*ε4 correlates with earlier and greater memory decline303–305 and progression to mild cognitive impairment or AD306. On the other hand, advanced education, active leisure activities and good vascular health are all likely to reduce the risk of APOE*ε4-mediated cognitive decline307. A sedentary lifestyle is associated with higher amyloid-β (Aβ) deposition in cognitively healthy individuals carrying APOE*ε4308. Thus, at least in the cognitively healthy population, APOE genotyping, when performed as part of the polygenic risk score analysis309,310 and/or combined with pathological biomarker status, might be helpful in assessing the potential risk of AD and age-related cognitive decline. The early detection of latent pathological processes is an integral part of precision medicine311 as it provides greater opportunities for effective intervention, including lifestyle changes. If the initiation of Aβ pathology is detected, different types of interventions could be considered depending on the individual’s APOE genotype (FIG. 5).
In several clinical studies, APOE*ε4 carriers and non-carriers responded differently to treatment for AD. In phase III trials of bapineuzumab, a humanized anti-Aβ monoclonal antibody, in individuals with mild-to-moderate AD, APOE*ε4 status was associated with differences in key biomarker outcomes including Aβ metabolism and tau pathology312. In APOE*ε4 carriers Aβ deposition detected by PET imaging remained unchanged in individuals treated with bapineuzumab (0.5 mg kg–1), whereas individuals receiving placebo showed increases in Aβ deposition over the course of 71 weeks. By contrast, in individuals not carrying APOE*ε4, no increase in Aβ deposition was observed in the placebo groups, and there was no difference between the placebo group and individuals receiving 0.5 mg/kg or 1.0 mg/kg bapinezumab. In addition, APOE genotype seems to influence the outcomes of treatment with intranasal insulin204–206,313,314 and cholinesterase inhibitors315 in individuals with AD. For example, APOE*ε4 non-carriers, but not APOE*ε4 carriers, show memory facilitation after intravenous insulin administration313. Acute administration of short-acting intranasal insulin improves verbal memory in APOE*ε4 non-carriers but not in APOE*ε4 carriers314. Interestingly, long-acting intranasal insulin treatment in patients with AD leads to memory improvement in APOE*ε4 carriers but worsening in APOE*ε4 non-carriers206. Further understanding the relationship between APOE genotype and treatment response will improve delivery of individualized AD treatment. As such, the APOE genotype could serve as a guide for precision medicine. Given its strong AD risk-determining effect, however, there are ethical considerations pertaining to human genomic-based medicine that need to be taken into consideration when testing the APOE genotype.
Figure 5: Model of precision medicine based on APOE genotype.
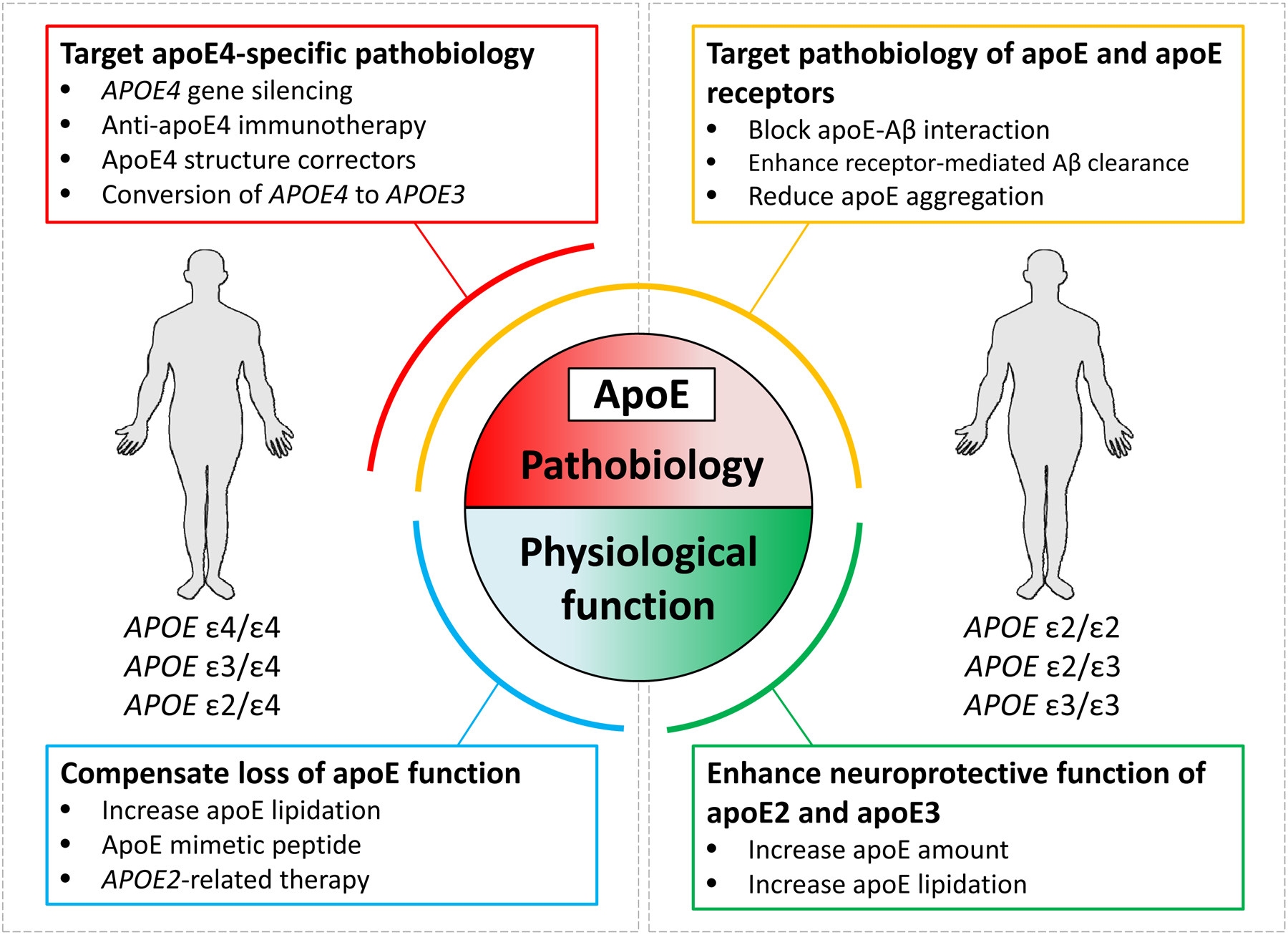
Although all apolipoprotein E (APOE) isoforms seem to promote amyloid-β (Aβ) deposition in the brain, the disease-driving effect is more pronounced with the presence of APOE*ε4 than of APOE*ε2 or APOE*ε3. Therefore, a precision medicine approach in which different treatment strategies are developed for individuals with different genotypes might be beneficial. The figure lists the potential therapeutic options according to the individuals who are most likely to benefit and whether the strategy attempts to correct APOE pathobiology or restore APOE physiological function. Modulation of Aβ pathology in individuals with different APOE genotypes is likely to require a combination of strategies.
