

SUMMARY
The periosteum is critical for bone maintenance and healing. However, the in vivo identity and specific regulatory mechanisms of adult periosteum-resident skeletal stem cells are unknown. Here, we report animal models that selectively and durably label postnatal Mx1+αSMA+ periosteal stem cells (P-SSCs) and establish that P-SSCs are a long-term repopulating, functionally distinct SSC subset responsible for lifelong generation of periosteal osteoblasts. P-SSCs rapidly migrate toward an injury site, supply osteoblasts and chondrocytes, and recover new periosteum. Notably, P-SSCs specifically express CCL5 receptors, CCR3 and CCR5. Real-time intravital imaging revealed that the treatment with CCL5 induces P-SSC migration in vivo and bone healing, while CCL5/CCR5 deletion, CCR5 inhibition, or local P-SSC ablation reduces osteoblast number and delays bone healing. Human periosteal cells express CCR5 and undergo CCL5-mediated migration. Thus, the adult periosteum maintains genetically distinct SSC subsets with a CCL5-dependant migratory mechanism required for bone maintenance and injury repair.
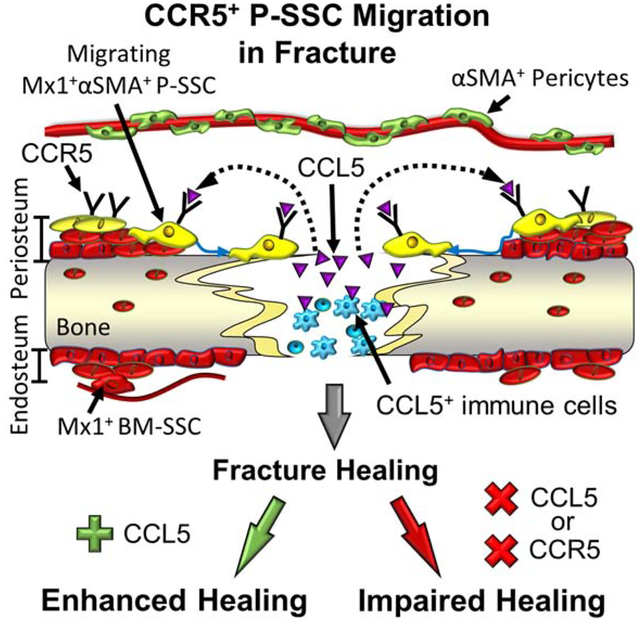
Graphical Abstract
eTOC
Ortinau et. al identified long-term repopulating, functionally distinct adult periosteal skeletal stem cells (P-SSCs) that can be marked by a combination of Mx1 and αSMA. These P-SSCs are critical for periosteal bone maintenance, specifically express CCL5 receptors, CCR5, and have a unique CCL5-dependant migratory mechanism required for injury repair.
INTRODUCTION
Skeletal stem cells (SSCs) are known to exist within the bone marrow (BM) and required for skeletal development, homeostasis, and repair (Bianco and Robey, 2015; Park et al., 2012). More recently, SSC populations have been identified within skeletal tissues other than BM (e.g. calvarial sutures and the periosteum), indicating that endogenous SSCs may have multiple tissue locations within adult bones (Roberts et al., 2015; Zhao et al., 2015). However, due to the lack of specific markers to distinguish rare P-SSCs from BM-SSCs in vivo, which SSCs recognize tissue injury and how they initiate the repair process remain fundamental questions.
Tissue-resident SSCs are a heterogeneous population with different locations. In the BM, long-term repopulating SSCs have been prospectively identified by their expression of CD140a (PDGFRα), V-cam1, and CD200 in mice (Chan et al., 2015; Morikawa et al., 2009) or CD146 and CD271 in humans (Buhring et al., 2009; Sacchetti et al., 2007). They are a subset of BM perivascular cells expressing hematopoietic stem cell (HSC) niche factors (Morrison and Scadden, 2014). Interestingly, a non-perivascular SSC population with in vivo osteogenic and chondrogenic potential that is distinctly different from perivascular SSCs is labeled by Gremlin 1 (Grem1) (Worthley et al., 2015). Similar to BM, osteo/chondrogenic progenitors have also been found in the periosteum (Duchamp de Lageneste et al., 2018; Olivos-Meza et al., 2010). These periosteal progenitors can contribute to outer bone shaping, cortical thickness, and fracture repair (Debnath et al., 2018; Ransom et al., 2016). In addition, periosteal cells are particularly important in the life-long regeneration of bones with limited or no BM, suggesting that SSC subsets reside in the adult periosteum. However, a significant drawback of these single genetic lineage tracing models is that there is a substantial heterogeneity within labeled cell populations and that they cannot distinguish labeled SSCs from their progeny. Therefore, the in vivo stem cell properties and physiological function of P-SSCs in adult bones remain elusive. Furthermore, the differential regulatory mechanisms influencing the activation and recruitment of specific SSC subtypes in the early repair process are essentially unknown.
To answer this fundamental question, we sought to develop a new method to label postnatal P-SSCs selectively, define their long-term repopulation and contribution to fracture repair in vivo, and identify specific factors that regulate P-SSC migration and proliferation under physiological conditions.
RESULTS
The Mx1 and αSMA combination selectively labels endogenous P-SSCs.
By using genetic mouse models, recent studies revealed that a subset of periosteal cells has skeletal progenitor function and plays an important role in intramembraneous bone formation and bone repair (Debnath et al., 2018; Duchamp de Lageneste et al., 2018). We also found that Mx1-labeled (hereafter Mx1+) progenitor cells are present in the periosteum (Park et al., 2012). However, one limitation of these models is the absence of specificity to distinguish P-SSCs from BM-SSCs and to track their long-term repopulation in vivo. Therefore, to further define a model enabling selective labeling of P-SSCs in adult bones, we crossed Mx1-Cre+Rosa26-Tomato+ mice with other skeletal stem/progenitor cell (SSPC) reporter mouse lines, including αSMA-GFP (Grcevic et al., 2012), Nestin-GFP (Mendez-Ferrer et al., 2010), and CXCL12-GFP (Omatsu et al., 2010). When these trigenic mice were 4–6 weeks old, polyinosinic:polycytidylic acid (pIpC: 25 mg/kg) was administered every other day for 10 days to activate the Mx1-Cre, leading to the tomato labeling of SSCs. Two weeks later, mice were irradiated (IR, 9.5 Gy) and transplanted with wild-type BM (106 cells/mouse, WT-BMT) to eliminate Mx1+ hematopoietic cells. Among the tested combinations, immunohistochemistry and in vivo imaging analysis of 3-month old trigenic Mx1-Cre+Rosa26-Tomato+αSMA-GFP+ (Mx1/Tomato/αSMA-GFP, pIpC at 4 weeks of age and IR+WT-BMT at 6 weeks of age) mice showed that the Mx1 and αSMA-GFP combination selectively labeled a subset of periosteal cells (Figures 1A, dot box, 1B, top), but double labeling was nearly undetectable in the BM (Figures 1A, 1B, bottom). Next, histological and in vivo image analysis of 7-month old Mx1/Tomato/αSMA-GFP (6 months after pIpC and IR+WT-BMT) mouse tibia and calvaria confirmed that most Mx1+αSMA+ (Tomato+GFP+) cells reside in the tibial periosteum (Figure S1A) and calvarial sutures (a known niche for craniofacial SSCs; S1C & S1D) (Zhao et al., 2015), while nearly all αSMA+ cells in the BM do not overlap with Mx1+ cells (Figures 1A, BM, S1B), suggesting that the selective Mx1/αSMA-GFP double labeling (Mx1+αSMA+) of periosteal cells is durable in adult mice. Fluorescence-activated cell sorting (FACS) analysis of the periosteum of 3-month old Mx1/Tomato/αSMA-GFP mice showed that periosteal cells formed a heterogeneous population and that ~1.7% (1.5±0.4, n=5) of CD45−CD31−Ter119− periosteal cells were Mx1 and αSMA-GFP positive (Figure 1C, red box). Subsequent SSC marker analysis revealed that most (~80%) of Mx1+αSMA+ cells expressed SSC immunophenotypic markers (CD105+CD140a+) with higher expression of SSC transcripts, including Runx2, Cxcl12, LepR, and Grem1 (Figure 1C). By contrast, only ~10% of Mx1−αSMA+ cells and ~27% of Mx1+αSMA− cells from the CD45−CD31−Ter119− fraction were CD105+CD140a+, with a ~5-fold lower expression of SSC transcripts (Figures S1E & 1C, respectively). When cultured at the single-cell level, Mx1+αSMA+ cells, but neither Mx1 nor αSMA-GFP single positive cells, formed colonies with osteogenic, chondrogenic, and adipogenic differentiation potential in vitro (Figure 1D), demonstrating that the Mx1 and αSMA-GFP combination further specifies endogenous P-SSCs with ex vivo SSC characteristics.
Figure 1. Mx1+αSMA+ periosteal cells highly express SSPC markers and contribute to bone injury repair.

A & B. The periosteal Mx1+αSMA+ population in the tibia of 3-month old Mx1/Tomato/αSMA-GFP mice (+pIpC at 4 weeks of age; IR+WT-BMT at 6 weeks of age) was analyzed by immunofluorescence staining (A) and intravital microscopy (B). Right panels show enlarged views of white dotted boxes. White arrows, Mx1+αSMA+ cells (A, Tomato+GFP+); green arrows, Mx1−αSMA+ cells (A & B, GFP+); Mx1+ Obs, Mx1+αSMA− osteoblasts (A, Tomato+); M, muscle; B, bone; BM, bone marrow. Anti-GFP staining was used for enhancing αSMA-GFP signals. Blue, DAPI. (n>5 males and females) C. Periosteal cells from Mx1/Tomato/αSMA-GFP mice were FACS-isolated into 5 indicated populations. Relative expression of indicated SSPC markers was determined by qRT-PCR (n=6). D. A representative single-cell-driven colony formation of Mx1+αSMA+ periosteal cells and osteogenic (Alizarin Red), adipogenic (Oil Red O), and chondrogenic (a pellet culture with Alcian blue) differentiation after 28 days culture of single cell-derived colonies with conditioned medium. Inserted image: Toluidine blue staining. (n=8–10 per each experiment). E. At 2 weeks after injury, the expansion of the Mx1+ (Tomato+) cells in the callus, repopulation of Mx1+αSMA+ (Tomato+GFP+) cells in the new periosteum (red arrow, New PO), and their contribution to the new osteoblasts (box a, lower left panel) and chondrocytes (box b, lower right panel) were assessed by anti-osteocalcin (left, white) and anti-aggrecan (right, white) immunostaining (n>5). F. Mx1+αSMA+ cell translocation (D7, arrow), repopulation (D15), and Mx1+ osteoblast differentiation (D21, Tomato+) at the calvarial injury sites of 4-month old Mx1/Tomato/αSMA-GFP mice were assessed by sequential in vivo imaging. Bone (blue) (n>5). G. 3-D reconstruction of in vivo z-stack images at 21 days after calvarial injury. Undifferentiated Mx1+αSMA+ cells in the periosteum (left arrow and top right a), Mx1+αSMA− (Tomato+) osteoblasts in the underneath injury callus and osteocytes (bottom right b, arrows) in the new bone (blue) were indicated.
