

ABSTRACT
Natural killer (NK) cells eliminate abnormal cells through the release of cytolytic granule contents. In this process, NK cells must adhere to target cells through integrin-mediated adhesion, which is highly dependent on the generation of F-actin. Ena/VASP-like (EVL) is an actin regulatory protein previously shown to regulate integrin-mediated adhesion in other cell types, but its role in NK cell biology is not known. Herein, we show that EVL is recruited to the NK cell cytotoxic synapse and is required for NK cell cytotoxicity. Significantly, EVL is involved in the generation of F-actin at the cytotoxic synapse, antibody-stimulated spreading, and NK cell-target cell adhesion. EVL interacts with WASP (also known as WAS) and VASP and is required for localization of both proteins to the synapse. Recruitment of EVL to points of cellular activation occurs through the receptor NKG2D–DAP10 (also known as KLRK1 and HCST, respectively) via a binding site previously implicated in VAV1 and Grb2 recruitment. Taken together, this study implicates DAP10-mediated Grb2 and VAV1 signaling in the recruitment of an EVL-containing actin regulatory complex to the cytotoxic synapse where it can promote F-actin nucleation leading to NK cell-mediated killing.
KEY WORDS: NK cell, Cytotoxicity, Actin polymerization, EVL
Summary: The NK activating receptor NKG2D–DAP10 recruits EVL to the cytotoxic synapse via a Grb2–VAV1 signaling node to promote F-actin generation, target cell adhesion and the development of cell-mediated cytotoxicity.
INTRODUCTION
Natural killer (NK) cells are innate immune cells responsible for removing abnormal cells on a cell-by-cell basis. During NK cell activation, the NK cell strongly adheres to target cells, a process that is dependent both on integrin activation (Zarcone et al., 1992; Mace et al., 2009) and on actin polymerization (Carpen et al., 1983; Hoffmann et al., 2011). This early step in NK cell cytotoxicity is a requirement for sustained signaling by activating and inhibitory receptors, and ultimately a decision process that ends in the polarization of the microtubule-organizing center (MTOC), lytic granule delivery to the immunological cytotoxic synapse (CS) and release of the granule contents to induce death of the target cell. The polymerization of F-actin at the NK cell synapse occurs downstream of both NK cell activating receptors (Brown et al., 2012) and integrins (Mace et al., 2010; Riteau et al., 2003).
Signaling from the NK cell activating receptor NKG2D–DAP10 (also known as KLRK1 and HCST, respectively) results in the recruitment of both Grb2–VAV1 and p85 (PIK3R1)-containing phosphoinositide 3-kinase (PI3K) (Billadeau et al., 2003; Upshaw et al., 2006) signaling complexes to drive F-actin generation, integrin-mediated adhesion and, ultimately, NK cell cytotoxicity. The generation of F-actin at the CS promotes NK cell adhesion to the target cell and stability of the synapse. One of the major mediators of this pathway is Wiskott–Aldrich syndrome protein (WASP, also known as WAS) (Orange et al., 2002), an effector of the small GTP-binding protein Cdc42. Binding of active Cdc42 with WASP allows its interaction with the ARP2/3 actin nucleator complex to promote branched F-actin nucleation (Blanchoin et al., 2000; Millard et al., 2004; Uruno et al., 2003). In the absence of WASP (Orange et al., 2002) or key components of the ARP2/3 complex (Butler and Cooper, 2009), actin polymerization does not occur, and NK cells lose their ability to effectively adhere to and kill target cells.
We have previously shown in NK cells that WASP is recruited to the CS through an interaction with the Cdc42 guanine nucleotide exchange factor DOCK8 (Ham et al., 2013), thus likely positioning WASP in the presence of activated Cdc42 for local generation of ARP2/3-generated branched F-actin. Recently, we have shown that DOCK8 also interacts with VASP (Wilton and Billadeau, 2018), a member of the Ena/VASP actin regulatory family, which includes the homologous proteins MENA and EVL. Previous studies in other cell types have shown a role for the Ena/VASP family in regulating the production of distinct cytoskeletal structures, including focal adhesions, stress fibers, lamellipodia and filopodia. In addition, actin-dependent adhesion in other cell types, including tight junction adhesions (Oldenburg et al., 2015), interactions during T cell migration (Estin et al., 2017), neutrophil adhesion (Deevi et al., 2010) and cell–cell contacts (Benz et al., 2008) have been shown to be dependent on actin polymerization mediated by Ena/VASP family members. Within T cells, VASP mediates the ability of T cells to form adhesions while crawling through tissues, and specifically during migration through blood vessel walls (diapedesis) (Estin et al., 2017). Somewhat surprisingly, we found that while VASP is recruited to the NK-target cell CS, it was dispensable for F-actin generation and integrin-mediated adhesion, but was critical in maintaining the convergence of lytic granules at the MTOC (Wilton and Billadeau, 2018). This result suggested that maybe other members of the Ena/VASP family of proteins are involved in regulating F-actin generation and integrin-mediated adhesion between NK cells and their targets.
Herein, we investigated the role of EVL in NK cell-mediated killing. Importantly, EVL is expressed in NK cells and localizes to the CS formed between NK cells and target cells, and depletion of EVL by siRNA results in defective NK cell killing. In contrast to VASP depletion, EVL is involved in mediating NK cell adhesion and synapse maturation. We further show that F-actin generation is defective in EVL-depleted cells and that EVL is needed for the recruitment of VASP and WASP to stimulatory contact sites in NK cells. Mechanistically, we found that NKG2D–DAP10 signaling through Grb2–VAV1 recruits EVL to the site of activation where it promotes F-actin generation.
RESULTS
EVL localizes to the NK cell synapse and promotes NK cell cytotoxicity
In order to ensure that, like VASP (Wilton and Billadeau, 2018), EVL was expressed within NK cells, samples from various NK cell lines (NKL and YTS), primary NK cells and other cell lines were lysed and immunoblotted for the presence of EVL. EVL was not expressed in the epithelial-derived pancreatic cancer cell lines L3.6 or BXPC3, but was expressed in the NK cell lines, NKL and YTS, and in primary NK cells, but notably at very low levels in the target cell line 721.221 (Fig. 1A). Of note, two bands are visible, particularly in the YTS cell line and primary T cells. These two bands have been previously identified to represent different isoforms and/or phosphorylation states of EVL, the exact functional difference of which has not been clearly defined (Lambrechts et al., 2000; Janssens et al., 2009).
Fig. 1.