Key points.
The apolipoprotein E (APOE) gene has three major allelic variants, ε2, ε3 and ε4; APOE*ε4 is associated with an increased risk and lower age of onset of Alzheimer disease (AD), whereas APOE*ε2 seems to confer protection against AD.
Increasing evidence suggests that the effect of APOE*ε4 on AD risk is exerted through inhibition of amyloid-β (Aβ) clearance and promotion of Aβ aggregation.
APOE influences tau pathology and tau-mediated neurodegeneration in an isoform-dependent manner, although the relevance of this observation to AD pathogenesis requires further investigation.
APOE4 also contributes to AD pathogenesis by impairing microglial responsiveness, lipid transport, synaptic integrity and plasticity, glucose metabolism, and cerebrovascular integrity and function; some of these effects are independent of Aβ-related pathways.
Current research into APOE-targeted AD therapeutic strategies aims to modulate APOE quantity and lipidation, APOE structural properties, APOE–Aβ interaction, and APOE receptor expression.
Acknowledgements
Support for work conducted in the authors’ laboratory was provided by the National Institutes of Health (NIH), the Cure Alzheimer’s Fund, the BrightFocus Foundation, the Alzheimer’s Association, the American Heart Association, the MetLife Foundation for Medical Awards Program, and the Mayo Foundation for Medical Education and Research. The authors also thank C. Stetler for critical reading and editing of the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lambert JC et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assoc A. s. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 14, 367–425, (2018). [Google Scholar]
- 3.Serrano-Pozo A, Frosch MP, Masliah E & Hyman BT Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med 1, a006189, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corder EH et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923, (1993). [DOI] [PubMed] [Google Scholar]
- 5.Saunders AM et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472, (1993). [DOI] [PubMed] [Google Scholar]
- 6.Farrer LA et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356, (1997). [PubMed] [Google Scholar]
- 7.Corder EH et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet 7, 180–184, (1994). [DOI] [PubMed] [Google Scholar]
- 8.Neu SC et al. Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease A Meta-analysis. JAMA Neurol. 74, 1178–1189, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genin E et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sando SB et al. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol. 8, 9, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu CC, Kanekiyo T, Xu H & Bu G Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol 9, 106–118, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi Y et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bras J et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum. Mol. Genet 23, 6139–6146, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerreiro R et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol. 17, 64–74, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuang D et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 70, 223–228, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, Chen P, Kaufer DI, Troster AI & Poole C Apolipoprotein E and dementia in Parkinson disease: a meta-analysis. Arch. Neurol 63, 189–193, (2006). [DOI] [PubMed] [Google Scholar]
- 17.Irwin DJ et al. Neuropathologic substrates of Parkinson disease dementia. Ann. Neurol 72, 587–598, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tropea TF et al. APOE, thought disorder, and SPARE-AD predict cognitive decline in established Parkinson’s disease. Mov. Disord 33, 289–297, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Josephs KA et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 127, 811–824, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wennberg AM et al. Association of Apolipoprotein E epsilon4 With Transactive Response DNA-Binding Protein 43. JAMA Neurol. 75, 1347–1354, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang H-S et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE ε4 haplotype status: a community-based cohort study. Lancet Neurol. 17, 773–781, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahley RW Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630, (1988). [DOI] [PubMed] [Google Scholar]
- 23.Xu Q et al. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci 26, 4985–4994, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang SS et al. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp Med 215, 2235–2245, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wahrle SE et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem 279, 40987–40993, (2004). [DOI] [PubMed] [Google Scholar]
- 26.Karten B, Campenot RB, Vance DE & Vance JE Expression of ABCG1, but not ABCA1, correlates with cholesterol release by cerebellar astroglia. J. Biol. Chem 281, 4049–4057, (2006). [DOI] [PubMed] [Google Scholar]
- 27.LaDu MJ et al. Nascent astrocyte particles differ from lipoproteins in CSF. J. Neurochem 70, 2070–2081, (1998). [DOI] [PubMed] [Google Scholar]
- 28.Bu G Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci 10, 333–344, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanekiyo T, Xu H & Bu G ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron 81, 740–754, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong J et al. Interaction of the N-terminal domain of apolipoprotein E4 with heparin. Biochemistry 40, 2826–2834, (2001). [DOI] [PubMed] [Google Scholar]
- 31.Segelke BW et al. Conformational flexibility in the apolipoprotein E amino-terminal domain structure determined from three new crystal forms: implications for lipid binding. Protein Sci. 9, 886–897, (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson C et al. Salt bridge relay triggers defective LDL receptor binding by a mutant apolipoprotein. Structure 2, 713–718, (1994). [DOI] [PubMed] [Google Scholar]
- 33.Dong LM et al. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J. Biol. Chem 269, 22358–22365, (1994). [PubMed] [Google Scholar]
- 34.Wilson C, Wardell MR, Weisgraber KH, Mahley RW & Agard DA Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 252, 1817–1822, (1991). [DOI] [PubMed] [Google Scholar]
- 35.Dong LM et al. Novel mechanism for defective receptor binding of apolipoprotein E2 in type III hyperlipoproteinemia. Nat. Struct. Biol 3, 718–722, (1996). [DOI] [PubMed] [Google Scholar]
- 36.Guttman M, Prieto JH, Handel TM, Domaille PJ & Komives EA Structure of the minimal interface between ApoE and LRP. J. Mol. Biol 398, 306–319, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, Li Q & Wang J Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl Acad. Sci. USA 108, 14813–14818, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richter JE et al. Protein molecular modeling shows residue T599 is critical to wild-type function of POLG and description of a novel variant associated with the SANDO phenotype. Hum. Genome Var 5, 18016, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puschmann A et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 140, 98–117, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kayode O et al. An Acrobatic Substrate Metamorphosis Reveals a Requirement for Substrate Conformational Dynamics in Trypsin Proteolysis. J. Biol. Chem 291, 26304–26319, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caulfield TR, Fiesel FC & Springer W Activation of the E3 ubiquitin ligase Parkin. Biochem. Soc. Trans 43, 269–274, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caulfield TR et al. Phosphorylation by PINK1 Releases the UBL Domain and Initializes the Conformational Opening of the E3 Ubiquitin Ligase Parkin. PLoS Comput. Biol 10, e1003935, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang YJ et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet 22, 3112–3122, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caulfield T & Devkota B Motion of transfer RNA from the A/T state into the A-site using docking and simulations. Proteins 80, 2489–2500, (2012). [DOI] [PubMed] [Google Scholar]
