

Abstract
The estrogen signaling pathway has been reported to modulate prostate cancer (PCa) progression through the activity of estrogen receptors α and β (ERα and ERβ). Given that selective estrogen receptor modulators (SERMs) are used to treat breast cancer, ERs have been proposed as attractive therapeutic targets in PCa. However, many inconsistencies regarding the expression of ERs and the efficacy of SERMs for PCa treatment exist, notably due to the use of ERβ antibodies lacking specificity and treatments with high SERM concentrations leading to off-target effects. To end this confusion, our objective was to study the impact of estrogenic and anti-estrogenic ligands in well-studied in vitro PCa models with appropriate controls, dosages, and ER subtype-specific antibodies. When using physiologically relevant concentrations of nine estrogenic/anti-estrogenic compounds, including five SERMs, we observed no significant modulation of PCa cell proliferation. Using RNA-seq and validated antibodies, we demonstrate that these PCa models do not express ERs. In contrast, RNA-seq from PCa samples from patients have detectable expression of ERα. Overall, our study reveals that commonly used PCa models are inappropriate to study ERs and indicate that usage of alternative models is essential to properly assess the roles of the estrogen signaling pathway in PCa.
Subject terms: Breast cancer, Prostate cancer, Cancer, Endocrinology, Molecular medicine, Oncology, Urology
Introduction
In the context of prostate cancer (PCa), the androgen receptor (AR) has many oncogenic functions such as increasing the proliferation and survival of cancer cells1. Furthermore, it is also now known that AR is an important regulator of metabolic pathways that sustain aberrant proliferation of PCa cells2–6. Accordingly, current hormonal treatments target this receptor with the objective of inhibiting its functions, with anti-androgens or androgen deprivation therapies1,7. However, despite a positive response to these treatments initially, progression of the disease to castration-resistant PCa (CRPC) is mostly inevitable1,7. This highlights the need to find novel approaches for the treatment of PCa.
Estrogens and the most active form estradiol (E2) are naturally produced from androgens through steroidogenesis and have been linked to PCa evolution, as reviewed recently by Dobbs et al.8. Indeed, it has been demonstrated in murine models and in human patients that increased levels of estrogens were positively correlated with the aggressiveness of PCa9–15, and that estrogen synthesis increases in cancer cells during disease progression16–20. This modulation of cancer evolution is thought to be caused by the activity of the estrogen receptors ERα and ERβ, two transcription factors that are both essential for the normal development of the prostate21–23. The actual model supports that ERα has oncogenic functions, as seen in murine models where its activation leads to an increased proliferation of cancer cells24–26; on the other hand, ERβ is thought to act as a tumor suppressor since its loss promotes prostate hyperplasia and the development of the disease21,27–30. Further supporting this model, ERα expression is reported to be increased and ERβ to be decreased during PCa progression31–37. These findings suggest that targeting the estrogen signaling pathway could be a viable therapeutic avenue for the management of PCa and CRPC.
Several anti-estrogen therapies are currently in use for the treatment of ERα-positive breast cancer tumors38. These drugs include tamoxifen, raloxifene, and toremifene, and are now known to be selective estrogen receptor modulators (SERMs). As their name implies, they have the interesting capacity to be antagonist or agonist of the ERs in a tissue-specific manner; for example, tamoxifen is an antagonist of ERα in the mammary gland and breast cancer cells, but is an agonist in other peripheric tissues, such as the bone, thus limiting adverse side-effects of estrogen blockade throughout the whole body39. Importantly in the breast cancer field, assessing ERα expression levels in patient tumors is always performed prior to the selection of anti-estrogen therapies, as the presence of the receptor is tightly linked to the patient’s response. In the context of PCa, a few clinical studies using these compounds were conducted with limited sample sizes, and both null and positive responses were observed7,40–45. However, no initial molecular characterization have been performed in these studies, such as ERα and ERβ expression or activity in prostate tumors, explaining—at least in part—the discrepancies and the heterogeneity in patient responses.
Moreover, not all in vitro PCa models are appropriate to study ER functions, but they have still been inconsistently used in this context. For example, it has been known for decades that LNCaP cells—the most widely used human PCa model—have a mutated AR that can be activated by E2 in addition to androgens46,47 and have low, if any, expression of both ERs48,49. Nevertheless, several groups used this model to study E2 impact on PCa cell proliferation and survival50–52. In addition, the lack of specific ERβ antibody, as clearly described recently48,53,54, has also lead to controvorsies in the literature regarding which PCa cell line models express or not ERβ. Finally, specific ligands for both ERs exist, such as PPT for ERα and DPN for ERβ. Yet, precise dosages have to be used to keep this specificity, as higher concentrations will lead to dual activation of ERs or modulation of other pathways. For example, the EC50 of DPN is of 66 nM and 0.85 nM for ERα and ERβ (Table 1), respectively, and has been used at 100 nM and 1000 nM in previous studies as an “ERβ-specific ligand”55–58. The same issue has occurred for the ERα agonist PPT, where its EC50 is of 0.2 nM and 82 nM for ERα and ERβ (Table 1), respectively, but has been used at a concentration of 100 nM57–59. Likewise, high concentrations used for SERMs treatment can have numerous impacts on other receptors than ERs. For example, 4-hydroxytamoxifen, an active metabolite of tamoxifen, has an IC50 of approximately 3.3 nM for ERα and ERβ (Table 1), but if used at concentrations higher than 90 nM, it also inhibits the estrogen-related receptor ERRγ, another transcription factor member of the nuclear receptor family60. It is thus essential to use appropriate drug dosages in order to solely modulate ERs’ activity.
Table 1.
Compound description with all the EC/IC50.
| Compounds | Action in PCa cells | EC50/IC50a | Treatment concentration |
|---|---|---|---|
| PPT | Agonist of ERα |
ERα: EC50 = 0.2 nM ERβ: EC50 = 82 nM |
1 nM |
| DPN | Agonist of ERβ |
ERα: EC50 = 66 nM ERβ: EC50 = 0.85 nM |
4.25 nM |
| Fulvestrant | Antagonist of both ERs |
ERα: IC50 = 0.47 nM ERβ: IC50 = 3.8 nM |
19 nM |
| 4-Hydroxytamoxifen (SERM) | Unsureb | ERs: IC50 = 3.3 nM | 10 nM |
| Raloxifene (SERM) | Unsureb | ERs: IC50 = 2.9–5.7 nM | 28.5 nM |
| Toremifene (SERM) | Unsureb | ERs: IC50 = 1,000 nM | 1,000 nM |
| Bazedeoxifene (SERM) | Unsureb |
ERα: IC50 = 26 nM ERβ: IC50 = 99 nM |
495 nM |
| Lasofoxifene (SERM) | Unsureb |
ERα: IC50 = 1.08 nM ERβ: IC50 = 4.41 nM |
22.05 nM |
aIC50 and EC50 were retrieved from supplier’s websites: Santa Cruz Biotechnology, Tocris, ApexBio, and MedChemExpress.
bSERMs are mostly believed to be antagonists in PCa cells, but this is mostly based on experiments performed in the same in vitro models as used in the current study.