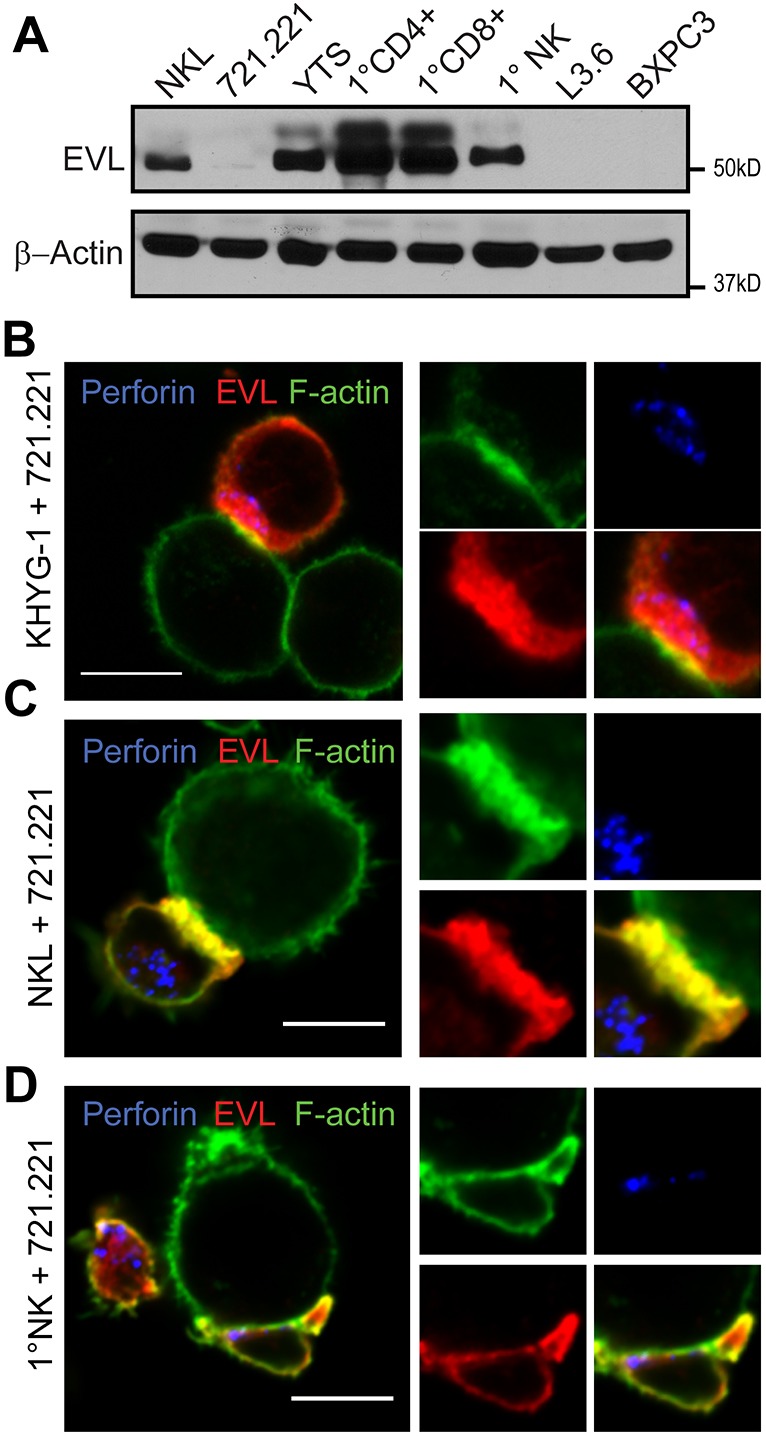
EVL localizes to the CS in NK cells. (A) Various cell lines and primary human cells were lysed and immunoblotted for the presence of EVL and β-actin. (B–D) Localization of EVL (red) was assessed in relation to the cell membrane (denoted by phalloidin staining of F-actin, green) and the lytic granules (denoted by perforin staining, blue) in KHYG-1 (B), NKL (C) and primary NK cells (D) which had been conjugated with 721.221 cells for 30 min.
To examine the localization of EVL upon NK-target interaction, cells representing three models of NK cells, KHYG-1, NKL and primary NK cells were allowed to form conjugates with the target 721.221 cell line. Cells were then fixed and stained for F-actin, to illustrate the perimeter of the cell and synapse, perforin, to indicate the position of the granules, and EVL. As shown in Fig. 1B–D, EVL was clearly visualized in all three models of NK cells, and interestingly, localized to the area around the CS with F-actin. Overall, this indicates that EVL is expressed in NK cells and localizes at the CS.
Although the sheer volume of EVL near the synapse is suggestive of an important role in NK cell cytolysis, it could be indicative of another cellular function. Thus, we next sought to assess the impact of EVL on NK cell cytolysis through siRNA-mediated knockdown (KD). At 72 h following siRNA-mediated knockdown of EVL (siEVL), NK cells were incubated with Cr51-stained 721.221 target cells and assessed for their ability to lyse the target cells. As shown in Fig. 2, EVL KD decreased cytolysis in KHYG-1 (Fig. 2A–C), NKL (Fig. 2D–F) and primary NK cells (Fig. 2G–I). Additionally, siRNA-mediated EVL KD impaired the ability of primary NK cells to kill anti-NKG2D- and anti-2B4 (hereafter anti-NKG2D/2B4; 2B4 is also known as CD244)-coated p815 cells (Fig. 2J–K), indicating decreased killing activity secondary to signaling depending on this receptor. Taken together, these data indicate that EVL participates in the regulation of NK cell cytotoxicity.
Fig. 2.

EVL KD inhibits NK cell cytotoxicity. KHYG-1 (A), NKL cells (D) or primary NK cells (G) were nucleofected with siRNAs targeting the EVL protein (siEVL #1 or #2), or with a control siRNA (siCtrl). Samples of cells were lysed and the presence of EVL and GAPDH was then assessed by immunoblotting for every experiment completed. Representative images are shown. Nucleofected cells were incubated with Cr51-labeled 721.221 cells in order to assess cytotoxicity through a chromium release assay. A representative experiment (B,E,H) and quantification at a specific effector to target (E:T) ratio (C,F,I) are shown for each NK cell model (KHYG-1, NKL and primary NK cells, respectively). In order to assess cytotoxicity through specific receptors, siRNA nucleofected primary NK cells were incubated with Cr51-labeled p815 cells, which were coated with antibodies against NKG2D and 2B4 (J–K), and assessed for chromium release. Representative experiments and quantification at a single effector to target ratio are shown for each. All experiments shown are representative of or quantified over a minimum of three independent experiments. Error bars indicate s.e.m. from the indicated mean. *P<0.05; **P<0.005; ***P<0.0005 [unpaired (B,E,H,J) or paired (C,F,I,K) Student's t-test]. The least significant P-value of two EVL-targeting siRNA groups is shown.
NK-cell–target-cell adhesion and actin polymerization, but not integrin signaling, are dependent on EVL
NK cell-mediated cytolysis is a complex process involving a large number of moving cytoskeletal components that result in the release of cytotoxic granules. Some of the first essential steps of this process are involved in the tight adhesion of the NK cell to the identified target cell. This sustained interaction is crucial to downstream signaling events, cytoskeletal reorganization and polarized delivery of the lytic granules. Therefore, the tight adhesion between NK cells and target cells is a key cellular mechanism to be investigated. To assess this, KHYG-1 (Fig. 3A), NKL (Fig. 3B) and primary NK cells (Fig. 3C) were stained with Calcein-AM and allowed to form conjugates for the indicated times with CMAC-stained 721.221 cells. The resulting cellular conjugates were then assessed by flow cytometry to quantify the extent to which EVL KD impacts NK–target-cell adhesion. In all cases, siRNA-mediated KD of EVL resulted in less conjugate formation with 721.221 cells. This phenotype of decreased adhesive ability was recapitulated using EVL KD NKL cells in an Fc-ICAM-binding assay (Fig. 3D), which requires both integrin binding and actin polymerization. Overall, these data indicate that EVL has a primary role in the promotion of NK cell adhesive forces and the stabilization of the interaction between NK cells and target cells.
Fig. 3.

EVL is required for NK cell-mediated adhesion. Calcein AM-stained KHYG-1 (A), NKL (B) or primary NK cells (C) nucleofected with one of two siRNAs targeting EVL (siEVL #1 or #2) or a control siRNA (siCtrl) were allowed to form conjugates with CMAC-stained 721.221 cells for the given amounts of time, vortexed, fixed and evaluated for their ability to form tight bound conjugates via two-color flow cytometry. (D) Calcein-AM stained NKL nucleofected with either one of two siRNAs targeting EVL or a control siRNA were evaluated for their ability to bind to a plate-bound Fc-ICAM. Shown are representative experiments chosen from a minimum of three independent experiments. Error bars indicate s.e.m. from the indicated mean. *P<0.05; **P<0.005; ***P<0.0005 (unpaired Student's t-test). The least significant P-value of two EVL-targeting siRNA groups is shown.
The interaction that allows NK-cell–target-cell conjugates involves multiple adhesive forces. After contact is made, signaling occurs through several receptors and the adhesion process begins. These receptors include activating receptors, like NKG2D and 2B4, which act to both induce actin polymerization and to promote the activation of integrins, including LFA-1 (the integrin αL–integrin β2 complex), through outside-in signaling. In addition, signaling through LFA-1 itself can promote actin polymerization and further adhesion.
In order to determine whether EVL was functioning primarily downstream of the NK cell activating receptors (NKG2D and 2B4) or downstream of LFA-1, the major integrin involved in target cell adhesion, NK cells were stimulated by anti-NKG2D/2B4 antibody crosslinking using goat anti-mouse-IgG, or by Fc-ICAM and goat anti-mouse-IgG for 20 min. Cells were then lysed and phosphotyrosine-containing complexes were immunoprecipitated by using the 4G10 monoclonal antibody. Although EVL appears in some activated complexes before stimulation, after 20 min, NKG2D/2B4-mediated stimulation induced a robust increase in the amount of EVL that was immunoprecipitated by 4G10 antibody (Fig. S1A). Interestingly, stimulation with Fc-ICAM resulted in a lower level of EVL in phosphotyrosine-containing complexes than in unstimulated cells. This may indicate that EVL is specifically excluded or removed from these complexes, or just that a large number of new complexes are formed, diluting the signal of any EVL protein that still remains in these complexes.
