

Abstract
Despite the benefits of chimeric antigen receptor (CAR)-T cell therapies against lymphoid malignancies, responses in solid tumors have been more limited, and off target toxicities more marked. Amongst the possible design limitations of CAR-T for cancer are unwanted tonic signaling and off target activation. Efforts to overcome these hurdles have been blunted by a lack of mechanistic understanding. Here, we show that single cell analysis employing time-course mass cytometry can provide a rapid means of assessing and diagnosing problems in CAR design. We compared signal transduction networks in expanded T-cells with or without expression of second-generation CARs, against fresh and non-expanded T-cells lacking CAR expression. We found that cell expansion enhances stimulatory responses, but also induces tonic signaling and reduces network plasticity, which are associated with expression of T-cell exhaustion markers. As this was most evident in pathways downstream from CD3ζ we performed similar analyses in chimeric costimulatory receptors (CCRs) lacking CD3ζ, expressed in ɣδT cells. ɣδT CCRs avoid tonic signaling but are enabled for full signaling and cytotoxic responses in the presence of both antigen and CCR stimuli in clinically relevant settings. We show how single-cell signaling analysis allows detailed characterization of CAR-T and CCR-T cell behavior, better contextualizing their functional properties. Furthermore we demonstrate that CCR-γδT cells may offer the potential to avoid on-target off-tumor toxicity and allo-reactivity in the context of myeloid malignancies.
One Sentence Summary:
Single cell signaling analysis of chimeric antigen receptor T cells contextualises rational design to overcome limitations of functional exhaustion and off target toxicity.
Introduction
Chimeric antigen receptors (CARs) represent a major technological advance in cancer therapeutics. Expression of CARs redirects effector T cells to become reactive against cell surface antigens in a manner that is independent of the major histocompatibility complex (MHC). This synthetic form of immunotherapy enables effective killing of immunologically “quiet” tumors and has achieved dramatic and encouraging successes against B lymphoid malignancies in particular(1–5). CARs are engineered to include an ectodomain, typically a single chain antibody (scFv), that determines tumor antigen specificity, and endodomains comprising signal transduction motifs from T-cell immunoreceptors. The most popular endodomain configuration employs the CD3ζ chain (which bypasses the MHC requirement in αβT cells, the prevalent subset of T cells) in combination with one or more costimulatory endodomains such as CD28 or 4–1BB. When activated, the CAR provides the equivalent of a T-cell receptor signal via CD3ζ in addition to the costimulation required for T-cell activation. The specific combination of the ectodomain and the costimulatory endodomain(s) included in the CAR influences the phenotype and longevity of the CAR-T product, and relative efficacy has been linked to differing phosphorylation kinetics in downstream signaling molecules(6, 7).
Two major limitations of CARs have emerged, especially as the field progresses from early demonstrations of efficacy in B cell disease to the more challenging solid tumors. Firstly, CARs based on single antibodies cannot discriminate between tumor and healthy cells that express the target antigen, raising the risk of on-target off-tumor toxicity(8–11) Secondly, antigen-independent CAR activation or leakiness, termed tonic signaling, can have deleterious effects on CAR-T cell phenotype and efficacy (12–14). The ability to fine-tune CAR signaling is thus critical to minimize tonic signaling and overcome these limitations. However, there is a paucity of detailed mechanistic analysis of CAR-T signaling, either as cells are generated and expanded ex vivo, or following encounter with tumor. Whilst recent reviews have favored research into novel antigens and combination therapies (15), if the fundamental signaling mechanics and behavior of CARs are not understood, changing antigens by itself may provide little advantage. Bulk phosphoproteomic analyses of CAR-T cell signaling have tried to address this(6, 7), but these lack the resolution to decipher molecular signaling relationships and their function within a cell. Techniques such as mass cytometry measure multiple molecular epitopes at single-cell resolution, and thus have potential to elucidate CAR-T cell signaling dynamics. We therefore sought to compare signaling at single-cell resolution in fresh and ex vivo expanded T-cells, to interrogate the effects of CAR expression on the intra-cellular signaling network, and to learn how this information can be used to guide CAR design.
