

A new approach of rationally designing an N449R mutation was developed to lock human muscarinic receptor M4 into an inactive form, and the crystal structure of the inactive M4 in a ligand-free form was determined.
Keywords: muscarinic acetylcholine receptors, G-protein-coupled receptors, M4, mutation design, ligand screening, Parkinson’s disease, Alzheimer’s disease, GPCRs
Abstract
Human muscarinic receptor M4 belongs to the class A subfamily of the G-protein-coupled receptors (GPCRs). M4 has emerged as an attractive drug target for the treatment of Alzheimer’s disease and schizophrenia. Recent results showed that M4-mediated cholinergic transmission is related to motor symptoms in Parkinson’s disease. Selective ligand design for the five muscarinic acetylcholine receptor (mAchR) subtypes currently remains challenging owing to the high sequence and structural similarity of their orthosteric binding pockets. In order to obtain M4-selective antagonists, a new approach was tried to lock M4 into an inactive form by rationally designing an N4497.49R mutation, which mimics the allosteric sodium binding in the conserved sodium site usually found in class A GPCRs. In addition, the crystal structure of the mutation-induced inactive M4 was determined. By comparative analysis with other mAchR structures, followed by functional assays, the N4497.49R mutation was shown to stabilize M4 into an inactive state. Virtual screening of a focused ligand library using the crystal structure showed that the inactive M4 prefers antagonists much more than agonists. This study provides a powerful mutation strategy to stabilize GPCRs in inactive states and facilitate their structure determination.
1. Introduction
Muscarinic acetylcholine receptors (mAchRs) are integral membrane proteins that belong to the class A G-protein-coupled receptors (GPCRs) and are activated by the neurotransmitter acetylcholine (Ach; Fredriksson et al., 2003 ▸). Among the five muscarinic receptor subtypes, the M1, M3 and M5 subtypes couple to the G protein Gq/11, activating phospholipase C and increasing cytosolic Ca2+, while the M2 and M4 subtypes couple to Gi/o, mediating the inhibition of adenylyl cyclase (Hulme et al., 1990 ▸). Each mAchR subtype also has a unique distribution throughout the peripheral or central nervous system in the human body, and they are attractive targets for the treatment of different pathophysiological conditions, including chronic obstructive pulmonary disease (COPD), overactive bladder and Sjögren’s syndrome (Eglen, 2012 ▸; Eglen et al., 1996 ▸).
The high sequence and structural similarities among mAchR subtypes make selective ligand design quite challenging, which precludes the precise modulation of mAchR for therapeutic benefits. In the periphery, M4 is expressed in various pre-junctional nerve endings, acting to inhibit parasympathetic and sympathetic transmissions, while in the central nervous system (CNS) M4 is distributed in the corpus striatum and co-localizes with dopamine receptors on striatal projecting neurons. M4 also plays an important role in various motor disorder diseases such as Parkinson’s disease and dystonia (Bernard et al., 1992 ▸; Eskow Jaunarajs et al., 2015 ▸; Ztaou et al., 2016 ▸). The majority of muscarinic antagonists, such as atropine and tiotropium, which is a commercial drug currently used for the treatment of COPD (Kato et al., 2006 ▸), are nonselective. The weak M4-selective antagonist tropicamide is used to dilate the pupil for eye examinations or other diagnostic procedures (Lam et al., 2010 ▸; Yazdani et al., 2018 ▸). Several muscarinic acetylcholine receptor structures have been solved, as shown in Table 1 ▸. Most of them are in inactive states with nonselective antagonists or inverse agonists.
Table 1. List of mAChRs with known structures.
| mAChR | Structure state | PDB code | Resolution (Å) | Ligand | Reference |
|---|---|---|---|---|---|
| M1 | Inactive | 5cxv | 2.7 | Tiotropium | Thal et al. (2016 ▸) |
| Active | 6oij | 3.3 | Iperoxo | Maeda et al. (2019 ▸) | |
| M2 | Inactive | 3uon | 3.0 | QNB† | Haga et al. (2012 ▸) |
| 5zkc | 2.3 | NMS‡ | Suno et al. (2018 ▸) | ||
| 5yc8 | 2.5 | NMS | Suno et al. (2018 ▸) | ||
| 5zkb | 2.95 | AF-DX 384 | Suno et al. (2018 ▸) | ||
| 5zk8 | 3.0 | NMS | Suno et al. (2018 ▸) | ||
| 5zk3 | 2.6 | QNB | Suno et al. (2018 ▸) | ||
| Active | 4mqt | 3.7 | Iperoxo, LY2119620 | Kruse et al. (2013 ▸) | |
| 4mqs | 3.5 | Iperoxo | Kruse et al. (2013 ▸) | ||
| 6oik | 3.6 | Iperoxo, LY2119620 | Maeda et al. (2019 ▸) | ||
| M3 | Inactive | 4daj | 3.4 | Tiotropium | Kruse et al. (2012 ▸) |
| 4u14 | 3.57 | Tiotropium | Kruse et al. (2012 ▸) | ||
| 4u15 | 2.8 | Tiotropium | Kruse et al. (2012 ▸) | ||
| 4u16 | 3.7 | NMS | Kruse et al. (2012 ▸) | ||
| 5zhp | 3.1 | 6o(BS46) | Liu et al. (2018 ▸) | ||
| M4 | Inactive | 5dsg | 2.6 | Tiotropium | Thal et al. (2016 ▸) |
R-(2)-3-quinuclidinyl benzilate.
N-Methylscopolamine.
It is well established that the activation of class A GPCRs is initiated by ligand binding, which induces conformational changes of transmembrane (TM) helices. Upon activation, one of the most obvious features is that the cytoplasmic side of TM6 swings away from the transmembrane bundle. Several conserved or unique residues participate in the activation process. Firstly, the side chain of the toggle switch W6.48 (superscripts indicate Ballesteros–Weinstein numbering for GPCRs; Ballesteros & Weinstein, 1995 ▸) in TM6 undergoes a conformational change which unleashes a series of conformational movements of the receptor. Consequently, the residue at 3.46 breaks the contact with the residue at 6.37 and forms a new contact with the rotated Y7.53 within the highly conserved NPxxY motif of TM7. The cytoplasmic ends of TM3 and TM6 are shown to disassociate owing to the breakage of a salt bridge between R3.50 at the end of TM3 (which is part of the conserved DRY motif) and E6.30 at the end of TM6.