- 45.Altschul SF et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402, (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hooft RW, Sander C, Scharf M & Vriend G The PDBFINDER database: a summary of PDB, DSSP and HSSP information with added value. Comput. Appl. Biosci 12, 525–529, (1996). [DOI] [PubMed] [Google Scholar]
- 47.Hooft RW, Vriend G, Sander C & Abola EE Errors in protein structures. Nature 381, 272, (1996). [DOI] [PubMed] [Google Scholar]
- 48.Krieger E et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 77 Suppl 9, 114–122, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frieden C, Wang H & Ho CMW A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain-domain interactions. Proc. Natl Acad. Sci. USA 114, 6292–6297, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mondal T et al. ApoE: In Vitro Studies of a Small Molecule Effector. Biochemistry 55, 2613–2621, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frieden C ApoE: the role of conserved residues in defining function. Protein Sci. 24, 138–144, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garai K, Verghese PB, Baban B, Holtzman DM & Frieden C The binding of apolipoprotein E to oligomers and fibrils of amyloid-beta alters the kinetics of amyloid aggregation. Biochemistry 53, 6323–6331, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frieden C & Garai K Concerning the structure of apoE. Protein Sci. 22, 1820–1825, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ji ZS, Dichek HL, Miranda RD & Mahley RW Heparan sulfate proteoglycans participate in hepatic lipaseand apolipoprotein E-mediated binding and uptake of plasma lipoproteins, including high density lipoproteins. J. Biol. Chem 272, 31285–31292, (1997). [DOI] [PubMed] [Google Scholar]
- 55.Hatters DM, Peters-Libeu CA & Weisgraber KH Apolipoprotein E structure: insights into function. Trends Biochem. Sci 31, 445–454, (2006). [DOI] [PubMed] [Google Scholar]
- 56.Phillips MC Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 66, 616–623, (2014). [DOI] [PubMed] [Google Scholar]
- 57.Lane-Donovan C et al. Genetic Restoration of Plasma ApoE Improves Cognition and Partially Restores Synaptic Defects in ApoE-Deficient Mice. J. Neurosci 36, 10141–10150, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nielsen HM et al. Peripheral apoE isoform levels in cognitively normal APOE epsilon3/epsilon4 individuals are associated with regional gray matter volume and cerebral glucose metabolism. Alzheimers Res. Ther 9, 5, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martinez-Morillo E et al. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 127, 633–643, (2014). [DOI] [PubMed] [Google Scholar]
- 60.Baker-Nigh AT et al. Human Central Nervous System (CNS) ApoE Isoforms Are Increased by Age, Differentially Altered by Amyloidosis, and Relative Amounts Reversed in the CNS Compared with Plasma. J. Biol. Chem 291, 27204–27218, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Christensen DZ, Schneider-Axmann T, Lucassen PJ, Bayer TA & Wirths O Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol. 119, 555–566, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kok E et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol 65, 650–657, (2009). [DOI] [PubMed] [Google Scholar]
- 63.Polvikoski T et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N. Engl. J. Med 333, 1242–1247, (1995). [DOI] [PubMed] [Google Scholar]
- 64.Schmechel DE et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl Acad. Sci. USA 90, 9649–9653, (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tiraboschi P et al. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 62, 1977–1983, (2004). [DOI] [PubMed] [Google Scholar]
- 66.Koffie RM et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain 135, 2155–2168, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rannikmae K et al. APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J. Neurol. Neurosurg. Psychiatry 85, 300–305, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shinohara M et al. Impact of sex and APOE4 on cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. 132, 225–234, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Serrano-Pozo A, Qian J, Monsell SE, Betensky RA & Hyman BT APOEepsilon2 is associated with milder clinical and pathological Alzheimer disease. Ann. Neurol 77, 917–929, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nelson PT et al. APOE-epsilon2 and APOE-epsilon4 correlate with increased amyloid accumulation in cerebral vasculature. J. Neuropathol. Exp. Neurol 72, 708–715, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nicoll JA et al. High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann. Neurol 41, 716–721, (1997). [DOI] [PubMed] [Google Scholar]
- 72.Biffi A et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann. Neurol 68, 934–943, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Love S et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am. J. Neurodegener. Dis 3, 19–32, (2014). [PMC free article] [PubMed] [Google Scholar]
- 74.Gonneaud J et al. Relative effect of APOE epsilon4 on neuroimaging biomarker changes across the lifespan. Neurology 87, 1696–1703, (2016). [DOI] [PubMed] [Google Scholar]
- 75.Kantarci K et al. APOE modifies the association between Abeta load and cognition in cognitively normal older adults. Neurology 78, 232–240, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sunderland T et al. Cerebrospinal fluid beta-amyloid1–42 and tau in control subjects at risk for Alzheimer’s disease: the effect of APOE epsilon4 allele. Biol. Psychiatry 56, 670–676, (2004). [DOI] [PubMed] [Google Scholar]
- 77.Morris JC et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol 67, 122–131, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jack CR Jr. et al. Age, Sex, and APOE epsilon4 Effects on Memory, Brain Structure, and beta-Amyloid Across the Adult Life Span. JAMA Neurol. 72, 511–519, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murphy KR et al. Mapping the effects of ApoE4, age and cognitive status on 18F-florbetapir PET measured regional cortical patterns of beta-amyloid density and growth. Neuroimage 78, 474–480, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fleisher AS et al. Apolipoprotein E epsilon4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol. Aging 34, 1–12, (2013). [DOI] [PubMed] [Google Scholar]
- 81.Mishra S et al. Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE epsilon4 genotype. Brain 141, 1828–1839, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lim YY & Mormino EC APOE genotype and early beta-amyloid accumulation in older adults without dementia. Neurology 89, 1028–1034, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grothe MJ, Villeneuve S, Dyrba M, Bartres-Faz D & Wirth M Multimodal characterization of older APOE2 carriers reveals selective reduction of amyloid load. Neurology 88, 569–576, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chiang GC et al. Hippocampal atrophy rates and CSF biomarkers in elderly APOE2 normal subjects. Neurology 75, 1976–1981, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jansen WJ et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 313, 1924–1938, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Berlau DJ, Corrada MM, Head E & Kawas CH APOE epsilon2 is associated with intact cognition but increased Alzheimer pathology in the oldest old. Neurology 72, 829–834, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shinohara M et al. APOE2 Eases Cognitive Decline during Aging: Clinical and Preclinical Evaluations. Ann. Neurol 79, 758–774, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tarasoff-Conway JM et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol 11, 457–470, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Castellano JM et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med 3, 89ra57, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fitz NF et al. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J. Neurosci 32, 13125–13136, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deane R et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Invest 118, 4002–4013, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Robert J et al. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. Elife 6, e29595, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ma Q et al. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener 13, 57, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin YT et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98, 1294, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koistinaho M et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med 10, 719–726, (2004). [DOI] [PubMed] [Google Scholar]