Overall, it is still not clear which PCa models represent a good model to study ERs functions, what is the impact of activating ERα and/or ERβ on PCa cell proliferation, and if SERMs and the pure antiestrogen fulvestrant can be used to block PCa cell proliferation. The aim of our study was to perform a systematic investigation of the impact of treatments with natural estrogen, specific ERα and ERβ ligands, and SERMs, at specific concentrations, on PCa cell proliferation.
Results
ERs mRNA and protein expression levels in breast cancer and PCa models
First, we assessed the protein expression levels of ERα and ERβ in our PCa models using recently validated antibodies48,61,62. We used as control the human breast cancer cell line MCF7, which showed high expression levels of ERα (as expected), no expression of ERβ and weak but detectable expression of AR (Fig. 1). All human AR-positive PCa cell lines (LNCaP, LAPC4 and 22Rv1) had high expression levels of AR, 22Rv1 also strongly expressed the AR-V7 splice variant (lower band). However, none of these cell lines had detectable expression of ERs. In the case of human AR-negative PCa cell lines (DU145 and PC3), they both showed no expression of AR and ERβ. However, longer exposure revealed weak but detectable expression of ERα in PC3 cells.
Figure 1.
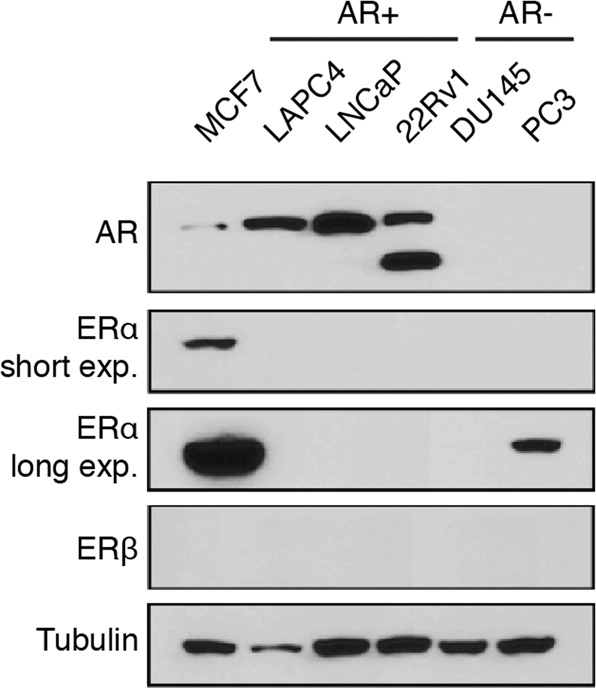
Weak estrogen receptors protein expression in PCa cell lines. Protein expression of AR, ERα, and ERβ in MCF7, LNCaP, LAPC4, 22Rv1, DU145 and PC3. α-tubulin was used as a loading control. No bands were detectable for ERβ at any exposure.
We also used RNA-seq data from 3 of these cell lines and from patient biopsies to analyze expression of the ESR1 and ESR2 genes, encoding respectively ERα and ERβ. As seen in Fig. 2A, there was no detectable expression of full length ESR1 mRNA in LNCaP, LAPC4, and 22Rv1 cell lines, either with or without R1881 treatment. Note that the only signal observed in these cells is at SYNE1, a gene located close to ESR1 (Fig. 2A). In comparison, ESR1 mRNA was detectable at low levels in three out of four patient samples tested in RNA-seq (Fig. 2B). ESR2 mRNA was undetectable in all our cancer cell lines and patient samples tested (data not shown).
Figure 2.

Comparison of mRNA expression of estrogen receptors between patient biopsies and PCa cell lines. (A) UCSC genome browser view of RNA-seq data at the ESR1 locus in LNCaP, LAPC4, and 22Rv1 cells, with and without 24 h treatment with the synthetic androgen R1881. (B) UCSC genome browser view of RNA-seq data at the ESR1 locus in 4 patients with PCa.
Given the lack of detectable expression of ERα and ERβ in most PCa in vitro models, it is hard to reconcile with previously published results that showed decreased proliferation and survival of these cell lines following treatments with various SERMs. These studies, as reviewed in the introduction, mostly used high concentrations of SERMs, often 100 to a 1000 times over their selectivity for ERs, suggesting that the observed effects were due to off-target effects. In that context, we wanted to specifically delineate the impact of estrogens and SERMs treatment on PCa cells by using specific ERα and ERβ ligands and SERMs at concentrations specific to ERs to mimize off-target effects.
Optimization of hormonal and SERM treatments using MCF7 cells
We first used MCF7 cells as controls to optimize treatments, as they are well-established in the breast cancer field to harbour high expression levels of ERα and no detectable expression of ERβ, and to be highly sensitive to estrogen stimulation48. These cells have been widely used to study the estrogen-dependent growth of breast cancer as well as to test anti-estrogen treatments. To validate that the different ERα ligand concentrations were biologically relevant, we used this model to optimize our proliferation assays using estradiol (E2), the ERα-specific ligand PPT, the ERβ-specific ligand DPN, five distinct SERMs that show anti-estrogen activity in breast cancer cells, and fulvestrant, a pure anti-estrogen that show antagonist functions of both receptors in all tissues tested.
In our settings, and as expected, treatment with E2 significantly increased MCF7 cell proliferation (Fig. 3). PPT was used at a lower concentration to avoid ERβ activation. Similar to estradiol, PPT significantly induced MCF7 proliferation rates (Fig. 3). Treatment with DPN was made at a concentration that could specifically activate the ERβ without activating ERα. As such, no significant changes in cell proliferation was noticeable for this compound.
Figure 3.
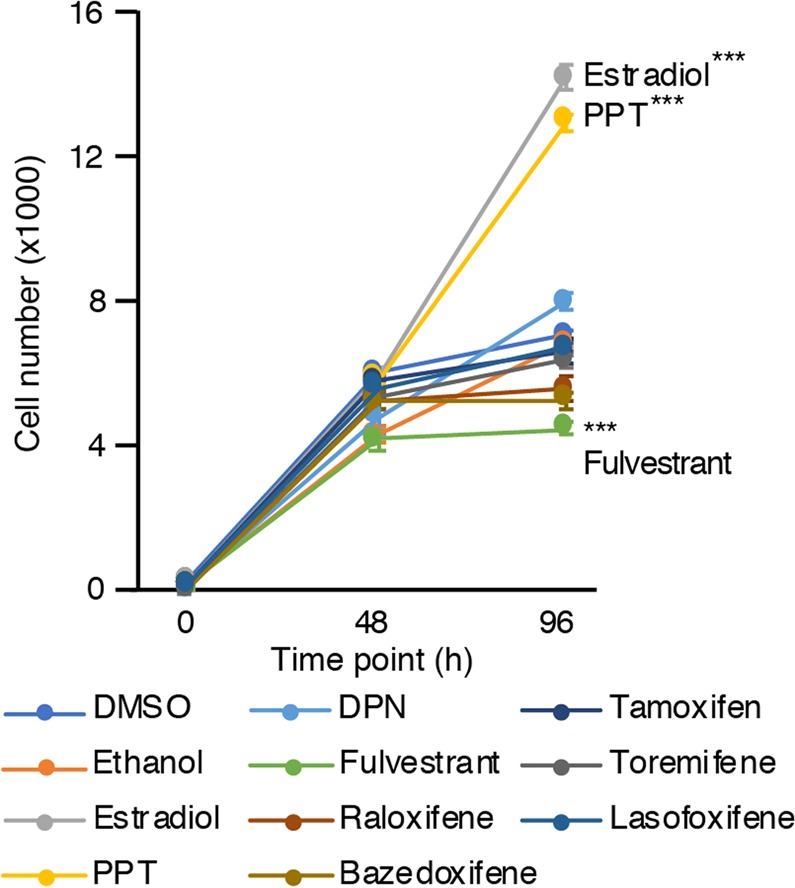
Proliferation protocol optimization using MCF7. Cell number of MCF7 after treatment with estrogenic and anti-estrogenic compounds at time point 0 h, 48 h and 96 h. Values were determined with crystal violet staining and are presented as the average ± standard error of the mean (n = 8 samples/treatment). Data from one representative experiment out of four independent experiments is shown. Asterisks (***p < 0.001) indicate that modulation of proliferation was significant in at least three out of four experiments.