Defective cytolysis and conjugate formation, as seen in the EVL KD NK cells, could be secondary to loss of activating receptor surface expression or failure of surface receptors to accumulate at the site of stimulation. As shown in Fig. S1B,C, cell surface expression of 2B4 and NKG2D was similar in siCtrl- and siEVL-nucleofected NKL cells. Similarly, 2B4 accumulated similarly at the activating-cell–bead contact site in siCtrl and siEVL KD cells (Fig. S1D,E). In all cases, accumulation of 2B4 at the activating contact site was higher than at the mouse (m)IgG–bead contact site indicating an activation-dependent accumulation. Gross signaling, as ascertained by phosphorylation of the MAP kinase ERK was also unaffected (Fig. S1F). In all, the accumulation of EVL in complexes downstream of NKG2D and 2B4 activating receptor signaling suggests that activating receptors likely drive the phenotype seen in these cells. In support of this, in EVL KD NKL cells, levels of LFA-1 integrin activation (Fig. S2A), surface LFA-1 expression (Fig. S2B) and Fc-ICAM-mediated GSK-3 inactivation (Zhang et al., 2014) (Fig. S2C,D) were all unchanged, as was gross actin polymerization as assessed by cytoskeletal pellet isolation (Fig. S2E).
Given the lack of a global defect in cytoskeletal actin, a more localized impact on the accumulation of actin at the NK-cell–target-cell CS seemed a likely role for EVL. To assess synaptic actin accumulation, NKL cells were nucleofected with a control siRNA or one of two siRNAs targeting EVL, and then allowed to form conjugates with latex beads coated with mIgG or anti-NKG2D/2B4 for a total of 30 min before being fixed and stained for microscopy. These antibody-coated beads were used in place of target cells in order to remediate the conjugate formation defect seen with EVL KD. Beads coated with mIgG served as a negative control. Nucleofection with siRNA targeting EVL resulted in a significant, though not complete (as indicated by conjugates using mIgG-coated beads), reduction in F-actin accumulation at the synapse (Fig. 4A,B; Fig. S3A,B). Of note, any cells that did not have decreased EVL expression in response to EVL-targeting siRNA were excluded from the analysis.
Fig. 4.

NK cell activating receptor-driven localized actin accumulation is dependent on EVL. (A) EVL (siEVL #1 or #2) or a control siRNA (siCtrl)-treated NKL cells were allowed to adhere to anti-NKG2D/2B4-coated latex beads and then fixed and imaged for the presence of F-actin via phalloidin staining. (B) Results from three independent experiments are quantified in comparison to the same cell groups conjugated with mIgG-coated beads as a negative control for F-actin accumulation (demarcated by dotted red line with full data in Fig. S3A,B). (C–E) NKL cells nucleofected with control siRNA or siEVL were allowed to adhere to anti-NKG2D/2B4-coated coverslips for 15 min, and then were fixed and imaged for the localization of EVL and F-actin via phalloidin staining (C). The surface area of the cell at the coverslip from three independent experiments interface is quantified in D and the amount of total EVL fluorescence for one representative experiment is quantified in E. (F,G) Primary NK cells nucleofected with control siRNA or siRNA targeting EVL were allowed to spread for 15 min on anti-NKG2D/2B4-coated coverslips, then fixed and imaged at the plane of the cell–coverslip interface (F). A representative quantification of a single experiment is shown in G and represents three independent experiments with independent NK cell donors. All data is representative of a minimum of three independent experiments. Error bars indicate s.e.m. from the indicated mean. ***P<0.0005 (Student's t-test). Scale bars: 10 µm.
Another method to test for activation-induced F-actin polymerization in NK cells is to allow NK cells to spread onto coverslips coated with stimulatory antibodies. In the absence of intact actin polymerization, the cells will remain spherical, and only touch the coverslip over a small surface area. In contrast, if actin polymerization is intact, these cells should spread into a large flat disc, increasing their surface area at the coverslip contact point, and thus increasing their interaction with the activating antibodies. Using this system, EVL KD NKL that were allowed to spread on anti-NKG2D/2B4-coated coverslips for 15 min showed a defect in actin-dependent spreading (Fig. 4C–E) consistent with the defect in actin accumulation at the cell–bead contact point (Fig. 4A,B). In the NKL actin-mediated cell spreading experiments, cells that failed to show EVL KD, as verified by immunofluorescence, were excluded based on a fluorescence threshold value, as shown in Fig. S3C. EVL KD primary NK cells (as confirmed by immunofluorescence co-staining with anti-EVL antibody, Fig. S3D) allowed to spread on anti-NKG2D/2B4-coated coverslips also showed a pronounced defect in actin-dependent cell spreading (Fig. 4F,G). Overall, this indicates that activation-induced signaling through the NKG2D and 2B4 receptors leads to F-actin polymerization that is dependent on EVL.
EVL is required for the localization of VASP and WASP to the NK cell contact site
Although direct EVL-mediated actin polymerization could be a potential mechanism for the observed actin defect, direct polymerization of actin by any of the Ena/VASP family members has been difficult to observe in vivo. However, several other proteins have been implicated in actin accumulation at the NK cell cytotoxic synapse, and therefore might interact with EVL. Specifically, DOCK8 (Fig. 5A), WASP (Fig. 5B) and VASP (Fig. 5B) have all been found to be constitutively in complex with EVL in resting NK cells. Of note, interactions between VASP, WASP and EVL all appeared to be stimulation independent (Fig. 5B), suggesting the presence of a stable multi-protein complex that appears to not be assembled or disassembled in response to activating receptor stimulation.
Fig. 5.

EVL is specifically required for the recruitment of WASP and VASP to the cytotoxic synapse. (A) NKL cells were lysed and immunoprecipitated (IP) with anti-DOCK8 antibody or rabbit IgG and immunoblotted for the indicated proteins. (B) NKL cells were stimulated over the indicated time course by ligation of NKG2D/2B4 receptors, and the indicated proteins were immunoprecipitated and immunoblotted. (C,E) NKL cells treated EVL siRNA (siEVL #1 or #2) or a control siRNA (siCtrl) were allowed to adhere to anti-NKG2D/2B4-coated latex beads for a total of 30 min, fixed and then imaged for the localization of (C) WASP and (E) VASP. Quantification of the MFI for (D) WASP and (F) VASP recruited to the cell–bead contact site from three independent experiments. Quantification is shown in relation to an average value for all three groups when conjugated to mIgG-coated latex beads, also in three independent experiments. Individual mIgG conjugate quantification can be seen in Fig. S4. Error bars indicate s.e.m. from the indicated mean **P<0.005; ***P<0.0005 (Student's t-test). Scale bars: 10 µm.
Since WASP is critical to both F-actin generation at the CS and NK cell adhesion, we next determined whether the loss of EVL might impact the recruitment of WASP to anti-NKG2D/2B4-coated beads. Significantly, WASP and VASP both accumulated at the stimulatory NK-cell–bead contact site in an EVL-dependent manner (Fig. 5C–F; Fig. S4). No accumulation was seen in the absence of stimulation (Fig. S4). As VASP and EVL are family members that tend to form stable tetramers (Riquelme et al., 2015), there remained a distinct possibility that the localization for the two proteins was co-dependent. To investigate this possibility, VASP was depleted using two independent siRNAs (Fig. 6A) and the stability and localization of EVL was investigated. As shown in Fig. 6B,C, the accumulation of EVL at the NKG2D/2B4-coated bead–NKL cell interface (Fig. 6C), relative to the mIgG bead–NKL interface (Fig. 6B), was unchanged by VASP knockdown (Fig. 6B–D). This indicates that VASP was not required for EVL localization to sites of NK cell activation. Taken together, these data indicate that EVL is required for the delivery of VASP and WASP to the sites of NKG2D/2B4 stimulation.
Fig. 6.