Next, to examine whether Mx1+αSMA+ periosteal cells contribute to injury repair in vivo, we generated drill-hole defects (~1 mm diameter) in tibia of 3-month old Mx1/Tomato/αSMA-GFP mice (2 months after pIpC and WT-BMT). We reasoned that a drill-hole defect can be a model of bone healing without compromising tissue architecture (endosteum vs periosteum) and achieve better quantification of stem cell responses compared to other fracture methods (He et al., 2011). Histological analysis of tibial injury at day 14 revealed that Mx1+αSMA+ periosteal cells repopulated the outer layer of the callus (Figure 1E, New PO) and supplied the majority of callus-forming cells (Figure 1E, Callus). We previously reported that Mx1+ BM-SSCs exhibit mainly osteogenic potential, with little adipogenic and no chondrogenic potential in postnatal bone maintenance (Park et al., 2012). Interestingly, however, these Mx1+αSMA+ periosteal cells contributed to 21±3% of aggrecan+ chondrocytes in the cartilage intermediate (Figure 1E, bottom right), and over 80% of new osteoblasts within the callus (Figure 1E, bottom left), implying that they differ from Mx1+ BM-SSCs. We next tested whether there is a divergence in the contribution of Mx1+αSMA+ periosteal cells in a fracture model. We consistently found that Mx1+αSMA+ periosteal cells mainly contributed to callus formation at 14 days after a transverse tibial fracture (Figure S1F). When Mx1+αSMA+ cells were tracked during bone repair of injured calvaria in vivo, these cells newly appeared at the injury site and repopulated on days 7 and 15 (Figures 1F & S1G, white arrows). In addition, 21 days after calvarial injury, undifferentiated Mx1+αSMA+ cells exclusively resided in the center space of the injury, while differentiated Mx1+αSMA− cells covered the outer space of the injury region with new bone formation (blue) (Figures 1F & S1G). Consistent with Figure 1E, 3D reconstitution of Z-stack images and optical sections of the periosteum (a) and underneath injury callus (b) at 21 days post-injury showed that Mx1+αSMA+ cells at the top surface of injury remained undifferentiated (Figure 1G, a, yellow) and formed new periosteum, while Mx1+αSMA+ cells in the middle callus of injury differentiated and became Mx1+αSMA− osteoblasts and osteocytes (Figure 1G, b, white arrows) imbedded in newly formed bones (Figure 1G, b, blue). Of note, Mx1+ BM cells near the injury site showed no observable activation, further demonstrating that Mx1+αSMA+ periosteal cells in adult bones include endogenous P-SSCs with a multi-lineage differentiation potential and supply the osteoblasts participating in bone healing.
Mx1+ periosteal cells are the major source of new osteoblasts in bone healing in vivo.
It is possible that the periosteum contains Mx1− P-SSCs that can contribute to new osteoblasts at injury sites. Therefore, to distinguish between P-SSCs and mature osteoblasts and to improve specific cell quantitation during bone healing, we generated trigenic Mx1-Cre+Rosa26-Tomato+osteocalcin-GFP+ (Mx1/Tomato/Ocn-GFP) mice. In this model, when Mx1+Ocn-GFP− (Mx1+Ocn−) P-SSCs differentiate into mature Mx1+Ocn-GFP+ osteoblasts, they express Tomato and GFP (Park et al., 2014) (Figures S2A). Anti-GFP immunofluorescence staining and in vivo imaging of Mx1/Tomato/Ocn-GFP mouse tibias at 2 weeks after pIpC treatment revealed that the induction of Mx1-Cre activity labeled distinct periosteal cells without detectable Ocn-GFP expression in the metaphysis and diaphysis (Figures 2A, 2B, & S2B, white arrows). In contrast to a widely mixed distribution of Mx1+Ocn− progenitors and Mx1+Ocn+ osteoblasts (Tomato+GFP+; ~50–60% of endosteal osteoblasts) in trabecular bones (Figures S2B & S2C), these Mx1+Ocn− periosteal cells exclusively existed in the cambial layer, a known location of skeletal progenitors (Bragdon and Bahney, 2018) (Figure S2B, CL). To determine that Mx1+Ocn− periosteal cells contribute to normal periosteal bone growth, we analyzed the percentage of Mx1+Ocn+ periosteal osteoblasts and osteocytes in the tibia of Mx1/Tomato/Ocn-GFP mice at 2, 8 and 16 weeks after pIpC treatment. We noted that the number of Mx1+Ocn+ periosteal osteoblasts and osteocytes (Figure S2C, Mx1+ Ocy) in cortical bone was markedly increased in 7-month old mice (from ~18% at 2 weeks to ~80% at 16 weeks post-pIpC) (Figures 2A, S2A, 2C, & S2C), supporting that Mx1+Ocn− periosteal cells are the major source of newly generated periosteal osteoblasts and osteocytes in postnatal bones. Consistent with Figure 1C, FACS analysis of collagenase-digested periosteal cells revealed that Mx1+Ocn− cells accounted for 48% of SSPC (CD105+CD140a+) population and formed clonogenic colonies with in vitro osteogenic differentiation potential (Figure S2D, right). Gene expression analysis with sorted Mx1−Ocn−, Mx1+Ocn−, and control cells from the periosteum revealed that the Mx1+Ocn−CD105+CD140a+ population expressed significantly higher levels of SSC markers, including Grem1, Cxcl12, and Runx2, but no detectable levels of Ocn (a mature osteoblastic marker) compared with other populations (Figures S2E & S2F).
Figure 2. Mx1+ progenitors in the periosteum supply the majority of osteoblasts in cortical bone injury.

A-C, Using 3-month old (A & B, 2 weeks after 5 doses of pIpC induction) and 6-month old (C, 16 weeks after pIpC induction) Mx1/Tomato/Ocn-GFP mice (IR and WT-BMT at 4 weeks of age), periosteal Mx1+ progenitor cells (white arrows) and endosteal osteoblasts (GFP+), new osteoblasts (Tomato+GFP+, green arrows) and osteocytes (red arrows) from Mx1+ progenitor cells (C) in the tibia were analyzed by anti-GFP staining (A & C) and intravital imaging (B) (n=3–5 per group). PO, periosteum; B, bone; BM, bone marrow. D. Percentage of Mx1+Ocn+ (Tomato+GFP+) periosteal osteoblasts and osteocytes in the metaphysis and diaphysis of the tibia 2 (A), 8 (data not shown), and 16 (C) weeks after pIpC induction (n=5). E & F. Representative tibia drill-hole defect (E) or periosteal defect (F) on cortical bone surface of Mx1/Tomato/Ocn-GFP mice. Contribution of Mx1+ periosteal progenitors (Tomato+) to the most of the new Mx1+Ocn+ osteoblasts (Tomato+GFP+) in the bone bridge (E) and to the majority of callus-forming osteoblasts (Mx1+Ocn+; white arrow) in the cortical defect (F) were assessed. White boxes in E and F represent the panels to the right. Graph shows percentage of Mx1+Ocn+ osteoblasts from uninjured periosteum (normal) and injured bone callus (Injury callus) (n=5).
To test whether Mx1+Ocn− periosteal cells supply new osteoblasts but remain undifferentiated in new periosteum during bone repair in vivo, we generated drill-hole defects in the tibial diaphysis of Mx1/Tomato/Ocn-GFP mice. Immunofluorescence analysis of the bone defect revealed that Mx1+Ocn− cells reconstituted the majority (~80%) of outer callus-forming cells (Tomato) within new periosteum (New PO), and >70% of the bone-forming osteoblasts (Tomato+GFP+) surrounding new bone bridges (Figures 2E & S2G). Next, we generated a bone defect in the periosteum and the outer surface of cortical bone, without BM damage, in the tibia of Mx1/Tomato/Ocn-GFP mice. Two weeks post-injury, we consistently observed robust callus formation with over 90% contribution of Mx1+ cells to new osteoblasts (Mx1+Ocn+) in the injury callus (Figure 2F). Taken together, these data show that Mx1 can label stem/progenitor cells in the periosteum and that the activation of Mx1+ periosteal cells upon cortical bone injury is sufficient to induce the osteogenesis and periosteal reconstitution.
Mx1+αSMA+ periosteal cells are long-term repopulating progenitors required for bone healing.
To test the long-term repopulation ability of Mx1+αSMA+ cells, we examined their contribution to bone repair after multiple rounds of injuries. Consecutive intravital imaging of the primary calvarial injury sites of Mx1/Tomato/αSMA-GFP mice at ~4 months of age (Figure 3A, First injury), and again at ~12 months of age, revealed complete bone and periosteum recovery with exclusive periosteum localization of Mx1+αSMA+ cells resembling pre-injury homeostatic condition (Figure 3A, bottom, Pre-injury). When Mx1+αSMA+ cells were further traced at the secondary injury sites, they rapidly repopulated and contributed to over 80% of osteoblasts, similar to their response to the primary injury (Figure 3A, bottom, D14). Compared to basal conditions (Figure 1C), the Mx1+αSMA+ population expanded ~3-fold (from 1.7% to 5.4% of CD45−CD31−Ter119− fraction) in response to multiple calvarial injuries (Figure 3B, red box). More importantly, Mx1+αSMA+ cells maintained a high expression of previously defined SSC markers such as CD200, CD140a, and Vcam-1 even after a second injury.
Figure 3. Mx1+αSMA+ P-SSCs are long-term repopulating cells and necessary for bone healing.

