

Abstract
Disseminated disease is present in ≈50% of colorectal cancer patients upon diagnosis, being responsible for most of cancer deaths. Addition of biological drugs, as Bevacizumab, to chemotherapy, has increased progression free survival and overall survival of metastatic colorectal cancer (mCRC) patients. However, these benefits have been only reported in a small proportion of patients. To date, there are not biomarkers that could explain the heterogeneity of this disease and would help in treatment selection. Recent findings demonstrated that microRNAs (miRNAs) play an important role in cancer and they can be encapsulated with high stability into extracellular vesicles (EVs) that are released in biological fluids. EVs can act as cell-to-cell communicators, transferring genetic information, such as miRNAs. In this context, we aimed to investigate serum EV associated miRNAs (EV-miRNAs) as novel non-invasive biomarkers for the diagnosis and prognosis of Bevacizumab-treated mCRC patients. We observed that baseline miRNA-21 and 92a outperformed carcinoembryonic antigen levels in the diagnosis of our 44 mCRC patients, compared to 17 healthy volunteers. In addition, patients who died presented higher levels of miRNA-92a and 222 at 24 weeks. However, in the multivariate Cox analysis, higher levels of miRNA-222 at 24 weeks were associated with lower overall survival. Altogether, these data indicate that EV-miRNAs have a strong potential as liquid biopsy biomarkers for the identification and prognosis of mCRC.
Subject terms: Tumour biomarkers, Tumour biomarkers, Colorectal cancer, Colorectal cancer
Introduction
Colorectal cancer (CRC) is the second most common cancer in women and the third most in men worldwide. Moreover, it accounts for 8.9% of all tumour-related mortality and is the second most common cause of cancer death1. Disseminated disease is present upon diagnosis in 50% of the patients [lymph nodes (35%) and distant organs (22%)] and half of the patients diagnosed as localized tumours will eventually develop it2. In recent years, the application of new targeted therapies, including anti-angiogenic drugs, has contributed to largely increase the overall survival (OS) of metastatic colorectal cancer mCRC patients, reporting median survivals of ≈30 months3. Bevacizumab, a humanized monoclonal antibody targeting vascular endothelial growth factor (VEGF) has demonstrated benefits in progression-free survival (PFS) and OS in combination with chemotherapy4,5. Despite these improvements in expectancy and quality of life and proven effectiveness of Bevacizumab, just 40% of patients exhibit favourable responses to the therapy6. Plasma/serum levels of VEGF-A7 and mutations in Ras/Raf/Mek/Erk pathway8, among others9, have been proposed as predictors of the efficacy of Bevacizumab. So far, controversial results have been reported10–13. Circulating tumour cells (CTCs), considered to be responsible for disease relapse, have been also evaluated as prognosis biomarker for mCRC patients under Bevacizumab therapy14–16, however more evidence is needed for clinical application.
This scenario creates an urgent need to discover and validate predictive biomarkers for prognosis and treatment response17. The use of liquid biopsy provides potential clinically-relevant non-invasive genomic and epigenomic signatures for cancer monitoring.
Due to their high abundance and their role as regulators of gene expression, circulating microRNAs (miRNAs), small non-coding RNAs (19–24 nucleotides long), have been proposed as potential markers in several cancer types18. Although, they are implicated in physiological processes, miRNAs take also part in cancer mechanisms including tumour growth, angiogenesis and metastasis. In cancer, miRNAs play two different roles that appear to be context specific, acting as onco-miRNAs when they inhibit the expression of tumour suppressor genes accelerating tumorigenesis or as tumour-suppressors when they prevent tumour progression by blocking oncogene expression19.
MiRNAs have been described in body fluids as circulating-free molecules20, associated with proteins21 or encapsulated in extracellular vesicles (EVs)22. Whereas circulating-free miRNAs isolated from body fluids include miRNAs released during cell death and damage23, miRNAs packaged in EVs are selectively released by cells for communication purposes. In addition, hypoxic conditions, that take place inside the tumour, enhance EVs release and modulate their content24. Thus, they might increase the amount of specific tumour miRNAs found in the blood25. Moreover, lipid membrane coverage protects miRNA from RNases degradation. As a result, EV-derived miRNAs present higher specificity and stability than circulating miRNAs, making them better liquid biopsy biomarkers for cancer diagnosis and prognosis.
This study aimed to compare the potential of a selectively designed panel of EV-miRNAs with the commonly used clinical or experimental biomarkers as CEA, CTCs or KRAS status in the diagnosis and prognosis of mCRC patients under first-line Bevacizumab combined chemotherapy treatment (Fig. 1).
Figure 1.

Graphical summary of the employed methodology during this study.
Results
This study included 44 mCRC patients, 23 (52.3%) of them with primary tumours located in the colon and 21 (47.7%) in the rectum. Median follow-up time for all patients was 27 months (range 1 – 67 months). Clinical characteristics of patients and healthy donors are summarized in Table 1, including baseline CEA values that were also available in our cohort of healthy donors.
Table 1.
Characteristics of metastatic colorectal cancer patients and healthy donors included in the study. Abbreviations: mCRC: metastatic colorectal cancer; 12w: 12 weeks; 24w: 24 weeks; CTCs: Circulating tumour cells.
| Characteristics N (%) | mCRC patients (N = 44) | Healthy Controls (N = 17) | |||
|---|---|---|---|---|---|
| Gender | Female | 14 (31.8) | 7 (41.2) | ||
| Male | 30 (68.2) | 10 (58.8) | |||
| Age (years) | <55 | 13 (29.5) | 15 (88.2) | ||
| > 55 | 31 (70.5) | 2 (11.8) | |||
| Primary tumour location | Colon | 23 (52.3) | |||
| Rectum | 21 (47.7) | ||||
| Metastasis location | Liver | 23 (54.7) | |||
| Lung | 13 (40) | ||||
| Other | 6 (14.3) | ||||
| Metachronous metastasis | Yes | 5 (11.4) | |||
| No | 39 (88.6) | ||||
| Metastasis surgery | Yes | 11 (27.5) | |||
| No | 29 (72.5) | ||||
| K-RAS status | Mutated | 19 (46.3) | |||
| Wild-Type | 22 (53.7) | ||||
| CEA levels | Baseline | 12w | 24w | Baseline | |
| Standard | 10 (22.7) | 15 (37.5) | 15 (38.5) | 16 (94.1%) | |
| High | 34 (77.3) | 25 (62.5) | 24 (61.5) | 1 (5.9%) | |
| CA 19.9 levels | Baseline | 12w | 24w | ||
| Standard | 19 (46.3) | 22 (55) | 22 (57.9) | ||
| High | 22 (53.7) | 18 (45) | 16 (42.1) | ||
| Response | 12w | 24w | |||
| Non-favourable | 17 (43.6) | 24 (60) | |||
| Favourable | 22 (56.4) | 16 (40) | |||
| Progression | Yes | 34 (79.1) | |||
| No | 9 (20.9) | ||||
| Death | Yes | 38 (86.4) | |||
| No | 6 (13.6) | ||||
| CTCs | Extraction | Baseline | 12w | 24w | |
| Mean (range) | 1.8 (0–17) | 3 (0–64) | 0.74 (0–6) | ||
| Negative | 27 (61.4) | 24 (61.5) | 27 (71.1)) | ||
| Positive | 17 (38.6) | 15 (38.5) | 11 (28.9) | ||
Isolation and characterization of EVs
The study of serum EVs from cancer patients has become a very promising tool in the liquid biopsy field. Before performing specific analysis, the characterization of EVs is a mandatory step. According to the recommendations of the International Society of Extracellular Vesicles (ISEV), there are minimal requirements to claim a proper isolation of EVs26. In order to assess the population heterogeneity, the ISEV recommends that the employment of electron or atomic force microscopy be paired with a single tracking method. It also recommends the identification of specific markers as transmembrane proteins (CD63), intracellular proteins associated to membrane proteins (Alix, Hsp70), and intracellular proteins non-associated to plasmatic membrane proteins (Calnexin) by Western blot analysis. Following these requirements, we performed a precise characterization of our EVs samples. First, the Nanoparticle Tracking Analysis (Nanosight, Marvel. UK) resulted in a concentration of 1.02×1011 particles/ml and a diameter of mode = 127 nm ± 6.5 nm (Fig. 2A). Second, similar size and typical morphology of EVs were observed by transmission electron microscopy (TEM) (Fig. 2B). Lastly, the Western blot demonstrated high expression of Alix and CD63 on EVs samples, similar expression of Hsp70 on both culture cells and EVs, and higher levels of calnexin in culture cells compared to EVs (Fig. 2C). These data suggest that the employed methodology is suitable to isolate EVs from serum samples. Original blots are available in Supplementary Fig. S1.