VASP knockdown does not perturb EVL stability or localization in NKL cells. NKL cells were nucleofected with one of two siRNAs targeting VASP (siVASP #1 or #2) or a control siRNA (siCtrl). (A) A sample of these cells from each experiment were lysed and evaluated by immunoblotting for the indicated proteins. (B,C) NKL cells were allowed to adhere to mIgG-coated latex beads (B) or anti-NKG2D/2B4-coated latex beads (C) for 30 min, fixed and evaluated by immunofluorescence for the localization of EVL. Representative images are shown. (D) Quantification of three independent experiments for each group. The red dotted line indicates the average value for all mouse IgG bead conjugates across the three-siRNA groups. Confocal scale bars indicate 10 µm unless otherwise indicated. Images represent three independent experiments. Dot graph shows all data points from three independent experiments. Error bars indicate s.e.m. from the indicated mean. ***P<0.0005 (Student's t-test).
DAP10-mediated VAV1 activation recruits EVL to sites of NK cell activation
Our data show that EVL is recruited to the CS, but the mechanism driving its recruitment is not known. In order to define the signaling pathway that facilitates EVL recruitment, we investigated the NKG2D-activating receptor itself. NKG2D associates with the transmembrane intracellular adaptor molecule DAP10, and upon NKG2D interaction with its cognate ligand (e.g. MICA) DAP10 is phosphorylated by Lck at a tyrosine residue that resides in a YINM motif (Billadeau et al., 2003). This tyrosine-containing motif can recruit both the Grb2–VAV1, and the p85–PI3K signaling complexes through SH2-mediated interaction with Grb2 and p85 (Upshaw et al., 2006). In order to determine whether DAP10 could recruit EVL to the site of cellular activation, we used a series of recombinant vaccinia viruses expressing Flag–CD4–DAP10 chimeric receptors in which the tyrosine residue was unchanged (WT), mutated to phenylalanine (YF), or uncoupled from p85–PI3K (M88Q) or Grb2–VAV1 (N87Q) (Upshaw et al., 2006). Infection of the KHYG-1 cell line showed that WT CD4.DAP10 was sufficient to drive EVL and F-actin accumulation at the anti-Flag-coated bead contact site, whereas the YF mutant could not (Fig. 7A–E). Interestingly, while the M88Q mutant was capable of driving EVL recruitment and F-actin polymerization toward anti-Flag-coated beads, the N87Q mutant, which does not interact with the Grb2–VAV1 complex, was unable to do so (Fig. 7A-E). Moreover, we found that NKG2D ligation results in an inducible association of VAV1 with EVL and VASP (Fig. 7F). Taken together, these data indicate that EVL is recruited to the site of NK cell activation via a DAP10-dependent Grb2–VAV1-associated signaling pathway.
Fig. 7.

DAP10-mediated activation of VAV1 recruits EVL to the site of activation. (A) KHYG-1 cells were infected at an MOI of 10:1 over 2 h with the indicated viruses and evaluated via flow cytometry for the presence of surface CD4, indicating the level of Flag–CD4–DAP10 chimeric receptor expression. (B,C) KHYG-1 cells infected with the indicated viruses were allowed to form conjugates with anti-Flag-coated latex beads over 30 min, and then fixed, permeabilized and stained for immunofluorescence. (D,E) The accumulation of EVL and F-actin to the point of cell–bead contact is quantified. (F) NKL cells were stimulated with anti-NKG2D over the indicated time course, lysed, immunoprecipitated with anti-VAV1 and immunoblotted for the indicated proteins. All images are representative of three independent experiments. Dot plots show all data points from three independent experiments analyzed together. Error bars indicate s.e.m. from the indicated mean. ***P<0.0005. Scale bars: 5 µm.
DISCUSSION
The polymerization of actin at the contact site between an NK cell and its target is required for effective NK cell-mediated cytolysis and is highly dependent on both WASP and the ARP2/3 complex (Orange et al., 2002; Blanchoin et al., 2000; Millard et al., 2004; Uruno et al., 2003). In this study, we further supplement that model with the knowledge that EVL, another protein with actin polymerization capabilities, is also located at the NK-cell–target-cell synapse and contributes to NK cell-mediated cytotoxicity by enhancing adhesion to the target cell, promoting the accumulation of F-actin at the CS and driving F-actin-dependent spreading in response to activating receptor engagement. Moreover, we found that EVL is required for the recruitment of WASP and VASP to the sites of NKG2D/2B4 activation and that the recruitment of EVL to the synapse is dependent on DAP10-mediated signaling, through its interaction with Grb2–VAV1. This leads to a model in which Lck-mediated phosphorylation of DAP10 prompts the association of Grb2 and VAV1, which in turn leads to the recruitment of a complex (or complexes) containing VASP, WASP, DOCK8 and EVL, which ultimately led to F-actin polymerization at the site of activation and enhanced integrin-mediated adhesion (Fig. 8).
Fig. 8.

Model depicting the recruitment of EVL complexes following NKG2D–DAP10 signaling. Binding of NKG2D to its ligand induces Lck-dependent tyrosine (Y85) phosphorylation of the YINM motif within the transmembrane signaling adaptor protein DAP10. Tyrosine phosphorylation leads to the recruitment of the Grb2–VAV1 complex, which facilitates the recruitment of EVL complexes containing VASP, WASP and probably DOCK8, leading to F-actin generation at the CS. F-actin at the synapse drives cell adhesion to target cells, formation of the cytotoxic synapse and, ultimately, NK cell cytotoxicity.
Adhesion between an NK cell and a target cell is dependent on two critical and interdependent systems – namely the polymerization of actin, and the activation and binding of integrins. These two mechanisms generally form a positive-feedback loop, where initial activating receptor signaling will increase actin polymerization (Brown et al., 2012), while also allowing for the activation of integrins such as LFA-1 (Bryceson et al., 2009; Brandsma et al., 2015). This activation allows a conformational change in the integrin to allow tighter binding to its ligand (Bryceson et al., 2009; Brandsma et al., 2015). This increases integrin-mediated signaling and further drives actin polymerization (Mace et al., 2010; Riteau et al., 2003), which in turn allows the rearrangement of activating receptors into microclusters at the synapse (Abeyweera et al., 2011) and preservation of the synapse for increased activating receptor signaling. Given the interdependent nature of this system, determining an exact mechanism of action can be difficult. In this case, EVL appeared to only join activated complexes after activating receptor signaling, and correlated with both more actin-dependent cell spreading and actin accumulation in an NKG2D/2B4 stimulation-dependent manner. The accumulation of EVL downstream of engagement of Flag–CD4–DAP10 also supports this model. Importantly, the recruitment of EVL and F-actin generation appear to be independent of VASP since EVL is still recruited to the site of cellular stimulation and F-actin still accumulates at the cytotoxic synapse after VASP depletion (Wilton and Billadeau, 2018). Given these findings, we believe that EVL has a more-prominent role than VASP in promoting actin polymerization downstream of NKG2D–DAP10-mediated activating receptor signaling in human NK cells.
The mechanisms driving WASP localization in NK cells are understudied. CRKL has been previously shown to be active in adhesion of NK cells to target cells (Nolz et al., 2008; Segovis et al., 2009), as well as in the localization of WASP to the T cell immunological synapse (Sasahara et al., 2002), though its impact on WASP localization in NK cells has not been investigated. In this context, it is interesting to note that both EVL (Lambrechts et al., 2000) and WASP-interacting protein (WIP, also known as WIPF1) (Sasahara et al., 2002) have been shown to bind to the SH3 domains of CRKL, without any requirement for the WIP domains that bind WASP (Ramesh et al., 1997; Ramesh et al., 2014; Volkman et al., 2002; Fried et al., 2014). This leaves interesting possibilities as to the interactions between EVL- and CRKL-mediated WASP localization pathways, and the possible conservation of CRKL-mediated WASP localization in NK cells. Another possible mechanism of recruitment of WASP to the NK CS is via its interaction with the Cdc42 GEF DOCK8 (Ham et al., 2013). Significantly, loss of DOCK8 in NK cells results in defective F-actin generation at the CS and diminished NK cell adhesion to target cells. Similarly, T cells lacking DOCK8 demonstrated defective F-actin generation in response to T cell receptor signaling (Janssen et al., 2016). Interestingly, we recently showed that VASP co-immunoprecipitates with DOCK8 in NK cells (Wilton and Billadeau, 2018). Given that VASP, EVL and WASP similarly co-immunoprecipitate with each other in NK cells (Fig. 6A), and each has been independently shown to also co-immunoprecipitate with DOCK8 (Ham et al., 2013; Wilton and Billadeau, 2018), it is possible that all of these proteins form a single large constitutively formed actin-regulatory complex that allows for rapid and robust actin accumulation to facilitate integrin-mediated adhesion and formation of the cytotoxic synapse.