High-resolution crystal structures have revealed that inactive class A GPCRs may harbour a conserved binding site for Na+ ions in the centre of their transmembrane domain (Fenalti et al., 2014 ▸; Miller-Gallacher et al., 2014 ▸; Zhang et al., 2012 ▸). The Na+ ion is coordinated by a salt bridge to D2.50, together with four additional polar interactions with side chains of receptors and water molecules in the high-resolution crystal structures. For example, the Na+ ion in the human A2A adenosine receptor (A2AAR) is coordinated by two highly conserved residues, D2.50 and S3.39, and three water molecules (Liu et al., 2012 ▸). Na+ ions are found to selectively reduce the affinity of agonists but not antagonists, which is consistent with the structural stabilization of the inactive state by ions (Suno et al., 2018 ▸). However, this Na+ ion-binding pocket is collapsed in active receptors. Mutations around the Na+ ion-binding site have a major impact on receptor function in most class A GPCRs, either completely abolishing G-protein coupling or resulting in constitutive ligand-independent or pathway-biased signalling (Suno et al., 2018 ▸; Fenalti et al., 2014 ▸; Huang et al., 2015 ▸).
In order to identify potential new selective antagonists for M4, we set out to take a different approach by creating an inactive M4 induced by the rationally designed mutation N4497.49R, which is involved in the potential Na+-binding pocket in the transmembrane domains, to stabilize the protein. With five more mutations to aid in the expression and protein yield of M4, we further determined the crystal structure of the inactivated M4 receptor. Through comparative analysis of our crystal structure and tiotropium-bound M4 (PDB entry 5dsg; Thal et al., 2016 ▸) and functional assays, the mutated M4 structure is shown to be in an inactive state. The virtual screening of a focused ligand library using our structure showed that antagonists are much more preferred than agonists, and that the mutation N4497.49R is the key element in preventing the activation of M4. Moreover, the inactivated mutation was so effective that a co-purifying, tightly bound ligand was trapped in the orthosteric site.
2. Experimental procedures
2.1. Construction, protein expression and purification
Wild-type M4 contains a long, probably poorly ordered third intracellular loop (ICL3) which is challenging for crystallization. To alleviate this problem, an M4-PGS (Pyrococcus abyssi glycogen synthase; PDB entry 2bfw; Horcajada et al., 2006 ▸) fusion protein construct (Yin et al., 2016 ▸) was generated using overlap PCR with six mutations: I932.65T, G1504.43A, I187ECL2A, S2195.62Y, N4497.49R and T4598.49E. The construct was cloned into a modified pFastBac1 vector containing an N-terminal FLAG epitope tag followed by a 10×His tag. The construct included an HRV 3C cleavage site between S21 and S22 in the N-terminus. Residues 228–389 of ICL3 were replaced by PGS. The modified M4-PGS protein was expressed in Spodoptera frugiperda (Sf9) Super 3 insect cells using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Sf9 Super 3 cells were infected at a cell density of 2–2.5 × 106 cells per millitre with high-titre viral stock at a multiplicity of infection (MOI) of 5.0. Cells were harvested by centrifugation for 48 h post-infection and stored at −80°C for future use.
Frozen cell pellets were thawed and lysed by repeated washing and centrifugation in hypotonic buffer consisting of 10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl and high osmotic buffer consisting of 10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl, 1.0 M NaCl with EDTA-free cOmplete protease-inhibitor cocktail tablets (Roche). The washed membranes were suspended in hypotonic buffer with 30% glycerol, flash-frozen with liquid nitrogen and stored at −80°C until further use. Purified membranes were thawed at room temperature and incubated with 2.0 mg ml−1 iodoacetamide (Sigma) and inhibitor cocktail at 4°C for 1 h. The membranes were solubilized in a buffer consisting of 30 mM HEPES pH 7.5, 750 mM NaCl, 0.75%(w/v) lauryl maltose neopentyl glycol (LMNG; Anatrace) and 0.15%(w/v) cholesterol hemisuccinate (CHS; Sigma–Aldrich) at 4°C for 3 h. The supernatant containing the solubilized M4 proteins was isolated by high-speed centrifugation and then incubated with TALON IMAC resin (Clontech) and 20 mM imidazole at 4°C overnight. The resin was washed with 15 column volumes (CV) of washing buffer I consisting of 20 mM HEPES pH 7.5, 500 mM NaCl, 10%(v/v) glycerol, 0.03%(w/v) LMNG, 0.006%(w/v) CHS, 30 mM imidazole and 5 CV of washing buffer II consisting of 20 mM HEPES pH 7.5, 200 mM NaCl, 10%(v/v) glycerol, 0.01%(w/v) LMNG, 0.002%(w/v) CHS, 50 mM imidazole. The protein was eluted using 3 CV of elution buffer consisting of 20 mM HEPES pH 7.5, 150 mM NaCl, 10%(v/v) glycerol, 0.01%(w/v) LMNG, 0.002%(w/v) CHS, 250 mM imidazole. A PD MiniTrap G-25 column (GE Healthcare) was used to remove imidazole. The protein was treated overnight with HRV 3C protease to cleave the N-terminal FLAG/His tags from the protein. Finally, the purified protein was concentrated to about 50 mg ml−1 using a 100 kDa cutoff concentrator (Sartorius) and used in crystallization trials. The protein yield and monodispersity were tested by analytical size-exclusion chromatography. No ligands were added during the entire procedures.
2.2. Crystallization in lipidic cubic phase
Crystallization was performed using the lipidic cubic phase (LCP) method (Caffrey & Cherezov, 2009 ▸). The concentrated M4-PGS was mixed with monoolein with 10%(w/w) cholesterol (Sigma) in a ratio of 2:3(w:w) using the syringe reconstitution method. The LCP mixture was dispensed in 35 nl droplets onto glass plates and overlaid with 800 nl precipitant solution using an NT8 robot (Formulatrix). The crystallization experiments were carried out in 96-well glass sandwich plates (Molecular Dimensions), which were subsequently stored in a Rock Imager (Formulatrix) at 20°C. Crystals of M4-PGS were obtained from precipitant conditions consisting of 300 mM diammonium hydrogen phosphate, 22–26% PEG 300, 0.1 M HEPES sodium pH 7.8 and reached a full size of 20–30 µm in 4–5 days, as shown in Supplementary Fig. S1.
2.3. Data collection and structure determination
X-ray diffraction data were collected on beamline 41XU at SPring-8 using an EIGER X 16M detector (X-ray wavelength 1.0000 Å). Diffraction images were processed using XDS (Kabsch, 2010 ▸) and scaled with utilities from the CCP4 suite (Winn et al., 2011 ▸). The structure was solved by molecular replacement with Phaser (McCoy et al., 2007 ▸) using the M4–tiotropium structure (PDB entry 5dsg; Thal et al., 2016 ▸) and the structure of the PGS domain (PDB entry 2bfw; Horcajada et al., 2006 ▸) as separate models for the M4 and PGS fusion proteins. Refinement, rebuilding and structure determination were carried out using Phenix (Liebschner et al., 2019 ▸), BUSTER (Smart et al., 2012 ▸) and Coot (Emsley et al., 2010 ▸). The structure was completed with R work and R free values of 0.231 and 0.264, respectively. The refinement statistics are summarized in Table 2 ▸.