Here, we present a strategy for using mass cytometry to diagnose problems in CAR design, with a focus on the longstanding challenges of tonic signaling and off target toxicity. We compare signaling networks in fresh αβT cells, cells expanded using CD3 and CD28 stimulatory antibodies, and cells expanded and transduced with second generation CARs containing the CD3ζ and CD28 endodomains. To quantify changes in terms of phospho-protein abundance and codependence in canonical signaling networks, we adapt analytical approaches including the earth mover’s distance (EMD) (16, 17) and density resampled estimate of mutual information (DREMI)(18), which provide a less biased assessment of information transfer between pairs of signaling molecules and enable robust comparisons across conditions. Our analysis pinpoints the source of tonic signaling from a common second generation CAR design, for three different antigen recognition domains, in the T-cell signaling network. We then demonstrate an alternative CAR design which lacks TCR signal transduction elements and provides an AND gate mechanism, and show it avoids on-target off-tumor toxicity when expressed in Vδ2+ γδT cells. For the first time, we show that despite using the same ectodomains as 28ζ CARs, chimeric costimulatory receptors (CCRs) expressed in γδT cells produce limited network perturbation and no tonic signaling. Mechanistic analysis of signaling through synthetic receptors offers a rapid and powerful way to improve engineering and overcome the limitations of CAR therapy.
Results
Stimulation and expansion increase co-dependency in T-cell signaling networks
Intracellular signaling networks involve protein phosphorylation cascades that govern the transmission of information, from the reception of environmental stimuli at the cell surface to changes in the nucleus. To understand this information flow, it is necessary to determine the influence of one network component on another in a data-driven manner. Using mass cytometry, we can quantitatively measure the abundance of multiple proteins in tens of thousands of individual cells in a single experiment. This scale provides statistical power to learn statistical dependencies between the measured phospho-proteins. We consider a network edge to be an interaction between two molecules, whose strength can be quantified from data using statistical dependency between the measured expression levels. However, conventional correlation metrics are biased towards densely sampled cell phenotypes and are thus insensitive to the entire dynamic range. To this end, the density rescaled visualization (DREVI) plot renormalizes the density of cells across the entire dynamic range of expression for any interacting protein pair, and the DREMI score quantifies the degree of influence between these proteins. The DREMI score for an edge (X→Y, with direction assigned a priori) thus indicates the degree of dependence of Y on X (Fig. 1A). DREMI is far more reliable than conventional correlation since it gives equal significance to sparser extremes of marker expression, which often encompass responding phenotypes (Fig 1B).
Figure 1. Codependence analysis of mass cytometry data can be used to interrogate T-cell receptor signaling.

A) To compute a DREMI score between molecules X and Y, joint density is first normalized over values of X (top left, colored by density), generating conditional densities that can be visualized on a DREVI plot (top right, colored by conditional density). The influence of X on Y for each equal bin of X values is then calculated and these are combined (bottom). A high DREMI score indicates that Y expression is highly dependent on X.
B) pSLP-76 and pERK expression in fresh CD8+ T-cells at resting state (US) or after 60 or 360s of CD3 stimulus. Conventinal biaxial scatter plots colored by joint density corresponding DREVI plots colored by conditional density are shown. Spearman correlation between pSLP-76 and pERK and DREMI scores for the pSLP-76→pERK edge are shown below the plots. All Spearman correlations are significant (p < 0.0001). Plots represent 22,000–26,000 cells.
C) DREMI scores and DREVI plots for edges in the canonical CD3 signaling pathway in fresh CD8+ T-cells (n = 4 biological replicates) and CD8+ T-cells expanded for 8 days (n = 3 biological replicates), before and after 360s stimulus with CD3 or CD3+CD28. DREMI scores represent mean ±s.e.m. Baseline scores are significantly higher in expanded cells for CD3→pSLP-76 and pSLP-76→pERK; DREMI scores after CD3 or CD3+CD28 stimulation follow the same pattern. *** indicates p = 0.0002 and **** indicates p<0.0001 by one-way ANOVA with Sidak’s correction.. DREVI plots (right) from representative donors illustrate the changes induced by expansion and stimulus on pSLP-76-pERK codependence.