A. After first calvarial injuries in 4-month old Mx1/Tomato/SMA-GFP mice (pIpC at 4 weeks of age and IR+WT-BMT at 6 weeks of age), long-term maintenance of Mx1+αSMA+ P-SSCs (Tomato+GFP+, Pre-injury) and their repopulation after second round of injuries (D14 injury) were assessed by sequential in vivo imaging at 12 months of age (8 months after the first injury). Bone (blue) (n=6). B. The expression of indicated SSC markers in the Mx1+αSMA+ P-SSCs (red line) and the αSMA-GFP+ cells (gray shading) from the periosteum after a second injury (n=4). C. Serial transplantation of Mx1+αSMA+ P-SSCs. Four weeks after transplantion of sorted Mx1+αSMA+ P-SSCs onto calvarial injury sites of C57BL/6 mice (~5,000 cells/mouse), repopulating Mx1+αSMA+ P-SSCs in the periosteum (PO, top left, Tomato+GFP+) and their osteogenic differentiation (top middle, white arrow) in newly generated bones (blue, middle dotted line) were analyzed by in vivo imaging. After re-sorting and transplantion of Mx1+αSMA+ P-SSCs (top right) into secondary injuries of C57BL/6 mice (~500 cells/mouse), their periosteal repopulation (PO, bottom left), osteogenic differentiation (bottom middle, white arrow) in new bones (blue), and SSC marker expression (CD105 and CD140a, bottom right) were analyzed by in vivo imaging and flow cytometry. As controls, transplantation of CD105+CD140a+Mx1+αSMA− or CD140a+Mx1−αSMA+ periosteal cells was also performed (Figure S3). (n=5 mice per group). D. Before injury, the periosteum of Mx1/tomato/iDTR mice was pretreated locally with a low-dose of diphtheria toxin (+DT; 20 μL at 1 μg/mL) or control (-DT; PBS) for seven days. After injury, the reduction in Mx1+ (Tomato+) cell number at the calvarial injury sites pretreated with DT (+DT) or PBS (-DT) was tracked at the indicated time points using in vivo imaging of the bone injury. Data represent at least three independent experiments. E. At 16 days after injury, representative μCT images (left) and bone volume/total volume (BV/TV) quantification (right) of calvarial defects treated with DT (+DT) or PBS (-DT) were assessed for the quantification of bone healing (n=5).
To better define the long-term repopulation characteristics and to exclude the possibility that Mx1+αSMA− periosteal cells include P-SSCs and become Mx1+αSMA+ cells in bone injury, we performed serial transplantations of these cells (Figure 3C). A total of 5000 Mx1+αSMA+CD140a+CD105+, Mx1+αSMA−, and Mx1−αSMA+ cells from the periosteal CD45−CD31−Ter119− fraction were sorted and transplanted with Matrigel onto the calvaria injury site of a wildtype mouse. We reasoned that this P-SSC transplantation onto a periosteal injury site is more physiologically relevant compared to the previously described SSC transplantation into kidney capsules (Chan et al., 2015) or mammary fat pads (Debnath et al., 2018). In vivo imaging and FACS analysis at 4 weeks after the first round of transplantation showed that the transplanted Mx1+αSMA+ cells proliferated and differentiated into Mx1+ osteoblasts and osteocytes that were imbedded within the newly formed bone tissue (Figure 3C, top arrow, New bone). A subset of the engrafted cells repopulated and maintained the Mx1+αSMA+ population in the periosteum (12–15% of all transplanted Mx1+ cells). The majority of the Mx1+αSMA+ cells (~72%) preserved the expression of SSPC surface markers (CD140a+CD105+) (Figure 3C, top). When these Mx1+αSMA+CD140a+CD105+ cells (~500 cells/mouse) were re-transplanted and analyzed at 4 weeks after the second transplantation, they displayed both intact repopulation of double positive cells (Figure 3C, bottom PO, ~15% of transplanted cells) with high CD140a+CD105+ expression (~74%) and their osteoblast and osteocyte differentiation (Figure 3C, bottom arrow, New bone). However, transplanted Mx1+αSMA− or Mx1−αSMA+ cells did not show such repopulation, and there was no detectable stem cell marker expression (Figures S3A & S3B), demonstrating that the key characteristics of SSCs—in vivo long-term repopulation and differentiation—are highly enriched in the Mx1+αSMA+ periosteal cells.
SSCs are known to be present in the BM and may contribute to bone healing if the periosteum is defective or removed. Therefore, we developed an inducible deletion model (Mx1-Cre+Rosa26-Tomato+Rosa26-DTR+) and tested the requirement for Mx1+ periosteal cells in bone healing. To restrict the conditional deletion of Mx1+ cells to the periosteum, we used local surface administration of the diphtheria toxin (DT) (20 μL, 1 μg/mL) to cover an area with ~1 cm of diameter at the junction of the sagittal and coronal sutures (Figure S4A). Periosteal DT treatment for seven days prior to injury reduced the number of Mx1+ periosteal cells in more than 90%, while Mx1+ cells in the BM and in the distal periosteum remained unchanged (Figure S4B). Under these conditions, sequential imaging of injury repair revealed that the DT-mediated local deletion of Mx1+ cells completely inhibited the recruitment and proliferation of Mx1+ osteogenic cells at the injury site (Figure 3D). Micro-computerized tomography (μCT) analysis showed that local DT treatment significantly delayed calvarial bone healing (Figure 3E). We observed a similar delay in tibial bone healing with local DT treatment (data not shown). Since Mx1+ osteoblasts and progenitors in the BM near the injury site remained intact, and no detectable changes in their population or position occurred during injury healing (data not shown), the new Mx1+ cells within the bone injury were highly likely to be derived from the periosteum. These data are consistent with Mx1+ periosteal cells being necessary for osteogenesis and bone healing.
Mx1+ periosteal cells, distinct from Nestin-GFP+ BM-SSCs, supply new osteoblasts in bone healing.
BM-SSCs were reported to localize in perivascular regions with Nestin-GFP expression (Mendez-Ferrer et al., 2010). Therefore, we developed the trigenic Mx1-Cre+Rosa26-Tomato+Nestin-GFP+ (Mx1/Tomato/Nestin-GFP) mouse and tested if Mx1 and Nestin double labeling can distinguish BM-SSCs from P-SSCs and their contributions to bone injury. In vivo imaging and FACS analysis of these mice revealed that there are two distinct cell populations (Mx1+Nestin+; ~27% and Mx1−Nestin+; ~73%) within Nestin+ perivascular cells in the BM, whereas Mx1+ periosteal cells are exclusively Nestin negative (Figure S5A & S5B). To determine if marker expression was different between P-SSCs and BM-SSCs, we conducted Affymetrix-based global gene expression analysis with FACS-isolated cells: (1) Mx1+ PCs (CD105+CD140a+Mx1+Ocn− periosteal cells) (Figure S2E, Mx1+), (2) Mx1+Nestin+, and (3) Mx1−Nestin+ cells from Mx1/Tomato/Nestin-GFP mouse BM (Figure S5B) and control cells, including (4) CD45+, (5) Osteocalcin+ (Ocn+), and (6) Osterix+ (Osx+) cells. Interestingly, the expression levels of SSC markers, including Grem1, LepR, and Cxcl12, were much higher in Mx1+Ocn− periosteal cells compared to Nestin+ BM perivascular cells and control cells (Figure S5C) (Deveza et al., 2018). We confirmed the highest expression level of Runx2 and Cxcl12 in Mx1+Ocn− periosteal cells by RT-PCR (Figure S5D). We next tracked Mx1+ (Tomato+) and Nestin+ (GFP+) cells at the bone injury site using intravital imaging and found that Mx1+Nestin− periosteal cells predominantly appeared at the injury site at 2–5 days (>95%) and subsequently increased in number. In marked contrast, few Mx1+Nestin+ (2–3%) or Mx1−Nestin+ (~1%) BM cells were observed at the injury site throughout the repair process (Figure S5E). Taken together, these results further support the idea that Mx1-labeled periosteal SSCs with αSMA-GFP expression (hereafter Mx1+αSMA+ P-SSCs) mainly respond to cortical bone injury and supply new osteoblasts.
Mx1+αSMA+ P-SSCs have a migratory mechanism regulated by CCR5 in vivo.
Skeletal progenitors have been proposed to have migratory or circulatory potential (Nitzsche et al., 2017). However, there is no in vivo evidence for the endogenous migratory mechanism of P-SSCs. To test whether P-SSCs migrate toward the injury site in vivo, we performed sequential Z-stack imaging of individual Mx1+αSMA+ P-SSCs and the detailed structures of the periosteum, bone matrix, and BM near the injury sites immediately (0), 24, and 48 hours after injury (Figure 4A). We noted that multiple Mx1+αSMA+ P-SSCs appeared to move toward the injury site (Figure 4A, arrows), and we confirmed these results by Z-stack imaging. However, in agreement with Figure S5E, the endosteal and vessel-associated Mx1+ BM cells were not observed to shift their position (Figure 4A, BM), indicating the presence of a migratory mechanism of Mx1+αSMA+ P-SSCs and suture cells.
Figure 4. Rapidly migrating Mx1+αSMA+ P-SSCs uniquely express CCR5 in vivo.

A. Using the 3-month old Mx1/Tomato/SMA-GFP mice from Figure 1A, the early injury response of Mx1+αSMA+ cells in the periosteum (PO) and suture, and bone marrow (BM) mesenchymal cells near the calvarial bone injury site, were imaged at the indicated times after injury (≥ 5 replicates). Arrows indicate migrating Mx1+αSMA+ P-SSCs. Bone (blue). B. List of cell migration genes that are most abundantly expressed in Mx1+Ocn− periosteal cells (Mx1+ PCs, cells from figure S2E) compared to control populations. C. Chemotaxis gene set analysis of Mx1+Ocn− periosteal cells (Mx1+ PCs) vs osteocalcin-GFP+ osteoblasts (Ocn+ obs). D. Relative cell surface expression of CCR5 (top histograms, graph) and CXCR4 (bottom histograms) in Mx1+αSMA+ P-SSCs (M+S+) from 3- to 4-month old Mx1/Tomato/SMA-GFP mice, compared to those of indicated cells and CD45+ cells. The graph represents mean ± SE from 3–5 mice (males and females) with three independent experiments. E. Relative cell surface expression of CCR5 in the indicated BM cells from the same mice in 4D. F. Relative cell surface expression of CCR5 in Prx1+ periosteal cells from 1-month-old Prx1CreER-GFP+ mice. G. Periosteal Prx1+ population in the calvaria of 2-month-old Prx1CreER-GFP mice (top) and Prx1+Mx1+ (bottom box, Tomato+GFP+) populations in the calvarial periosteum of Mx1/tomato/Prx1-GFP mice without tamoxifen induction (pIpC at 3 weeks of age, WT-BMT at 5 weeks of age) were examined by intravital microscopy.