We also treated MCF7 cells with fulvestrant. At a concentration where both receptors are blocked, 19 nM, treatment with this anti-estrogen significantly decreased cell proliferation compared to controls, probably reflecting blockade of low levels of residual estrogens in our experiment setting. In addition, five different SERMs were used, namely tamoxifen, bazedoxifene, raloxifene, toremifene, and lasofoxifene. As expected, in absence of estrogens, none of them had a significant impact on MCF7 cell proliferation.
Treatments of AR-negative PCa cell lines
We then studied two AR-negative PCa cell lines, namely PC3 and DU145 cells. Contrary to MCF7 cells, E2 or PPT had no significant impact on DU145 cells (Fig. 4A), in line with undetectable levels of ERα in these cells. Consequently, none of the SERMs nor fulvestrant had any significant impact on cell proliferation neither. In the case of PC3 (Fig. 4B), we observed the same pattern as for DU145. None of the estrogenic compounds activating ERα (PPT), ERβ (DPN) or both (E2) nor the anti-estrogenic compounds had a significant impact of PC3 cell proliferation and survival after 4 days of treatments when compared to vehicles. Our results indicate that even if ERα is expressed at low levels in PC3 cells, it does not contribute to cell proliferation in standard in vitro culture conditions.
Figure 4.

No significant modulation of proliferation upon hormonal treatment in AR-negative PCa cell lines (PC3 and DU145). Cell number of DU145 (A) and PC3 (B) cells after treatment with estrogenic and anti-estrogenic compounds detailed in the legend at time point 0 h, 48 h and 96 h. Values were determined with crystal violet staining and are presented as the average ± standard error of the mean (n = 8 samples/treatment). Data from one representative experiment out of three independent experiments is shown. There was no reproducible significant modulation of proliferation.
Treatments of AR-positive PCa cells
Then, we studied three AR-positive PCa cell lines, namely LNCaP, LAPC4, and 22Rv1 PCa cells. They were all isolated from patients with castration-resistant PCa63–65. LNCaP cells have high AR expression and depend on its activity for proliferation, while LAPC4 and 22Rv1 are considered as androgen-sensitive cell lines, i.e. that do not depend on AR for proliferation but that are positively stimulated by androgens.
In absence of androgens, we observed no changes in LNCaP cell numbers, but a significant increase in proliferation after treatment with R1881 (Fig. 5A). LNCaP cells have been used in several previous studies on the effect of estrogens in PCa, even though their AR is mutated and can be activated by E2 besides androgens46,47. Thus, as expected, E2 significantly increased LNCaP cell proliferation compared to controls (Fig. 5A). However, no significant difference was observed when the cells were co-treated with R1881, suggesting that this response is purely AR-dependent. Consistent with this idea, treatment with specific ER agonists PPT or DPN had no significant impact on PCa cell proliferation (Fig. 5B,C). In the case of the five SERMs tested, such as tamoxifen (Fig. 5D), raloxifene (Fig. 5E), bazedoxifene (Fig. 5F), lasofoxifene (Fig. 5G), and toremifene (Fig. 5H), no significant increase or decrease of proliferation was noted after treatment, with or without the presence of androgens. After being treated with a pure antagonist of both estrogen receptors, fulvestrant, cell proliferation had once again no significant modulation (Fig. 5I) regardless of co-treatment with androgens. These results indicate that in LNCaP cells, E2 is solely acting through AR activation and not through modulation of ERs activity. It also suggests that these SERMs cannot modulate mutated AR activity.
Figure 5.

Proliferation of LNCaP cells is increased by E2 only in an AR-dependent manner. LNCaP cell number after treatment, with and without R1881, of either E2 (A), PPT (B), DPN (C), tamoxifen (D), raloxifene (E), bazedoxifene (F), lasofoxifene (G), toremifene (H), and fulvestrant (I). Values were determined with crystal violet staining and are presented as the average ± standard error of the mean (n = 8 samples/treatment). Data from one representative experiment out of six independent experiments is shown. Asterisks (***p < 0.001) indicate that modulation of proliferation is significant in at least four out of six independent experiments.
The second AR-positive cell line tested was LAPC4, a cell line that does not depend on AR for proliferation but that is still sensitive to androgens. Similar to AR-negative PCa cell lines (Fig. 4), E2 did not modulate significantly LAPC4 cell proliferation, and neither did PPT or DPN (Fig. 6A,B) regardless of the presence of androgens in the culture media. Consequently, treatment with the anti-estrogen fulvestrant or any SERMs did not reveal any significant impact on LAPC4 cell proliferation (Fig. 6A,B). This is consistent with lack of detectable expression of both ERs in these cells (Figs. 1 and 2A).
Figure 6.

No significant modulation of proliferation upon estrogenic/anti-estrogenic treatment in LAPC4 cell number after treatment with estrogenic and anti-estrogenic compounds, without (A) and with co-treatment with R1881 (B). Values were determined with crystal violet staining and are presented as the average ± standard error of the mean (n = 8 samples/treatment). Data from one representative experiment out of six independent experiments is shown. There was no reproducible significant modulation of proliferation.
We also studied the AR-positive PCa cell line 22Rv1, which is characterized by high expression of AR-V7, a splice variant of AR that lacks the ligand binding domain66. As for LAPC4 cells, 22Rv1 cells are not dependent on androgens for proliferation but are nevertheless sensitive to it. When treated with agonist ligands of ERs, with and without co-treatment with androgens, 22Rv1 did not have their proliferation significantly modulated (Fig. 7A,B). As for other cell lines, the anti-estrogen fulvestrant and most SERMs did not have any significant impact on 22Rv1 cell proliferation, regardless of co-treatment with R1881 (Fig. 7A,B). Interestingly, treatment with the SERM toremifene induced a small but significant decrease of 22Rv1 cell proliferation in absence of androgens (Fig. 7A), which was observed in all our independent experiments.