Figure 2.

Characterization of EVs: (A) Nanoparticle Tracking Analysis analysis of extracellular vesicles from serum of healthy donors: Concentration 1.02×1011 ± 1.38×1010 and mode 127 nm ± 16 nm in size. (B) Transmission Electron Microscopy (TEM) image of isolated EVs from patients after purification via size exclusion chromatography (SEC), in order to reduce protein contamination in the staining. (C) Western blot image from cell control and serum EVs lysates, showing higher levels of calnexin, lower of Alix and lower of CD63 in cell compared to EV lysates. In addition, similar levels of Hsp70 could be observed between the 3 samples.
EV-miRNAs as biomarkers in mCRC identification
To evaluate the potential of the selected miRNAs to identify mCRC patients, we compared the baseline (B) serum expression of the 10 selected EVs-miRNAs in our cohort of 44 mCRC patients to the 17 healthy volunteers. The selection of these 10 miRNAs was based on previously described roles in the pathogenesis or having a CRC associated gene as a target. miRNAs analysed were miR-126, 155, 19b, 194, 20a, 200b, 21, 222, 552, and 92a (Supplementary Table S1).
We identified statistically significant differences in 7 miRNAs, detecting higher levels of miR-19b (p = 0.008), miR-20a (p < 0.001), miR-200b (p < 0.001), miR-21 (p < 0.001), miR-222 (p < 0.001), miR-552 (p = 0.038) and miR-92a (p < 0.001) in mCRC patients (Mann-Whitney U test). However, miR-126 (p = 0.054), miR-194 (p = 0.343) and miR-155 (p = 0.131) showed no statistically significant differences between two groups (Fig. 3).
Figure 3.
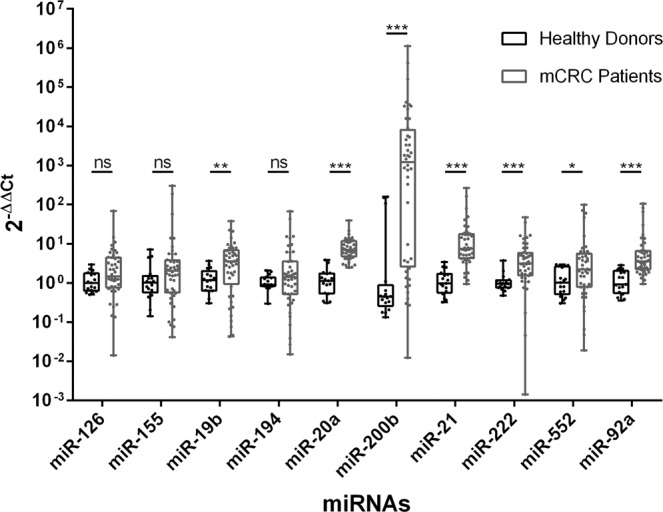
miRNA expression in mCRC patients and healthy donors: Extracellular vesicles (EVs) from metastatic colorectal cancer (mCRC) patients showed higher expression of miR-19b, 200b, 21, 222 and 92a in comparison to those of healthy donors. Data are presented as a box and whiskers plots (min to max). Mann-Whitney U test was used: *p < 0.05, **p < 0.01, ***p < 0.001, ns: no significant differences.
The univariate logistic binary regressions, adjusted by age and gender, showed that miR-19b (p = 0.029), miR-21 (p = 0.005), miR-222 (p = 0.026) and miR-92a (p = 0.005) were independent diagnostic factors of mCRC (Supplementary Table S2). We also evaluated their respective ROC curves to check the sensitivity and specificity in comparison with the current clinical biomarker CEA (Fig. 4A). The AUC of miR-21 and miR-92a were significantly higher than CEA AUC (p < 0.0001), while miR-222 and miR-19b presented a similar AUC (p = 0.147) and lower AUC (p = 0.0084) respectively (Fig. 4B) (Supplementary Table S2).
Figure 4.

ROC curves and AUC of EV-miRNA expression in mCRC identification: (A) Receiver Operating Characteristics (ROC) curves and AUC of baseline EVs miR-19b, 21, 222, 92a and CEA, adjusted by age and gender, according to metastatic colorectal cancer (mCRC) identification. Shaded areas show the increased sensitivity and specificity of each miRNA vs. the CEA curve. (B) Histogram representing the AUC of each miRNA and their statistical differences with baseline CEA. miR-21 and miR-92 were the best predictors in the identification of mCRC with higher sensitivity and specificity than baseline CEA, while miR-222 showed no statistical differences and 19b had lower sensitivity and specificity than CEA. Data are presented as histogram of the mean with the standard error bars. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns: no significant differences.
EV-miRNAs vs clinical characteristics and CTCs
We compared the clinical characteristics and CTCs of our 44 mCRC patients with the expression of the 4 cancer-related EV-miRNAs (Table 2). The correlation with the other 6 EV-miRNAs has been presented in Supplementary table S3. We observed that high miR-222 (B) levels correlated with older patients (p = 0.017). K-RAS mutated patients showed low-levels of miR-92a (B) (p = 0.010) (Supplementary Figure S2). Furthermore, patients with elevated levels of CEA (B) expressed higher levels of miR-126 (p = 0.041) and miR-200b (p = 0.035) at baseline (data not shown). Low levels of miR-222 (B), were associated with high levels of Ca 19.9 at 24 weeks (24w), during second response assessment (p = 0.042), but no association was found between EV-miRNAs and other clinical variables. More interestingly, when we compared the expression of EVs-miRNAs and CTCs presence during the follow-up, we found a correlation between high levels of miR-21 (B) and the presence of CTCs (24w) (p = 0.023) (Table 2). Similarly, high levels of miR-92a at 12 weeks (12w), during first response assessment, were associated with presence of CTCs (12w) (p = 0.031) (data not shown).
Table 2.
Association between baseline EV miRNA expression and mCRC patient characteristics. Mann-Whitney U and Kruskal-Wallis tests were used. Abbreviations: CTCs: Circulating tumour cells; miR: miRNA; B: Baseline; 12w: 12 weeks; 24w: 24weeks; *p < 0.05.