- 96.Basak JM, Verghese PB, Yoon H, Kim J & Holtzman DM Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. J. Biol. Chem 287, 13959–13971, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Verghese PB et al. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc. Natl Acad. Sci. USA 110, E1807–1816, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jiang Q et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee CYD, Tse W, Smith JD & Landreth GE Apolipoprotein E Promotes beta-Amyloid Trafficking and Degradation by Modulating Microglial Cholesterol Levels. J. Biol. Chem 287, 2032–2044, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Saido T & Leissring MA Proteolytic degradation of amyloid beta-protein. Cold Spring Harb. Perspect. Med 2, a006379, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miners JS et al. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol 65, 1012–1021, (2006). [DOI] [PubMed] [Google Scholar]
- 102.Cook DG et al. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon4 allele. Am. J. Pathol 162, 313–319, (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weller RO, Subash M, Preston SD, Mazanti I & Carare RO Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hawkes CA et al. Disruption of arterial perivascular drainage of amyloid-beta from the brains of mice expressing the human APOE epsilon4 allele. PloS one 7, e41636, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Louveau A et al. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Invest 127, 3210–3219, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Achariyar TM et al. Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol. Neurodegener 11, 74, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Arosio P, Knowles TP & Linse S On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys 17, 7606–7618, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim J, Basak JM & Holtzman DM The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu CC et al. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 96, 1024–1032 e1023, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huynh TPV et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of beta-amyloidosis. Neuron 96, 1013-+, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.O’Brien RJ & Wong PC Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci 34, 185–204, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang YA, Zhou B, Wernig M & Sudhof TC ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 168, 427–441 e421, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang C et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med 24, 647–657, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nuriel T et al. The Endosomal-Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front. Neurosci 11, 702, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang Y & Mandelkow E Tau in physiology and pathology. Nat. Rev. Neurosci 17, 5–21, (2016). [DOI] [PubMed] [Google Scholar]
- 116.Leyns CEG & Holtzman DM Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener 12, 50, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Farfel JM, Yu L, De Jager PL, Schneider JA & Bennett DA Association of APOE with tau-tangle pathology with and without beta-amyloid. Neurobiol. Aging 37, 19–25, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Crary JF et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tsuboi Y, Josephs KA, Cookson N & Dickson DW APOE E4 is a determinant for Alzheimer type pathology in progressive supranuclear palsy. Neurology 60, 240–245, (2003). [DOI] [PubMed] [Google Scholar]
- 120.Tabaton M et al. Apolipoprotein E epsilon 4 allele frequency is not increased in progressive supranuclear palsy. Neurology 45, 1764–1765, (1995). [DOI] [PubMed] [Google Scholar]
- 121.Ikeda K et al. A subset of senile dementia with high incidence of the apolipoprotein E epsilon2 allele. Ann. Neurol 41, 693–695, (1997). [DOI] [PubMed] [Google Scholar]
- 122.Zhao N et al. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat. Commun 9, 4388, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ossenkoppele R et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139, 1551–1567, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brier MR et al. Tau and Abeta imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med 8, 338ra366, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Josephs KA et al. Beta-amyloid burden is not associated with rates of brain atrophy. Ann. Neurol 63, 204–212, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Arriagada PV, Growdon JH, Hedley-Whyte ET & Hyman BT Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639, (1992). [DOI] [PubMed] [Google Scholar]
- 127.Brecht WJ et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci 24, 2527–2534, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Federoff M, Jimenez-Rolando B, Nalls MA & Singleton AB A large study reveals no association between APOE and Parkinson’s disease. Neurobiol. Dis 46, 389–392, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Spatola M & Wider C Genetics of Parkinson’s disease: the yield. Parkinsonism Relat. Disord 20 Suppl 1, S35–38, (2014). [DOI] [PubMed] [Google Scholar]
- 130.Spillantini MG et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840, (1997). [DOI] [PubMed] [Google Scholar]
- 131.Lashuel HA, Overk CR, Oueslati A & Masliah E The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci 14, 38–48, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Apaydin H, Ahlskog JE, Parisi JE, Boeve BF & Dickson DW Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch. Neurol 59, 102–112, (2002). [DOI] [PubMed] [Google Scholar]
- 133.McKeith IG et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89, 88–100, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Olichney JM et al. Cognitive decline is faster in Lewy body variant than in Alzheimer’s disease. Neurology 51, 351–357, (1998). [DOI] [PubMed] [Google Scholar]
- 135.Selkoe DJ & Hardy J The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 8, 595–608, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gallardo G, Schluter OM & Sudhof TC A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat. Neurosci 11, 301–308, (2008). [DOI] [PubMed] [Google Scholar]
- 137.Dickson DW et al. APOE epsilon4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 91, e1182–e1195, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sugiyama T et al. A novel low-density lipoprotein receptor-related protein mediating cellular uptake of apolipoprotein E-enriched beta-VLDL in vitro. Biochemistry 39, 15817–15825, (2000). [DOI] [PubMed] [Google Scholar]
- 139.Brodeur J et al. LDLR-related protein 10 (LRP10) regulates amyloid precursor protein (APP) trafficking and processing: evidence for a role in Alzheimer’s disease. Mol. Neurodegener 7, 31, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Quadri M et al. LRP10 genetic variants in familial Parkinson’s disease and dementia with Lewy bodies: a genome-wide linkage and sequencing study. Lancet Neurol. 17, 597–608, (2018). [DOI] [PubMed] [Google Scholar]
- 141.Neumann M et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133, (2006). [DOI] [PubMed] [Google Scholar]
- 142.Itagaki S, McGeer PL, Akiyama H, Zhu S & Selkoe D Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol 24, 173–182, (1989). [DOI] [PubMed] [Google Scholar]