Table 2. Data-collection and structure-refinement statistics for mutation-induced inactive M4.
Values in parentheses are for the highest resolution shell.
| No ligand | HMDB0010212 docked | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.000 | |
| Resolution range (Å) | 50.00–3.00 (3.08–3.00) | |
| Space group | P212121 | |
| a (Å) | 56.10 | |
| b (Å) | 61.32 | |
| c (Å) | 203.74 | |
| Observed reflections | 398969 | |
| Unique reflections | 14718 | |
| Multiplicity | 27.1 (6.0) | |
| Completeness (%) | 99.7 (97.0) | |
| Mean I/σ(I) | 19.3 (2.3) | |
| Wilson B factor (Å2) | 89.34 | |
| R merge † | 0.132 (0.586) | |
| CC1/2 ‡ | 0.996 (0.379) | |
| Refinement | ||
| Resolution range (Å) | 49.14–3.00 | |
| Reflections (work/test) | 13985/731 | |
| R work/R free § | 0.231/0.264 | |
| No. of atoms | ||
| Total | 3711 | 3734 |
| Macromolecules | 3711 | 3711 |
| Ligands | 0 | 23 |
| Solvent | 0 | 0 |
| No. of protein residues | 474 | 477 |
| R.m.s.d., bonds (Å) | 0.008 | 0.014 |
| R.m.s.d., angles (°) | 0.94 | 1.67 |
| Ramachandran statistics¶ | ||
| Favoured (%) | 96.39 | 94.48 |
| Allowed (%) | 3.41 | 5.10 |
| Outliers (%) | 0.42†† | 0.42†† |
| Clashscore | 2.42 | 3.75 |
| PDB code | 6kp6 | |
R
merge = 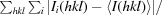
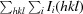 , where Ii(hkl) is the intensity of observation i of reflection hkl.
, where Ii(hkl) is the intensity of observation i of reflection hkl.
As defined by Karplus & Diederichs (2012 ▸).
R = 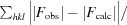
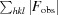 for all reflections, where F
obs and F
calc are the observed and calculated structure factors, respectively. R
free is calculated analogously for the test reflections, which were randomly selected and excluded from the refinement.
for all reflections, where F
obs and F
calc are the observed and calculated structure factors, respectively. R
free is calculated analogously for the test reflections, which were randomly selected and excluded from the refinement.
As defined by MolProbity (Chen et al., 2010 ▸).
Glycine residues 1075 and 1144 are in sharp-turn domains of the PGS fusion protein.
2.4. Protein stability measurement using CPM assays
Protein thermostability was measured using a microscale fluorescent thermostability assay as described previously (Alexandrov et al., 2008 ▸). For the thermostability assay, CPM [7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin] dye was dissolved in DMSO to 4 mg ml−1 as a stock solution and diluted 1:40 in buffer [20 mM HEPES pH 7.5, 200 mM NaCl, 10%(v/v) glycerol, 0.001%(w/v) LMNG, 0.0002%(w/v) CHS] before use. A total of 1 µl diluted CPM dye was added to the same buffer with approximately 0.5–2 µg M4 in a final volume of 50 µl. The samples were then incubated on ice for 15 min. The thermal denaturation assay was performed in a Rotor-Gene real-time PCR cycler (Qiagen). The excitation wavelength was 365 nm and the emission wavelength was 460 nm. All assays were performed over a temperature range from 25 to 99°C using a temperature rate of 2.0°C min−1. The stability data were processed with GraphPad Prism.
2.5. Split luciferase biosensor cAMP assay
Human M4-expressing cell lines were generated using HEK293T cells. To validate the effects of the mutations on the M4 Gi protein signalling pathway, the split luciferase-based GloSensor cAMP biosensor technology (Promega) was employed followed by mutagenesis experiments. One day prior to assay, 4 µg M4 DNA and 5 µg GloSensor cAMP DNA (Promega) were co-transfected into HEK293T cells using Lipofectamine 2000 (Life Technologies). The cells grew for 5 h and half of the culture medium was replaced by equilibration medium. The cells were then added into 384-well white poly-d-lysine-coated plates (Greiner) with Dulbecco’s Modified Eagle Medium (DMEM; Life Technologies) supplemented with 1% dialyzed fetal bovine serum (dFBS) at a density of 15 000–20 000 cells in 40 µl medium per well and incubated overnight (20–24 h) at 37°C in 5% carbon dioxide. The following day, the culture medium was removed from the cell plates. The wells were loaded with 20 µl 2 mg ml−1 d-luciferin sodium salt prepared in Hanks’ balanced salt solution (HBSS) pH 7.4 and incubated for 1 h at 37°C. All of the following steps were carried out at room temperature. To measure the Ach activity at M4, 10 µl 3× Ach solution was added with a final concentration ranging from 1 nM to 3 µM and reacted for 15 min. The plates for the agonist assay were diluted by adding 10 µl isoproterenol (Sigma) at a final concentration of 200 nM, paused for 15 min and followed by measuring the luminescence using an EnVision plate reader (Perkin Elmer). Data were analysed using GraphPad Prism.
2.6. Molecular docking
Prediction of ligand binding to M4 was performed with Schrödinger Suite 2018-4 (Schrödinger). The protein structures were processed with the Protein Preparation Wizard and the structures of ligands were prepared by LigPrep. Molecular docking was carried out with Glide.