| n | miR-19b | miR-21 | miR-222 | miR-92a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| median | p | median | p | median | p | median | p | |||
| Gender | Male | 30 | 3.756 | 0.364 | 7.191 | 0.529 | 2.848 | 0.087 | 3.696 | 0.940 |
| Female | 14 | 3.146 | 8.588 | 4.437 | 3.079 | |||||
| Age (years) | <55 | 13 | 3.546 | 0.495 | 5.759 | 0.463 | 1.748 | 0.017* | 2.886 | 0.389 |
| >55 | 31 | 4.455 | 7.831 | 4.762 | 3.616 | |||||
| Primary tumour location | Colon | 23 | 3.569 | 0.916 | 3.329 | 0.647 | 3.226 | 0.245 | 5.518 | 0.169 |
| Rectum | 21 | 3.546 | 7.809 | 3.106 | 3.203 | |||||
| Metastasis location | Liver | 23 | 3.020 | 0.087 | 9.561 | 0.687 | 4.155 | 0.519 | 3.571 | 0.280 |
| Lung | 13 | 4.986 | 7.155 | 2.661 | 2.439 | |||||
| Other | 6 | 0.343 | 7.422 | 2.709 | 5.748 | |||||
| Metachronous metastasis | No | 39 | 3.820 | 0.386 | 7.809 | 0.216 | 3.478 | 0.802 | 3.571 | 0.858 |
| Yes | 5 | 2.800 | 5.080 | 3.106 | 3.203 | |||||
| Metastasis surgery | No | 29 | 3.546 | 0.811 | 7.831 | 0.591 | 3.019 | 0.185 | 3.342 | 0.835 |
| Yes | 11 | 3.569 | 5.759 | 5.101 | 3.185 | |||||
| K-RAS status | Wild-type | 22 | 2.789 | 0.053 | 7.724 | 0.347 | 4.768 | 0.143 | 3.696 | 0.010* |
| Mutated | 19 | 5.061 | 5.462 | 3.035 | 2.340 | |||||
| CEA (B) | Standard | 10 | 2.319 | 0.534 | 7.025 | 0.128 | 2.588 | 0.534 | 2.560 | 0.312 |
| High | 34 | 3.882 | 13.151 | 3.352 | 3.696 | |||||
| CA 19.9 (B) | Standard | 19 | 3.271 | 0.855 | 7.227 | 0.448 | 3.117 | 0.734 | 2.681 | 0.182 |
| High | 22 | 4.138 | 11.242 | 3.123 | 3.696 | |||||
| CEA (12w) | Standard | 15 | 3.943 | 0.699 | 6.822 | 0.376 | 4.155 | 0.192 | 2.681 | 0.720 |
| High | 25 | 3.546 | 13.378 | 3.019 | 3.571 | |||||
| CA 19.9 (12w) | Standard | 22 | 3.420 | 0.527 | 7.242 | 0.840 | 3.817 | 0.240 | 3.194 | 0.840 |
| High | 18 | 3.6883 | 10.377 | 2.481 | 3.524 | |||||
| CEA (24w) | Standard | 15 | 3.943 | 0.539 | 7.329 | 0.658 | 4.155 | 0.270 | 2.681 | 0.539 |
| High | 24 | 3.557 | 13.151 | 3.027 | 3.673 | |||||
| CA 19.9 (24w) | Standard | 22 | 3.695 | 0.455 | 7.473 | 0.737 | 4.346 | 0.042* | 3.035 | 0.781 |
| High | 16 | 4.001 | 6.114 | 2.185 | 3.673 | |||||
| Response (12w) | Non-favourable | 17 | 3.569 | 0.747 | 7.227 | 0.547 | 3.035 | 0.510 | 3.775 | 0.362 |
| Favourable | 22 | 3.146 | 7.219 | 3.817 | 2.667 | |||||
| Response (24w) | Non-favourable | 24 | 3.408 | 0.318 | 8.696 | 0.420 | 3.076 | 0.672 | 4.052 | 0.795 |
| Favourable | 16 | 4.428 | 6.142 | 3.631 | 3.194 | |||||
| Progression | No | 9 | 4.455 | 0.353 | 6.822 | 0.268 | 5.681 | 0.471 | 2.681 | 0.736 |
| Yes | 34 | 3.420 | 7.820 | 3.112 | 3.594 | |||||
| Death | No | 6 | 3.728 | 0.934 | 5.115 | 0.193 | 2.822 | 0.677 | 3.983 | 0.652 |
| Yes | 38 | 3.557 | 7.713 | 3.171 | 3.524 | |||||
| CTC (B) | Negative | 27 | 3.943 | 0.392 | 12.923 | 0.477 | 4.155 | 0.604 | 3.616 | 0.539 |
| Positive | 17 | 3.020 | 7.227 | 3.106 | 2.681 | |||||
| CTC (12w) | Negative | 24 | 3.557 | 0.943 | 7.473 | 0.875 | 4.246 | 0.146 | 2.9333 | 0.383 |
| Positive | 15 | 4.455 | 6.822 | 2.301 | 3.571 | |||||
| CTC (24w) | Negative | 27 | 3.569 | 0.505 | 5.462 | 0.023* | 3.019 | 0.251 | 2.439 | 0.308 |
| Positive | 11 | 6.855 | 17.722 | 5.101 | 3.342 | |||||
EV-miRNAs, CTCs, CEA and Ca19.9 in RECIST response prediction
RECIST responses (12w) were evaluated in 39 patients, 17 (43.6%) of them developed non-favourable responses while 22 (56.4%) patients exhibited favourable responses. RECIST response (24w) was evaluated in 40 patients, 24 (60%) patients presented non-favourable responses while 16 (40%) exhibited favourable responses (Table 1). We compared our clinical (CEA and Ca 19.9) and experimental biomarkers (EV-miRNAs and CTCs) with RECIST response during the follow-up to evaluate which was the best biomarker at predicting the response at 12 and 24 weeks. At baseline time, no clinical or experimental biomarkers were related to RECIST responses (p > 0.05). However, CEA (12w) was statistically correlated with the RECIST response (24w), resulting in higher CEA levels in those patients with non-favourable responses (p = 0.049).
Progression-free survival
Thirty-four of the 44 patients progressed during the study (77.3%). Median PFS of patients who progressed was 11 months (range: 1–63), compared to 34 (range: 7–66) months in those who did not progress. We evaluated the association between the clinicopathological characteristics, including our experimental biomarkers, and tumour-progression. This analysis showed that among all of these variables, only higher levels of miR-92a (12w) were associated with tumour-progression, although it was not statistically significant (p = 0.073) (Mann-Whitney U test). In the same way, when the miR values were dichotomized, the Kaplan-Meier analysis correlated higher risk of progression risk to patients with higher miR-92a (12w) levels (log-rank test p = 0.076) (Fig. 5A). In the univariate Cox’s regression analysis, we observed that the primary tumour location, the number of CTCs at baseline status and the expression of miR-92 (12w) were the only variables to be included in the model. However, in the multivariate analysis, none of them were an independent factor associated with PFS (p > 0.05) (Table 3). On the other hand, we analysed the role of these biomarkers as independent prognostic factors for PFS in wild-type (N = 22) and in mutated KRAS (N = 19) patients (Supplementary Table S4).
Figure 5.

Kaplan-Meier curves of PFS and OS according to miR-92a and miR-222 expression: (A) Progression-free survival (PFS) probability curves according to lower and higher expression of miR-92 at 12 weeks (12w) showing increased DFS in the lower expression group but no statistical differences. (B) Overall survival (OS) probability curves according to lower and higher expression of miR-222 at 24 weeks (24w) are statistically different. (C) Overall survival (OS) probability curves according to lower and higher expression of miR-92 (24w) showing 2 separated curves but with non-statistically significant differences. Log-rank test was used. p = p-value.
Table 3.