- 143.Song WM & Colonna M The identity and function of microglia in neurodegeneration. Nat. Immunol 19, 1048–1058, (2018). [DOI] [PubMed] [Google Scholar]
- 144.Deczkowska A et al. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 173, 1073–1081, (2018). [DOI] [PubMed] [Google Scholar]
- 145.Rangaraju S et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener 13, 24, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Krasemann S et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581 e569, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ulrich JD et al. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med 215, 1047–1058, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Keene CD, Cudaback E, Li X, Montine KS & Montine TJ Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr. Opin. Neurobiol 21, 920–928, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rodriguez GA, Tai LM, LaDu MJ & Rebeck GW Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J. Neuroinflamm 11, 111, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shi Y & Holtzman DM Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yeh FL, Wang Y, Tom I, Gonzalez LC & Sheng M TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 91, 328–340, (2016). [DOI] [PubMed] [Google Scholar]
- 152.Atagi Y et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem 290, 26043–26050, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bailey CC, DeVaux LB & Farzan M The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J. Biol. Chem 290, 26033–26042, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jonsson T et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med 368, 107–116, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Guerreiro R et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368, 117–127, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pimenova AA, Marcora E & Goate AM A Tale of Two Genes: Microglial Apoe and Trem2. Immunity 47, 398–400, (2017). [DOI] [PubMed] [Google Scholar]
- 157.Jay TR, von Saucken VE & Landreth GE TREM2 in Neurodegenerative Diseases. Mol. Neurodegener 12, 56, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Efthymiou AG & Goate AM Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener 12, 43, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Parhizkar S et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci 22, 191–204, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Arendt T et al. Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein epsilon 4 allele. J. Neurosci 17, 516–529, (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ji Y et al. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuroscience 122, 305–315, (2003). [DOI] [PubMed] [Google Scholar]
- 162.Sweet RA et al. Apolipoprotein E*4 (APOE*4) Genotype Is Associated with Altered Levels of Glutamate Signaling Proteins and Synaptic Coexpression Networks in the Prefrontal Cortex in Mild to Moderate Alzheimer Disease. Mol. Cell. Proteomics 15, 2252–2262, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Love S et al. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol. Aging 27, 797–803, (2006). [DOI] [PubMed] [Google Scholar]
- 164.Liraz O, Boehm-Cagan A & Michaelson DM ApoE4 induces Abeta42, tau, and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol. Neurodegener 8, 16, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yong SM, Lim ML, Low CM & Wong BS Reduced neuronal signaling in the ageing apolipoprotein-E4 targeted replacement female mice. Sci. Rep 4, 6580, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dumanis SB et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci 29, 15317–15322, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang C et al. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol. Dis 18, 390–398, (2005). [DOI] [PubMed] [Google Scholar]
- 168.Klein RC, Mace BE, Moore SD & Sullivan PM Progressive loss of synaptic integrity in human apolipoprotein E4 targeted replacement mice and attenuation by apolipoprotein E2. Neuroscience 171, 1265–1272, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen Y, Durakoglugil MS, Xian X & Herz J ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl Acad. Sci. USA 107, 12011–12016, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lane-Donovan C & Herz J The ApoE receptors Vldlr and Apoer2 in central nervous system function and disease. J. Lipid Res 58, 1036–1043, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Andrews-Zwilling Y et al. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci 30, 13707–13717, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li G et al. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 5, 634–645, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tensaouti Y, Stephanz EP, Yu TS & Kernie SG ApoE Regulates the Development of Adult Newborn Hippocampal Neurons. eNeuro 5, ENEURO.0155–0118.2018, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kara E et al. Isoform- and cell type-specific structure of apolipoprotein E lipoparticles as revealed by a novel Forster resonance energy transfer assay. J. Biol. Chem 292, 14720–14729, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fu Y et al. Apolipoprotein E lipoprotein particles inhibit amyloid-beta uptake through cell surface heparan sulphate proteoglycan. Mol. Neurodegener 11, 37, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhao J et al. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet 26, 2690–2700, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Heinsinger NM, Gachechiladze MA & Rebeck GW Apolipoprotein E Genotype Affects Size of ApoE Complexes in Cerebrospinal Fluid. J. Neuropathol. Exp. Neurol 75, 918–924, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yassine HN et al. ABCA1-Mediated Cholesterol Efflux Capacity to Cerebrospinal Fluid Is Reduced in Patients With Mild Cognitive Impairment and Alzheimer’s Disease. J. Am. Heart Assoc 5, e002886, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hanson AJ et al. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol. 70, 972–980, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rebeck GW The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res 58, 1493–1499, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fryer JD et al. The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J. Biol. Chem 280, 25754–25759, (2005). [DOI] [PubMed] [Google Scholar]
- 182.Riddell DR et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci 28, 11445–11453, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ulrich JD et al. In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol. Neurodegener 8, 13, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Petersen RC et al. Association of Elevated Amyloid Levels With Cognition and Biomarkers in Cognitively Normal People From the Community. JAMA Neurol. 73, 85–92, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jack CR Jr. & Holtzman DM Biomarker modeling of Alzheimer’s disease. Neuron 80, 1347–1358, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bertens D, Knol DL, Scheltens P & Visser PJ Temporal evolution of biomarkers and cognitive markers in the asymptomatic, MCI, and dementia stage of Alzheimer’s disease. Alzheimers Dement. 11, 511–522, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Willette AA et al. Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA Neurol. 72, 1013–1020, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Craft S Alzheimer disease: Insulin resistance and AD--extending the translational path. Nat. Rev. Immunol 8, 360–362, (2012). [DOI] [PubMed] [Google Scholar]
- 189.Craft S, Cholerton B & Baker LD Insulin and Alzheimer’s disease: untangling the web. J. Alzheimers Dis 33 Suppl 1, S263–275, (2013). [DOI] [PubMed] [Google Scholar]
- 190.Hoyer S Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol 490, 115–125, (2004). [DOI] [PubMed] [Google Scholar]
- 191.Steen E et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis 7, 63–80, (2005). [DOI] [PubMed] [Google Scholar]