2.7. Molecular-dynamics simulation of M4 in complex with agonists and antagonists
The processed crystal structure of M4 in complex with either an agonist or an antagonist was embedded in a bilayer composed of 140 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipids using the CHARMM-GUI Membrane Builder (Jo et al., 2008 ▸; Wang et al., 2004 ▸). The protein orientation in the membrane is referenced to M4 (PDB entry 5dsg) in the Orientations of Proteins in Membranes (OPM) database (Lomize et al., 2012 ▸). The protein–membrane system was solvated in a periodic 0.15 M NaCl TIP3P water box with a minimum water height of 20.0 Å at the top and bottom of the system. All simulations were performed on a GPU cluster using the CUDA version of PMEMD (particle mesh Ewald molecular dynamics) in Amber18 (University of California, San Francisco). The protein was modelled with the ff14SB protein force field (Maier et al., 2015 ▸), ligands with the GAFF2 force field (Wang et al., 2004 ▸) and lipids with the AMBER Lipid17 force field. The constructed system was first energy-minimized for 10 000 steps; the first 5000 of these were performed using the steepest-descent method and the remaining 5000 used the conjugate-gradient method. The simulation system was then heated from 0 to 100 K using Langevin dynamics with a constant box volume. Restraints were applied to protein, ligands and lipids with a constant force of 10 kcal mol Å−2. Subsequently, the temperature was increased to 310 K, where the periodic box was coupled accordingly using anisotropic Berendsen control in order to maintain the pressure at around 1 bar. The particle mesh Ewald (PME; Darden et al., 1993 ▸) method was used to treat all electrostatic interactions beyond a cutoff of 9 Å. The SHAKE algorithm (Ryckaert et al., 1977 ▸) was used to record the length of bonds involving hydrogen during the simulation, with an integration time step of 2 fs. Further equilibration was then carried out at 310 K with harmonic restraints applied to the protein starting at 5 kcal mol Å−2 and reduced in a stepwise fashion every 2 ns for 10 ns, followed by 0.1 kcal mol Å−2 restraints for 20 ns to give a total of 30 ns of equilibration. 2 µs production simulations with no restraints were then performed at 310 K and 1 bar in the NPT ensemble for each system for three independent runs, and these trajectories were used for analysis with Visual Molecular Dynamics (VMD; Humphrey et al., 1996 ▸) and CPPTRAJ (Roe & Cheatham, 2013 ▸).
3. Results
3.1. Rationally designed mutation-induced inactive M4
Point mutations have been shown to be an effective method to improve the expression and the thermostability of GPCRs (Peng et al., 2018 ▸; Popov et al., 2018 ▸, 2019 ▸). After rational design and intensive screening, six mutations were introduced to improve the expression of M4 (S2195.62Y, G1504.43A, T4598.49E, I187ECL2A, I932.65T and N4497.49R). Among these, S2195.62Y, G1504.43A and T4598.49E increase the thermostability of M4, while I187ECL2A and I932.65T have been reported to increase the binding affinity of ligands and the cooperativity of positive allosteric modulators (Chan et al., 2008 ▸; Thal et al., 2016 ▸). N4497.49R was rationally designed to play a similar role to the sodium ion in order to lock M4 into an inactive state. In the functional assay assessing the regulation of adenylyl cyclase, the first five mutations only showed a less than tenfold increased or decreased signalling activity compared with wild-type M4 receptor [Figs. 1 ▸(a) and 1 ▸(b)]. However, the N4497.49R mutation almost abolished the signalling and led to M4 that was completely inactive towards Ach, and the construct with these six mutations also showed no activity in the functional assay [Figs. 1 ▸(c) and 1 ▸(d)]. More importantly, the N4497.49R mutation significantly increased the thermostability of the receptor by 8°C compared with the construct without the N4497.49R mutation [Figs. 1 ▸(e) and 1 ▸(f)].
Figure 1.

Pharmacological characterization and thermostability assay of mutants of M4. (a, b) Dose–response studies of agonist Ach activity for each mutant compared with wild-type M4 (M4-WT). The EC50 values (mean ± SEM) of Ach are 1.059 ± 0.1489, 1.036 ± 0.139, 1.777 ± 0.6448, 2.314 ± 1.012, 0.9343 ± 0.1192 and 6.357 ± 1.262 nM for the M4-WT, T4598.49E, G1504.43A, S2195.62Y, I932.65T and I187ECL2A constructs, respectively. (c, d) Gi activation assays of M4 with the key point mutation N4497.49R and M4 with six mutations as a function of Ach compared with that of M4-WT. (e) Thermostability assay of the crystallization construct with six mutations (M4_6 mutations) and a construct with the other five mutations apart from N4497.49R (M4_5 mutations), where the fusion protein used in the constructs is PGS. (f) Thermostability assay of M4-N4497.49R and M4-WT with the fusion protein modified T4 lysozyme. The N4497.49R mutant showed an increase in the melting temperature (T m) by about 8.1 ± 1.1°C.
3.2. Crystal structure determination of the mutation-induced inactive M4
To facilitate crystallization, the M4 sequence was further modified by inserting a soluble fusion protein within the third intracellular loop 3 (ICL3), which is long and poorly ordered. In this work, PGS (P. abyssi glycogen synthase) was identified as a stabilizing fusion partner. The M4-PGS construct was expressed in Spodoptera frugiperda (Sf9) Super 3 insect cells using the Bac-to-Bac Baculovirus Expression System (Invitrogen). The construct yielded about 1.5 mg of M4 protein per litre from Sf9 Super 3 cells. The protein yield was improved greatly with the key mutation N4497.49R. The M4 crystals were formed in lipidic cubic phase (Caffrey & Cherezov, 2009 ▸) and diffracted to 3.0 Å resolution, as shown in Table 2 ▸. Unexpectedly, unknown electron density was observed in the orthosteric binding pocket even though no ligands were added from cell growth to crystallization. The unknown density position also differs from that of PEG 300, which was present in the allosteric binding pocket in inactive M3 and M4 structures (Liu et al., 2018 ▸; Thal et al., 2016 ▸). As the purified protein showed a high T m value without the addition of any ligands, we proposed that the unknown molecule may bind to the receptor during expression.
In order to identify the unknown binding molecule, we tried the molecular-docking method and virtually screened 114 157 metabolite entries from the Human Metabolome Database (HMDB, http://www.hmdb.ca/), which covers most of the eukaryotic cell metabolites. Three fatty acids (HMDB0010212, HMDB0034295 and HMDB0010217) showed relatively high potential for binding with M4. We then modelled the three fatty acids into the electron density and performed structure refinement. The results showed that all of them fit well into the density (Supplementary Fig. S4), and the R work and R free decreased after refinement (Supplementary Table S2).
In addition, previous studies have shown that high n-6 polyunsaturated fatty acid (PUFA) diets can influence the binding affinity of muscarinic receptors (Farkas et al., 2002 ▸; Flynn & Mash, 1993 ▸; Freund et al., 1986 ▸). The n-6 PUFA group displayed a reduction in M2/M4 binding in the caudate putamen, anterior cingulate cortex and hippocampus (du Bois et al., 2005 ▸). The unknown density observed in this study may provide some evidence that M4 might potentially bind to endogenous polyunsaturated fatty acids.