Univariate and multivariate Cox proportional-hazards regression analysis for progression-free survival. Abbreviations: PFS: Progression-free survival; HR: Hazard Ratio; CI: Confidence Interval; p: p-value; CTCs: Circulating tumour cells; miR: miRNA; B: Baseline; 12w: 12 weeks; 24w: 24weeks; *p < 0.05.
| Univariate analysis | Multivariate analysis | |||||||
|---|---|---|---|---|---|---|---|---|
| Characteristics | Median PFS (months) | HR | 95% CI | p | HR | 95% CI | p | |
| All patients | 11.5 | |||||||
| Gender | Female | 11 | 1.17 | 0.55–2.47 | 0.688 | 1.62 | 0.60–4.33 | 0.339 |
| Male | 14 | |||||||
| Age (years) | >55 | 13.5 | 0.75 | 0.36–1.56 | 0.436 | 0.49 | 0.16–1.46 | 0.198 |
| <55 | 10 | |||||||
| Primary tumour location | Colon | 14 | 0.34 | 0.16–0.74 | 0.006* | 0.47 | 0.18–1.25 | 0.131 |
| Rectum | 10 | |||||||
| Metastasis location | Liver | 13 | 0.509 | |||||
| Lung | 12 | 0.94 | 0.43–2.09 | 0.885 | ||||
| Other | 7 | 1.73 | 0.64–4.68 | 0.281 | ||||
| Metachronous metastasis | Yes | 12 | 1.67 | 0.63–4.42 | 0.301 | |||
| No | 11.5 | |||||||
| Metastasis surgery | Yes | 14 | 0.82 | 0.36–1.85 | 0.630 | |||
| No | 11.5 | |||||||
| K-RAS status | Mutated | 11.5 | 0.89 | 0.43–1.81 | 0.743 | |||
| Wild-Type | 13.5 | |||||||
| CEA (B) | High | 11 | 1.33 | 0.58–3.06 | 0.509 | |||
| Standard | 13 | |||||||
| Ca 19.9 (B) | High | 14 | 0.66 | 0.33–1.31 | 0.235 | |||
| Standard | 10 | |||||||
| CEA (12w) | High | 11 | 1.46 | 0.69–3.09 | 0.324 | |||
| Standard | 14 | |||||||
| Ca 19.9 (12w) | High | 13 | 0.66 | 0.31–1.38 | 0.267 | |||
| Standard | 13 | |||||||
| CEA (24w) | High | 11 | 1.48 | 0.71–3.09 | 0.302 | |||
| Standard | 14 | |||||||
| Ca 19.9 (24w) | High | 14 | 0.65 | 0.30–1.41 | 0.279 | |||
| Standard | 13 | |||||||
| Response (12w) | Favourable | 12.5 | 0.61 | 0.30–1.25 | 0.181 | |||
| Non-favourable | 9.5 | |||||||
| Response (24w) | Favourable | 14 | 0.67 | 0.33–1.36 | 0.263 | |||
| Non-favourable | 10 | |||||||
| Number of CTCs (B) | 1.15 | 1.02–1.31 | 0.029* | |||||
| Number of CTCs (12w) | 1.02 | 0.98–1.05 | 0.357 | |||||
| Number of CTCs (24w) | 0.98 | 0.75–1.28 | 0.896 | |||||
| miR-92a (12w) | High | 12 | 2.45 | 0.83–7.23 | 0.104 | 2.36 | 0.76–7.32 | 0.138 |
| Low | 14 | |||||||
Overall survival
Thirty-eight patients died during the follow-up (86.4%), having a median OS of 23.5 months (range: 1–67), compared to 34 months (range: 28–66) of the 7 patients who survived. The association between the survival and the clinical variables, including our liquid biopsy biomarkers, was also analysed. Thus, the Mann-Whitney U test showed that higher levels of miR-222 (24w) and miR-92a (24w) were associated with the death of these patients (p = 0.009 & p = 0.024, respectively) (data not shown). As represented in Fig. 5B, when miR levels were dichotomized, there was a clear worse prognosis in patients with high levels of miR-222 (24w) (p = 0.015). On the contrary, despite visible differences in the curves, no statistical association was found between higher levels of this miRNA and a worse prognosis (p = 0.166) (Fig. 5C). In the Cox’s univariate analysis, miR-92a (24w) was not statistically associated with OS. However, high levels of miR-222 (24w), as well as the number of CTCs (12w), the clinical CEA levels (12w & 24w), and RECIST responses (12w & 24w) were all associated with worse prognosis (Table 4). The multivariate analysis presented the RECIST response and the miR-222 expression at 24 weeks as the only statistically associated variables with OS (p = 0.017 & p = 0.023, respectively) (Table 4). On the other hand, we analysed the role of these biomarkers as independent prognostic factors for OS in wild-type (N = 22) and in mutated KRAS (N = 19) patients (Supplementary Table S5).
Table 4.
Univariate and multivariate Cox proportional-hazards regression analysis for overall survival. Abbreviations: OS: Overall survival; HR: Hazard Ratio; CI: Confidence Interval; p: p-value; CTCs: Circulating tumour cells; miR: miRNA; B: Baseline; 12w: 12 weeks; 24w: 24weeks; *p < 0.05.
| Univariate analysis | Multivariate analysis | |||||||
|---|---|---|---|---|---|---|---|---|
| Characteristics | Median OS (months) | HR | 95% CI | p | HR | 95% CI | p | |
| All patients | 27 | |||||||
| Gender | Female | 23.5 | 1.04 | 0.52–2.09 | 0.918 | 1.35 | 0.60–3.04 | 0.467 |
| Male | 27 | |||||||
| Age (years) | >55 | 28 | 0.93 | 0.46–1.89 | 0.842 | 0.55 | 0.22–1.41 | 0.213 |
| <55 | 22 | |||||||
| Primary tumour location | Colon | 27 | 0.95 | 0.49–1.84 | 0.883 | |||
| Rectum | 27.5 | |||||||
| Metastasis location | Liver | 29 | 0.328 | |||||
| Lung | 26 | 1.48 | 0.67–3.26 | 0.334 | ||||
| Other | 18.5 | 2.07 | 0.74–5.78 | 0.167 | ||||
| Metachronous metastasis | Yes | 27 | 1.22 | 0.47–3.17 | 0.684 | |||
| No | 27.5 | |||||||
| Metastasis surgery | Yes | 45 | 0.55 | 0.25–1.23 | 0.146 | |||
| No | 27 | |||||||
| K-RAS status | Mutated | 30 | 0.73 | 0.36–1.47 | 0.374 | |||
| Wild-Type | 27 | |||||||
| CEA (B) | High | 25 | 1.31 | 0.61–2.82 | 0.490 | |||
| Standard | 29 | |||||||
| Ca 19.9 (B) | High | 26 | 1.07 | 0.54–2.11 | 0.848 | |||
| Standard | 29 | |||||||
| CEA (12w) | High | 24.5 | 2.34 | 1.04–5.26 | 0.041* | |||
| Standard | 37 | |||||||
| Ca 19.9 (12w) | High | 24.5 | 1.58 | 0.76–3.29 | 0.222 | |||
| Standard | 35 | |||||||
| CEA (24w) | High | 22 | 2.85 | 1.25–6.51 | 0.013* | |||
| Standard | 37 | |||||||
| Ca 19.9 (24w) | High | 26 | 1.62 | 0.75–3.45 | 0.220 | |||
| Standard | 35 | |||||||
| Response (12w) | Favourable | 30.5 | 0.38 | 0.18–0.78 | 0.008* | |||
| Non-favourable | 15 | |||||||
| Response (24w) | Favourable | 36 | 0.356 | 0.17–0.76 | 0.007* | 0.32 | 0.13–0.81 | 0.017* |
| Non-favourable | 18 | |||||||
| Number of CTCs (B) | 1.12 | 0.98–1.28 | 0.089 | |||||
| Number of CTCs (12w) | 1.04 | 1.00–1.07 | 0.045* | |||||
| Number of CTCs (24w) | 1.13 | 0.90–1.42 | 0.285 | |||||
| miR-222 (24w) | High | 25 | 2.55 | 1.16–5.58 | 0.02* | 2.65 | 1.15–6.13 | 0.023* |
| Low | 34 | |||||||
| miR-92 (24w) | High | 27 | 1.63 | 0.81–3.32 | 0.174 | |||
| Low | 30 | |||||||
Proteinase K and size exclusion chromatography controls
One of the major concerns of this work was the purity of EVs from serum samples. Serial centrifugation methods might also lead to the co-isolation of lipoproteins and proteins complex. More interestingly, these protein aggregates could be co-precipitated with serum circulating free-miRNAs. For this reason, to ensure that our results of miRNA expression were mostly related to EVs content, we performed two controls. The first control included a proteinase K treatment while the second consisted in the isolation of EVs by Size Exclusion Chromatography (SEC).