Ena/VASP family proteins are highly regulated by phosphorylation events, which in some cases appear to determine their location. For instance, phosphorylation of the VASP Ser157 residue is generally found on VASP that is membrane localized – although a specific mechanism is not known (Doppler and Storz, 2013). A synonymous site on EVL, Ser156 may prove to be critical to this pathway (Doppler and Storz, 2013). Clearly, EVL harbors, or is required for the production of, the crucial localization signal that facilitates the movement of the EVL, VASP and WASP complexes to the CS, since VASP is not localized to the site of NK cell activation in the absence of EVL, but EVL still localizes in the absence of VASP. Future studies into phosphorylation events on EVL and VASP may elucidate a more complete mechanism by which they are regulated during NK cell activation.
The interaction and roles of EVL, VASP and WASP in the polymerization of F-actin at the cytotoxic synapse can be inferred. EVL and VASP serve as elongating factors for preformed F-actin filaments (Bachmann et al., 1999; Breitsprecher, 2011). WASP serves as a nucleation-promoting factor for the ARP2/3 complex, which creates new branched F-actin filaments (Derivery and Gautreau, 2010; Luan et al., 2018; Millard et al., 2004; Rodnick-Smith et al., 2016). All three appear to play a role, although only EVL and WASP are needed for adhesion. The role of EVL is further complicated by its role in recruiting the other actin polymerization factors to the synapse. Given that EVL is a stronger nucleation inducer in vitro (Lambrechts et al., 2000), and is regulated differently by phosphorylation (Krause et al., 2003), it is likely that EVL functions as both a recruiter of other actin regulators to the site of activation and a promoter of F-actin-driven cytoskeletal events at the NK cell CS.
Overall, this study has shown that EVL is required for NK cell adhesion, and that EVL contributes to actin polymerization and function at the site of activation. This phenotype is likely dependent on the ability of EVL to drive localization of itself and other actin polymerization factors, including VASP and WASP, to the synapse through its interaction with VAV1.
MATERIALS AND METHODS
Cells, reagents and antibodies
NKL cells (obtained from Dr Michael Robertson, Indiana University Cancer Center, Indianapolis, IN) and KHYG-1 cells (Leibniz Institute DSMZ, Braunschweig, Germany) were maintained in RPMI 1640 (hereafter RPMI; Gibco, Grand Island, NY) with penicillin and streptomycin (Pen-Strep; 10,000 U/ml), 200 mM L-glutamine, 100 mM sodium pyruvate and 0.01 mM MEM non-essential amino acids (all Corning, Manassass, VA) and 10% FBS (Sigma-Aldrich, St. Louis, MO or Atlanta Biologicals, Flowery Branch, GA) supplemented with IL-2 (Peprotech, Rocky Hill, NJ). Target cell lines 721.221, K562 and p815 (American Type Culture Collection, Rockville, MD) were maintained in RPMI with 10% FBS (Sigma-Aldrich, St Louis, MO or Atlanta Biologicals, Flowery Branch, GA) and Pen-Strep (Corning). All cell lines were routinely tested for mycoplasma. Primary NK cells were isolated from blood donor waste products using a modified version of a previously published protocol (Phatarpekar et al., 2016) using the Rosette Separation NK cell isolation kit (Stem Cell Technologies, Cambridge, MA), which has been previously described (Wilton and Billadeau, 2018). In brief, the blood product was diluted 1:1 with sterile PBS and layered over Ficoll–Hypaque (GE Healthcare, Uppsala, Sweden) at room temperature. The layered solution was then spun at 400 g for 30 min with no brake at room temperature. The resulting peripheral blood mononuclear cell (PBMC) layer was removed and washed three times with PBS. A portion of the PBMC sample was set aside for flow analysis and the remaining sample was mixed with 100-fold excess RBCs from a separate donor and centrifuged for 5 min at 400 g. The resulting mixture was then supplemented with 1 µl of NK cell separation antibody mixture per million PBMCs and mixed gently by swirling. The mixture was then allowed to incubate at room temperature for 20 min with intermittent mixing before being layered over Ficoll–Hypaque and spun at 400 g for 30 min with no brake at room temperature. The resulting NK cell layer was then removed and washed three times with PBS. A small sample was then taken for flow cytometry-based purity analysis, and the remaining cells were placed in culture with recombinant human IL-2.
Purified primary NK cells were used without expansion for all immunoblot-based assays, confocal assays and cytotoxicity assays. These cells were cultured with 20% FBS in lieu of the standard 10% FBS in order to increase viability. Some of the primary NK cells used for the conjugate assays were expanded using the previously described K562 cell line genetically modified to express membrane bound IL-21 (Somanchi and Lee, 2016). Antibodies used in this study for immunoblotting, immunofluorescence and cell stimulation are described in detail in Table S1.
Small interfering RNA constructs and nucleofection
KHYG-1, NKL, primary NK cells and expanded primary NK cells were all nucleofected using the standard Lonza protocol. Cells were pelleted at 100 g for 10 min, with no brake and then washed with serum-free RPMI and pelleted at 100 g for 10 min with no brake. Cells were then resuspended in Lonza nucleofection V supplemented with 300 pMol per nucleofection of appropriate siRNA, as indicated in Table S2. The samples were then nucleofected using Amaxa Nucleofector and then placed in serum-free medium for 2 h before being further supplemented with FBS. Cells were later (3–24 h after nucleofection) pelleted at 100 g for 10 min with no brake and resuspended in complete RPMI with recombinant human IL-2. Unexpanded primary NK cells were supplemented with 20% FBS in order to maintain their viability.
CD4–DAP10 chimeric receptors and viral infection
Recombinant vaccinia viruses expressing chimeric Flag–CD4–DAP10 receptors were previously described (Upshaw et al., 2006; Billadeau et al., 2003). KHYG-1 cells were infected at a multiplicity of infection (MOI) of 10:1 in serum-free medium for 2 h at 37°C before being harvested for conjugate formation, slide preparation and microscopy. After viral infection, samples of all groups were taken for CD4 surface analysis by flow cytometry to confirm viral infection.
NK cell stimulations
All KHYG-1, NKL and primary NK cell stimulations were performed in RPMI with 0.5% BSA. Cells were washed twice to remove FBS from the sample, and then incubated with primary stimulating antibodies for 15 min on ice. The cells were pelleted and resuspended in 100 µl of 1:50 goat anti-mouse immunoglobulin G serum (MP Biomedicals, LLC, Solon, OH), vortexed and place in a 37°C water bath for the indicated amount of time. Samples were then quenched with ice-cold PBS, pelleted (∼1000–1500 g for 1 min) and processed for the appropriate experiment. Stimulations were completed using 1 µg/ml mouse anti-NKG2D antibody (cat. MAB139, R&D Systems, Minneapolis, MN), 1 µg/ml mouse anti-2B4 antibody (clone C1.7, from Hybridoma), 1 µg/ml mouse anti-CD16 antibody (clone 3G8, from Hybridoma), 1 µg/ml recombinant human Fc-ICAM (vat. 720-IC, R&D Systems, Minneapolis, MN) or a combination as indicated in the experiment.
Cytotoxicity assays
Cytotoxicity assays were standard chromium release assays. In brief, 721.221 target cells were incubated with Cr51 (Perkin Elmer, Boston, MA) for a minimum of 1 h and then washed to remove excess chromium. Nucleofected KHYG-1, NKL or primary NK cells were plated in a 96-well plate at the indicated dilutions and then incubated with Cr51-labeled 721.221 cells for 3 to 4 h to allow interaction and subsequent cytotoxicity. The plate was spun down and the medium supernatant was removed onto a Luma-Plate 96 (Perkin Elmer, Boston, MA) and allowed to dry overnight before being sealed with Top-Seal A Plus (Perkin Elmer, Boston, MA) and read on a Top Count NXT Microplate Scintillation and Luminescence Counter. All values shown are relative to a maximum lysis of 721.221 cells (MAX), as defined by Cr51 release in 0.5% Triton X-100 (Sigma-Aldrich) lysis solution, and a spontaneous lysis (MIN), as defined by Cr51 release in complete RPMI in the absence of cytotoxic cells. Specific Cr51 release was calculated for each sample by: percentage specific Cr51 release=(experimental value−MIN)/(MAX−MIN)×100.