Figure 7.

Proliferation of 22Rv1 cells is decreased with toremifene (SERM) only when androgens are absent. 22Rv1 cell number after treatment with estrogenic and anti-estrogenic compounds, without (A) and with co-treatment with R1881 (B). Values were determined with crystal violet staining and are presented as the average ± standard error of the mean (n = 8 samples/treatment). The average of two experiments out of five independent experiments is shown. 22Rv1 cell number after treatment with high concentrations of tamoxifen (C) and raloxifene (D) at 10 µM and 28.5 µM, respectively. Data from one representative experiment out of three independent experiments is shown. Asterisks (*p < 0.05, **p < 0.01, ***p < 0.001) indicate that modulation of proliferation is significant compared to controls.
Finally, to study if the results obtained in previous studies were ERs-independent off-target effects due to treatments at high concentrations, we also tested high concentrations of the two most studied SERMs in in vitro PCa models, namely tamoxifen and raloxifene. In 22Rv1 cells, even though E2, PPT, and DPN had no significant impact on proliferation at physiological levels (Fig. 7A,B), treatments with tamoxifen, and raloxifene at 10 uM and 28.5 uM, respectively, significantly impaired cancer cell proliferation (Fig. 7C,D). These results demonstrate that SERMs, at high concentrations, can block PCa cell proliferation in an ERs-independent manner.
Discussion
The goal of the current study was to perform a systematic investigation of estrogens and anti-estrogens impact on PCa cell proliferation with appropriate controls, dosages, and in multiple well-studied in vitro PCa models. Globally, we observed that estrogens and anti-estrogens do not have a significant impact on proliferation at dosages where these molecules bind specifically to ERα, ERβ, or both. Consistent with an absence of effect of these compounds on PCa cell proliferation, mRNA and protein expression data indicate that for the majority of these models, ERs’ expression levels are undetectable. Therefore, our study highlights that these models are inappropriate to study ERs functions in PCa cells, and further suggests the usage of alternative PCa models to properly assess the roles of the estrogen signaling pathway in PCa. Importantly, our study also indicates that most previous studies most probably reported off-target effects of SERMs in context of PCa.
Certainly the most widely used in vitro model of PCa, LNCaP cells have been isolated from a patient with castration-resistant PCa63. One of the mechanisms that explain this resistance is an AR mutation in its ligand binding domain, which allows AR to bind and be activated by other steroids, including E246,47. Thus, studying ERs functions by using only E2 will inevitably lead to misinterpretations and biasing the real contribution of the estrogen signaling pathway in PCa cell proliferation50–52. Our results demonstrate that the significant increase of proliferation induced by E2 is mediated through mutated-AR activation and not through ERs activation. Indeed, the specific agonists of each estrogen receptor, PPT and DPN, do not modulate proliferation compared to E2, suggesting low, if any, contribution of ERs on LNCaP cell proliferation. Furthermore, it is rather clear now using validated ERα and ERβ antibodies that these cells do not express these receptors48,53,54. It is thus hard to reconciliate the inhibitory effect of tamoxifen and raloxifene on LNCaP cell proliferation in an ERα-dependent manner67, and the high dosages used, sometimes up to 10−4M, probably explain this inhibitory off-target effect. Results obtained with 22Rv1 cells, showing that estrogens and SERMs have no impact on proliferation at ERs-specific concentrations but decreased proliferation at high concentrations (Fig. 7), support this conclusion.
The high concentrations previously employed have most probably also led to positive effects on proliferation in other cell lines independently of ERs expression levels and activity. Indeed, LNCaP, PC3, and DU145 cells have been studied in several previous publications, using notably tamoxifen and raloxifene at high concentrations and without co-testing estrogenic positive controls such as E267–69 to ensure proper ERs activation or inhibition through SERMs. Our results showed undetectable protein expression levels of both ERs in DU145 and LNCaP cells and weak, but detectable, levels of ERα in PC3 cells. Yet, activation of this receptor with E2 or PPT had no significant modulation of proliferation in the latter cell line, and neither did the anti-estrogens. In that context, we believe that inhibitory effects of tamoxifen and other SERMS reported in these models at high concentrations are mostly due to off-target effects, and not through modulation of ERs activity. In line, using high dosages of SERMs in 22Rv1 does recapitulate previous reports on decreased PCa cell proliferation, supporting off-targets effects in that context. Importantly, it was recently reported that several SERMs, such as tamoxifen, at high concentrations in the µM range, are microtubule modulators that can decrease cancer cell proliferation70. Such ERs-independent effects most probably explain the previously observed results. In addition, we cannot exclude that, in PC3 cells, ERα contributes to the cancer phenotype, such as in regulating invasion or migration. Overall, our study highlights the importance of using appropriate agonist ligands along with specific dosages and anti-estrogenic compounds to study more precisely the ERs activity in PCa models.
An important point raised in the last 2 years, as exemplified in the current study, is either or not ERs are expressed at all in some of the most commonly used PCa models. Indeed, there was no consensus as to whether LNCaP, DU145, and PC3 express ERβ69,71,72 or not56,69,73. Most of these discrepencies came from usage of unspecific antibodies. To counter this issue, several groups published systematic validation of ERβ antibodies48,53,54. Importantly, it was confirmed that LNCaP cells do not express ERβ. Yet, despite state-of-the-art validation using mass spectrometry, it is still debated if ERβ is expressed or not in the human prostate and PCa tissues48,53,54. Furthermore, results from genetically engineered mouse model disrupting Esr2 are also conflictual74,75. A similar problem is observed for ERα expression in some PCa cell line models, notably in LNCaP cells25,50,71,73, although to a lesser extend due to the important work done on this receptor in the breast cancer field. The general consensus is that ERα is expressed in prostate stromal cells, as well as in human PCa and castration-resistant PCa tissues24,34,35,76,77. This is supported by our reanalysis of RNA-seq from PCa tumours in which ESR1 was expressed in most samples (Fig. 2B).
The G protein-coupled estrogen receptor 1 (GPER), located at the plasma membrane and not part of the nuclear receptor family like the ERs, was not investigated in this study. Although it can also be activated by E2, it is probably not involved in proliferation of the PCa cell lines tested herein, at least in standard cell culture conditions. Indeed, if GPER was involved in regulating PCa cell proliferation, a significant modulation of proliferation would have been observed upon treatment with E2.
One interesting result we observed is the significant decrease of proliferation in 22Rv1 cells by toremifene, a SERM known to be an agonist of ERs in bone. While PCa patients follow an anti-androgenic therapy, one of the major side effects is bone loss that can lead to osteoporosis78–80. A few clinical trials have tested toremifene in that context, which revealed well-kept bone mineralization in patients taking this SERM combined with anti-androgen therapy81–84. Interestingly, the significant decrease of proliferation induced by this SERM only occurred in 22Rv1 cells, which express high levels of AR-V7. Considering that E2 had no significant effect on proliferation (Fig. 7) and Western blots results indicating a lack of expression of both ERs (Fig. 1), one possibility is that toremifene effect on proliferation is mediated by AR-V7. Yet, additional mechanistic studies are required to test this hypothesis.