In the proteinase K control, neither miR-21 nor miR-222 showed statistical differences between treated vs. control paired samples in the Wilcoxon matched-pairs signed rank test (p = 0.25 & p = 1.00, respectively) (Supplementary Figure S3). Similarly, in the comparison between paired samples obtained by ultracentrifugation and by SEC, no statistical differences were found regarding miR-21 nor miR-222 expression (p = 0.5 & p = 0.25, respectively) (Supplementary Figure S4).
Discussion
CRC is one of the most frequent causes of cancer death in the world1. In the last years, screening techniques have been improved, with special emphasis on blood based biomarkers27. Nevertheless, disseminated disease is still present at the time of diagnosis in a large percentage of patients, being responsible for most of these deaths. On the other hand, miRNAs have important roles on biological and pathological process in all the cell types. Particularly in cancer cells, specific miRNAs are actively encapsulated in EVs that are released to the circulation. In addition, due to the hypoxic microenvironment and/or alterations on endo-lysosomal trafficking, EVs production rates are relatively increased in tumour cells28. Considering the benefits of liquid biopsy as non-invasive with potential multiple sampling over time, especially important in the follow-up of metastatic patients, we thought that serum EV-derived miRNAs could serve as reliable and specific tumour biomarkers. The aim of the present study was to evaluate the disseminated disease identification and prognostic potential of our EV-miRNA panel in mCRC patients treated with anti-angiogenic therapy in combination with a FOLFOX regimen. This study included a homogeneous mCRC population treated with Bevacizumab-FOLFOX-6m, the standard first-line combination for those patients.
Our designed panel consisted of 10 miRNAs that had been previously described as markers associated with diagnosis (miR-155, 20a, 200b, 21, 222, 92a), prognosis (miR-155, 194, 20a, 21, 222, 552 and 92a), metastasis location (miR-19b, 194, 200b, 20a and 552), chemotherapy response (miR-126, 200b, 21), bevacizumab treatment (miR-126 and 155), and clinical stage (miR-21, 552 and 92a) (Supplementary Table 1). Our results showed the metastatic detection and prognostic value of each of these EV-miRNAs in our cohort of patients.
Based on our experiments, we were able to claim that our methodology successfully characterized miRNA expression of EVs cargo, with minimal contamination from circulating miRNAs that could be associated to EVs in protein aggregates.
EV-miRNA expression analysis from healthy subjects and mCRC patients showed miR-19b, miR-21, miR-222 and miR-92a as independent diagnostic factors with high specificity and sensitivity. The clear differences between the two groups demonstrate the potential use of these 4 miRNAs as biomarkers for early prediction of metastatic disease in CRC patients. In addition, miR-21 and 19b presented higher sensitivity and specificity than the routine clinical biomarker CEA.
Our results fall in line with previous studies on CRC where plasma or serum were employed as a sample for miRNA study. Chen et al. detected higher levels of serum miR-222 in CRC patients using Solexa sequencing29, and Giráldez, et al. found that miR-19b were significantly up-regulated in plasma from CRC patients compared to healthy donors30. As we observed in our results, the meta-analysis performed by Yang, et al. defined plasma miR-92a as a diagnostic marker for CRC31. Furthermore, Ogata-Kawata, et al. proved that serum exosome-derived miR-21 was an independent diagnostic factor32 and predictive factor for OS in all stages of CRC33. Our results reinforce those from Ostenfeld et al., that reported a significant higher expression of miR-222 in EpCAM+ EVs from CRC compared to healthy donors34, and from Fangfang et al. that correlated exosomal miR-92a expression to clinical stage of CRC patients35. Interestingly, despite including similar cohorts of patients, each of these studies identified a different miRNA as a diagnostic biomarker. These differences could suggest that the miRNA signalling depends on the disease status and/or the type of sample extracted (EV-miRNAs, EPCAM+ Exo miRNAs or free miRNAs).
The observed over-expression of miR-20a, miR-21, miR-222 and miR-92a are all in concordance with multiple evidence that demonstrate the importance of these miRNAs as tumour promoters. For example, it has been well documented that miRNA-21 targets a great number of key proteins: activated protein 1 (AP-1), nuclear factor I (NFI), maspin, fas ligand, programmed cell death protein 4 (PDCD4), tropomyosin 1(TPM1), tissue inhibitor of metalloproteinase 3 (TIMP3), acid nuclear phosphoprotein 32 A (ANP32A), phosphate and tensin homologue (PTEN), signal transducer and activator of transcription 3 (STAT3), c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), mitogen associated protein kinase (MAPK),s and many others proteins with important roles in cancer36. However, due to the high homogeneity of our population and the high range of complex gene interactions of each miRNAs, we did not observe many significant associations between miRNA levels and clinical data. Other researchers investigated KRAS-dependence for miRNA selection as EVs cargo, where miR-10b was preferentially enriched in wild-type KRAS-derived exosomes, while miR-100 was enriched in mutant KRAS-derived exosomes37. Here, we observed lower levels of EVs-derived miR-92a in KRAS mutated compared with wild-type patients. This can be explained by the study conducted by Mackenzie et al.; their results showed that KRAS mutation reduces packaging of specific miRNAs into EVs through the reduction on Ago2 phosphorylation38.
Additionally, our data report an association between miRNA levels and the presence or absence of CTCs. During the last 20 years, CTCs have become an stronger marker for prognosis and response to the treatment39,40. CTCs are implicated in disease dissemination and they have been proposed as indicators of minimal residual disease41, which cannot be detected by standard imaging methodologies in the clinical practice42,43. miRNAs profiles have been related to the metastatic spread of the tumour, characterized by the presence of CTCs44. However, the methods for CTCs identification have not been completely standardized, including several expensive and time-consuming isolation and characterization protocols45. Our study, with a shorter turnaround time, proves that EV-miRNAs could become an important liquid biopsy tool to replace or complement the clinical relevance of CTCs.
Finally, in accordance with previous studies, our results report high expression of EV-miR-92a in patients with shorter PFS and OS, although not reaching enough statistical significance, probably due to the low number of patients. On the other hand, we observed that patients with higher EV-miR-222 levels presented shorter OS with clear statistical differences. Other studies have reported that the overexpression of miR-222 is related to poor survival in several types of cancer, including colorectal cancer46, as it has been associated to enhanced migration and invasion of cancer cells. In addition, our multivariate analysis presented miR-222 (24w) as a better prognostic factor for the overall survival than the clinical biomarkers CEA and Ca 19.9, as well as the CTCs that lost statistical significance during the analysis.
In conclusion, our data prove the importance of EV-miRNAs as potential liquid biopsy biomarkers in metastatic disease detection and the prognosis of overall survival of mCRC patients, complementing or even replacing the current clinical biomarkers CEA and Ca 19.9 and the CTCs.
Limitations of the study
This study has two considerable limitations. First, our cohort included only 44 patients, what makes necessary to confirm these results in a larger case series. Second, we restricted our analysis to a specific panel of EV-miRNAs on the basis of literature and bioinformatics target results, however, we cannot exclude the possibility that other miRNAs may also have clinical relevance in our population.
Methods
Study design
This prospective longitudinal study included 44 mCRC patients who underwent first-line treatment with FOLFOX-6m (Oxaliplatin 85 mg/m2, Leucovorin 400 mg/m2, 5-fluorouracil (FU) 400 mg/m2 bolus and 5-FU 2400 mg/m2) over 46 h and Bevacizumab (5 mg/kg) every 2 weeks until disease progression, at the Department of Oncology, San Cecilio University Hospital in Granada (Spain), between April 2011 and November 2015. None of the patients had previously received any other type of biological treatment. A cohort of 17 blood donors with no history of malignant disease was recruited from the University of Balearic Islands, Mallorca (Spain). The study was conducted in accordance with the Declaration of Helsinki and approved by the ethical Committee of the Hospital. Written informed consent was obtained from every enrolled subject.