- 192.Ekblad LL et al. Midlife insulin resistance, APOE genotype, and late-life brain amyloid accumulation. Neurology 90, e1150–e1157, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jagust WJ & Landau SM Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J. Neurosci 32, 18227–18233, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhao N et al. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 96, 115–129 e115, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Reiman EM et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N. Engl. J. Med 334, 752–758, (1996). [DOI] [PubMed] [Google Scholar]
- 196.Peila R, Rodriguez BL & Launer LJ Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 51, 1256–1262, (2002). [DOI] [PubMed] [Google Scholar]
- 197.Small GW et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273, 942–947, (1995). [PubMed] [Google Scholar]
- 198.Reiman EM et al. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc. Natl Acad. Sci. USA 102, 8299–8302, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Small GW et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl Acad. Sci. USA 97, 6037–6042, (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mosconi L et al. MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET. Neurology 63, 2332–2340, (2004). [DOI] [PubMed] [Google Scholar]
- 201.Drzezga A et al. Cerebral glucose metabolism in patients with AD and different APOE genotypes. Neurology 64, 102–107, (2005). [DOI] [PubMed] [Google Scholar]
- 202.Mosconi L et al. Brain metabolic decreases related to the dose of the ApoE e4 allele in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 75, 370–376, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pardo JV & Lee JT Atypical Localization and Dissociation between Glucose Uptake and Amyloid Deposition in Cognitively Normal APOE*E4 Homozygotic Elders Compared with Patients with Late-Onset Alzheimer’s Disease. eNeuro 5, ENEURO.0396–0317.2018, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Reger MA et al. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol. Aging 27, 451–458, (2006). [DOI] [PubMed] [Google Scholar]
- 205.Claxton A et al. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease. J. Alzheimers Dis 35, 789–797, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Claxton A et al. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis 44, 897–906, (2015). [DOI] [PubMed] [Google Scholar]
- 207.Geijselaers SLC et al. Association of Cerebrospinal Fluid (CSF) Insulin with Cognitive Performance and CSF Biomarkers of Alzheimer’s Disease. J. Alzheimers Dis 61, 309–320, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Morris JK et al. Effect of APOE epsilon4 Genotype on Metabolic Biomarkers in Aging and Alzheimer’s Disease. J. Alzheimers Dis 58, 1129–1135, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wu L, Zhang X & Zhao L Human ApoE Isoforms Differentially Modulate Brain Glucose and Ketone Body Metabolism: Implications for Alzheimer’s Disease Risk Reduction and Early Intervention. J. Neurosci 38, 6665–6681, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Johnson LA et al. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J. Cereb. Blood Flow Metab, 271678X17746186, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Liu CC et al. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci 35, 5851–5859, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sun JH et al. Genetics of Vascular Dementia: Systematic Review and Meta-Analysis. J. Alzheimers Dis 46, 611–629, (2015). [DOI] [PubMed] [Google Scholar]
- 213.Davidson Y et al. Apolipoprotein E epsilon4 allele frequency in vascular dementia. Dement. Geriatr. Cogn. Disord 22, 15–19, (2006). [DOI] [PubMed] [Google Scholar]
- 214.Sudre CH et al. APOE epsilon4 status is associated with white matter hyperintensities volume accumulation rate independent of AD diagnosis. Neurobiol. Aging 53, 67–75, (2017). [DOI] [PubMed] [Google Scholar]
- 215.Schilling S et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology 81, 292–300, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Halliday MR et al. Relationship Between Cyclophilin A Levels and Matrix Metalloproteinase 9 Activity in Cerebrospinal Fluid of Cognitively Normal Apolipoprotein E4 Carriers and Blood-Brain Barrier Breakdown. JAMA Neurol. 70, 1198–1200, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Halliday MR et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab 36, 216–227, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Alata W, Ye Y, St-Amour I, Vandal M & Calon F Human apolipoprotein E varepsilon4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J. Cereb. Blood Flow Metab 35, 86–94, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bell RD et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Koizumi K et al. Apoepsilon4 disrupts neurovascular regulation and undermines white matter integrity and cognitive function. Nat. Commun 9, 3816, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bien-Ly N et al. Lack of Widespread BBB Disruption in Alzheimer’s Disease Models: Focus on Therapeutic Antibodies. Neuron 88, 289–297, (2015). [DOI] [PubMed] [Google Scholar]
- 222.Iturria-Medina Y et al. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun 7, 11934, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Janelidze S et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology 91, e867–e877, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yamazaki Y et al. Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain 142, 1077–1092, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Nation DA et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med 25, 270–276, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Ulrich V et al. Genetic variants of ApoE and ApoER2 differentially modulate endothelial function. Proc. Natl Acad. Sci. USA 111, 13493–13498, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Tai LM et al. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 131, 709–723, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Groot C et al. Clinical phenotype, atrophy, and small vessel disease in APOE epsilon 2 carriers eth Alzheimer disease. Neurology 91, e1851–e1859, (2018). [DOI] [PubMed] [Google Scholar]
- 229.Hong C & Tontonoz P Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat. Rev. Drug Discov 13, 433–444, (2014). [DOI] [PubMed] [Google Scholar]
- 230.Burns MP et al. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J. Neurochem 98, 792–800, (2006). [DOI] [PubMed] [Google Scholar]
- 231.Donkin JJ et al. ATP-binding Cassette Transporter A1 Mediates the Beneficial Effects of the Liver X Receptor Agonist GW3965 on Object Recognition Memory and Amyloid Burden in Amyloid Precursor Protein/Presenilin 1 Mice. J. Biol. Chem 285, 34144–34154, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Koldamova RP et al. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J. Biol. Chem 280, 4079–4088, (2005). [DOI] [PubMed] [Google Scholar]
- 233.Riddell DR et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell. Neurosci 34, 621–628, (2007). [DOI] [PubMed] [Google Scholar]
- 234.Vanmierlo T et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 32, 1262–1272, (2011). [DOI] [PubMed] [Google Scholar]
- 235.Cramer PE et al. ApoE-Directed Therapeutics Rapidly Clear beta-Amyloid and Reverse Deficits in AD Mouse Models. Science 335, 1503–1506, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Fitz NF, Cronican AA, Lefterov I & Koldamova R Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 340, 924–c, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Boehm-Cagan A & Michaelson DM Reversal of apoE4-Driven Brain Pathology and Behavioral Deficits by Bexarotene. J. Neurosci 34, 7293–7301, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Tachibana M et al. Rescuing effects of RXR agonist bexarotene on aging-related synapse loss depend on neuronal LRP1. Exp. Neurol 277, 1–9, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Veeraraghavalu K et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 340, 924-f, (2013). [DOI] [PubMed] [Google Scholar]
- 240.Tesseur I et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 340, 924-e, (2013). [DOI] [PubMed] [Google Scholar]