We next examined molecules involved in P-SSC migration by Affymetrix-based global gene expression analysis of Mx1+Ocn− periosteal cells (Figure 4B and S5C). Interestingly, the expression of two specific CC chemokine receptors, CCR3 and CCR5, was ~4-fold higher in Mx1+Ocn− periosteal cells compared to Nestin+ BM cells and control cells (Figure 4B). Mx1+Ocn− periosteal cells also had increased expression of genes involved in cell migration and proliferation, including Integrin α3, Icam1, and Fgf5. Consistently, Gene Set Enrichment Analysis (GSEA) showed a significant upregulation of the filopodia-associated migratory gene set in Mx1+Ocn− periosteal cells compared to more mature osteoblasts (Figure 4C). Subsequent FACS analysis determined that almost all Mx1+αSMA+ P-SSCs expressed CCR5 (92%, Figure 4D) and CCR3 (90%, data not shown) on the cell surface, while more differentiated Mx1+ cells (Mx1+SMA-), αSMA+ cells (Mx1-SMA+), and CD45+ hematopoietic cells had much lower CCR5 expression (Figure 4D). By contrast, within the BM we did not observe a distinct Mx1+αSMA+ population (~0.01% of CD45−CD31−Ter119− stromal cells), and there was no significant upregulation of CCR5 in Mx1+ cells (26% of Mx1+140a+) and αSMA+ cells (~20% of SMA+, n=5) in the BM (Figure 4E). Furthermore, there was no observable differential expression of CXCR4 (CXCL12 receptor, a known chemoattractant for BMSCs) in Mx1+αSMA+ P-SSCs, suggesting the presence of a CCR5-mediated regulatory mechanism for P-SSC migration.
It may be possible that Mx1 does not label all SSCs in the BM and that Mx1− BM-SSCs can express CCR3/5. Using LepR-Cre+Rosa26-Tomato+ mice to label postnatal BM-SSCs (Zhou et al., 2014), we tested if LepR-Cre-labeled (herein LepR+) cells in the BM express CCR5 and contribute to osteolineage cells in injury repair. Notably, we observed that the LepR+ cells are present in both the periosteum and the BM (Figure S6A), and that LepR+ periosteal cells increased the contribution to periosteal osteoblasts and osteocytes in growing bones (Figure S6B). In addition, the majority of LepR+ periosteal cells expressed CCR5 (74%) with CD140a (82%), while only 20% of LepR+ cells from the BM expressed CCR5 (Figure S6C), further supporting the selective expression of CCR5 in endogenous P-SSCs. Since both Mx1+ and LepR+ periosteal cells express CCR5, we reasoned that a subset of LepR+ periosteal cells can be P-SSCs with αSMA-GFP expression. Thus, we generated LepR-Cre+Rosa26-Tomato+αSMA-GFP+ mice and found that a subset of LepR+ periosteal cells are αSMA-GFP positive (Figure S6D, top) but LepR+ BM cells have no such expression (Figure S6D, bottom). When LepR+αSMA+ cells were tracked in the calvarial injuries, we consistently observed the repopulation of LepR+αSMA+ cells and their contribution to bone-forming osteoblasts (LepR+ Obs) at injury sites (Figures S6E & S6F). These data demonstrate that both Mx1-Cre and LepR-Cre can mark a common postnatal P-SSC population with CCR5 and αSMA-GFP expression and that they are required for bone healing osteoblasts.
Prx1+ cells in the periosteum were reported to have progenitor characteristics and contribute to injury repair (Duchamp de Lageneste et al., 2018; Ouyang et al., 2013). Therefore, to exclude any possible contamination of CCR5+ hematopoietic cells in the Mx1+αSMA+ P-SSC fraction, we performed FACS analysis of periosteal cells from Prx1-CreER-GFP mice and confirmed that Prx1-GFP+ (Prx1+) periosteal cells have high cell surface expression of CCR5 and CD140a (86%; Figure 4F). Intravital imaging of Mx1/Tomato/Prx1-GFP mice without tamoxifen induction revealed that nearly all Prx1+ periosteal cells overlap with Mx1+ P-SSCs (Tomato+) in vivo (Figure 4G), supporting that Mx1+ P-SSCs with Prx1-GFP expression have specific CCR5 expression that may regulate early P-SSC migration toward injury sites in vivo.
CCL5, but not TNFα or CXCL12, induces the in vivo migration of P-SSCs.
To determine which growth factors and cytokines stimulate the migration of P-SSCs, we performed in vitro transwell migration assay (12 hours) with various bone growth factors, followed by four-day culture of migrating Mx1+Ocn− periosteal cells. We found that tumor necrosis factor alpha (TNFα) and CCL5 (a common ligand for CCR3 and CCR5) (Blanpain et al., 2001) significantly enhance the migration capability of Mx1+Ocn− periosteal cells (Figure 5A). To test if CCL5 induces the migration of P-SSCs toward injury sites in vivo, we performed in vivo imaging of Mx1+αSMA+ P-SSCs one hour after the administration of CCL5 (2 μL, 10 ng/μL in Matrigel) at the injury site. Surprisingly, we could visualize in real time the in vivo migration of Mx1+αSMA+ P-SSCs by administering CCL5 locally (Figures 5B & 5C bottom, Movie S3), whereas various doses of TNFα or CXCL12 (2 μL,10–50 ng in Matrigel) did not induce their migration in vivo (Figures 5B & 5C top, Movie S1 & S2). We found that CCL5 induced the directional migration of adjacent Mx1+αSMA+ P-SSCs toward the injury site while many distal P-SSCs remained stationary, with little movement (Movie S3 & S4). When the total distance (i.e. multidimensional movement) of individual migrating cells (~20 cells/group) was assessed for 2 hours (1 minute/frame), we observed that their average migration speed under CCL5 activation was 57 μm/h, while there was no distinct movement with TNFα and CXCL12 treatment (Figures 5B & 5C). We also found that CCL5 induced the directional migration of Prx1+ P-SSCs (GFP+) toward the injury site with a similar migration speed (Figure 5D, Movie S5), supporting that these CCL5-mediated migrating cells are the non-hematopoietic Mx1+αSMA+ P-SSC fraction. Despite a wide variation in the migration of each P-SSCs due to a potential difference in CCL5 concentration at discrete locations at the time of imaging, our data indicate that P-SSCs are indeed dynamic and migrate upon CCL5 activation.
Figure 5. Migration of P-SSC is induced by CCL5 in vitro and in vivo.

A. The effects of TNF-α (10 ng/mL), CCL5 (20 ng/mL), PDGF-β (20 ng/mL), BMP (20 ng/mL), and TGF-β (10 ng/mL) on the migration of sorted Mx1+Ocn− periosteal cells (CD105+CD140a+Mx1+Ocn-GFP− cells from Figure S2E; 5,000/well) were examined using a transwell assay (8-μm pore) for 12 hours (left), followed by colony formation (>50 cells) for 4 days (right) (≥ 3 replicates). *P<0.05; **P<0.01. B. The average in vivo migration distance of individual Mx1+αSMA+ P-SSCs induced by TNFα, CXCL12, or CCL5 (all, 2 L at 10 ng/μL of Matrigel) was measured in Mx1/Tomato/αSMA-GFP mice from Figure 1A. C. In vivo real-time imaging shows that CCL5, but not TNFα or CXCL12, at the injury site (dotted line) induced the migration of Mx1+αSMA+ P-SSCs (Tomato+GFP+). The numbers indicate the time of imaging. Representative of 3–5 mice per group. D. CCL5 induces in vivo real-time migration of Prx1+ P-SSCs (GFP+) in Prx1CreERGFP mice treated with CCL5 (2 μL at 10 ng/μL of Matrigel) or Matrigel alone (2 μL, Control). Bottom images represent sequential images of Prx1+ P-SSC migration over 16 minutes (1 image per minute, representative of 5 mice). Scale bar is 50 μm.
CCL5 is required for bone healing and local provision of CCL5 accelerates P-SSC recruitment and bone healing.
To test the physiologic importance of CCL5 in P-SSC migration and in injury repair, we assessed the bone healing of Ccl5-deficient mice compared to that of wild-type littermates. While there were no noticeable differences in bone parameters (BV/TV, travecular numbers, and trabecular thickness) of 3-month old Ccl5-deficient mice (data not shown), external callus mineralization (External BV/TV) and new bone formation (BV/TV) at defect sites 10 days after injury were significantly reduced (~35%, P<0.01) in Ccl5-deficient mice compared to wild-type controls (Figure 6A). Next, to test whether immune cells are the source of CCL5 for P-SSC migration and bone healing, wild-type mice were irradiated (9.5 Gy) and transplanted with Ccl5-deficient BM (106 cells/mouse, Ccl5−/− BMT) or wild-type BM as a control (WT-BMT). Five weeks after transplantation, we induced a tibial drill-hole defect, and assessed bone healing 10 days after injury. We observed partial but significant delay in bone healing of Ccl5−/− BMT mice compared to WT-BMT mice (Figure 6B). Consistent with previous reports showing CCL5 expression in multiple cell types upon injury (Aldinucci and Colombatti, 2014; Kovacic et al., 2010), these results imply that immune cells and other tissue cells express CCL5 at the injury site and contribute to P-SSC migration and bone healing. Next, bone injuries were induced in mouse tibia at ~3–4 months of age, and CCL5 (+CCL5) mixed with Matrigel (2 μL at 10 ng/μL) or Matrigel alone (CON) were locally treated at the site of mechanical injury on days 0, 2, and 4. We found that the treatment of tibial injuries with CCL5 significantly increased early bone mineralization (Figure 6C, D7). However, this effect was not observed 14 days post-injury, implying that CCL5 is necessary and beneficial for early bone healing. We further assessed the P-SSC recruitment and therapeutic potential of CCL5 compared to that of CXCL12 (a known chemoattractant for BMSCs). Bone injuries were induced in mouse calvaria of pIpC-induced, BM-replaced Mx1/Tomato/αSMA-GFP mice at ~4 months of age. Next, we performed local treatment with CCL5 (+CCL5) or CXCL12 (+CXCL12) mixed with Matrigel (2 μL at 10 ng/μL) or Matrigel alone (CON) at the site of injury on days 0, 2, and 4. We observed that treatment with CCL5 induced rapid recruitment of Mx1+αSMA+ P-SSCs at day 5 and substantially increased their number at day 10 with accelerated bone mineralization (Figures 6D & E). By contrast, CXCL12 treatment did not show such increase in P-SSC recruitment and bone healing, indicating that high CCL5 in defect sites is beneficial and specific to P-SSCs. Taken together, these results suggest that local, early CCL5 expression after an injury is required for the migration of endogenous P-SSCs toward injury sites and early bone healing.