3.3. Comparison with known mAchR structures
To date, crystal structures of M1, M3 and M4 bound to the antagonist tiotropium have been determined, as well as a QNB-bound M2 structure, as shown in Table 1 ▸. The ligands share almost identical binding poses in the orthosteric binding pockets [Fig. 2 ▸(d)]. The overall structure of mutation-induced inactive M4 is similar to the structures of antagonist-bound mAChRs, with an especially high similarity between TM3, TM6 and TM7. For example, the root-mean-square deviation (r.m.s.d.) between mutation-induced inactive M4 and the M4–titropium complex structure (PDB entry 5dsg) is only 0.5–0.7 Å (Supplementary Table S1). Compared with the solved inactive mAChR structures, the mutation-induced inactive M4 structure has a more open extracellular domain owing to the outward movement of the extracellular region of TM1, TM6, TM7 and ECL3 [Figs. 2 ▸(a) and 2 ▸(b)]. The electron-density maps for the seven transmembrane helices and typical key residues are shown in Supplementary Fig. S5.
Figure 2.

Comparison of the mutation-induced inactive M4 structure with structures of M2 and M4 in inactive and active states. (a, b, c) M2 and M4 are aligned with the mutation-induced inactive M4 structure (teal blue). The M4–tiotropium structure is shown as an orange cartoon and the inactive and active M2 structures are shown as pale yellow and magenta cartoons, respectively. (d) Extracellular region comparison of mutation-induced inactive M4 and M4–tiotropium (PDB entry 5dsg) structures. (e) The highly conserved residues in orthosteric binding pockets for tiotropium and QNB. (f) The ‘tyrosine lid’ is formed by three tyrosines: Y6.51, Y3.33 and Y7.39. There is a 110° rotation of Y7.39 compared with that in M4–tiotropium; the arrow shows the rotation of Y4397.39 in the mutation-induced inactive M4 structure. (g) The different rotations of W6.48 in the mutation-induced inactive M4, active M2 and M4–tiotropium structures. (h, i) Comparison of the NPxxY and D(E)RY motifs between the inactive and active mAchR structures.
The orthosteric binding pocket is mainly composed of the D1123.32, Y1133.33, S1163.36, N4176.52 and Y4397.39 residues in mAChR structures. Compared with the M4–tiotropium complex structure, the side chains of these residues in the mutation-induced inactive M4 structure show outward movements. Additionally, the mutation-induced inactive M4 structure shows several unique features; for example, the side chain of Y4397.39 in the conserved tyrosine lid rotates about 110°, which forms the aromatic cage enclosing the amine and forms a lid over the orthosteric pocket [Fig. 2 ▸(e)]. With the rotation of Y4397.39, the network of hydrogen-bond interactions collapses, creating a largely hydrophobic binding cavity between the orthosteric and allosteric binding pockets. The N4497.49R substitution allowed an unknown hydrophobic ligand to be trapped in a hydrophobic pocket of the orthosteric site, which further stabilized the inactive state. This rotation of Y4397.39 also occurs in the docking results for some other M4-selective agonists, in which Y4397.39 adopted an ‘open’ conformation in all selected templates, including the agonist iperoxo (Yang et al., 2019 ▸), which differs from what was observed in the reported X-ray structure of the activated form of M2 (Kruse et al., 2013 ▸). The conformations of the orthosteric and allosteric sites are coupled such that the presence of a ligand in one site affects the shape of the other (Burger et al., 2018 ▸).
The side chain of W4136.48, the ‘toggle switch’ residue, rotates about 45° compared with the M4–tiotropium structure, but differs from that in the active M2 structure (PDB entry 4mqs; Kruse et al., 2013 ▸) [Fig. 2 ▸(f)]. In addition, the salt bridge between D3.49 and R3.50 in the conserved D(E)RY motif is still intact, further indicating that the structure is in the inactive state [Figs. 2 ▸(g) and 2 ▸(h)].
3.4. The mechanism of mutation-induced inactivation of M4
D2.50 is highly conserved among the class A GPCRs and is important for coordinating the binding of the allosteric sodium ion in many cases (Vickery et al., 2018 ▸; Katritch et al., 2014 ▸). In the recent M2–AF-DX 384 structure, the point mutation S1103.39R was designed based on the structure of the A2A adenosine receptor (PDB entry 4eiy; Liu et al., 2012 ▸) and the S1103.39R mutation acts like the sodium cation and forms a salt bridge with D2.50 (Suno et al., 2018 ▸) [Fig. 3 ▸(b)]. In our mutation-induced M4 structure, N4497.49R in the NPxxY motif forms a salt bridge with D782.50, as well as a hydrogen bond to S1193.39 [Fig. 3 ▸(b)]. The ionic network, especially the interaction between D782.50 and the mutated R4497.49, locks the M4 structure into an inactive state [Fig. 3 ▸(d)]. Notably, on comparison with the structure of the A2A receptor with sodium ion (PDB code 4eiy), the positive charge of R4497.49 in the mutation-induced inactive M4 has the same function as the allosteric sodium ion in the A2A structure [Fig. 3 ▸(d)]. In class A GPCRs, polar amino acids are present with a 98% probability at N7.49 of the NPxxY motif, with a 78% probability of asparagine (Supplementary Fig. S2). The probabilities of serine at 3.39 and aspartic acid at 2.50 are 70% and 96%, respectively. Thus, the N7.49R mutation approach should have a high success rate in stabilizing most class A GPCRs in an inactive form to facilitate structure determination.
Figure 3.

The ionic networks in the mutation-induced inactive M4 structure. (a) Alignment of the structures of M2–AF-DX 384 (PDB entry 5zkb; grey cartoon) and mutation-induced inactive M4 (teal blue cartoon). (b) In the mutation-induced inactive M4 structure (top), S1193.39, D782.50 and R4497.49 form an ionic network, while in the M2 structure (bottom) D692.50, S4337.46 and R1103.39 form a salt-bridge interaction. (c) The distance between CG of D782.50 and CZ of R4497.49 during MD simulation of Ach-bound or tiotropium-bound M41 (top) and M46 (bottom). The detailed information is from the computational analysis of mutation-induced inactive M4. (d) Comparison of the sodium-binding site in the ZM241385–A2A structure (PDB entry 4eiy; lemon) and the ionic network around R4497.49 in mutation-induced inactive M4.
