
Table 2. Data-collection and structure-refinement statistics for mutation-induced inactive M4.
Values in parentheses are for the highest resolution shell.
| No ligand | HMDB0010212 docked | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.000 | |
| Resolution range (Å) | 50.00–3.00 (3.08–3.00) | |
| Space group | P212121 | |
| a (Å) | 56.10 | |
| b (Å) | 61.32 | |
| c (Å) | 203.74 | |
| Observed reflections | 398969 | |
| Unique reflections | 14718 | |
| Multiplicity | 27.1 (6.0) | |
| Completeness (%) | 99.7 (97.0) | |
| Mean I/σ(I) | 19.3 (2.3) | |
| Wilson B factor (Å2) | 89.34 | |
| R merge † | 0.132 (0.586) | |
| CC1/2 ‡ | 0.996 (0.379) | |
| Refinement | ||
| Resolution range (Å) | 49.14–3.00 | |
| Reflections (work/test) | 13985/731 | |
| R work/R free § | 0.231/0.264 | |
| No. of atoms | ||
| Total | 3711 | 3734 |
| Macromolecules | 3711 | 3711 |
| Ligands | 0 | 23 |
| Solvent | 0 | 0 |
| No. of protein residues | 474 | 477 |
| R.m.s.d., bonds (Å) | 0.008 | 0.014 |
| R.m.s.d., angles (°) | 0.94 | 1.67 |
| Ramachandran statistics¶ | ||
| Favoured (%) | 96.39 | 94.48 |
| Allowed (%) | 3.41 | 5.10 |
| Outliers (%) | 0.42†† | 0.42†† |
| Clashscore | 2.42 | 3.75 |
| PDB code | 6kp6 | |
†
R
merge = 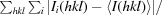
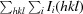 , where Ii(hkl) is the intensity of observation i of reflection hkl.
, where Ii(hkl) is the intensity of observation i of reflection hkl.
‡
As defined by Karplus & Diederichs (2012 ▸).
§
R = 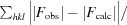
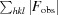 for all reflections, where F
obs and F
calc are the observed and calculated structure factors, respectively. R
free is calculated analogously for the test reflections, which were randomly selected and excluded from the refinement.
for all reflections, where F
obs and F
calc are the observed and calculated structure factors, respectively. R
free is calculated analogously for the test reflections, which were randomly selected and excluded from the refinement.
¶
As defined by MolProbity (Chen et al., 2010 ▸).
††
Glycine residues 1075 and 1144 are in sharp-turn domains of the PGS fusion protein.