Figure 6. CCL5 is necessary for Mx1+αSMA+ P-SSC recruitment and activation leading to accelerated bone healing.

A. Representative μCT images of proximal tibial injuries (left) of age and sex-matched WT (n=12) or 2- to 4-month old CCL5−/− mice 10 days post-injury (n=13). BV/TV of external callus (left) and injury sites (right) was assessed. B. WT-BL6 mice (n=10 per group) were irradiated (9.5 Gy) and transplanted with 106 WT (WT-BMT) or Ccl5−/− BM mononuclear cells (Ccl5−/− BMT). Five weeks later, BV/TV of injury sites was assessed at 10 days after tibial drill-hole injuries. C. Representative μCT images (top) and BV/TV quantification (bottom) of tibia injuries (WT-BL6 mice) treated locally with CCL5 (+CCL5; 10 μL at 1 ng/μL Matrigel) or under control conditions (10 μL Matrigel, CON) on Days 0, 2, and 4 post-injury (n=5–7). D. Sequential in vivo imaging of 3- to 4-month old Mx1/Tomato/αSMA-GFP mice from Fig. 1 treated with supplemental CCL5, CXCL12 (2 μL at 10 ng/μL of Matrigel), or Matrigel alone (2 μL, CON) at the indicated times after calvarial injury. E. Representative μCT images (left) and BV/TV (right) analysis of calvarial injuries (from 6D) treated with CCL5, or CXCL12, 10 days post-injury (left).
The deletion and inhibition of CCR5 delay P-SSC migration and bone repair in vivo.
CCL5 is known to have high affinity to CCR1 and CCR5 and weak affinity to CCR3 (Blanpain et al., 2001). We found that Mx1+αSMA+ P-SSCs differentially expressed CCR5 but not CCR1 (Figure 4). Therefore, to evaluate the function of CCR5 in bone healing, a tibial drill-hole defect (~1 mm) model was used to assess bone healing at 10 days post-injury in Ccr5-deficient mice. Ccr5-deficient mice displayed a significant reduction in external callus formation (TV) and bone healing (BV/TV) of defect sites than wild-type littermate controls (Figure 7A). To further evaluate the specific function of CCR5 in Mx1+αSMA+ P-SSCs migration during early bone healing, calvarial injuries were induced in Mx1/Tomato/αSMA-GFP mice (2 months after pIpC and WT-BMT) and treated with a pharmacological CCR5 inhibitor (Maravoric) every other day for 10 days. Inhibition of CCR5 substantially reduced the recruitment of Mx1+αSMA+ P-SSCs 5 and 10 days post-injury (Figure 7B). Furthermore, a significant reduction of bone healing (BV/TV) was observed at 7 days after injury (Figure 7C). These data indicate that CCL5-CCR5 signaling is an important component of P-SSC migration in response to injury.
Figure 7. CCR5-CCL5 influences periosteal cell migration and bone healing.

A. The external callus volume (left), BV/TV (right), and μCT images of proximal tibial bone injuries in WT or Ccr5−/− mice at 10 days post-injury were used to assess bone healing. B-C. Sequential in vivo imaging of Mx1/Tomato/αSMA-GFP mouse (3-month-old) calvarial injuries treated with a CCR5 inhibitor (+R5 Inhibitor; 10 μL at 2.5 μg/μL Maraviroc in Matrigel) or gel control (10 μL of Matrigel) for 10 days post-injury (B). Representative μCT images on Day 7 post-injury (C, left) and BV/TV analysis (C, right graph) were used for the quantification of bone healing. D. Primary cells isolated from human periosteal tissue (Human PO) were analyzed for expression of CCR5 and indicated SSC markers. Dotted line, isotype controls. E. Single-cell driven colonies from human periosteal cells were cultured for 7–10 days (P1), then analyzed for expression of CCR5 and SSC markers (CD105 and CD140a). Blue line, isotype controls. F. A scratch assay was used to determine the effects of 10 ng/mL of CCL5 or TNFα on human periosteal cell migration (10,000 cells/well). G. A transwell migration assay (8-μm pore) was used to determine the effects of CCL5 (5 ng/mL) or Cxcl12 (5 ng/mL) on human periosteal migration (P1, 15,000/well) migration for 24 hours.
To confirm the translational implications of our findings, primary cells isolated from collagenase-digested human periosteal tissue were analyzed for the expression of human SSC markers and CCR5. We found that CD45−CD31−CD235a−CD140a+ human periosteal cells expressed CCR5 as well as previously defined human SSC markers such as CD105, CD146, and CD271 (Figure 7D). When single-cell driven colonies from human periosteal tissues were cultured for 7 days (passage 1), all clonogenic periosteal cells homogeneously expressed CCR5 and human SSC markers, implicating that clonogenic progenitor cells in the human periosteum express CCR5 (Figure 7E, P1). Further consistent with Figure 6D, treatment with CCL5, but not with CXCL12, significantly induced human periosteal cell migration in vitro (Figure 7F) and in a transwell migration assay (Figure 7G). These data indicate that local treatment with CCL5 enhances the recruitment of both mouse and human P-SSCs and may contribute to the recovery of fractured bones.
DISCUSSION
The in vivo identity and functional difference of SSCs present at multiple tissue locations have long been a source of controversy (Caplan and Correa, 2011). Our data indicate that the Mx1 and αSMA combination selectively labels adult P-SSCs that maintain in vivo stem cell function, as exemplified by their long-term repopulation capability and by their contribution to the majority of bone-forming osteoblasts, even after multiple injuries and serial transplantations (Figure 3). The Mx1+αSMA+ P-SSCs are predominantly osteolineage-restricted with injury-induced chondrogenic potential, which is more consistent with the characteristics of osteochondrogenic stem cells (Ransom et al., 2016) than broadly defined BM-MSCs (Bianco et al., 2008). Whether the chondrocyte intermediates derived in vivo from bipotential Mx1+αSMA+ P-SSCs or from a distinct subset within the Mx1+ pool has to be elucidated.
BM-SSCs labeled by LepR (Zhou et al., 2014) or Grem1 (Worthley et al., 2015) are known to contribute to bone injury repair. However, many of these studies have used fracture models in which the measurement of the proportional contribution of P-SSCs and BM-SSCs is limited. In fact, Grem1+ BM-SSCs are reported to contribute only ~28% of osteoblasts to injury repair (Worthley et al., 2015), while Mx1+ P-SSCs have a far greater (over 80%) contribution (Figure 2). Further, we show here that the distinct LepR+ periosteal cell subset is αSMA-GFP positive (Figure S6), and these cells contribute to bone healing rather than LepR+ BMSCs (Figures S6E & F), which implies that many Mx1+αSMA+ P-SSCs functionally overlap with LepR+αSMA+ periosteal cells with CCR5 expression (Figure S6C). Given that LepR+ cells first appear in the periosteum and BM at E17.5 and contribute to postnatal osteoblasts and BM stromal cells (Zhou et al., 2014), it is likely that Mx1+αSMA+ periosteal cells acquire a distinct P-SSC function after the segregation of the bone compartment and throughout the bone growing process. Conversely, it is possible that different injury models at different ages and locations might give a different result and that the BM-SSCs may have a greater contribution if trabecular bones are injured. Taken together, our data support the conclusion that, in adult bone, P-SSCs are a distinct subset of long-term repopulating stem cells responsible for cortical bone regeneration and repair, and that they differ significantly from BM-SSCs that are necessary for trabecular bone maintenance, bone resorption and hematopoietic stem cell (HSC) microenvironment.
P-SSCs are reported to display endochondral ossification and intramembranous bone formation, while BM-SSCs only participate in the latter process (Colnot, 2009; Colnot et al., 2006). However, the molecular and cellular mechanisms that can explain these functional differences remain to be determined. Notably, our data showed for the first time that Mx1+ P-SSCs rapidly respond to bone injury through a newly identified migration mechanism, which we did not observe in Mx1+, Nestin-GFP+, nor in LepR+ BM cells in vivo. In addition, P-SSCs have distinct molecular signatures compared to BM-SSCs, which is exemplified by the specific expression of CCR5 (a receptor for CCL5) and its regulation of P-SSCs rather than BM-SSCs (Figures 4B & D). Our data also revealed that P-SSCs rapidly respond to injury by local translocation without detectable stem cell circulation, as reported by others (Otsuru et al., 2008). Intravital imaging of endogenous stem cells revealed that CCL5, but not TNFα or CXCL12, induces the migration of P-SSCs and increases bone healing in vivo (Figures 5B, & C). To the best of our knowledge, this is the first visualization of endogenous SSC migration toward bone injury sites in real time and the first identification of a selective signal necessary for P-SSC migration and activation in both mouse and human. Furthermore, CCL5- and CCR5-deficient mice displayed significant reduction of new bone formation and external callus volume at defect sites after injury (Figure 6A & 7A, respectively). Thus, our data support the notion that there are both intrinsic and extrinsic cues that regulate the behaviors and functions of specific stem cell subsets. Furthermore, the periosteum might have an essential role in facilitating and maintaining a distinct SSC population with a distinct regulatory mechanism under stress conditions.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILTY
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dongsu Park (dongsu.park@bcm.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
Six- to eight-week old C57BL/6, Mx1-Cre (Kuhn et al., 1995), Rosa26-loxP-stop-loxP-tdTomato (Rosa-Tom) (Srinivas et al., 2001), and Rosa26-loxP-stop-loxP-DTR (iDTR) (Buch et al., 2005) mice were purchased from The Jackson laboratory. Osteocalcin-GFP, αSMA-GFP (Matthews et al., 2016), and Nestin-GFP (Mignone et al., 2004) (C57/BL6 background) mice were kindly provided by Drs. Ivo Kalajzic and Henry Kronenberg. Genotyping of all Cre-transgenic mice was performed by PCR (GenDEPOT) using primers detecting the Cre sequence. Genotyping of the Rosa locus was performed according to The Jackson laboratory’s protocols. Ccr5−/− and Ccl5−/− mice were obtained from The Jackson laboratory, and genotyping was performed according to The Jackson laboratory’s protocols. For Mx1-Cre induction, mice were injected intraperitoneally with 25 mg/kg of pIpC (Sigma) every other day for 10 days. For tracing of Mx1-labeled cells at the injury sites, Mx1 mice were lethally irradiated with 9.5 Gy one day before intravenous transplantation of 106 whole bone marrow mononuclear cells from wild-type C57BL/6 mice (WT-BMT). After six to eight weeks of recovery (when the host’s hematopoietic cells are less than 5%), mice were subjected to bone injury experiments. For the inducible ablation of Mx1/Tom/iDTR+ progenitors in the periosteum, mice were injected subcutaneously (i.e. calvaria) daily for 7 days with 30–50 μL of diphtheria toxin (1 μg/mL PBS). For Ccl5−/− BM replacement studies, wild-type C57BL/6 syngeneic mice (4–6 weeks old) were lethally irradiated with 9.5 Gy at one day before intravenous transplantation of 106 whole bone marrow mononuclear cells from Ccl5−/− mice (Ccl5−/− BMT) or WT mice (WT-BMT); five weeks later, mice were subjected to bone injury experiments. All mice were maintained in pathogen-free conditions, and all procedures were approved by Baylor College of Medicine’s Institutional Animal Care and Use Committee (IACUC) and Human IRB Committee.