D) Stimulation of fresh CD8+ T-cells generates a smaller pAKT response than stimulation of expanded CD8+ T-cells. Histograms (left) display results for representative CD8+ cells stained with 152Sm-pAKT. The bar graph (right) provides EMD scores for the full stimulation time course, for 4 fresh and 3 expanded donors, and for all stimuli (CD28, CD3 or CD3+CD28) compared to time- and donor-matched unstimulated controls. **** indicates p<0.0001 by one-way ANOVA with Sidak’s correction.
When CAR-T cells are manufactured, they are typically subjected to ex vivo expansion using a stimulus such as a combination of CD3 and CD28 antibodies. To investigate whether stimulation and expansion alter signaling responses and network states, we used mass cytometry together with DREMI analysis. We measured canonical T-cell signaling phospho-proteins in the PI3K, MAPK/ERK and p38/MAPK pathways(19–21) (fig. S1) of freshly isolated T cells, T cells expanded for 8 days using CD3+CD28 stimulus, and T cells that had been expanded and transduced with a CD19–28ζ CAR. Cells from each group were stimulated by cross-linking αCD3, αCD28, αCD3+αCD28 or αCAR antibodies for 60, 180 or 360s at 37°C before analysis. In mass cytometry data pooled from experimental replicates and different antibody stimuli in CD8+ cells, we observed that, in both freshly isolated and expanded cells, the stimulus led to increased codependence in canonical TCR signaling (CD3→pSLP-76 and pSLP76→pERK edges), indicating that stimulus generated greater information transfer (Fig. 1C). Moreover, expanded CD8+ cells had higher DREMI scores compared to freshly isolated cells, both in resting state and following antibody stimulation. Baseline DREMI scores for expanded CD8+ cells often exceeded the peak post-stimulation DREMI scores of fresh cells (Fig. 1C).
In addition to the insights from DREMI analysis, a population-level estimation of the changes in phospho-protein abundance would give a more comprehensive overview of signaling events. Because of the many conditions, markers and levels of background phospho-protein between donors, it is is unwieldy and impractical to visualize expression differences globally using conventional histograms. We therefore explored succinct techniques for describing differences between stimulated cells and matched unstimulated controls. Mean or median signal intensity, whilst commonly used in flow cytometry, neglects the single-cell resolution of the dataset by collapsing values to averages. Instead, distribution based methods such as the Kolmogorov Smirnov test can be used to assess the differences in marker expression in different conditions. However, KS-test has been heavily criticized in the past for its sensitivity towards outliers(22, 23). To this end, Earth Movers’ Distance (EMD)(16) provides an alternative for robustly determining signal. EMD describes change in signal strength based on difference in probability distribution, with a higher EMD denoting a larger change; it has previously been used with mass cytometry data to describe changes between groups controlling for unequal size between groups(17). The use of EMD to describe changes in signaling expression allows multiple biological replicates to be characterized with a high degree of consistency, without collapsing data to mean or median values. Given that stimulation tends to increase the expression of a marker, a strongly positive EMD score can be reduced by an increase in baseline marker expression, a decrease in expression after stimulation, or both. Similar to the DREMI analysis, we observed difference in EMD between fresh and expanded CD8+ cells, with significantly higher EMD for pAKT (Fig. 1D), pSLP-76 and pERK in expanded cells (fig. S2A). A similar pattern was observed for CD4+ T-cells (fig. S2B). Together, our results show that expanded T cells respond to stimulus with a greater magnitude of phosphorylation and more information flow through T-cell signaling pathways.