All experimental groups were tested as in triplicate or quadruplicate replicates for each of a minimum of three independent experiments. Spontaneous and maximum lysis was determined as the mean of 12 replicates for each. Reverse antibody-dependent cytotoxicity experiments were completed in nucleofected primary NK cells with the p815 target cell line. The p815 target cells were coated with mouse anti-CD16 stimulatory antibody (clone 3G8) or a combination of mouse anti-NKG2D (cat. MAB139, R&D Systems, Minneapolis, MN) and mouse anti-2B4 (clone C1.7) stimulatory antibodies.
Conjugate assay
In order to assess the ability of NK cell models to form tight conjugates with the 721.221 target cell line, NKL cell models were first stained with Calcein-AM (Invitrogen, Eugene, OR) for a minimum of 1 h at 37°C with mixing every 15 mins. The target cell line 721.221 was stained with 7-amino-4-chloromethylcoumarin (CMAC, Invitrogen, Eugene, OR) for 30 min in serum-free RPMI at 37°C, then allowed to recover for a minimum of 15 min in RPMI with Pen-Strep and 10% FBS. Calcein-AM-stained NK cell models and CMAC-stained 721.221s were then washed twice with serum-free RPMI and placed on ice for a minimum of 15 min. The Calcein-AM-stained NK models and the CMAC-stained 721.221 were then mixed and centrifuged at 9 g for 5 min, with no brake at 4°C. After centrifugation, the tubes were placed in a 37°C water bath for the indicated amounts of time before being vortexed on maximum for 10 s and then subsequently fixed with the 1:1 addition of 4% paraformaldehyde. Samples were then run on a BD FACSCanto II flow cytometer and analyzed using BD FACSDiva Software (version 6.1.1). All samples were run in triplicate. Gates were defined by single-color-stained controls. Analysis was performed in FlowJo software (TreeStar, Inc Version 8.8.7) by computing the percentage Calcein-AM+ cells that were also CMAC+ over the total number of Calcein-AM+ cells.
Fc-ICAM-binding assay
Fc-ICAM-binding assays were modified from a previously described protocol for T cells (Nolz et al., 2008). Briefly, a 96-well plate was coated overnight at 4°C with the indicated amounts of recombinant human Fc-ICAM (cat. 720-IC, R&D Systems, Minneapolis, MN) in each well. The plate was then washed twice with sterile PBS and blocked with 2.5% BSA in sterile PBS for 2 h at 37°C. Nucleofected NKL cells were stained with Calcein-AM (Invitrogen) for 1 h at 37°C. The cells were then pre-stimulated with anti-NKG2D/2B4 antibodies and placed into the 96-well plate. The entire plate was then placed on ice for 30 min before being placed at 37°C for 15 min. After the incubation at 37°C, the plate was read on a SpectraMax M3 fluorescence plate reader (Molecular Devices, San Jose, CA) with Softmax Pro (Version 7.0.2, Molecular Devices, San Jose, CA) to determine the maximum level of fluorescence. The plate was then read ten additional times, each after a subsequent wash with PBS. All reads were analyzed in reference to the maximum, and screened to determine the indicated wash point that would allow for the detection of different affinities to Fc-ICAM, as indicated by the Fc-ICAM dilution. All groups were tested as a minimum of triplicate repeats in at least three independent experiments.
Immunoprecipitation, cytoskeletal isolation and immunoblotting
All lysis conditions included the addition of protease inhibitors, including 1 mM PMSF, 5 μg/ml aprotinin and 10 μg/ml leupeptin and 1 mM sodium orthovanadate. After lysis, all samples were quantified using a standard Bradford assay and lysed with SDS at 100°C for 15 min. Samples were then separated by acrylamide gel electrophoresis and analyzed via immunoblotting.
Cytoskeletal pellet isolation was completed after cellular stimulation, as defined in the NK cell stimulations methods section. Cell stimulation was terminated by quenching with 1 ml of ice-cold PBS and then spinning at 3824 g for 30 s at 4°C. The pellet was then immediately lysed with 80 mM PIPES pH 6.9 with 1% Triton X-100, 1 mM EGTA, 1 mM MgCl2 and 5 mM NaF with protease inhibitors. After resuspension of the pellet, the sample was immediately centrifuged at 5057 g for 5 min at 4°C. After centrifugation, the supernatant was removed and placed on ice. The pellet was then washed with cold PBS with protease inhibitors and centrifuged at 3824 g for 3 min at 4°C. The pellet was then resuspended in a high-salt buffer containing 10 mM HEPES pH 7.9 with 1 mM EDTA 5 mM NaF, 400 mM NaCl and protease inhibitors, and vortexed using a Thermomixer (Eppendorf, Hauppauge, NY) at 4°C for an hour. The cytoskeletal pellet samples were then centrifuged for 10 min at 16,000 g at 4°C. The resulting supernatant was then removed and all samples were quantified via a Bradford assay, lysed with SDS and further separated by PAGE for immunoblot analysis.
For immunoprecipitation, NKL cells were stimulated as defined in the NK cell stimulations section and then immediately lysed with 1M HEPES (pH 7.2) with 50 mM potassium acetate, 1 mM EDTA, 200 mM D-sorbitol and 0.1% Triton X-100 with protease inhibitors for 30 min on ice. The resulting lysate was then quantified via the Bradford assay and immunoprecipitated using the indicated antibodies, rabbit IgG or mouse IgG (whole molecule) antibodies coated on agarose beads (Sigma-Aldrich) or Protein A–Sepharose Beads (Sigma-Aldrich) for a minimum of an hour at 4°C. The resulting bead-bound protein complexes were washed three times and resuspended in 4× SDS Laemmli sample buffer.
All other immunoblot preparations were completed by washing cells with PBS, followed by lysis in 10 mM Tris-HCl pH 7.4, with 50 mM NaCl, 5 mM EDTA, 50 mM NaF, 30 mM Na4P2O7, and 1% Triton X-100 with protease inhibitors (10 μg/ml leupeptin, EMD Millipore Cat # 18975, and 5 μg/ml apoprotinin, EMD Millipore Cat # 616370) for 30 min at 4°C. The lysates were then centrifuged at 20,817 g for 10 min at 4°C, quantified via Bradford assay and lysed with SDS Laemmli sample buffer.
Confocal microscopy and analysis
Immunofluorescence analysis of NK cell models was completed in three ways. The first involved the conjugation of NKL cell models with 721.221 target cells. In this case, both cell types were washed with serum-free medium and then incubated on ice for a minimum of 15 min. The two populations of cells were then combined and centrifuged at 9 g for 5 min at 4°C with no brake. After centrifugation, the cells were placed at 37°C for 15 min and then transferred to poly-L-lysine (PLL)-coated coverslips.
The second method involved the conjugation of NKL cells to sulfate latex beads (Molecular Probes, Invitrogen, Eugene, OR) coated in antibody, either mouse immunoglobulin G (mIgG), anti-NKG2D/2B4 antibodies, or anti-Flag antibody. Specifically, anti-NKG2D/2B4- and anti-Flag-coated beads were used as a positive stimulus to induce cytoskeletal changes in the NK cell and to monitor recruitment of key proteins or F-actin polymerization at the cell-bead contact site. In contrast, mouse IgG-coated beads were used as a negative control to determine the baseline level of protein recruitment to the cell–bead contact site in a non-stimulatory situation. NKL cells were washed in serum-free medium and then incubated on ice for 15 min. The relevant beads were then added to the tubes, which were centrifuged at 9 g for 5 min at 4°C with no brake. The tubes were then placed in a 37°C water bath for 15 min, and then transferred to PLL-coated coverslips. Coverslips were incubated in a humidification container at 37°C for 15 min.
The third method involves the placement of NK cell models onto antibody-coated coverslips in order to induce actin-dependent NK cell spreading. PLL-coated coverslips were washed with PBS and then incubated overnight with 1 µg anti-CD16 or 1 µg anti-NKG2D and 1 µg anti-2B4 antibodies per coverslip. The coverslips were then washed, and nucleofected NK cells in serum-free medium were placed on the coverslips and allowed to spread for 15 min at 37°C in a humidification container.