3.5. Computational analysis of mutation-induced inactive M4
To verify whether the mutation-induced M4 structure could be used as an inactive template for in silico screening of potential antagonists, we performed molecular docking of known mAchR ligands using the new structure. The computational analysis was divided into two steps. In the first step, in order to further probe whether the N4497.49R mutation is the key factor that induces the M4 into an inactive state, docking and molecular-dynamics (MD) simulations were performed using four different forms of M4, as shown in Table 3 ▸: (i) the structure of M4 bound to the antagonist tiotropium (PDB entry 5dsg) as a positive control, (ii) M46, the crystal structure of mutation-induced M4 (with six mutations) in this study, (iii) M41, a modified M4 with the N4497.49R mutation only, and (iv) M40, a modified M4 without any mutations. Ach and tiotropium were selected as a representative agonist and antagonist, respectively. Their initial docking poses were obtained by superimposing iperoxo-bound M2 (PDB entry 4mqs) or tiotropium-bound M4 (PDB entry 5dsg) onto the four M4 structures that were just described. The composed Ach-bound or tiotropium-bound M4 template structures were embedded in a lipid bilayer and each MD simulation system was fully equilibrated and relaxed; three independent 2 µs production simulations with no restraints were then performed at 310 K and 1 bar in a constant total number of particles, pressure and temperature (NPT) ensemble for each case (Table 3 ▸). After the MD runs, the final binding poses of Ach and tiotropium, including the M4 structures, are shown in Fig. 4 ▸. Not surprisingly, Ach has diffused away from its initial binding site and has conformationally fluctuated quite significantly in terms of r.m.s.d. values, while the r.m.s.d. values for the antagonist tiotropium were obviously lower. Remarkably, for M46 and M41, which contain the N4497.49R mutation, Ach is more mobile throughout the entire simulation compared with tiotropium-bound M4 (PDB entry 5dsg) and M40. Accordingly, tiotropium is stable in M46 and M41 template structures, which is consistent with PDB entry 5dsg (Fig. 4 ▸). This further confirms that the M46 structure is indeed in an inactive state and that its orthosteric pocket prefers to bind antagonists.
Table 3. Molecular-dynamics simulation of the agonist Ach and the antagonist tiotropium with different forms of M4.
| System | Mutation details | Ligand | Simulation time (µs) × No. of runs |
|---|---|---|---|
| PDB entry 5dsg | None | Ach | 2 × 3 |
| Tiotropium | 2 × 3 | ||
| M40 | None | Ach | 2 × 3 |
| Tiotropium | 2 × 3 | ||
| M41 | N4497.49R | Ach | 2 × 3 |
| Tiotropium | 2 × 3 | ||
| M46 | I932.65T, G1504.43A, I187ECL2A, S2195.62Y, N4497.49R, T4598.49E | Ach | 2 × 3 |
| Tiotropium | 2 × 3 |
Figure 4.

Molecular-dynamics simulations of different forms [M4–tiotropium (PDB entry 5dsg), M40, M41 and M46] of M4. The last frames from the trajectories of the protein with Ach (a) and tiotropium (b) were aligned to show the locations of the ligands in M46 (purple), M41 (blue), M40 (red) and M4–tiotropium (PDB entry 5dsg; dark colour). (c) R.m.s.d. of the agonist Ach (top) and the antagonist tiotropium (bottom) with respect to the protein and its binding pocket during the simulations. Tiotropium is stable in the M46 and M41 templates when compared with Ach in the binding pocket.
To further investigate the performance of the mutation-induced M4 structure in the virtual screening of potential antagonists with a broader ligand library, we carried out molecular docking against a focused muscarinic receptors compound library selected from the IUPHAR database (http://www.guidetopharmacology.org). The selected library is composed of 90 ligands, including agonists, antagonists and allosteric modulators for muscarinic receptors. All of the compounds in this library showed efficacies against certain muscarinic receptors in both functional and binding assays. The docking was performed with Schrödinger Suite 2018-4 (Schrödinger) and the results are listed in descending order by docking scores along with the experimental pK i values (Fig. 5 ▸). In agreement with our results above, the top-ranked compounds in the list are antagonists. Five M4 antagonists, tiotropium, atropine, proprantheline, QNB and umeclidinium, were selected as representatives of different scaffolds for further analysis and their predicted binding poses are shown in Fig. 5 ▸. These five antagonists share similar binding poses, mainly interacting with D1123.32, Y1133.33 and Y4166.51.
Figure 5.

Molecular docking of muscarinic receptor ligands using the mutation-induced inactive M4 structure. (a) The results are listed with compounds in descending order of docking score along with the experimental pK i value. Antagonists and agonists are shown as blue and red dots, respectively. (b–f) The chemical structures and predicted binding poses of antagonists obtaining high scores in (a). (b) Tiotropium (green sticks), (c) atropine (yellow sticks), (d) umeclidinium (grey sticks), (e) propantheline (purple sticks) and (f) QNB (orange sticks).
4. Discussion
Recent structural studies on muscarinic acetylcholine receptors provided important structural insights into the receptor–ligand interactions and ligand-binding poses in orthosteric or allosteric binding pockets. In this study, we demonstrate how to obtain a GPCR structure in its inactive state via key point mutations. In this specific case, we took M4 as an example and created a single point mutation, N4497.49R, to stabilize M4 in its inactive form. We further crystallized and determined the crystal structure of the mutation-induced inactive M4. In a structural comparison with agonist-bound and antagonist-bound mAchRs structures, the mutation-induced inactive M4 structure is obviously similar to the antagonist-bound M4 structure. However, our mutation-induced M4 trapped an unidentified, co-purifying ligand that bound to M4 like a high-affinity antagonist. In addition, we observed that the positively charged N4497.49R residue mimics the allosteric sodium ion binding to the conserved residues D782.50 and S1193.39, which lock M4 into an inactive state. Also, the intracellular domains of the mutation-induced inactive M4 tend to fold into a tighter structural conformation compared with the M4–titropium structure by forming hydrophilic interactions among R1303.50, T3996.34, R144ICL2 and E3956.30. Taken together, the sodium-mimicking ionic lock between TM3 and TM6 and the ionic interaction between R1303.50 and E3956.30 stabilize M4 in an inactive state. The N4497.49R mutation and the switches of TM3 and TM6 were so effective in stabilizing the inactive state that a co-purifying hydrophobic molecule was trapped. Through further computational analysis using molecular docking and MD simulations of M4 structures of different forms, the N4497.49R mutation is shown to be the key mutation which makes M4 prefer to bind antagonists compared with agonists.
GPCRs are important drug targets and are involved in virtually every biological process. In this study, we experimentally and computationally validated that the mutation N4497.49R effectively inactivated M4. The designed ionic bond network is formed by S3.39, D2.50 and N7.49R, which is similar to the effect of the S3.39R mutation in mimicking allosteric Na+ binding in the conserved Na+ ion-binding site in class A GPCRs, as previously reported. Thus, either the N7.49R or S3.39R mutation may apply to other class A GPCRs to stabilize the receptor in an inactive state and to facilitate structure determination in complex with an antagonist or in an apo state.