Human Periosteum
For human periosteal cell isolation, normal periosteal tissue was obtained from femurs and tibias of five patients (mean age, 15 years; range, 10–22 years), who provided informed consent when undergoing fracture surgery. The two males were aged 10 and 11 years; the three females were aged 17, 21 and 22 years (IRB: H-40670).
METHOD DETAILS
Live animal imaging pre- and post-injury
For live in vivo imaging of osteogenic cells at injury sites, mice were anesthetized and prepared for visualization under a customized two-photon and confocal hybrid microscope (Leica TCS SP8MP with DM6000CFS) specifically designed for live animal imaging, as described in our previous report (Park et al., 2012). Briefly, a small incision was made on the scalp of Mx1/Tomato/iDTR, Mx1/Tomato/αSMA-GFP, Mx1/Tomato/Nestin-GFP or LepR/Tomato mice, and two separate drilled-holes were generated with a 27G needle on the surface of calvaria, near the intersection of sagittal and coronal sutures. The mice were then mounted on a 3-D axis motorized stage (Anaheim Automation Anaheim, CA), and the calvarial bones and BM structures were visualized by second harmonic generation (440nm SHG by femto-second titanium:sapphire laser pulses: 880 nm) from bone collagen fibers to identify the injury sites and the intersection of the sagittal and coronal sutures. GFP-expressing cells (488 nm excitation, 505–550 nm detection) and Tomato-expressing cells (561 nm excitation, 590–620 nm detection) were simultaneously imaged using confocal spectral fluorescence. All images were recorded with their distances to the intersection of the sagittal and coronal sutures to define their precise location. Each image was recorded by Z-stacks with 50–100 mm depth from the bone surface at a 2-mm interval. A PCI-based image capture board (Snapper, Active Silicon) was used to acquire up to three channels simultaneously using the Leica Application Suite software (Version 3.3). After in vivo imaging, the scalp was closed using a VICRYL plus suture (Ethicon), and post-operative care was provided as previously described (Park et al., 2012). 3D Images were reconstructed using the Leica Application Suite software, and osteoblasts were counted. A two-tailed, unpaired t-test was used to identify significant differences (P<0.05) between groups.
In vivo imaging was performed on Mx1/Tomato/iDTR mice, after seven days of treatment with DT, at the time a calvarial injury was generated. Injuries were sequentially imaged on days 0, 5, 11, and 16 post-injury. In a separate experiment, injuries were made to the calvaria of Mx1/Tomato/αSMA-GFP mice, which were then locally treated with 20–100 ng of murine CCL5 or CXCL12 (10 ng/μL of Matrigel) on days 0, 2, and 4 post-injury. For this cohort, calvarial injuries were imaged on days 0, 2, and 10 post-injury. For in vivo real-time imaging of SSC migration in living animals, injury sites were treated with 20 ng of CCL5, CXCL12, or TNFα with Matrigel at the injury sites, and one hour later stem cell migration near the injury sites was continuously imaged for 2–4 hours by capturing snapshots every minute. Movies were generated by reconstructing these images using the Leica Application Suite software (~6 frames/s).
Tibial injury and μCT analysis
All tibial injuries were performed using aseptic technique. A small (<1 cm) incision was made on the anterior side of the hind limb below the knee to visualize the proximal tibia. A 20G needle was used to create an injury with ~1 mm in diameter. The skin was closed using sutures, and a small amount of triple antibiotic ointment was applied to the surrounding skin and sutures using a sterile cotton swab. For treatment of tibial injuries with CCL5 (Biolegend), 3–4-month old WT-BL6 mice were treated locally with CCL5 (+CCL5; 10 μL at 1 ng/μL Matrigel) or under control conditions (10 μL Matrigel) on Days 0, 2, and 4 post-injury. For inhibition of CCR5, tibial injuries of 3–4-month old WT-BL6 mice were treated immediately with 4.9 mM Maraviroc (Sigma-Aldrich), and a local injection was given daily for one week. Injury healing was assessed 7 days post-injury. Tibial injuries were also performed on 2-month old WT-BL6 mice as well as age-and sex-matched Ccl5−/− or Ccr5−/− mice. Injury healing was assessed 7- and 14-days post-injury for CCL5 treatment, 7 days post-injury for Maraviroc treatment, and 10 days post-injury for Ccl5−/− and Ccr5−/− mice.
Tibial cortical bone healing was assessed using μCT (Scanco μCT 40, SCANCO Medical AG, Brüttisellen, Switzerland). Scans were performed with an X-ray setting of 55 kVp voltage and 145 uA current with a 200-ms exposure at a high resolution. Calibration images were collected prior to data acquisition. Scans were performed with an effective voxel size of 10 μm3. For image analysis, a global upper threshold of 255 and lower threshold of 120 (μCT grey scale value) were used for all samples to separate the bone from the background and soft tissue. CTan version 1.14.1 (SkyScan, Bruker-microCT) was used to determine bone volume over total volume.
Isolation and flow cytometry analysis of mouse SSCs
To isolate periosteal cells, dissected femurs, tibias, pelvis, and calvaria from mice were placed in PBS, and the overlying skin, fascia, and muscle were carefully removed. The bones with periosteum were incubated in ice-cold PBS with 1% FBS for 30 min, and the loosely associated periosteum was peeled off using forceps, scalpel, and dissecting scissors. The soft floating periosteal tissues collected with a 40-μm strainer were then incubated with 5–10 mL of 0.2% collagenase and 10% FBS in PBS at 37°C for one hour, and the dissociated periosteal cells were washed with PBS, filtered with a 40-μm strainer, and resuspended at ~1×107 cells/mL. To isolate cells from bones and BM, dissected femurs, tibias, and pelvis bones after periosteum removal were crushed with a pestle and rinsed three times to remove and collect BM cells. The remaining bones were fragmented with a scalpel and dissecting scissors, and then the bones and bone marrow were incubated with 10 mL of 0.2% collagenase and 10% FBS in PBS at 37°C for one hour, according to the previously desc ribed protocol (Worthley et al., 2015). The dissociated cells were washed with PBS, filtered with a 40-μm strainer, and resuspended at ~1×107 cells/mL. To analyze or isolate SSCs and osteogenic cells, cells were stained with CD105-PE-Cy7 (clone: MJ7/18), CD140a-APC (clone: APA5), CD45-pacific blue (clone: 30-F11), Sca-1-Alexa Fluor 647 (clone: E13–161.7), Ter119-APC-Cy7 (clone: TER-119), and CD31-eFlour 450 (clone: 390) in combination with CCR3-PE-Cy7 (clone: J073E5) or CCR5-PE-Cy7 (clone: HM-CCR5). Antibodies were purchased from eBioscience unless otherwise stated. Propidium iodide was used for viable cell gating. Flow cytometric experiments and sorting were performed using the LSRII and FACS Aria cytometer (BD Biosciences, San Jose, CA). Data were analyzed with the FlowJo software (TreeStar, Ashland, OR) and represented as histograms, contour, or dot plots of fluorescence intensity.
Cell Transplantation
Periosteal cell isolation and antibody staining for FACs sorting of Mx1/Tomato/αSMA-GFP dual reporter mice were performed as stated above. Three populations of periosteal cells were collected: 1) CD45−CD31−Ter119−CD105+CD140+Mx1+αSMA+ (Mx1+αSMA+), 2) CD45−CD31− Ter119−CD105+CD140+Mx1+αSMA− (Mx1+αSMA−), and 3) CD45−CD31−Ter119−CD105+ CD140+Mx1−αSMA+ (Mx1−αSMA+). Injuries were made to the calvaria of C57BL/6 recipient mice with a 27G needle, and ~5,000 cells in Matrigel (10uL) were transplanted into the injury sites. Four weeks after transplantation, intravital microscopy was used to confirm the engraftment of Mx1+αSMA+ cells. The next day, the calvaria of Mx1+αSMA+ mice were collected, fragmented with a scalpel or dissecting scissors, and dissociated with 10 mL of 0.2% collagenase and 10% FBS in PBS at 37°C. FACs analys is and cell sorting were performed and the Mx1+αSMA+ cell population was collected for secondary transplantation. Mx1+αSMA+ cells (~500 cells) in Matrigel were transplanted the calvarial injury sites of C57BL/6 mice. In vivo imaging and FACs analysis were performed 4 weeks after secondary transplantation. For Mx1+αSMA− and Mx1−αSMA+ cells, ~5,000 cells in Matrigel were transplanted into the calvaria injury sites of C56BL/6 mice. Transplanted cells were then tracked using intravital microscopy 2, 4, and 8 weeks post transplantation. Four weeks after transplantation, the transplanted cells were analyzed by FACs.
Microarray analysis
Cell isolation and antibody staining for FACs sorting of Mx1/Tomato/Nestin, Mx1/Tom/Ocn-GFP, and Osterix-GFP (Osx) reporter mice were performed as stated above. Six populations of cells were collected from two or three male and female mice: 1) CD45+, 2) CD45−CD31−Ter119−Ocn+ (Ocn+), 3) CD45−CD31−Ter119−Osx+ (Osx+), 4) bone marrow CD45−CD31−Ter119−CD105+CD140a+Mx1−Nestin+ (BM Mx-N+), 5) bone marrow CD45−CD31−Ter119−CD105+CD140a+Mx1+Nestin+ (BM Mx+N+), and 6) Periosteum CD45−CD31−Ter119−CD105+CD140a+Mx1+Ocn− (Mx1+PCs). Sorted cell populations were used to isolate RNA using the RNeasy Micro kit (Qiagen), according to the manufacturer’s instructions. Purified RNA was reverse-transcribed, amplified, and labeled with the Affymetrix Gene Chip whole transcript sense target labeling kit. Labeled cDNA (2 biological repeats) from the indicated cells was analyzed using Affymetrix mouse A430 microarrays, according to the manufacturer’s instructions, performed at the Baylor College of Medicine Genomic and RNA profiling core. CEL files (containing raw expression measurements) were imported to Partek GS, and data were preprocessed and normalized using the RMA (Robust Multichip Average) algorithm. Gene Set enrichment analysis was performed as previously described (Subramanian et al., 2005).