Computed tomography of the chest, abdomen and pelvis was performed at baseline, at 12 weeks (12w), at 24 weeks (24w) and finally each 12 weeks until death. Image interpretation was performed using Response Evaluation Criteria in Solid Tumours (RECIST) version 1.147 to classify the patient evolution as complete response, partial response, stable disease, considered favourable responses, or as progressive disease, considered non-favourable response. Data were collected for the following clinical variables: age, gender, primary tumour location, metastasis surgery, primary tumour surgery, synchronous metastasis, K-RAS status, baseline, 12 weeks and 24 weeks Carcinoembryonic antigen (CEA) and Carbohydrate antigen 19.9 (Ca 19.9), RECIST response at 12 and 24 weeks, progression and survival. Clinical outcomes were evaluated in terms of PFS and OS. PFS was defined as the elapsed time from the start of the treatment to progression or death. OS was defined as the elapsed time from the start of the treatment to death.
Blood samples and CTC isolation
Peripheral blood samples (10 ml in EDTA Vacutainer® tubes for CTCs and 5 ml in BD Vacutainer® SST™ II Advance tubes for serum) were extracted before the initiation of therapy (B) and subsequently at 12 weeks and 24 weeks after, coinciding with the RECIST response assessment. Five ml of peripheral blood were also collected from healthy donors. CTC enrichment and detection were performed according to protocols previously established by our group48,49. Briefly, samples were processed by density gradient centrifugation with Histopaque-1119 (Sigma-Aldrich, UK) at 700 ×g for 45 min to isolate the mononuclear cell fraction containing the CTCs. This fraction was incubated with the multi-cytokeratin-specific antibody microbeads (CK3–11D5) (that binds to clones 7, 8, 18 and 19) (Miltenyi Biotec, Germany) and the FITC-anti-cytokeratin antibody (CK3-6H5) (Miltenyi Biotec, Germany). Cells were passed through the MACS Cell Separation magnetic columns (Miltenyi Biotec, Germany) and the enriched cytokeratin positive cells were spun down onto polylysine-coated glass slides for subsequent fluorescent microscopy visualization and enumeration.
EVs isolation
Blood samples were centrifuged at 1500 ×g for 15 min for serum collection. Then, serum was centrifuged at 10.000 ×g during 30 min to eliminate cellular debris. Supernatants were ultracentrifuged in 6 ml polyallomer ultracentrifuge tubes (Thermo Scientific, UK) in TFT 80.4 Rotor (Thermo Scientific, UK) at 100.000 ×g for 1 h at 4 °C. After that, supernatants were removed and EVs pellets were directly resuspended into the tube, either by adding RIPA lysis buffer, for Western blot protein controls, or homogenization buffer from the Maxwell 16 miRNA Tissue kit (Promega, USA), for subsequent miRNA analysis in patients. Samples were stored at −80 °C until further processing.
Cell culture
MCF-7 cells were cultured in DMEM high glucose Glutamax (Gibco, Germany) supplemented with 10% of fetal bovine serum (Gibco, Germany), 100 U/ ml penicillin and 100 ng/ ml streptomycin (Gibco, Germany) in a humidified incubator with 5% CO2 at 37 °C.
Nanoparticle tracking analysis
EV size distribution and concentration were measured using a NanoSight NS300 system equipped with an LM14 405 nm violet laser unit (Malvern Instruments, UK). EVs were diluted in PBS (1:500) for appropriate analysis and visualized at camera level 16 under control of a script, which included acquisition of 3 movies for 1 min at a fixed temperature of 22 °C. Analysis was performed using NTA 3.1 software. Detection threshold was set at 5 and other settings were kept at default.
Transmission electron microscopy (TEM) analysis
First, EV suspensions were purified using SEC to eliminate protein background. Then, they were adsorbed on active-carbon coated grids for 10 min, washed and fixed for 15 min in a 2% paraformaldehyde and 0.2% glutaraldehyde solution. Grids were briefly rinsed with water and immediately transferred to drops of uranyl methyl cellulose pH 4.0 on a cooled metal plate for 5 min, picked up and dried at room temperature. Finally, grids were introduced in a FEI Tecnai™ F20 (ThermoFisher, USA) TEM for imaging.
Western blot analysis
EV proteins were extracted adding 50 µl of RIPA lysis buffer (Sigma Aldrich, Germany) to the obtained EVs pellets or MCF-7 cells for cell controls. Lysates were centrifuged at 14.000 ×g and supernatants were collected and stored for protein determination. Protein concentration was quantified by BCA Protein Assay Kit (Thermo Scientific, UK), according with manufacturer’s instructions. Same amount of protein from each sample were loaded into precast SDS-PAGE gel (GE life science, UK) and run at 100 mV until optimal separation was obtained. Then, proteins were transferred to a PDVF membrane at 30 mV overnight. Plotted membranes were cut and incubated with primaries antibodies [anti-CD63 (clone MEM-259) (Abcam, UK), anti-Hsp70 (clone BB70) (ENZO Life Science, USA), anti-calnexin (clone AF18) and anti-Alix (clone 3A9) (Thermo Scientific, UK)] overnight at 4 °C and later with secondary IRDye 800CW anti-mouse antibody (LICOR, Germany) for 1 h at room temperature. Finally, membranes were revealed with an Odyssey infrared scanner.
RNA extraction, reverse-transcription and qRT-PCR
EV-miRNAs were extracted and analysed according previous publications of our group44. Briefly, EVs miRNAs extraction was performed using the Maxwell® 16 miRNA Tissue Kit (Promega, USA). Complementary DNA was synthesized with the TaqMan™ Advanced miRNA cDNA Synthesis Kit (Applied Biosystems, UK) and miRNA expression levels were analysed in triplicate using TaqMan™ MicroRNA assay probes (Supplementary Table S1) and TaqMan™ Universal PCR Master Mix (Applied Biosystems, UK) according to manufacturer recommendations in an Applied Biosystems 7900HT Fast Real-Time PCR System (Applied Biosystems, UK).
miRNA panel selection
We designed a panel of 10 miRNAs (126, 155, 19b, 194, 20a, 200b, 21, 222, 552, 92a) and miR-16 as the endogenous control. Normalization was performed according to the 2-ΔΔCt method50. miRNAs were selected according to previously described association with migration, angiogenesis, metastasis, diagnosis and prognosis in CRC. They were also selected based on their reported mRNA targets of genes associated with bevacizumab, CRC and mCRC as VEGF-1, VEGF-2, KRAS, APC, TGFBR, PI3KCA and TP5351, using the starBase v3.0 database52 (Supplementary Table S1).
Proteinase K treatment
First, three serum samples of 4 ml were divided in two aliquots of 2 ml each. They were centrifuged at 10.000 ×g during 30 min and the obtained supernatant was centrifuged at 100.000 ×g for 3 h. After that, the supernatant was discarded and the pellets were resuspended in 100 µl of digestion buffer (50 mM Tris-HCL; pH 8, 0,1 mM CaCl2, 3 mM DTT and 2.0 M Urea). Second, an aliquot of each sample was treated with proteinase K (Qiagen, Germany) to a final concentration of 0.25 mg/ml while the other was considered the untreated control. All aliquots were incubated at 37 °C during 30 min according for digestion, and at 70 °C during 15 min for Proteinase K inactivation. Finally, treated and untreated tubes were washed with PBS and centrifuged again at 100.000 ×g during 3 h. Exosome pellets were obtained and used for subsequent miRNA analysis.
Size exclusion chromatography (SEC)
As in the proteinase K treatment, two aliquots of 2 ml from 3 serum samples were employed. All samples were centrifuged at 10.000 ×g during 30 min and then, supernatants were employed to EVs isolation; one aliquot from each sample by SEC and another by ultracentrifugation. In the SEC, 2 ml supernatant was injected into a SEC Hiprep™ 16/60 Sephacryl® S-400 HR column (GE Healthcare, USA) at a flow rate of 0.5 ml PBS/min as previously described53. Aliquots of 5 ml were collected and EVs-containing fractions were pulled down by 100.000 ×g centrifugation during 3 h. Aliquots for ultracentrifugation followed our previously described protocol. Exosome pellets were employed for miRNAs analysis.