The coverslips were then incubated at 37°C, fixed with 4% paraformaldehyde, affixed with 0.15% Surfact-Amps (Thermo Scientific, Rockford, IL) in PBS, washed with PBS, blocked and then stained for the relevant antibodies. Secondary reagents included phalloidin, donkey anti-mouse-IgG and donkey anti-rabbit-IgG conjugated to Alexa Fluor 488, Alexa Fluor 568, Alexa Fluor 647 or Rhodamine (Invitrogen). Coverslips were mounted onto glass slides using SlowFade (Invitrogen, Eugene, OR). Images were taken on a C-Acromat 63×/1.2w DIC water lens equipped on an LSM-710 laser scanning confocal microscope (Carl Zeiss, Thornwood, NY) using the LSM software package (Carl Zeiss, Thornwood, NY).
Image analysis was performed in ImageJ (version 1.50i; NIH) on unadjusted images taken to be in the dynamic range of the staining, which was not always in a range readily analyzed by the human eye. As such, representative images were further adjusted to allow for easier comparison with consistent adjustment made across all experimental groups within an experiment. Conjugate-based immunofluorescence was analyzed by defining the mean fluorescence intensity (MFI) of the contact region divided by the MFI of the total cell. Analysis of cells spread onto coverslips was undertaken by imaging at the cell–coverslip contact point and measuring the two-dimensional surface area that the cell had spread out to, as defined by the outermost staining of phalloidin. In addition, the MFI in this plane was also measured. In cases where co-stain with EVL was possible, any cells in the siEVL groups that did not appear to have decreased expression of EVL were excluded from analyses.
Flow cytometry
Samples were washed and stained in 2% FBS in PBS. Antibodies used for integrin analysis included anti-CD18 PE (eBioscience, Carlsbad, CA and Invitrogen), and anti-activated LFA-1 antibody (clone M24) conjugated to Alexa Fluor 488 or FITC (Biolegend, San Diego, CA). Antibodies against NKG2D and 2B4 were purchased from BD Biosciences (San Jose, CA). Primary NK cell purity analysis was completed using anti-CD4, anti-CD8, anti-CD3 and anti-CD56 antibodies conjugated to FITC, PE, PercP and APC (BD Biosciences, San Jose, CA). All primary NK cell purity analysis was undertaken using a minimum of three colors including antibodies to CD56, CD3 and CD8. Conjugate assay flow analysis did not require any antibody-mediated staining, but instead was completed by using two-color flow cytometry for Calcein-AM and CMAC. All flow cytometry was completed on a BD FACSCanto II flow cytometer using multi-color compensation and appropriate one-color controls. Data was collected using BD FACSDiva Software (version 6.1.1) and analyzed using FlowJo software (TreeStar, Inc., Version 8.8.7).
Statistics and analysis
Analysis and graphing was completed using Prism GraphPad Prism version 5.01 software. Individual experiment analysis was completed using Student's t-test. The analysis of combined experiments used a paired Student's t-test in order to accommodate experimental variation. All confocal analysis was quantified as a meta-analysis and compared using a Student's t-test without pairing.
Supplementary Material
Acknowledgements
The authors would likely to acknowledge the technical help of Kenneth Uy in the preparation of this manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: K.M.W., D.D.B; Methodology: K.M.W., B.L.O, D.D.B. Data collection: K.M.W., B.L.O., D.D.B. Formal analysis: K.W. Data curation: K.M.W., D.D.B.; Writing - original draft: K.M.W.; Writing - review & editing: K.M.W., B.L.O, D.D.B.; Supervision: D.D.B.; Funding acquisition: D.D.B.
Funding
This work was supported by the Mayo Foundation and National Institute of Allergy and Infectious Diseases Grant R01-AI120949 (to D.D.B.). K.M.W. was supported by the National Institutes of General Medical Sciences (grant T32-GM-65841) and the Mayo Clinic College of Medicine's Medical Scientist Training Program. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.230508.supplemental
References
- Abeyweera T. P., Merino E. and Huse M. (2011). Inhibitory signaling blocks activating receptor clustering and induces cytoskeletal retraction in natural killer cells. J. Cell Biol. 192, 675-690. 10.1083/jcb.201009135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann C., Fischer L., Walter U. and Reinhard M. (1999). The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding, and actin bundle formation. J. Biol. Chem. 274, 23549-23557. 10.1074/jbc.274.33.23549 [DOI] [PubMed] [Google Scholar]
- Benz P. M., Blume C., Moebius J., Oschatz C., Schuh K., Sickmann A., Walter U., Feller S. M. and Renne T. (2008). Cytoskeleton assembly at endothelial cell-cell contacts is regulated by alphaII-spectrin-VASP complexes. J. Cell Biol. 180, 205-219. 10.1083/jcb.200709181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billadeau D. D., Upshaw J. L., Schoon R. A., Dick C. J. and Leibson P. J. (2003). NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 4, 557-564. 10.1038/ni929 [DOI] [PubMed] [Google Scholar]
- Blanchoin L., Amann K. J., Higgs H. N., Marchand J. B., Kaiser D. A. and Pollard T. D. (2000). Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature 404, 1007-1011. 10.1038/35010008 [DOI] [PubMed] [Google Scholar]
- Brandsma A. M., Jacobino S. R., Meyer S., Ten Broeke T. and Leusen J. H. (2015). Fc receptor inside-out signaling and possible impact on antibody therapy. Immunol. Rev. 268, 74-87. 10.1111/imr.12332 [DOI] [PubMed] [Google Scholar]
- Breitsprecher D. (2011). Molecular mechanism of Ena/VASP-mediated actin-filament elongation. EMBO J. 30, 456-467. 10.1038/emboj.2010.348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C., Dobbie I. M., Alakoskela J. M., Davis I. and Davis D. M. (2012). Super-resolution imaging of remodeled synaptic actin reveals different synergies between NK cell receptors and integrins. Blood 120, 3729-3740. 10.1182/blood-2012-05-429977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryceson Y. T., Ljunggren H. G. and Long E. O. (2009). Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood 114, 2657-2666. 10.1182/blood-2009-01-201632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler B. and Cooper J. A. (2009). Distinct roles for the actin nucleators Arp2/3 and hDia1 during NK-mediated cytotoxicity. Curr. Biol. 19, 1886-1896. 10.1016/j.cub.2009.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpen O., Virtanen I., Lehto V. P. and Saksela E. (1983). Polarization of NK cell cytoskeleton upon conjugation with sensitive target cells. J. Immunol. 131, 2695-2698. [PubMed] [Google Scholar]
- Deevi R. K., Koney-Dash M., Kissenpfennig A., Johnston J. A., Schuh K., Walter U. and Dib K. (2010). Vasodilator-stimulated phosphoprotein regulates inside-out signaling of beta2 integrins in neutrophils. J. Immunol. 184, 6575-6584. 10.4049/jimmunol.0903910 [DOI] [PubMed] [Google Scholar]
- Derivery E. and Gautreau A. (2010). Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. BioEssays 32, 119-131. 10.1002/bies.200900123 [DOI] [PubMed] [Google Scholar]
- Doppler H. and Storz P. (2013). Regulation of VASP by phosphorylation: consequences for cell migration. Cell Adh. Migr. 7, 482-486. 10.4161/cam.27351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estin M. L., Thompson S. B., Traxinger B., Fisher M. H., Friedman R. S. and Jacobelli J. (2017). Ena/VASP proteins regulate activated T-cell trafficking by promoting diapedesis during transendothelial migration. Proc. Natl. Acad. Sci. USA 114, E2901-E2910. 10.1073/pnas.1701886114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried S., Reicher B., Pauker M. H., Eliyahu S., Matalon O., Noy E., Chill J. and Barda-Saad M. (2014). Triple-color FRET analysis reveals conformational changes in the WIP-WASp actin-regulating complex. Sci. Signal. 7, ra60 10.1126/scisignal.2005198 [DOI] [PubMed] [Google Scholar]