Overall, our results demonstrate that most in vitro PCa models actually lack detectable expression of both ERs in standard culture conditions. Furthermore, they suggest that previous studies using these models most probably reported ERs-independent effects of SERMs due to the high concentrations used. We cannot exclude that lack of ERs expression in vitro is an artifact of these model systems, as ERs, and particularly ERα, are most probably expressed in vivo and relevant to PCa biology. In conclusion, our results indicate that classic PCa models are not appropriate to study the estrogen-signaling pathway and highlight the necessity to find appropriate PCa models that express biologically relevant levels of ERα and ERβ.
Materials and Methods
Cell culture
Six cell lines were used for proliferation assays: one breast cancer cell line serving as an ERα positive control (MCF7 [RRID: CVCL_0031]), two AR-negative PCa cell lines (PC3 [RRID: CVCL_0035] and DU145 [RRID: CVCL_0105]) and three AR-positive PCa cell lines (LNCaP [RRID: CVCL_1379], 22Rv1 [RRID: CVCL_1045], LAPC4 [RRID: CVCL_4744]). All of them were initially obtained from ATCC, were kept in culture for no more than 3 months after resuscitation and were tested every 4 months for mycoplasma presence. They were grown in phenol red RPMI media supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin, and sodium pyruvate, and kept in incubators at 37 °C and 5% CO2. The media was changed every two days and confluence in plates was kept below 75%.
Western blots
To evaluate the protein expression of the receptors, cell lysates of all cell lines were harvested in Buffer K supplemented with protease and phosphatase inhibitors as previously described5 before analysis by Western Blots. The primary antibodies used were to detect α-tubulin (11H10 [RRID: AB_10695471], Cell Signaling Technology, dilution 1:1,000), AR (441 [RRID: AB_626671], Santa Cruz Biotechnology, dilution 1:1,000), ERα (F-10 [RRID: AB_627558], Santa Cruz Biotechnology, dilution 1:1,000) and ERβ (CWK-F12 [RRID: AB_2722105], DSHB, dilution 1:1,000). Complete Western Blot figures are shown in Supplemental Fig. S1.
RNA-seq analysis
RNA-seq data we previously generated were used to study mRNA expression of ERα, encoded by the ERS1 gene in LNCaP and LAPC4 cells85. Briefly, cells were seeded for 48 h in RPMI 1640 media without phenol red and with 5% CSS for steroid deprivation. They were then treated for 24 h with 10 nM R1881 or vehicle (96% EtOH) before being harvested for RNA purification and sequencing. We used the same protocol for 22Rv1 cells and after sequencing they were processed as described previously to obtain transcripts per million (TPM) values. In short, FastQC was used for raw sequencing data quality control86 and Trimmomatic was used to trim adaptor and over-represented sequences87. Pseudoalignement to the transcriptome (hg38) was then performed using Kallisto88 and samples were visualized using the UCSC genome browser. Results were then converted to bedgraphs before being loaded in the UCSC genome browser for visualization.
Hormonal treatments
Cells were firstly seeded in 96-well plates with phenol red-free RMPI 1640 supplemented with 5% charcoal-stripped serum (CSS) and incubated for 48 h to allow steroid deprivation. Media was then renewed (100 μL per well) and treatment was added. The chosen treatment concentration for estrogenic/anti-estrogenic compounds and SERMs correspond to 5-fold the EC50/IC50 specific to each compound, except for tamoxifen and toremifene (see Table 1 for more details). The compounds used for treatments were: DMSO (vehicle; Sigma), ethanol 96% (vehicle; Greenfield Global), 17β-estradiol (10 nM; Sigma), PPT (1,3,5-tris(4-hydroxyphenyl)-4-propyl-1H-pyrazole; Santa Cruz Biotechnology), DPN (2,3-bis(4-hydroxyphenyl) propionitrile; Santa Cruz Biotechnology), fulvestrant (ICI 182,780; Santa Cruz Biotechnology), 4-OH-tamoxifen (Santa Cruz Biotechnology), raloxifene hydrochloride (Tocris), toremifene citrate (Santa Cruz Biotechnology), bazedoxifene (Tocris) and lasofoxifene (Santa Cruz Biotechnology). AR-positive PCa cell lines were also co-treated or not with the synthetic androgen R1881 (10 nM; Steraloids). The media and treatments were renewed every 48 h until the end of the proliferation assays (either after 96 h or 168 h in total, depending on the cell lines).
Quantification of cellular proliferation with crystal violet
The crystal violet assay developed by Feoktistova et al.89 was used to stain the cells in order to determine cell number after treatment. Media was firstly removed, then 50 μL of a 0.5% crystal violet solution was added in each well. The plates were left on a rocking bench for 30 mins at room temperature, rinsed thoroughly 4 times with water, and left to dry at least 48 h (protected from light). The stained cells were then lysed by adding 200 μL of SDS 2% in each well, and the plates were left on a rocking bench for 4 h at room temperature. The plates had their optical density (OD) measured at 570 nm using a spectrophotometer. OD values were converted into cell numbers by using standard curves specific to each cell line.
Quantification of cellular proliferation with CyQUANT
Given weaker adherence of LAPC4 and LNCaP cells, the CyQUANT Cell Proliferation Assay Kit (Catalog #: C7026, Invitrogen) was also used to confirm proliferation assays. After treatment, media was removed from wells, then the plates were frozen at −20 °C at least 24 hours before analysis using the manufacturer’s instructions. The plates were then thawed, and a solution of CyQUANT was added in each well containing cells. The plates were incubated in the dark for 5 minutes, and the fluorescence was measured at 480 nm of excitation and 520 nm of emission using a spectrophotometer. Fluorescence values were converted into cell numbers by using standard curves specific for each cell line.
Statistical analysis for proliferation assays
Proliferation assays were done at least 3 times independently for each cell line, with 4–8 biological samples per group per experiment. To test for statistical significance of estrogenic, anti-estrogenic, and androgenic treatments, one-way ANOVA with Tukey HSD and Dunnett’s post-hoc tests were performed using XL STATS.
Supplementary information
{kind=link}
Acknowledgements
This work was supported by funding to EAW from the Canadian Institutes for Health Research (CIHR; PJT159530), the Fonds de recherche du Québec - Santé - Chercheur-Boursier junior 1 salary award, and supported by the 2018 AACR-Bayer Innovation and Discovery Grant. CL has a scholarship from the Fondation du CHU de Québec.
Author contributions
C.L. realized most of the experiments, wrote partially the manuscript, designed the figures, and reviewed the manuscript. L.G. worked on RNA-seq data generated, wrote partially the manuscript and reviewed it. C.W. performed some of the proliferation assays and reviewed the manuscript. E.A.W. designed the project, designed the figures, wrote partially the manuscript and reviewed it.