Supplementary Material
PDB reference: muscarinic receptor M4, 6kp6
Supplementary tables and figures. DOI: 10.1107/S2052252520000597/jt5041sup1.pdf
Acknowledgments
The synchrotron-radiation experiments were performed on BL41XU at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; proposal Nos. 2019A2522 and 2019A2722). We thank the Cloning, Cell Expression, Protein Purification and Assay Core Facilities of the iHuman Institute for their support. We thank Jianjun Cheng, Liang Tan, Wenqing Shui, Bingjie Zhang and Qingtong Zhou for helpful discussion and suggestions.
Funding Statement
This work was funded by National Natural Science Foundation of China grants 31870744 and 31971178. ShanghaiTech University grant .
References
- Alexandrov, A. I., Mileni, M., Chien, E. Y. T., Hanson, M. A. & Stevens, R. C. (2008). Structure, 16, 351–359. [DOI] [PubMed]
- Ballesteros, J. A. & Weinstein, H. (1995). Methods Neurosci. 25, 366–428.
- Bernard, V., Normand, E. & Bloch, B. (1992). J. Neurosci. 12, 3591–3600. [DOI] [PMC free article] [PubMed]
- Bois, T. M. du, Bell, W., Deng, C. & Huang, X.-F. (2005). J. Chem. Neuroanat. 29, 282–288. [DOI] [PubMed]
- Burger, W. A. C., Sexton, P. M., Christopoulos, A. & Thal, D. M. (2018). J. Gen. Physiol. 150, 1360–1372. [DOI] [PMC free article] [PubMed]
- Caffrey, M. & Cherezov, V. (2009). Nat. Protoc. 4, 706–731. [DOI] [PMC free article] [PubMed]
- Chan, W. Y., McKinzie, D. L., Bose, S., Mitchell, S. N., Witkin, J. M., Thompson, R. C., Christopoulos, A., Lazareno, S., Birdsall, N. J. M., Bymaster, F. P. & Felder, C. C. (2008). Proc. Natl Acad. Sci. USA, 105, 10978–10983. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B. III, Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Darden, T., York, D. & Pedersen, L. (1993). J. Chem. Phys. 98, 10089–10092.
- Eglen, R. M. (2012). Handb. Exp. Pharmacol. 208, 3–28. [DOI] [PubMed]
- Eglen, R. M., Hegde, S. S. & Watson, N. (1996). Pharmacol. Rev. 48, 531–565. [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Eskow Jaunarajs, K. L., Bonsi, P., Chesselet, M. F., Standaert, D. G. & Pisani, A. (2015). Prog. Neurobiol. 127–128, 91–107. [DOI] [PMC free article] [PubMed]
- Farkas, E., de Wilde, M. C., Kiliaan, A. J., Meijer, J., Keijser, J. N. & Luiten, P. G. (2002). Brain Res. 954, 32–41. [DOI] [PubMed]
- Fenalti, G., Giguere, P. M., Katritch, V., Huang, X.-P., Thompson, A. A., Cherezov, V., Roth, B. L. & Stevens, R. C. (2014). Nature, 506, 191–196. [DOI] [PMC free article] [PubMed]
- Flynn, D. D. & Mash, D. C. (1993). Synapse, 14, 283–296. [DOI] [PubMed]
- Fredriksson, R., Lagerström, M. C., Lundin, L.-G. & Schiöth, H. B. (2003). Mol. Pharmacol. 63, 1256–1272. [DOI] [PubMed]
- Freund, G., Brophy, T. R. & Scott, J. D. (1986). Exp. Gerontol. 21, 37–44. [DOI] [PubMed]
- Haga, K., Kruse, A. C., Asada, H., Yurugi-Kobayashi, T., Shiroishi, M., Zhang, C., Weis, W. I., Okada, T., Kobilka, B. K., Haga, T. & Kobayashi, T. (2012). Nature, 482, 547–551. [DOI] [PMC free article] [PubMed]
- Horcajada, C., Guinovart, J. J., Fita, I. & Ferrer, J. C. (2006). J. Biol. Chem. 281, 2923–2931. [DOI] [PubMed]
- Huang, W., Manglik, A., Venkatakrishnan, A. J., Laeremans, T., Feinberg, E. N., Sanborn, A. L., Kato, H. E., Livingston, K. E., Thorsen, T. S., Kling, R. C., Granier, S., Gmeiner, P., Husbands, S. M., Traynor, J. R., Weis, W. I., Steyaert, J., Dror, R. O. & Kobilka, B. K. (2015). Nature, 524, 315–321. [DOI] [PMC free article] [PubMed]
- Hulme, E. C., Birdsall, N. J. & Buckley, N. J. (1990). Annu. Rev. Pharmacol. Toxicol. 30, 633–673. [DOI] [PubMed]
- Humphrey, W., Dalke, A. & Schulten, K. (1996). J. Mol. Graph. 14, 33–38. [DOI] [PubMed]
- Jo, S., Kim, T., Iyer, V. G. & Im, W. (2008). J. Comput. Chem. 29, 1859–1865. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Karplus, P. A. & Diederichs, K. (2012). Science, 336, 1030–1033. [DOI] [PMC free article] [PubMed]
- Kato, M., Komamura, K. & Kitakaze, M. (2006). Circ. J. 70, 1658–1660. [DOI] [PubMed]
- Katritch, V., Fenalti, G., Abola, E. E., Roth, B. L., Cherezov, V. & Stevens, R. C. (2014). Trends Biochem. Sci. 39, 233–244. [DOI] [PMC free article] [PubMed]
- Kruse, A. C., Hu, J., Pan, A. C., Arlow, D. H., Rosenbaum, D. M., Rosemond, E., Green, H. F., Liu, T., Chae, P. S., Dror, R. O., Shaw, D. E., Weis, W. I., Wess, J. & Kobilka, B. K. (2012). Nature, 482, 552–556. [DOI] [PMC free article] [PubMed]
- Kruse, A. C., Ring, A. M., Manglik, A., Hu, J., Hu, K., Eitel, K., Hübner, H., Pardon, E., Valant, C., Sexton, P. M., Christopoulos, A., Felder, C. C., Gmeiner, P., Steyaert, J., Weis, W. I., Garcia, K. C., Wess, J. & Kobilka, B. K. (2013). Nature, 504, 101–106. [DOI] [PMC free article] [PubMed]
- Lam, P. T. H., Chan, C. K. M., Rao, S. K., Fan, D. S. P. & Lam, D. S. C. (2010). J. Optom. 3, 37–43.