- Ham H., Guerrier S., Kim J., Schoon R. A., Anderson E. L., Hamann M. J., Lou Z. and Billadeau D. D. (2013). Dedicator of cytokinesis 8 interacts with talin and Wiskott-Aldrich syndrome protein to regulate NK cell cytotoxicity. J. Immunol. 190, 3661-3669. 10.4049/jimmunol.1202792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S. C., Cohnen A., Ludwig T. and Watzl C. (2011). 2B4 engagement mediates rapid LFA-1 and actin-dependent NK cell adhesion to tumor cells as measured by single cell force spectroscopy. J. Immunol. 186, 2757-2764. 10.4049/jimmunol.1002867 [DOI] [PubMed] [Google Scholar]
- Janssen E., Tohme M., Hedayat M., Leick M., Kumari S., Ramesh N., Massaad M. J., Ullas S., Azcutia V., Goodnow C. C. et al. (2016). A DOCK8-WIP-WASp complex links T cell receptors to the actin cytoskeleton. J. Clin. Invest. 126, 3837-3851. 10.1172/JCI85774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens K., De Kimpe L., Balsamo M., Vandoninck S., Vandenheede J. R., Gertler F. and Van Lint J. (2009). Characterization of EVL-I as a protein kinase D substrate. Cell. Signal. 21, 282-292. 10.1016/j.cellsig.2008.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M., Dent E. W., Bear J. E., Loureiro J. J. and Gertler F. B. (2003). Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol. 19, 541-564. 10.1146/annurev.cellbio.19.050103.103356 [DOI] [PubMed] [Google Scholar]
- Lambrechts A., Kwiatkowski A. V., Lanier L. M., Bear J. E., Vandekerckhove J., Ampe C. and Gertler F. B. (2000). cAMP-dependent protein kinase phosphorylation of EVL, a Mena/VASP relative, regulates its interaction with actin and SH3 domains. J. Biol. Chem. 275, 36143-36151. 10.1074/jbc.M006274200 [DOI] [PubMed] [Google Scholar]
- Luan Q., Zelter A., Maccoss M. J., Davis T. N. and Nolen B. J. (2018). Identification of Wiskott-Aldrich syndrome protein (WASP) binding sites on the branched actin filament nucleator Arp2/3 complex. Proc. Natl. Acad. Sci. USA 115, E1409-e1418. 10.1073/pnas.1716622115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace E. M., Monkley S. J., Critchley D. R. and Takei F. (2009). A dual role for talin in NK cell cytotoxicity: activation of LFA-1-mediated cell adhesion and polarization of NK cells. J. Immunol. 182, 948-956. 10.4049/jimmunol.182.2.948 [DOI] [PubMed] [Google Scholar]
- Mace E. M., Zhang J., Siminovitch K. A. and Takei F. (2010). Elucidation of the integrin LFA-1-mediated signaling pathway of actin polarization in natural killer cells. Blood 116, 1272-1279. 10.1182/blood-2009-12-261487 [DOI] [PubMed] [Google Scholar]
- Millard T. H., Sharp S. J. and Machesky L. M. (2004). Signalling to actin assembly via the WASP (Wiskott-Aldrich syndrome protein)-family proteins and the Arp2/3 complex. Biochem. J. 380, 1-17. 10.1042/bj20040176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolz J. C., Nacusi L. P., Segovis C. M., Medeiros R. B., Mitchell J. S., Shimizu Y. and Billadeau D. D. (2008). The WAVE2 complex regulates T cell receptor signaling to integrins via Abl- and CrkL-C3G-mediated activation of Rap1. J. Cell Biol. 182, 1231-1244. 10.1083/jcb.200801121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg J., Van Der Krogt G., Twiss F., Bongaarts A., Habani Y., Slotman J. A., Houtsmuller A., Huveneers S. and De Rooij J. (2015). VASP, zyxin and TES are tension-dependent members of Focal Adherens Junctions independent of the alpha-catenin-vinculin module. Sci. Rep. 5, 17225 10.1038/srep17225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange J. S., Ramesh N., Remold-O'donnell E., Sasahara Y., Koopman L., Byrne M., Bonilla F. A., Rosen F. S., Geha R. S. and Strominger J. L. (2002). Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc. Natl. Acad. Sci. USA 99, 11351-11356. 10.1073/pnas.162376099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatarpekar P. V., Lee D. A. and Somanchi S. S. (2016). Electroporation of siRNA to silence gene expression in primary NK cells. Methods Mol. Biol. 1441, 267-276. 10.1007/978-1-4939-3684-7_22 [DOI] [PubMed] [Google Scholar]
- Ramesh N., Anton I. M., Hartwig J. H. and Geha R. S. (1997). WIP, a protein associated with wiskott-aldrich syndrome protein, induces actin polymerization and redistribution in lymphoid cells. Proc. Natl. Acad. Sci. USA 94, 14671-14676. 10.1073/pnas.94.26.14671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh N., Massaad M. J., Kumar L., Koduru S., Sasahara Y., Anton I., Bhasin M., Libermann T. and Geha R. (2014). Binding of the WASP/N-WASP-interacting protein WIP to actin regulates focal adhesion assembly and adhesion. Mol. Cell. Biol. 34, 2600-2610. 10.1128/MCB.00017-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme D. N., Meyer A. S., Barzik M., Keating A. and Gertler F. B. (2015). Selectivity in subunit composition of Ena/VASP tetramers. Biosci. Rep. 35 10.1042/BSR20150149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riteau B., Barber D. F. and Long E. O. (2003). Vav1 phosphorylation is induced by beta2 integrin engagement on natural killer cells upstream of actin cytoskeleton and lipid raft reorganization. J. Exp. Med. 198, 469-474. 10.1084/jem.20021995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnick-Smith M., Luan Q., Liu S.-L. and Nolen B. J. (2016). Role and structural mechanism of WASP-triggered conformational changes in branched actin filament nucleation by Arp2/3 complex. Proc. Natl. Acad. Sci. USA 113, E3834-E3843. 10.1073/pnas.1517798113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasahara Y., Rachid R., Byrne M. J., De La Fuente M. A., Abraham R. T., Ramesh N. and Geha R. S. (2002). Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol. Cell 10, 1269-1281. 10.1016/S1097-2765(02)00728-1 [DOI] [PubMed] [Google Scholar]
- Segovis C. M., Schoon R. A., Dick C. J., Nacusi L. P., Leibson P. J. and Billadeau D. D. (2009). PI3K links NKG2D signaling to a CrkL pathway involved in natural killer cell adhesion, polarity, and granule secretion. J. Immunol. 182, 6933-6942. 10.4049/jimmunol.0803840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanchi S. S. and Lee D. A. (2016). Ex vivo expansion of human NK cells using K562 engineered to express membrane bound IL21. Methods Mol. Biol. 1441, 175-193. 10.1007/978-1-4939-3684-7_15 [DOI] [PubMed] [Google Scholar]
- Upshaw J. L., Arneson L. N., Schoon R. A., Dick C. J., Billadeau D. D. and Leibson P. J. (2006). NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 7, 524-532. 10.1038/ni1325 [DOI] [PubMed] [Google Scholar]
- Uruno T., Liu J., Li Y., Smith N. and Zhan X. (2003). Sequential interaction of actin-related proteins 2 and 3 (Arp2/3) complex with neural Wiscott-Aldrich syndrome protein (N-WASP) and cortactin during branched actin filament network formation. J. Biol. Chem. 278, 26086-26093. 10.1074/jbc.M301997200 [DOI] [PubMed] [Google Scholar]
- Volkman B. F., Prehoda K. E., Scott J. A., Peterson F. C. and Lim W. A. (2002). Structure of the N-WASP EVH1 domain-WIP complex: insight into the molecular basis of Wiskott-Aldrich Syndrome. Cell 111, 565-576. 10.1016/S0092-8674(02)01076-0 [DOI] [PubMed] [Google Scholar]
- Wilton K. M. and Billadeau D. D. (2018). VASP regulates NK cell lytic granule convergence. J. Immunol. 201, 2899-2909. 10.4049/jimmunol.1800254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarcone D., Viale O., Cerruti G., Tenca C., Malorni W., Arancia G., Iosi F., Galandrini R., Velardi A., Moretta A. et al. (1992). Antibodies to adhesion molecules inhibit the lytic function of MHC-unrestricted cytotoxic cells by preventing their activation. Cell. Immunol. 143, 389-404. 10.1016/0008-8749(92)90035-N [DOI] [PubMed] [Google Scholar]
- Zhang M., March M. E., Lane W. S. and Long E. O. (2014). A signaling network stimulated by beta2 integrin promotes the polarization of lytic granules in cytotoxic cells. Sci. Signal. 7, ra96 10.1126/scisignal.2005629 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.