RT-qPCR
Cell mRNA was extracted using the Qiagen RNeasy Mini Kit, according to the manufacturer’s instructions. mRNA was quantified and checked for purity using a Nanodrop spectrophotometer (DeNovix, Wilmington, DE). cDNA was generated from 1 μg of mRNA using the cDNA Reverse Transcription Kit (Applied Biosystems). RT-qPCR was performed using PWR SYBR Green (LightCycler 96, Roche). Fold changes were calculated as 2−ΔΔCT with GAPDH used as the endogenous control. A list of primers can be found in Table S1.
Colony-forming efficiency assay and ex vivo differentiation assay
For the CFU-F assay with unfractionated periosteal cells, cells were plated on 6-well plates at a concentration of ~102–104 cells/well in culture medium (αMEM with 20% FBS) for 14 days. For the CFU-F assay with Mx1+SMA+ periosteal cells, FACs-sorted cells were plated on 6-well plates at a concentration of 10 cells/cm2 in culture medium (αMEM with 20% FBS), ensuring that colonies formed at clonal density. The presence of more than 50 cells in a cluster was counted as a colony. To assess osteogenic differentiation, expanded cells from single cell-derived colonies were plated at 2×104 cells/well in a 12-well plate (BD Biosciences) at 33°C. The next day, the medium was replaced with osteogenic induction medium: αMEM with 20% FBS modified with glycerol 2-phosphate (2.16 mg/mL), 2-phospho-l-ascorbic acid (0.05 mg/mL), and dexamethasone (10 nM) (Sigma-Aldrich, G6251, 49752 and D1756, respectively). After 14 days of differentiation, Alizarin Red staining was carried out according to the manufacturer’s instructions. For adipogenic differentiation, 2×104 cells/well were cultured with adipogenic induction medium containing DMEM with 1% antibiotic/antimycotic, 10% FBS, 10 μg/mL insulin, 1 μM dexamethasone, 0.5 μM isobutylmethylxanthine, and 60 μM indomethacin (all reagents purchased from Sigma-Aldrich). After 2 weeks, the cells were washed, fixed in formalin, and stained with Oil Red O (according to the manufacturer’s protocol) to detect lipids. For chondrogenic differentiation, 2×105 cells were centrifuged to form a pellet and the medium was carefully aspirated and replaced with DMEM containing 1% antibiotic/antimycotic, 10% FBS, 1 μM dexamethasone, 20 ng/ml TGF-β3 (R&D Systems), and 200 μM ascorbic acid. After 4 weeks, cells were stained with Alcain blue or Toluidine blue (according to the manufacturer’s protocol). In all experiments, control medium was DMEM containing 10% FBS with 1% antibiotic/antimycotic.
Flow cytometry analysis and in vitro cell migration assay of human periosteal cells
Periosteal tissues were transported to the laboratory in Earl’s balanced salt solution at 4°C. After cutting periosteal tissues into small pieces (0.5–1 cm2) and washing them with PBS to remove blood cells, the pieces were incubated with 5–10 mL of 0.1% collagenase and 10% FBS in PBS at 37°C for one hour, and the dissociated pe riosteal cells were washed with PBS and filtered with a 40-μm strainer. To analyze the expression of human SSC markers, the digested cell suspension was stained with human CD146-APC (clone: SHM-57), CD271-APC (clone: ME20.4), human CD105-APC (clone: 43A3), CCR3-PE (clone: NP-6G4) or CCR5-PE (clone: 5E8) in combination with human CD140a-PE-Cy7 (clone: 16A), human CD45-pacific blue (clone: HI30), human CD235a-pacific blue (clone: H1264), and CD31-Pacific Blue (clone: WM59). Non-immune isotype controls were used as negative controls. For the migration assay, expanded cells from single-cell driven colonies from collagenase-digested human periosteum (P1, ~10 colonies/sample) were plated at 3×104 cells/well in a 12-well plate and incubated at 37°C. The next day, the cell monolayer was scraped in a straight line with a 200 μL pipette tip, as previously described (Liang et al., 2007). Cells were then washed with DPBS, and the medium was replaced. Baseline images were taken, and cells were then treated with either CCL5 or TNFα at a concentration of 10 ng/mL. Cells were incubated at 37°C and re-imaged eight hours after cytokine treatment. For the transwell migration assay, 1.5×104 human periosteal cells (200 μL DMEM with 10% FBS) from single-cell driven colonies (P1) were seeded in the top chamber of an 8-μm pore polycarbonate transwell plate (Corning) and incubated with human CCL5 (10 ng/mL) or human CXCL12 (10 ng/mL) in 800 μL of DMEM plus 10% FBS in bottom chambers for 24 hours. DMEM with 10% FBS alone was used as a negative control. Nonmigrated cells were removed from the upper side of the membrane with a cotton swap, and migrated cells on the bottom side of the membrane and in bottom chamber were counted.
Immunofluorescence
Frozen sections from bones fixed in 4% paraformaldehyde and decalcified were stained with goat or rabbit polyclonal anti-GFP/YFP antibody (Abcam), rabbit anti-Osteocalcin (Abcam), rabbit anti-perilipin (Sigma), and anti-CD31 biotin-conjugated antibody (BD Pharmingen) according to the manufacturer’s instructions. Non-immune rabbit or goat IgG was used negative controls (Abcam). Anti-goat or anti-rabbit Alexa Fluor 488, and anti-rabbit Alexa Flour 595 or 647 were used as secondary antibodies (Invitrogen). Vectashield (Vector Labs) containing DAPI nuclear counterstain was used to mount the sections. Bone and BM structures were visualized by overlaying DAPI staining and polarized transmission lights. Images were acquired with a Nikon Eclipse Ti epifluorescence microscope equipped with a Q-imaging Micropublisher digital CCD color camera, or with a Zeiss LSM 780 confocal microscope in the Optical Imaging and Vital Microscopy Core at BCM. Images were processed with the Nikon NID-elements software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Microarray Analysis
Microarray data was pre-processed for normalization using a robust multichip average (RMA) technique. Statistical differences were calculated with the limma package in R. Cluster 3.0 software was used for post-processing cluster analysis and plotted using Java TreeView software. We assessed pairwise comparisons between each population of sorted cells. We evaluated the number of statistically different genes by changing the p-value statistical criteria for acceptance. We determined an acceptance criterion of P<0.05.
In vivo imaging, immunohistochemistry, and μCT analysis
For comparison of 3 or more groups, differences were analyzed by one-way analysis of variance (ANOVA) with a significant main effect of P<0.05 followed by a Tukey’s multiple comparisons test to detect differences between groups at a significance of P<0.05. Differences between two experimental groups were determined with an unpaired two tail t-test at a significance of P<0.05. Outcomes are reported as means ± standard error. Number of samples per group are indicated in the figure legends.
DATA AND CODE AVAILABILTY
The NIH GEO accession number for the microarray analysis data included in this paper is GFE107798.
Supplementary Material
Time-lapse intravital imaging of Mx1+αSMA+ P-SSCs (red and green) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with TNFα (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Arrow indicates the site of TNFα treatment. Playback, 6 fps; scale bar = 50 μm.
Time-lapse intravital imaging of Mx1+αSMA+ P-SSCs (red and green) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with CXCL12 (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Arrow indicates the site of CXCL12 treatment. Playback, 6 fps; scale bar = 50 μm.
Time-lapse intravital imaging of proximal Mx1+αSMA+ P-SSCs (<500 μm from injury) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with CCL5 (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Arrow indicates the site of CCL5 treatment. Playback, 6 fps; scale bar = 25 μm.
Time-lapse intravital imaging of distal Mx1+αSMA+ P-SSCs (~1 mm from injury) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with CCL5 (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Playback, 6 fps; scale bar = 50 μm.
Time-lapse intravital imaging of distal Prx1+ P-SSCs (~1 mm from injury) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with CCL5 (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Playback, 6 fps; scale bar = 50 μm.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-Cy7 anti-mouse CD105 | BioLegend | Cat#120409 |
| APC anti-mouse CD140a | eBioscience | Cat#17-1401-81 |
| Pacific blue anti mouse CD45 | BioLegend | Cat#103126 |
| Alexa Fluor 647 anti-mouse Sca-1 | BioLegend | Cat#122517 |
| APC-Cy7 anti-mouse Ter119 | BioLegend | Cat#116223 |
| eFlour 450 anti-mouse CD31 | eBioscience | Cat#48-0311-80 |
| PE-Cy7 anti-mouse CCR3 | BioLegend | Cat#144513 |
| PE-Cy7 anti-mouse CCR5 | BioLegend | Cat#107017 |
| APC anti-mouse CD200 | Biolegend | Cat#123809 |
| APC anti-mouse VCAM1 | BioLegend | Cat#105717 |
| PerCP-Cy5.5 anti-mouse CXCR4 | BioLegend | Cat#146509 |
| APC anti-human CD105 | BioLegend | Cat#3323207 |
| Pacific blue anti-human CD45 | BioLegend | Cat#304021 |
| Pacific blue anti-human CD31 | BioLegend | Cat#303113 |
| PE anti-human CCR3 | BioLegend | Cat#310705 |
| PE anti-human CCR5 | eBioscience | Cat#12-1956-41 |
| PE-Cy7 anti-human CD140a | BioLegend | Cat#323507 |
| APC anti-human CD146 | BioLegend | Cat#342011 |
| APC anti-human CD271 | BioLegend | Cat#345107 |
| APC anti-human CD200 | BioLegend | Cat#329307 |
| Pacific Blue anti-human CD235 | BioLegend | Cat#319108 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Polyinosinic:polycytidylic acid (pIpC) | Sigma | |
| Collagenase, Type I, powder | Sigma | C0130 |
| Recombinant Murine CCL5 (RANTES) | Biolegend | 594206 |
| Recombinant Murine SDF-1 (CXCL12) | Biolegend | 578702 |
| Recombinant Human TNFα | Gibco | PHC3016 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Microarray analysis data | NIH GEO | GFE107798 |
| Experimental Models: Cell Lines | ||
| Primary human periosteal cells | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.Cg-Tg(Mx1-cre)1Cgn/J | The Jackson Laboratory | JAX: 003556 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX: 007914 |
| Mouse: C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J | The Jackson Laboratory | JAX: 007900 |
| Mouse: Osteocalcin-GFP | Drs. Ivo Kalajzic and Henry Kronenberg | NA |
| Mouse: αSMA-GFP36 | Drs. Ivo Kalajzic and Henry Kronenberg | NA |
| Mouse: Nestin-GFP37 | Drs. Ivo Kalajzic and Henry Kronenberg | NA |
| Mouse: B6.129(Cg)-Leprtm2(cre)Rck/J | The Jackson Laboratory | JAX: 008320 |
| Mouse: B6.Cg-Tg(Prrx1-cre/ERT2,-EGFP)1Smkm/J | The Jackson Laboratory | JAX: 029211 |
| Mouse: C57BL/6 | The Jackson Laboratory | JAX: 000664 |
| Mouse: B6.129P2-Ccr5tm1kuz/J | The Jackson Laboratory | JAX: 005427 |
| Mouse: B6.129P2-Ccl5tm1Hso/J | JAX: 005090 | |
| Oligonucleotides | ||
| Refer to Table S1 | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| Fiji/Imagej/BoneJ | Fiji.sc/ | N/A |
| Leica Application Suite software (Version 3.3) | Leica Microsystems | N/A |
| FlowJo | Tree Star | N/A |
| GraphPad Prism | GraphPad Software | N/A |
| Other | ||
Highlights.