Effects of CAR transduction on T-cell signaling
Synthetic signaling molecules such as CARs have not evolved within the natural cellular context, and their introduction could have unexpected effects on the carefully regulated intracellular environment. In pursuit of precise and effective cancer immunotherapy, they allow an entire polyclonal T-cell population to be re-directed against a single target epitope, bypassing MHC restricted epitope-TCR interactions which allow only a minority of T-cells to recognize the tumor. Previous studies have shown that second generation CARs (providing both TCR stimulus and CD28 costimulation) are prone to tonic signaling that can inhibit CAR-T cell function (increased functional exhaustion)(12–14). We therefore investigated the signaling phenotype of expanded T cells transduced with second generation CARs and compared it to that of identically expanded controls that were not transduced.
T cells from healthy donors were expanded for 8 days in the presence of IL-2 using an initial αCD3+αCD28 stimulation. A set of these were transduced on day 3 to express CD19–28ζ (employing FMC63 scFv), a second-generation CAR that has exhibited great clinical efficacy(2–5). Whilst there is wide variation in basal phospho-protein expression between donors, samples transduced with CD19–28ζ consistently demonstrated higher basal phosphorylation of various proteins, including SLP-76, ERK, RelA(p65) and MAPKAPK2 (Fig. 2A). To quantify this basal “leakiness”, we determined EMD scores between matched transduced and untransduced samples for pSLP-76, pERK and pRelA, which we found to be significantly correlated with transduction efficiency, consistent with tonic signaling from the CAR. The strongest correlation was seen in pERK whereas weak correlation was seen with pAKT and pMAPKAPK2 (Fig. 2B). We next compared the EMD scores of CD19–28ζ transduced and untransduced cells for each marker following stimulus with αCD3, αCD28 or αCD3+αCD28 (pooled data from all antibody stimuli). The responses of pSLP-76, pERK, pRelA and pMAPKAPK2 were reduced (lower EMD scores between unstimulated and stimulated) in transduced compared to untransduced cells, as might be expected from the higher basal phosphorylation. This