Data availability
RNA-seq data generated during this study is available from the Gene Expression Omnibus (GSE128749 for LNCaP and LAPC4 cells; GSE128201 for 22 Rv1) and are described elsewhere85. Data from biopsies come from SRA access number SRR1164789, SRR1164790, SRR1164791 and SRR116479290.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-60844-3.
References
- 1.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nature Reviews Cancer. 2015;15(12):701–711. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonthier, K., Poluri, R. T. K. & Audet-Walsh, E. Functional genomic studies reveal the androgen receptor as a master regulator of cellular energy metabolism in prostate cancer. Journal of Steroid Biochemistry and Molecular Biology. 191 (2019). [DOI] [PubMed]
- 3.Audet-Walsh E, et al. Androgen-Dependent Repression of ERR gamma Reprograms Metabolism in Prostate Cancer. Cancer Research. 2017;77(2):378–389. doi: 10.1158/0008-5472.CAN-16-1204. [DOI] [PubMed] [Google Scholar]
- 4.Audet-Walsh E, et al. Inverse Regulation of DHT Synthesis Enzymes 5 alpha-Reductase Types 1 and 2 by the Androgen Receptor in Prostate Cancer. Endocrinology. 2017;158(4):1015–1021. doi: 10.1210/en.2016-1926. [DOI] [PubMed] [Google Scholar]
- 5.Audet-Walsh E, et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes & Development. 2017;31(12):1228–1242. doi: 10.1101/gad.299958.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Audet-Walsh E, et al. SREBF1 Activity Is Regulated by an AR/mTOR Nuclear Axis in Prostate Cancer. Molecular Cancer Research. 2018;16(9):1396–1405. doi: 10.1158/1541-7786.MCR-17-0410. [DOI] [PubMed] [Google Scholar]
- 7.Fujimura, T. et al. Estrogen and Androgen Blockade for Advanced Prostate Cancer in the Era of Precision Medicine. Cancers (Basel), 10(2) 2018. [DOI] [PMC free article] [PubMed]
- 8.Dobbs RW, et al. Estrogens and prostate cancer. Prostate Cancer and Prostatic Diseases. 2019;22(2):185–194. doi: 10.1038/s41391-018-0081-6. [DOI] [PubMed] [Google Scholar]
- 9.Bosland MC, Ford H, Horton L. Induction at high incidence of ductal prostate adenocarcinomas in NBL/Cr and Sprague-Dawley Hsd:SD rats treated with a combination of testosterone and estradiol-17 beta or diethylstilbestrol. Carcinogenesis. 1995;16(6):1311–7. doi: 10.1093/carcin/16.6.1311. [DOI] [PubMed] [Google Scholar]
- 10.Bosland MC, Mahmoud AM. Hormones and prostate carcinogenesis: Androgens and estrogens. J Carcinog. 2011;10:33. doi: 10.4103/1477-3163.90678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McPherson SJ, et al. Elevated androgens and prolactin in aromatase-deficient mice cause enlargement, but not malignancy, of the prostate gland. Endocrinology. 2001;142(6):2458–67. doi: 10.1210/endo.142.6.8079. [DOI] [PubMed] [Google Scholar]
- 12.Jarred RA, et al. Prostate phenotypes in estrogen-modulated transgenic mice. Trends Endocrinol Metab. 2002;13(4):163–8. doi: 10.1016/S1043-2760(02)00575-1. [DOI] [PubMed] [Google Scholar]
- 13.Salonia A, et al. Circulating estradiol, but not testosterone, is a significant predictor of high-grade prostate cancer in patients undergoing radical prostatectomy. Cancer. 2011;117(22):5029–38. doi: 10.1002/cncr.26136. [DOI] [PubMed] [Google Scholar]
- 14.Giton F, et al. Estrone sulfate (E1S), a prognosis marker for tumor aggressiveness in prostate cancer (PCa) J Steroid Biochem Mol Biol. 2008;109(1-2):158–67. doi: 10.1016/j.jsbmb.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Ozten N, et al. Role of Estrogen in Androgen-Induced Prostate Carcinogenesis in NBL Rats. Hormones & Cancer. 2019;10(2-3):77–88. doi: 10.1007/s12672-019-00360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montgomery RB, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Research. 2008;68(11):4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellem SJ, et al. Local aromatase expression in human prostate is altered in malignancy. Journal of Clinical Endocrinology & Metabolism. 2004;89(5):2434–2441. doi: 10.1210/jc.2003-030933. [DOI] [PubMed] [Google Scholar]
- 18.Gianfrilli, D. et al. Sex Steroid Metabolism in Benign and Malignant Intact Prostate Biopsies: Individual Profiling of Prostate Intracrinology. Biomed Research International, 2014. [DOI] [PMC free article] [PubMed]
- 19.Miftakhova R, et al. Cyclin A1 and P450 Aromatase Promote Metastatic Homing and Growth of Stem-like Prostate Cancer Cells in the Bone Marrow. Cancer Research. 2016;76(8):2453–2464. doi: 10.1158/0008-5472.CAN-15-2340. [DOI] [PubMed] [Google Scholar]
- 20.Boibessot C, Toren P. Sex steroids in the tumor microenvironment and prostate cancer progression. Endocrine-Related Cancer. 2018;25(3):R179–R196. doi: 10.1530/ERC-17-0493. [DOI] [PubMed] [Google Scholar]
- 21.Warner M, Huang B, Gustafsson JA. Estrocen Receptor beta as a Pharmaceutical Target. Trends in Pharmacological Sciences. 2017;38(1):92–99. doi: 10.1016/j.tips.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 22.McPherson, S. J. et al. The role of ER alpha and ER beta in the prostate: Insights from genetic models and isoform-selective ligands. In International Symposium on Tissue-Specific Estrogen Action. Berlin, GERMANY (2006). [DOI] [PubMed]
- 23.Bonkhoff H. Estrogen receptor signaling in prostate cancer: Implications for carcinogenesis and tumor progression. Prostate. 2018;78(1):2–10. doi: 10.1002/pros.23446. [DOI] [PubMed] [Google Scholar]
- 24.Takizawa I, et al. Estrogen receptor alpha drives proliferation in PTEN-deficient prostate carcinoma by stimulating survival signaling, MYC expression and altering glucose sensitivity. Oncotarget. 2015;6(2):604–616. doi: 10.18632/oncotarget.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakravarty D, et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nature Communications. 2014;5:5383. doi: 10.1038/ncomms6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furic L, Lawrence MG, Risbridger GP. Pro-tumorigenic role of ER alpha in prostate cancer cells. Aging-Us. 2015;7(6):356–357. doi: 10.18632/aging.100769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mak P, et al. Prostate Tumorigenesis Induced by PTEN Deletion Involves Estrogen Receptor beta Repression. Cell Reports. 2015;10(12):1982–1991. doi: 10.1016/j.celrep.2015.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu WF, et al. Estrogen receptor beta, a regulator of androgen receptor signaling in the mouse ventral prostate. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(19):E3816–E3822. doi: 10.1073/pnas.1702211114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McPherson SJ, et al. Estrogen receptor-beta activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNF alpha mediated. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):3123–3128. doi: 10.1073/pnas.0905524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McPherson SJ, et al. Essential role for estrogen receptor beta in stromal-epithelial regulation of prostatic hyperplasia. Endocrinology. 2007;148(2):566–574. doi: 10.1210/en.2006-0906. [DOI] [PubMed] [Google Scholar]