Mx1 and αSMA combination selectively labels SSCs resident in adult periosteum (PSSC).
Mx1+αSMA+ P-SSCs are long-term repopulating and functionally distinct SSCs.
Mx1+αSMA+ P-SSCs and human P-SSCs specifically express the CCL5 receptor, CCR5.
CCL5 induces P-SSC migration in vivo and its loss delays bone healing.
ACKNOWLEDGEMENTS
This work was supported by the Bone Disease Program of Texas Award and The Caroline Wiess Law Fund Award to D.P., supported by the NIAMS of the National Institutes of Health under award numbers 1K01AR061434 and 1R01AR072018 to D.P and U54 AR068069 to B.L, and by NIDDK of the NIH to D.T.S. We are grateful to B. Garrison and M. Kim for advice and technical assistance, and to Patricia Fonseca for editing the manuscript. We also thank J. Lee and former members of the Park laboratory, Lee laboratory, and Scadden laboratory for fruitful discussions. We also thank M. E. Dickinson, T. J. Vadakkan in the BCM Optical Imaging and Vital Microscopy Core, and J. Sederstrom in the BCM Cytometry and Cell Sorting Core.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
LCO and DP disclose a pending patent entitled “Periosteal Skeletal Stem Cells in Bone Repair” (BAYM.P0264US.P1). The remaining authors declare no competing interests.
REFERENCES
- Aldinucci D, and Colombatti A (2014). The inflammatory chemokine CCL5 and cancer progression. Mediators of inflammation 2014, 292376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P, and Robey PG (2015). Skeletal stem cells. Development 142, 1023–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P, Robey PG, and Simmons PJ (2008). Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell 2, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Buser R, Power CA, Edgerton M, Buchanan C, Mack M, Simmons G, Clapham PR, Parmentier M, and Proudfoot AE (2001). A chimeric MIP-1alpha/RANTES protein demonstrates the use of different regions of the RANTES protein to bind and activate its receptors. Journal of leukocyte biology 69, 977–985. [PubMed] [Google Scholar]
- Bragdon BC, and Bahney CS (2018). Origin of Reparative Stem Cells in Fracture Healing. Curr Osteoporos Rep 16, 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, and Waisman A (2005). A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods 2, 419–426. [DOI] [PubMed] [Google Scholar]
- Buhring HJ, Treml S, Cerabona F, de Zwart P, Kanz L, and Sobiesiak M (2009). Phenotypic characterization of distinct human bone marrow-derived MSC subsets. Ann N Y Acad Sci 1176, 124–134. [DOI] [PubMed] [Google Scholar]
- Caplan AI, and Correa D (2011). The MSC: An Injury Drugstore In Cell Stem Cell, pp. 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CK, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, Tevlin R, Seita J, Vincent-Tompkins J, Wearda T, et al. (2015). Identification and specification of the mouse skeletal stem cell. Cell 160, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C (2009). Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J Bone Miner Res 24, 274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C, Huang S, and Helms J (2006). Analyzing the cellular contribution of bone marrow to fracture healing using bone marrow transplantation in mice. Biochemical and Biophysical Research Communications 350, 557–561. [DOI] [PubMed] [Google Scholar]
- Debnath S, Yallowitz AR, McCormick J, Lalani S, Zhang T, Xu R, Li N, Liu Y, Yang YS, Eiseman M, et al. (2018). Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveza L, Ortinau L, Lei K, and Park D (2018). Comparative analysis of gene expression identifies distinct molecular signatures of bone marrow- and periosteal-skeletal stem/progenitor cells. PLoS One 13, e0190909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchamp de Lageneste O, Julien A, Abou-Khalil R, Frangi G, Carvalho C, Cagnard N, Cordier C, Conway SJ, and Colnot C (2018). Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nature communications 9, 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grcevic D, Pejda S, Matthews BG, Repic D, Wang L, Li H, Kronenberg MS, Jiang X, Maye P, Adams DJ, et al. (2012). In vivo fate mapping identifies mesenchymal progenitor cells. Stem Cells 30, 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YX, Zhang G, Pan XH, Liu Z, Zheng LZ, Chan CW, Lee KM, Cao YP, Li G, Wei L, et al. (2011). Impaired bone healing pattern in mice with ovariectomy-induced osteoporosis: A drill-hole defect model. Bone 48, 1388–1400. [DOI] [PubMed] [Google Scholar]
- Kovacic JC, Gupta R, Lee AC, Ma M, Fang F, Tolbert CN, Walts AD, Beltran LE, San H, Chen G, et al. (2010). Stat3-dependent acute Rantes production in vascular smooth muscle cells modulates inflammation following arterial injury in mice. J Clin Invest 120, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, and Rajewsky K (1995). Inducible gene targeting in mice. Science 269, 1427–1429. [DOI] [PubMed] [Google Scholar]
- Liang CC, Park AY, and Guan JL (2007). In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nature protocols 2, 329–333. [DOI] [PubMed] [Google Scholar]
- Matthews BG, Torreggiani E, Roeder E, Matic I, Grcevic D, and Kalajzic I (2016). Osteogenic potential of alpha smooth muscle actin expressing muscle resident progenitor cells. Bone 84, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, and Frenette PS (2010). Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, and Enikolopov G (2004). Neural stem and progenitor cells in nestin-GFP transgenic mice. The Journal of comparative neurology 469, 311–324. [DOI] [PubMed] [Google Scholar]
- Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E, Suzuki S, Miyauchi-Hara C, Nagoshi N, Sunabori T, et al. (2009). Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med 206, 2483–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, and Scadden DT (2014). The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitzsche F, Muller C, Lukomska B, Jolkkonen J, Deten A, and Boltze J (2017). Concise Review: MSC Adhesion Cascade-Insights into Homing and Transendothelial Migration. Stem cells 35, 1446–1460. [DOI] [PubMed] [Google Scholar]
- Olivos-Meza A, Fitzsimmons JS, Casper ME, Chen Q, An KN, Ruesink TJ, O’Driscoll SW, and Reinholz GG (2010). Pretreatment of periosteum with TGF-beta1 in situ enhances the quality of osteochondral tissue regenerated from transplanted periosteal grafts in adult rabbits. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society 18, 1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, and Nagasawa T (2010). The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33, 387–399. [DOI] [PubMed] [Google Scholar]
- Otsuru S, Tamai K, Yamazaki T, Yoshikawa H, and Kaneda Y (2008). Circulating bone marrow-derived osteoblast progenitor cells are recruited to the bone-forming site by the CXCR4/stromal cell-derived factor-1 pathway. Stem Cells 26, 223–234. [DOI] [PubMed] [Google Scholar]
- Ouyang Z, Chen Z, Ishikawa M, Yue X, Kawanami A, Leahy P, Greenfield EM, and Murakami S (2013). Prx1 and 3.2kb Col1a1 promoters target distinct bone cell populations in transgenic mice. Bone 58, 136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, Lin CP, Kronenberg HM, and Scadden DT (2012). Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 10, 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Spencer JA, Lin CP, and Scadden DT (2014). Sequential in vivo imaging of osteogenic stem/progenitor cells during fracture repair. Journal of visualized experiments : JoVE 87, e51289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom RC, Hunter DJ, Hyman S, Singh G, Ransom SC, Shen EZ, Perez KC, Gillette M, Li J, Liu B, et al. (2016). Axin2-expressing cells execute regeneration after skeletal injury. Sci Rep 6, 36524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SJ, van Gastel N, Carmeliet G, and Luyten FP (2015). Uncovering the periosteum for skeletal regeneration: the stem cell that lies beneath. Bone 70, 10–18. [DOI] [PubMed] [Google Scholar]
- Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M, et al. (2007). Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131, 324–336. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, and Costantini F (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, Levin D, Schwartz MG, Uygur A, Hayakawa Y, et al. (2015). Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Feng J, Ho TV, Grimes W, Urata M, and Chai Y (2015). The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nature cell biology 17, 386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BO, Yue R, Murphy MM, Peyer JG, and Morrison SJ (2014). Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 15, 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse intravital imaging of Mx1+αSMA+ P-SSCs (red and green) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with TNFα (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Arrow indicates the site of TNFα treatment. Playback, 6 fps; scale bar = 50 μm.
Time-lapse intravital imaging of Mx1+αSMA+ P-SSCs (red and green) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with CXCL12 (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Arrow indicates the site of CXCL12 treatment. Playback, 6 fps; scale bar = 50 μm.
Time-lapse intravital imaging of proximal Mx1+αSMA+ P-SSCs (<500 μm from injury) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with CCL5 (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Arrow indicates the site of CCL5 treatment. Playback, 6 fps; scale bar = 25 μm.
Time-lapse intravital imaging of distal Mx1+αSMA+ P-SSCs (~1 mm from injury) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with CCL5 (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Playback, 6 fps; scale bar = 50 μm.
Time-lapse intravital imaging of distal Prx1+ P-SSCs (~1 mm from injury) on the outer bone surface (blue, collagen SHG) 1 hour after treatment with CCL5 (20 ng/2 μL with Matrigel) at the injury site. Video records were obtained by sequential snapshots captured at 1-minute intervals for ~2 hours. Playback, 6 fps; scale bar = 50 μm.