- 241.Price AR et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 340, 924-d, (2013). [DOI] [PubMed] [Google Scholar]
- 242.Tai LM et al. Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem 289, 30538–30555, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Cummings JL et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimers Res. Ther 8, 4, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ghosal K et al. A randomized controlled study to evaluate the effect of bexarotene on amyloid-beta and apolipoprotein E metabolism in healthy subjects. Alzheimers Dement (N Y). 2, 110–120, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Lalloyer F et al. Rexinoid bexarotene modulates triglyceride but not cholesterol metabolism via gene-specific permissivity of the RXR/LXR heterodimer in the liver. Arter. Thromb. Vasc. Biol 29, 1488–1495, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Finan GM et al. Bioactive Compound Screen for Pharmacological Enhancers of Apolipoprotein E in Primary Human Astrocytes. Cell Chem. Biol 23, 1526–1538, (2016). [DOI] [PubMed] [Google Scholar]
- 247.Dodart JC et al. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 102, 1211–1216, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zhao L et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 44, 159–172, (2016). [DOI] [PubMed] [Google Scholar]
- 249.Hudry E et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med 5, 212ra161, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Hu J et al. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol. Neurodegener 10, 6, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Bien-Ly N, Gillespie AK, Walker D, Yoon SY & Huang Y Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci 32, 4803–4811, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kim J et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J. Neurosci 31, 18007–18012, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Youmans KL et al. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem 287, 41774–41786, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Koldamova R, Staufenbiel M & Lefterov I Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem 280, 43224–43235, (2005). [DOI] [PubMed] [Google Scholar]
- 255.Wahrle SE et al. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem 280, 43236–43242, (2005). [DOI] [PubMed] [Google Scholar]
- 256.Wahrle SE et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest 118, 671–682, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Liao F et al. Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J. Neurosci 34, 7281–7292, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Kim J et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J. Exp. Med 209, 2149–2156, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Liao F et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Invest 128, 2144–2155, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Vitek MP et al. APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer’s disease transgenics. Neurodegener. Dis 10, 122–126, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Handattu SP et al. In vivo and in vitro effects of an apolipoprotein e mimetic peptide on amyloid-beta pathology. J. Alzheimers Dis 36, 335–347, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ghosal K et al. The apolipoprotein-E-mimetic COG112 protects amyloid precursor protein intracellular domain-overexpressing animals from Alzheimer’s disease-like pathological features. Neurodegener. Dis 12, 51–58, (2013). [DOI] [PubMed] [Google Scholar]
- 263.Mahley RW Central Nervous System Lipoproteins ApoE and Regulation of Cholesterol Metabolism. Arter. Thromb. Vasc. Biol 36, 1305–1315, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Mahley RW & Huang Y Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein E4-associated neuropathology. J. Med. Chem 55, 8997–9008, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Chen HK et al. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem 286, 5215–5221, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Chen HK et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J. Biol. Chem 287, 5253–5266, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Brodbeck J et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem 286, 17217–17226, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Sadowski M et al. A synthetic peptide blocking the apolipoprotein E/beta-amyloid binding mitigates beta-amyloid toxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am. J. Pathol 165, 937–948, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Sadowski MJ et al. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer’s disease. Proc. Natl Acad. Sci. USA 103, 18787–18792, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Liu S et al. Blocking the apolipoprotein E/amyloid beta interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid beta and tau pathology. J. Neurochem 128, 577–591, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Kuszczyk MA et al. Blocking the interaction between apolipoprotein E and Abeta reduces intraneuronal accumulation of Abeta and inhibits synaptic degeneration. Am. J. Pathol 182, 1750–1768, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Pankiewicz JE et al. Blocking the apoE/Abeta interaction ameliorates Abeta-related pathology in APOE epsilon2 and epsilon4 targeted replacement Alzheimer model mice. Acta Neuropathol. Commun 2, 75, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Zhao N, Liu CC, Qiao W & Bu G Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol. Psychiatry 83, 347–357, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Cao D, Fukuchi K, Wan H, Kim H & Li L Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice. Neurobiol. Aging 27, 1632–1643, (2006). [DOI] [PubMed] [Google Scholar]
- 275.Katsouri L & Georgopoulos S Lack of LDL receptor enhances amyloid deposition and decreases glial response in an Alzheimer’s disease mouse model. PloS one 6, e21880, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kim J et al. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron 64, 632–644, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Kanekiyo T et al. Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J. Neurosci 33, 19276–19283, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Liu CC et al. Astrocytic LRP1 Mediates Brain Abeta Clearance and Impacts Amyloid Deposition. J. Neurosci 37, 4023–4031, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kanekiyo T, Liu CC, Shinohara M, Li J & Bu G LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. J. Neurosci 32, 16458–16465, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Qosa H, Abuznait AH, Hill RA & Kaddoumi A Enhanced Brain Amyloid-beta Clearance by Rifampicin and Caffeine as a Possible Protective Mechanism Against Alzheimer’s Disease. J. Alzheimers Dis 31, 151–165, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Shinohara M et al. Reduction of Brain beta-Amyloid (A beta) by Fluvastatin, a Hydroxymethylglutaryl-CoA Reductase Inhibitor, through Increase in Degradation of Amyloid Precursor Protein C-terminal Fragments (APP-CTFs) and A beta Clearance. J. Biol. Chem 285, 22091–22102, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Mak AC et al. Effects of the absence of apolipoprotein e on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol. 71, 1228–1236, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Centeno EGZ, Cimarosti H & Bithell A 2D versus 3D human induced pluripotent stem cell-derived cultures for neurodegenerative disease modelling. Mol. Neurodegener 13, 27, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Karch CM & Goate AM Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Carmona S, Hardy J & Guerreiro R The genetic landscape of Alzheimer disease. Handb Clin Neurol 148, 395–408, (2018). [DOI] [PubMed] [Google Scholar]
- 286.Alzgene. Meta-analysis of all published AD association studies (case-control only) APOE_E2/3/4. http://www.alzgene.org/meta.asp?geneID=83, (2010).