Statistical methods
Statistical analyses and graphs were performed using SPSS [SPSS Statistics for Windows, Version 22.0 (IBM Corp., USA)] and GraphPad Prism [Version 7.00 (GraphPad Software, USA)]. EV-miRNAs and CTCs were assessed as quantitative and dichotomous (miRNA: low/high and CTC: presence/absence) variables. miRNAs expression cut-offs were calculated with the Cutoff Finder web application54. The association between miRNAs and clinical characteristics or CTCs was evaluated using non-parametric Mann-Whitney U and Kruskal-Wallis tests. Paired samples controls were analysed by Wilcoxon matched-pair signed rank test. Logistic binary regression, receiver-operating characteristics (ROC) curves and the Area Under the Curve (AUC) were performed to test the sensibility and specificity of the miRNAs to identify mCRC patients. Categorical variables were compared by the Fisher’s exact test and correlations were measured by Spearman’s rank correlation. PFS and OS analyses were performed by log-rank test and by univariate and multivariate Cox Proportional-Hazards Regression. We applied the criterion of more than a 10% change in the coefficient estimate and p < 0.1555 for the selection of variables to be included in the multivariate model. p < 0.05 values were considered statistically significant.
Supplementary information
Acknowledgements
We would like to extend our gratitude to the all the patients and the healthy volunteers who participated in the study, as well as the University of Granada, Biomedicine PhD program. This work was supported by Roche Spain, the PhD grant from the University of Granada (DdMP) (2014) and the PhD grant from the Spanish Government (ARM) (FPU) 2014, REF FPU14/05461.
Author contributions
M.J.S., J.A.L., F.G.O.S. and J.L.P. conceived and designed the project. M.D., F.G.O.S and J.L.P. performed patient selection. D.d.M.P., A.R.M., A.O.P., A.R.R. and F.G.O.S. performed the experiments and analysed the data. D.d.M.P., ARM and FGOS wrote the manuscript. M.J.S., J.E.H. and J.A.L. provided critical revisions. All authors read and approved the final manuscript.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Diego de Miguel Pérez and Alba Rodriguez Martínez.
Contributor Information
Francisco Gabriel Ortega Sánchez, Email: franciscogabriel.ortega@ssib.es.
Ma Jose Serrano, Email: mjose.serrano@genyo.es.
Supplementary information
is available for this paper at 10.1038/s41598-020-60212-1.
References
- 1.Ferlay, J. et al. Global Cancer Observatory: Cancer Today. Lyon, France: International Agency for Research on Cancer. https://Gco.Iarc.Fr/Today144, 1941–1953 (2018).
- 2.Smyth EC, et al. Gastric cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016;27:v38–v49. doi: 10.1093/annonc/mdw350. [DOI] [PubMed] [Google Scholar]
- 3.Van Cutsem E, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016;27:1386–1422. doi: 10.1093/annonc/mdw235. [DOI] [PubMed] [Google Scholar]
- 4.Hurwitz HI, et al. Efficacy and Safety of Bevacizumab in Metastatic Colorectal Cancer: Pooled Analysis From Seven Randomized Controlled Trials. Oncologist. 2013;18:1004–1012. doi: 10.1634/theoncologist.2013-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.M. K, N. F. Vascular endothelial growth factor signaling pathways: Therapeutic perspective. Clin. Cancer Res. 2006;12:5018–22. doi: 10.1158/1078-0432.CCR-06-1520. [DOI] [PubMed] [Google Scholar]
- 6.M. W, et al. Efficacy and safety of first-line chemotherapy plus bevacizumab in patients with metastatic colorectal cancer: a meta-analysis. Chin. Med. J. (Engl). 2014;127:538–546. [PubMed] [Google Scholar]
- 7.Zhao L, et al. High VEGF-A level at baseline predicts poor treatment effect of bevacizumab-based chemotherapy in metastatic colorectal cancer: a meta-analysis. Panminerva Med. 2016;58:48–58. [PubMed] [Google Scholar]
- 8.Wang XW, Zhang YJ. Targeting mTOR network in colorectal cancer therapy. World J. Gastroenterol. 2014;20:4178–4188. doi: 10.3748/wjg.v20.i15.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuurbier L, et al. Apelin: A putative novel predictive biomarker for bevacizumab response in colorectal cancer. Oncotarget. 2017;8:42949–42961. doi: 10.18632/oncotarget.17306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiala O, et al. G12V and G12A KRAS mutations are associated with poor outcome in patients with metastatic colorectal cancer treated with bevacizumab. Tumor Biol. 2016;37:6823–6830. doi: 10.1007/s13277-015-4523-7. [DOI] [PubMed] [Google Scholar]
- 11.Pathak S, et al. Review on comparative efficacy of bevacizumab, panitumumab and cetuximab antibody therapy with combination of FOLFOX-4 in KRAS-mutated colorectal cancer patients. Oncotarget. 2018;9:7739–7748. doi: 10.18632/oncotarget.22471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabart M, et al. Le statut KRAS n’influence pas l’efficacité de l’oxaliplatine ou de l’irinotécan, en association au bévacizumab, dans le traitement de première ligne du cancer colorectal métastatique. Bull. Cancer. 2016;103:541–551. doi: 10.1016/j.bulcan.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Sun DC, et al. KRAS mutation and primary tumor location do not affect efficacy of bevacizumab-containing chemotherapy in stagae IV colorectal cancer patients. Sci. Rep. 2017;7:14368. doi: 10.1038/s41598-017-14669-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sastre Javier, Vidaurreta Marta, Gómez Auxiliadora, Rivera Fernando, Massutí Bartomeu, López Margarita Reboredo, Abad Albert, Gallen Manuel, Benavides Manuel, Aranda Enrique, Rubio Eduardo Díaz. Prognostic Value of the Combination of Circulating Tumor Cells Plus KRAS in Patients With Metastatic Colorectal Cancer Treated With Chemotherapy Plus Bevacizumab. Clinical Colorectal Cancer. 2013;12(4):280–286. doi: 10.1016/j.clcc.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Krebs Matthew G., Renehan Andrew G., Backen Alison, Gollins Simon, Chau Ian, Hasan Jurjees, Valle Juan W., Morris Karen, Beech Janette, Ashcroft Linda, Saunders Mark P., Dive Caroline. Circulating Tumor Cell Enumeration in a Phase II Trial of a Four-Drug Regimen in Advanced Colorectal Cancer. Clinical Colorectal Cancer. 2015;14(2):115-122.e2. doi: 10.1016/j.clcc.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 16.Delgado-Ureña, M. et al. Circulating tumor cells criteria (CyCAR) versus standard RECIST criteria for treatment response assessment in metastatic colorectal cancer patients. J. Transl. Med. 16, (2018). [DOI] [PMC free article] [PubMed]
- 17.Maru D, Venook AP, Ellis LM. Predictive biomarkers for bevacizumab: Are we there yet? Clin. Cancer Res. 2013;19:2824–2827. doi: 10.1158/1078-0432.CCR-12-3409. [DOI] [PubMed] [Google Scholar]
- 18.Stiegelbauer V, et al. MicroRNAs as novel predictive biomarkers and therapeutic targets in colorectal cancer. World J. Gastroenterol. 2014;20:11727–11735. doi: 10.3748/wjg.v20.i33.11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding, L. et al. The Dual Role of MicroRNAs in Colorectal Cancer Progression. Int. J. Mol. Sci. 19, (2018). [DOI] [PMC free article] [PubMed]
- 20.Ferracin, M. et al. Absolute quantification of cell-free microRNAs in cancer patients. Oncotarget6, (2015). [DOI] [PMC free article] [PubMed]