- Liebschner, D., Afonine, P. V., Baker, M. L., Bunkóczi, G., Chen, V. B., Croll, T. I., Hintze, B., Hung, L.-W., Jain, S., McCoy, A. J., Moriarty, N. W., Oeffner, R. D., Poon, B. K., Prisant, M. G., Read, R. J., Richardson, J. S., Richardson, D. C., Sammito, M. D., Sobolev, O. V., Stockwell, D. H., Terwilliger, T. C., Urzhumtsev, A. G., Videau, L. L., Williams, C. J. & Adams, P. D. (2019). Acta Cryst. D75, 861–877.
- Liu, H., Hofmann, J., Fish, I., Schaake, B., Eitel, K., Bartuschat, A., Kaindl, J., Rampp, H., Banerjee, A., Hübner, H., Clark, M. J., Vincent, S. G., Fisher, J. T., Heinrich, M. R., Hirata, K., Liu, X., Sunahara, R. K., Shoichet, B. K., Kobilka, B. K. & Gmeiner, P. (2018). Proc. Natl Acad. Sci. USA, 115, 12046–12050. [DOI] [PMC free article] [PubMed]
- Liu, W., Chun, E., Thompson, A. A., Chubukov, P., Xu, F., Katritch, V., Han, G. W., Roth, C. B., Heitman, L. H., IJzerman, A. P., Cherezov, V. & Stevens, R. C. (2012). Science, 337, 232–236. [DOI] [PMC free article] [PubMed]
- Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. (2012). Nucleic Acids Res. 40, D370–D376. [DOI] [PMC free article] [PubMed]
- Maeda, S., Qu, Q., Robertson, M. J., Skiniotis, G. & Kobilka, B. K. (2019). Science, 364, 552–557. [DOI] [PMC free article] [PubMed]
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E. & Simmerling, C. (2015). J. Chem. Theory Comput. 11, 3696–3713. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Miller-Gallacher, J. L., Nehmé, R., Warne, T., Edwards, P. C., Schertler, G. F., Leslie, A. G. W. & Tate, C. G. (2014). PLoS One, 9, e92727. [DOI] [PMC free article] [PubMed]
- Peng, Y., McCorvy, J. D., Harpsøe, K., Lansu, K., Yuan, S., Popov, P., Qu, L., Pu, M., Che, T., Nikolajsen, L. F., Huang, X.-P., Wu, Y., Shen, L., Bjørn-Yoshimoto, W. E., Ding, K., Wacker, D., Han, G. W., Cheng, J., Katritch, V., Jensen, A. A., Hanson, M. A., Zhao, S., Gloriam, D. E., Roth, B. L., Stevens, R. C. & Liu, Z.-J. (2018). Cell, 172, 719–730. [DOI] [PMC free article] [PubMed]
- Popov, P., Kozlovskii, I. & Katritch, V. (2019). Curr. Opin. Struct. Biol. 55, 25–33. [DOI] [PubMed]
- Popov, P., Peng, Y., Shen, L., Stevens, R. C., Cherezov, V., Liu, Z.-J. & Katritch, V. (2018). eLife, 7, e34729. [DOI] [PMC free article] [PubMed]
- Roe, D. R. & Cheatham, T. E. (2013). J. Chem. Theory Comput. 9, 3084–3095. [DOI] [PubMed]
- Ryckaert, J. P., Ciccotti, G. & Berendsen, H. J. C. (1977). J. Comput. Phys. 23, 327–341.
- Smart, O. S., Womack, T. O., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., Vonrhein, C. & Bricogne, G. (2012). Acta Cryst. D68, 368–380. [DOI] [PMC free article] [PubMed]
- Suno, R., Lee, S., Maeda, S., Yasuda, S., Yamashita, K., Hirata, K., Horita, S., Tawaramoto, M. S., Tsujimoto, H., Murata, T., Kinoshita, M., Yamamoto, M., Kobilka, B. K., Vaidehi, N., Iwata, S. & Kobayashi, T. (2018). Nat. Chem. Biol. 14, 1150–1158. [DOI] [PMC free article] [PubMed]
- Thal, D. M., Sun, B., Feng, D., Nawaratne, V., Leach, K., Felder, C. C., Bures, M. G., Evans, D. A., Weis, W. I., Bachhawat, P., Kobilka, T. S., Sexton, P. M., Kobilka, B. K. & Christopoulos, A. (2016). Nature, 531, 335–340. [DOI] [PMC free article] [PubMed]
- Vickery, O. N., Carvalheda, C. A., Zaidi, S. A., Pisliakov, A. V., Katritch, V. & Zachariae, U. (2018). Structure, 26, 171–180. [DOI] [PMC free article] [PubMed]
- Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. (2004). J. Comput. Chem. 25, 1157–1174. [DOI] [PubMed]
- Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A. & Wilson, K. S. (2011). Acta Cryst. D67, 235–242. [DOI] [PMC free article] [PubMed]
- Yang, Q., Lachapelle, E. A., Kablaoui, N. M., Webb, D., Popiolek, M., Grimwood, S., Kozak, R., O’Connor, R. E., Lazzaro, J. T., Butler, C. R. & Zhang, L. (2019). ACS Med. Chem. Lett. 10, 941–948. [DOI] [PMC free article] [PubMed]
- Yazdani, N., Sadeghi, R., Momeni-Moghaddam, H., Zarifmahmoudi, L. & Ehsaei, A. (2018). J. Optom. 11, 135–143. [DOI] [PMC free article] [PubMed]
- Yin, J., Babaoglu, K., Brautigam, C. A., Clark, L., Shao, Z., Scheuermann, T. H., Harrell, C. M., Gotter, A. L., Roecker, A. J., Winrow, C. J., Renger, J. J., Coleman, P. J. & Rosenbaum, D. M. (2016). Nat. Struct. Mol. Biol. 23, 293–299. [DOI] [PubMed]
- Zhang, C., Srinivasan, Y., Arlow, D. H., Fung, J. J., Palmer, D., Zheng, Y., Green, H. F., Pandey, A., Dror, R. O., Shaw, D. E., Weis, W. I., Coughlin, S. R. & Kobilka, B. K. (2012). Nature, 492, 387–392. [DOI] [PMC free article] [PubMed]
- Ztaou, S., Maurice, N., Camon, J., Guiraudie-Capraz, G., Kerkerian-Le Goff, L., Beurrier, C., Liberge, M. & Amalric, M. (2016). J. Neurosci. 36, 9161–9172. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: muscarinic receptor M4, 6kp6
Supplementary tables and figures. DOI: 10.1107/S2052252520000597/jt5041sup1.pdf