- 31.Horvath LG, et al. Frequent loss of estrogen receptor-beta expression in prostate cancer. Cancer Research. 2001;61(14):5331–5335. [PubMed] [Google Scholar]
- 32.Bonkhoff H, Berges R. The Evolving Role of Oestrogens and Their Receptors in the Development and Progression of Prostate Cancer. European Urology. 2009;55(3):533–542. doi: 10.1016/j.eururo.2008.10.035. [DOI] [PubMed] [Google Scholar]
- 33.Fixemer T, Remberger K, Bonkhoff H. Differential expression of the estrogen receptor beta (ER beta) in human prostate tissue, premalignant changes, and in primary, metastatic, and recurrent prostatic adenocarcinoma. Prostate. 2003;54(2):79–87. doi: 10.1002/pros.10171. [DOI] [PubMed] [Google Scholar]
- 34.Bonkhoff H, et al. Estrogen receptor expression in prostate cancer and premalignant prostatic lesions. American Journal of Pathology. 1999;155(2):641–647. doi: 10.1016/S0002-9440(10)65160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw GL, et al. The Early Effects of Rapid Androgen Deprivation on Human Prostate Cancer. European Urology. 2016;70(2):214–218. doi: 10.1016/j.eururo.2015.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji Q, et al. Frequent loss of estrogen and progesterone receptors in human prostatic tumors determined by quantitative real-time PCR. Molecular and Cellular Endocrinology. 2005;229(1-2):103–110. doi: 10.1016/j.mce.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 37.Bonkhoff H, et al. Progesterone receptor expression in human prostate cancer: Correlation with tumor progression. Prostate. 2001;48(4):285–291. doi: 10.1002/pros.1108. [DOI] [PubMed] [Google Scholar]
- 38.Traboulsi T, et al. Antiestrogens: structure-activity relationships and use in breast cancer treatment. Journal of Molecular Endocrinology. 2017;58(1):R15–R31. doi: 10.1530/JME-16-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel HK, Bihani T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacology & Therapeutics. 2018;186:1–24. doi: 10.1016/j.pharmthera.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 40.Blesa JM, Candel VA. PSA decrease with fulvestrant acetate in a hormone-resistant metastatic prostate cancer patient. Onkologie. 2010;33(1-2):57–9. doi: 10.1159/000264612. [DOI] [PubMed] [Google Scholar]
- 41.Fechon A, Droz JP. Do we really need new trials on fulvestrant in prostate cancer? Onkologie. 2010;33(1–2):12–3. doi: 10.1159/000271605. [DOI] [PubMed] [Google Scholar]
- 42.Gasent Blesa JM, et al. Experience with fulvestrant acetate in castration-resistant prostate cancer patients. Ann Oncol. 2010;21(5):1131–2. doi: 10.1093/annonc/mdq010. [DOI] [PubMed] [Google Scholar]
- 43.Gasent Blesa JM, Candel VA. PSA Decrease with Fulvestrant Acetate in a Hormone-Resistant Metastatic Prostate Cancer Patient: A Case Report. Case Rep Oncol. 2009;2(1):72–76. doi: 10.1159/000214838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chadha MK, et al. Phase II study of fulvestrant (Faslodex) in castration resistant prostate cancer. Prostate. 2008;68(13):1461–6. doi: 10.1002/pros.20813. [DOI] [PubMed] [Google Scholar]
- 45.Ho TH, et al. A Study of Combination Bicalutamide and Raloxifene for Patients With Castration-Resistant Prostate Cancer. Clin Genitourin Cancer. 2017;15(2):196–202 e1. doi: 10.1016/j.clgc.2016.08.026. [DOI] [PubMed] [Google Scholar]
- 46.Tan J, et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11(4):450–9. doi: 10.1210/mend.11.4.9906. [DOI] [PubMed] [Google Scholar]
- 47.Veldscholte J, et al. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J Steroid Biochem Mol Biol. 1992;41(3–8):665–9. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- 48.Nelson AW, et al. Comprehensive assessment of estrogen receptor beta antibodies in cancer cell line models and tissue reveals critical limitations in reagent specificity. Molecular and Cellular Endocrinology. 2017;440(C):138–150. doi: 10.1016/j.mce.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robinson JL, et al. Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J. 2011;30(15):3019–27. doi: 10.1038/emboj.2011.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adjakly M, et al. Comparative Effects of Soy Phytoestrogens and 17 beta-Estradiol on DNA Methylation of a Panel of 24 Genes in Prostate Cancer Cell Lines. Nutrition and Cancer-an International Journal. 2014;66(3):474–482. doi: 10.1080/01635581.2014.884236. [DOI] [PubMed] [Google Scholar]
- 51.Castagnetta LA, et al. Growht of LNCaP human prostate-cancer cells is stimulated by estradiol via its own receptor. Endocrinology. 1995;136(5):2309–2319. doi: 10.1210/endo.136.5.7536668. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi Y, et al. 17 beta-estradiol differentially regulates androgen-responsive genes through estrogen receptor-beta- and extracellular-signal regulated kinase-dependent pathways in LNCaP human prostate cancer cells. Molecular Carcinogenesis. 2007;46(2):117–129. doi: 10.1002/mc.20254. [DOI] [PubMed] [Google Scholar]
- 53.Andersson S, et al. Insufficient antibody validation challenges oestrogen receptor beta research. Nature Communications. 2017;8:12. doi: 10.1038/s41467-017-00025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gustafsson, J. A., Strom, A. & Warner, M., Update on ERbeta. Journal of Steroid Biochemistry and Molecular Biology. 191 (2019). [DOI] [PubMed]
- 55.Motawi TK, et al. Combinatorial strategy of epigenetic and hormonal therapies: A novel promising approach for treating advanced prostate cancer. Life Sciences. 2018;198:71–78. doi: 10.1016/j.lfs.2018.02.019. [DOI] [PubMed] [Google Scholar]
- 56.Pravettoni A, et al. Estrogen receptor beta (ERbeta) and inhibition of prostate cancer cell proliferation: Studies on the possible mechanism of action in DU145 cells. Molecular and Cellular Endocrinology. 2007;263(1–2):46–54. doi: 10.1016/j.mce.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 57.Verma V, et al. Designed modulation of sex steroid signaling inhibits telomerase activity and proliferation of human prostate cancer cells. Toxicology and Applied Pharmacology. 2014;280(2):323–334. doi: 10.1016/j.taap.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 58.Kumar R, et al. A precisely substituted benzopyran targets androgen refractory prostate cancer cells through selective modulation of estrogen receptors. Toxicology and Applied Pharmacology. 2015;283(3):187–197. doi: 10.1016/j.taap.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 59.Silva RD, et al. Activation of estrogen receptor beta (ER beta) regulates the expression of N-cadherin, E-cadherin and beta-catenin in androgen-independent prostate cancer cells. International Journal of Biochemistry & Cell Biology. 2018;96:40–50. doi: 10.1016/j.biocel.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 60.Tremblay GB, Bergeron D, Giguere V. 4-hyroxytamoxifen is an isoform-specific inhibitor of orphan estrogen-receptor-related (ERR) nuclear receptors beta and gamma. Endocrinology. 2001;142(10):4572–4575. doi: 10.1210/endo.142.10.8528. [DOI] [PubMed] [Google Scholar]