- 21.Arroyo JD, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. 2011;108:5003–5008. doi: 10.1073/pnas.1019055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valadi H, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–9. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 23.Turchinovich A, Tonevitsky AG, Burwinkel B. Extracellular miRNA: A Collision of Two Paradigms. Trends in Biochemical Sciences. 2016;41:883–892. doi: 10.1016/j.tibs.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Choudhry Hani, Harris Adrian L. Advances in Hypoxia-Inducible Factor Biology. Cell Metabolism. 2018;27(2):281–298. doi: 10.1016/j.cmet.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Park JE, et al. Hypoxic Tumor Cell Modulates Its Microenvironment to Enhance Angiogenic and Metastatic Potential by Secretion of Proteins and Exosomes. Mol. Cell. Proteomics. 2010;9:1085–1099. doi: 10.1074/mcp.M900381-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lötvall Jan, Hill Andrew F., Hochberg Fred, Buzás Edit I., Di Vizio Dolores, Gardiner Christopher, Gho Yong Song, Kurochkin Igor V., Mathivanan Suresh, Quesenberry Peter, Sahoo Susmita, Tahara Hidetoshi, Wauben Marca H., Witwer Kenneth W., Théry Clotilde. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. Journal of Extracellular Vesicles. 2014;3(1):26913. doi: 10.3402/jev.v3.26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Issa IA, NouredDine M. Colorectal cancer screening: An updated review of the available options. World Journal of Gastroenterology. 2017;23:5086–5096. doi: 10.3748/wjg.v23.i28.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao, C. et al. Role of hypoxia-induced exosomes in tumor biology. Molecular Cancer17, (2018). [DOI] [PMC free article] [PubMed]
- 29.Chen X, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 30.Giráldez María Dolores, Lozano Juan José, Ramírez Georgina, Hijona Elizabeth, Bujanda Luis, Castells Antoni, Gironella Meritxell. Circulating MicroRNAs as Biomarkers of Colorectal Cancer: Results From a Genome-Wide Profiling and Validation Study. Clinical Gastroenterology and Hepatology. 2013;11(6):681-688.e3. doi: 10.1016/j.cgh.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 31.Yang Xin, Zeng Zongyue, Hou Yixuan, Yuan Taixian, Gao Chao, Jia Wei, Yi Xiaoyan, Liu Manran. MicroRNA-92a as a Potential Biomarker in Diagnosis of Colorectal Cancer: A Systematic Review and Meta-Analysis. PLoS ONE. 2014;9(2):e88745. doi: 10.1371/journal.pone.0088745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogata-Kawata, H. et al. Circulating exosomal microRNAs as biomarkers of colon cancer. PLoS One9, (2014). [DOI] [PMC free article] [PubMed]
- 33.Tsukamoto M, Iinuma H, Yagi T, Matsuda K, Hashiguchi Y. Circulating Exosomal MicroRNA-21 as a Biomarker in Each Tumor Stage of Colorectal Cancer. Oncol. 2017;92:360–370. doi: 10.1159/000463387. [DOI] [PubMed] [Google Scholar]
- 34.Ostenfeld Marie Stampe, Jensen Steffen Grann, Jeppesen Dennis Kjølhede, Christensen Lise-Lotte, Thorsen Stine Buch, Stenvang Jan, Hvam Michael Lykke, Thomsen Anni, Mouritzen Peter, Rasmussen Mads Heilskov, Nielsen Hans Jørgen, Ørntoft Torben Falck, Andersen Claus Lindbjerg. miRNA profiling of circulating EpCAM+extracellular vesicles: promising biomarkers of colorectal cancer. Journal of Extracellular Vesicles. 2016;5(1):31488. doi: 10.3402/jev.v5.31488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu F, Jiang W, Zhou L, Chen Z. Circulating Exosomal miR-17-5p and miR-92a-3p Predict Pathologic Stage and Grade of Colorectal Cancer. Transl. Oncol. 2018;11:221–232. doi: 10.1016/j.tranon.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng Y-H, Tsao C-J. Emerging role of microRNA-21 in cancer. Biomed. Reports. 2016;5:395–402. doi: 10.3892/br.2016.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cha DJ, et al. KRAS-dependent sorting of miRNA to exosomes. Elife. 2015;4:e07197. doi: 10.7554/eLife.07197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKenzie AJ, et al. KRAS-MEK Signaling Controls Ago2 Sorting into Exosomes. Cell Rep. 2016;15:978–987. doi: 10.1016/j.celrep.2016.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Negin BP, Cohen SJ. Circulating tumor cells in colorectal cancer: Past, present, and future challenges. Current Treatment Options in Oncology. 2010;11:1–13. doi: 10.1007/s11864-010-0115-3. [DOI] [PubMed] [Google Scholar]
- 40.Hardingham JE, et al. Detection and Clinical Significance of Circulating Tumor Cells in Colorectal Cancer—20 Years of Progress. Mol. Med. 2015;21:S25–S31. doi: 10.2119/molmed.2015.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong, H. et al. The propensity of invasive circulating tumor cells (iCTCs) in metastatic progression and therapeutic responsiveness. Cancer Med. 10.1002/cam4.2218 (2019) [DOI] [PMC free article] [PubMed]
- 42.Pantel Klaus, Alix-Panabières Catherine. Liquid biopsy and minimal residual disease — latest advances and implications for cure. Nature Reviews Clinical Oncology. 2019;16(7):409–424. doi: 10.1038/s41571-019-0187-3. [DOI] [PubMed] [Google Scholar]
- 43.Delgado-Ureña, M. et al. Circulating tumor cells criteria (CyCAR) versus standard RECIST criteria for treatment response assessment in metastatic colorectal cancer patients. J. Transl. Med. 16, (2018). [DOI] [PMC free article] [PubMed]
- 44.Rodríguez-Martínez, A. et al. Exosomal miRNA profile as complementary tool in the diagnostic and prediction of treatment response in localized breast cancer under neoadjuvant chemotherapy. Breast Cancer Res. 21, (2019). [DOI] [PMC free article] [PubMed]
- 45.Alvarez Cubero, M. J. et al. In Methods in Molecular Biology1634, 283–303 (2017). [DOI] [PubMed]
- 46.Song, J. et al. Potential value of miR-221/222 as diagnostic, prognostic, and therapeutic biomarkers for diseases. Frontiers in Immunology8, (2017). [DOI] [PMC free article] [PubMed]
- 47.Eisenhauer EA, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur. J. Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 48.Nadal, R. et al. Biomarkers characterization of circulating tumour cells in breast cancer patients. Breast Cancer Res. 14, (2012). [DOI] [PMC free article] [PubMed]
- 49.Ortega, F. G. et al. MiRNA in situ hybridization in circulating tumor cells - MishCTC. Sci. Rep. 5, (2015). [DOI] [PMC free article] [PubMed]
- 50.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 51.Saridaki, Z. & Souglakos, J. Genetic alterations in colorectal cancer in older patients. Manag. Color. Cancers Older People 9–20 (2013).
- 52.Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. StarBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, (2014). [DOI] [PMC free article] [PubMed]
- 53.Ortega Francisco G., Roefs Marieke T., de Miguel Perez Diego, Kooijmans Sander A., de Jong Olivier G., Sluijter Joost P., Schiffelers Raymond M., Vader Pieter. Interfering with endolysosomal trafficking enhances release of bioactive exosomes. Nanomedicine: Nanotechnology, Biology and Medicine. 2019;20:102014. doi: 10.1016/j.nano.2019.102014. [DOI] [PubMed] [Google Scholar]
- 54.Budczies Jan, Klauschen Frederick, Sinn Bruno V., Győrffy Balázs, Schmitt Wolfgang D., Darb-Esfahani Silvia, Denkert Carsten. Cutoff Finder: A Comprehensive and Straightforward Web Application Enabling Rapid Biomarker Cutoff Optimization. PLoS ONE. 2012;7(12):e51862. doi: 10.1371/journal.pone.0051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maldonado G, Greenland S. Simulation study of confounder-selection strategies. Am. J. Epidemiol. 1993;138:923–936. doi: 10.1093/oxfordjournals.aje.a116813. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.