- 287.Mattsson N et al. Prevalence of the apolipoprotein E epsilon4 allele in amyloid beta positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement. 14, 913–924, (2018). [DOI] [PubMed] [Google Scholar]
- 288.Cacace R, Sleegers K & Van Broeckhoven C Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 12, 733–748, (2016). [DOI] [PubMed] [Google Scholar]
- 289.van Duijn CM et al. Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer’s disease. Nat. Genet 7, 74–78, (1994). [DOI] [PubMed] [Google Scholar]
- 290.Chartier-Harlin MC et al. Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: analysis of the 19q13.2 chromosomal region. Hum. Mol. Genet 3, 569–574, (1994). [DOI] [PubMed] [Google Scholar]
- 291.Houlden H et al. ApoE genotype is a risk factor in nonpresenilin early-onset Alzheimer’s disease families. Am. J. Med. Genet 81, 117–121, (1998). [DOI] [PubMed] [Google Scholar]
- 292.Sorbi S et al. Epistatic effect of APP717 mutation and apolipoprotein E genotype in familial Alzheimer’s disease. Ann. Neurol 38, 124–127, (1995). [DOI] [PubMed] [Google Scholar]
- 293.Pastor P et al. Apolipoprotein Eepsilon4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann. Neurol 54, 163–169, (2003). [DOI] [PubMed] [Google Scholar]
- 294.Wijsman EM et al. APOE and other loci affect age-at-onset in Alzheimer’s disease families with PS2 mutation. Am. J. Med. Genet. B Neuropsychiatr. Genet 132B, 14–20, (2005). [DOI] [PubMed] [Google Scholar]
- 295.Velez JI et al. APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. Mol. Psychiatry 21, 916–924, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Ryman DC et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 83, 253–260, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Altmann A, Tian L, Henderson VW & Greicius MD Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol 75, 563–573, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Buckley RF et al. Sex, amyloid, and APOE epsilon4 and risk of cognitive decline in preclinical Alzheimer’s disease: Findings from three well-characterized cohorts. Alzheimers Dement. 14, 1193–1203, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Deming Y et al. Sex-specific genetic predictors of Alzheimer’s disease biomarkers. Acta Neuropathol. 136, 857–872, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Hohman TJ et al. Sex-Specific Association of Apolipoprotein E With Cerebrospinal Fluid Levels of Tau. JAMA Neurol. 75, 989–998, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Buckley RF et al. Sex Differences in the Association of Global Amyloid and Regional Tau Deposition Measured By Positron Emission Tomography in Clinically Normal Older Adults. JAMA Neurol. 76, 542–551, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Sundermann EE, Tran M, Maki PM & Bondi MW Sex differences in the association between apolipoprotein E epsilon4 allele and Alzheimer’s disease markers. Alzheimers Dement. 10, 438–447, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Caselli RJ et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N. Engl. J. Med 361, 255–263, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Caselli RJ et al. Longitudinal changes in cognition and behavior in asymptomatic carriers of the APOE e4 allele. Neurology 62, 1990–1995, (2004). [DOI] [PubMed] [Google Scholar]
- 305.Caselli RJ et al. Longitudinal modeling of frontal cognition in APOE epsilon4 homozygotes, heterozygotes, and noncarriers. Neurology 76, 1383–1388, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Bonham LW et al. Age-dependent effects of APOE epsilon4 in preclinical Alzheimer’s disease. Ann. Clin. Transl. Neurol 3, 668–677, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Ferrari C et al. How can elderly apolipoprotein E epsilon 4 carriers remain free from dementia? Neurobiol. Aging 34, 13–21, (2013). [DOI] [PubMed] [Google Scholar]
- 308.Head D et al. Exercise Engagement as a Moderator of the Effects of APOE Genotype on Amyloid Deposition. Arch. Neurol 69, 636–643, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Stocker H, Mollers T, Perna L & Brenner H The genetic risk of Alzheimer’s disease beyond APOE epsilon4: systematic review of Alzheimer’s genetic risk scores. Transl. Psychiatry 8, 166, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Escott-Price V, Myers AJ, Huentelman M & Hardy J Polygenic risk score analysis of pathologically confirmed Alzheimer disease. Ann. Neurol 82, 311–314, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Montine TJ & Montine KS Precision medicine: Clarity for the clinical and biological complexity of Alzheimer’s and Parkinson’s diseases. J. Exp. Med 212, 601–605, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Salloway S et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med 370, 322–333, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Craft S et al. Insulin effects on glucose metabolism, memory, and plasma amyloid precursor protein in Alzheimer’s disease differ according to apolipoprotein-E genotype. Ann. N Y Acad. Sci 903, 222–228, (2000). [DOI] [PubMed] [Google Scholar]
- 314.Reger MA et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers Dis 13, 323–331, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Petersen RC et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med 352, 2379–2388, (2005). [DOI] [PubMed] [Google Scholar]