- 61.Formisano L, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019;10(1):1373. doi: 10.1038/s41467-019-09068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herold S, et al. Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature. 2019;567(7749):545–549. doi: 10.1038/s41586-019-1030-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Horoszewicz JS, et al. LNCaP model of human prostatic-carcinoma. Cancer Research. 1983;43(4):1809–1818. [PubMed] [Google Scholar]
- 64.Klein KA, et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nature Medicine. 1997;3(4):402–408. doi: 10.1038/nm0497-402. [DOI] [PubMed] [Google Scholar]
- 65.Sramkoski RM, et al. A new human prostate carcinoma cell line, 22R upsilon 1. In Vitro Cellular & Developmental Biology-Animal. 1999;35(7):403–409. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 66.Dehm SM, et al. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Research. 2008;68(13):5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim IY, et al. Raloxifene, a selective estrogen receptor modulator, induces apoptosis in androgen-responsive human prostate cancer cell line LNCaP through an androgen-independent pathway. Cancer Research. 2002;62(13):3649–3653. [PubMed] [Google Scholar]
- 68.Piccolella M, et al. Modulators of estrogen receptor inhibit proliferation and migration of prostate cancer cells. Pharmacological Research. 2014;79:13–20. doi: 10.1016/j.phrs.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 69.Kim IY, et al. Raloxifene, a mixed estrogen agonist/antagonist, induces apoptosis in androgen-independent human prostate cancer cell lines. Cancer Research. 2002;62(18):5365–5369. [PubMed] [Google Scholar]
- 70.Lo YC, et al. Pocket similarity identifies selective estrogen receptor modulators as microtubule modulators at the taxane site. Nat Commun. 2019;10(1):1033. doi: 10.1038/s41467-019-08965-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu, Q. X. et al. ZFHX3 is indispensable for ER beta to inhibit cell proliferation via MYC downregulation in prostate cancer cells. Oncogenesis8, 2019. [DOI] [PMC free article] [PubMed]
- 72.Liu XX, Arnold JT, Blackman MR. Dehydroepiandrosterone Administration or G alpha q Overexpression Induces beta-Catenin/T-Cell Factor Signaling and Growth via Increasing Association of Estrogen Receptor-beta/Dishevelled2 in Androgen-Independent Prostate Cancer Cells. Endocrinology. 2010;151(4):1428–1440. doi: 10.1210/en.2009-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mishra S, et al. Estrogen and estrogen receptor alpha promotes malignancy and osteoblastic tumorigenesis in prostate cancer. Oncotarget. 2015;6(42):44388–44402. doi: 10.18632/oncotarget.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Antal MC, et al. Sterility and absence of histopathological defects in nonreproductive organs of a mouse ERbeta-null mutant. Proc Natl Acad Sci USA. 2008;105(7):2433–8. doi: 10.1073/pnas.0712029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maneix L, et al. Estrogen receptor beta exon 3-deleted mouse: The importance of non-ERE pathways in ERbeta signaling. Proc Natl Acad Sci USA. 2015;112(16):5135–40. doi: 10.1073/pnas.1504944112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grindstad T, et al. Estrogen receptors alpha and beta and aromatase as independent predictors for prostate cancer outcome. Sci Rep. 2016;6:33114. doi: 10.1038/srep33114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nelson AW, et al. Estrogen receptor beta in prostate cancer: friend or foe? Endocr Relat Cancer. 2014;21(4):T219–34. doi: 10.1530/ERC-13-0508. [DOI] [PubMed] [Google Scholar]
- 78.Greenspan SL, et al. Bone loss after initiation of androgen deprivation therapy in patients with prostate cancer. Journal of Clinical Endocrinology & Metabolism. 2005;90(12):6410–6417. doi: 10.1210/jc.2005-0183. [DOI] [PubMed] [Google Scholar]
- 79.Alibhai SMH, et al. Fracture Types and Risk Factors in Men With Prostate Cancer on Androgen Deprivation Therapy: A Matched Cohort Study of 19,079 Men. Journal of Urology. 2010;184(3):918–923. doi: 10.1016/j.juro.2010.04.068. [DOI] [PubMed] [Google Scholar]
- 80.Poulsen MH, et al. Osteoporosis and prostate cancer; a 24-month prospective observational study during androgen deprivation therapy. Scandinavian Journal of Urology. 2019;53(1):34–39. doi: 10.1080/21681805.2019.1570328. [DOI] [PubMed] [Google Scholar]
- 81.Steiner MS, et al. Toremifene citrate versus placebo for treatment of bone loss and other complications of androgen deprivation therapy in patients with prostate cancer. Journal of Clinical Oncology. 2004;22(14):406S–406S. [Google Scholar]
- 82.Smith MR, et al. Toremifene increases bone mineral density in men receiving androgen deprivation therapy for prostate cancer: Interim analysis of a multicenter phase 3 clinical study. Journal of Urology. 2008;179(1):152–155. doi: 10.1016/j.juro.2007.08.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith MR, et al. Toremifene Decreases Vertebral Fractures in Men Younger Than 80 Years Receiving Androgen Deprivation Therapy for Prostate Cancer. Journal of Urology. 2011;186(6):2239–2244. doi: 10.1016/j.juro.2011.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smith MR, et al. Toremifene to Reduce Fracture Risk in Men Receiving Androgen Deprivation Therapy for Prostate Cancer. Journal of Urology. 2013;189(1):S45–S50. doi: 10.1016/j.juro.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 85.Poluri RTK, et al. RNA sequencing data of human prostate cancer cells treated with androgens. Data in brief. 2019;25:1–4. doi: 10.1016/j.dib.2019.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andrews, S. FastQC - A quality control tool for high throughput sequence data; Available from, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, 2010.
- 87.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bray NL, et al. Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology. 2016;34(5):525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 89.Feoktistova M, Geserick P, Leverkus M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harbor Protocols. 2016;2016(4):pdb.prot087379. doi: 10.1101/pdb.prot087379. [DOI] [PubMed] [Google Scholar]
- 90.Long Q, et al. Global Transcriptome Analysis of Formalin-Fixed Prostate Cancer Specimens Identifies Biomarkers of Disease Recurrence. Cancer Research. 2014;74(12):3228–3237. doi: 10.1158/0008-5472.CAN-13-2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data generated during this study is available from the Gene Expression Omnibus (GSE128749 for LNCaP and LAPC4 cells; GSE128201 for 22 Rv1) and are described elsewhere85. Data from biopsies come from SRA access number SRR1164789, SRR1164790, SRR1164791 and SRR116479290.