differential responsiveness can be quantified as the difference in slope between the linear regression and the equivalence (y=x) lines in a plot of transduced EMD vs. untransduced EMD, and it is most significant in the canonical TCR signaling pathway containing pSLP-76, pERK and pRelA. There was no difference in pAKT responsiveness, which suggests that the CD3ζ but not the CD28 endodomain predominantly contributes to tonic signaling (Fig. 2C).
Figure 2. CAR expression leads to tonic activation of signaling networks.
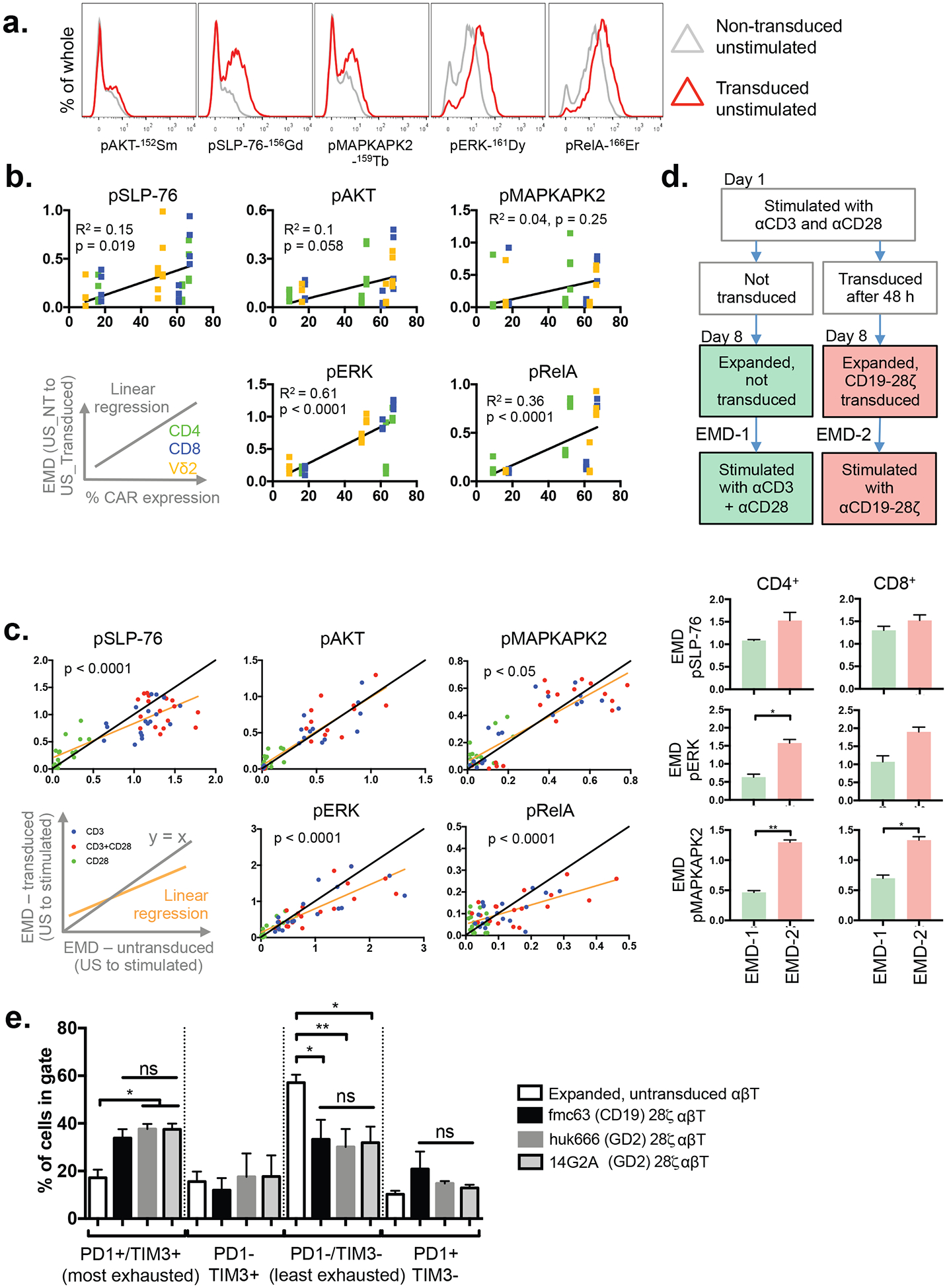
A) Baseline phospho-protein expression in CD8+ T-cells transduced to express the CD19–28ζ CAR compared to untransduced controls. Representative data from one of three independent donors.
B) EMD scores for baseline (unstimulated) phospho-protein expression calculated between T-cells (CD4+, CD8+ or CD4−/CD8−/Vδ2+) transduced with CD19–28ζ and donor-matched untransduced controls from the same T-cell subset, and plotted against transduction efficiency (% of transduced cells expresssing CAR). Each point represents an EMD score for a T-cell subset in an individual donor (n=3). The points are colored to indicate the T-cells subset (CD4: green, CD8: blue, Vδ2: yellow). Pearson R2 values and p-values for the Pearson correlation are provided. US: unstimulated, NT: non-transduced.
C) Comparison of phospho-protein abundance upon CAR transduction. Expanded T-cells transduced with CD19–28ζ and donor-matched expanded, untransduced controls were stimulated by αCD3, αCD28 or αCD3+αCD28 for 60s, 180s or 360s. Donor, stimulus, T-cell subset and time-matched EMD scores for transduced and untransduced samples are plotted against each other. The color of the points indicates the stimulus provided (blue: CD3, red: CD3+CD28, green: CD28). The yellow line shows the linear regression; black line, y = x. p-values indicate significant differences in slope determined by ANOVA. US, unstimulated. Data from 3 independent donors.
D) Signal strengths of responses produced from CD3+CD28 stimulus in untransduced cells and from CD19–28ζ CAR stimulus in donor-matched transduced T-cells. Untransduced cells were used as the comparator in this case because higher baseline phospho-protein levels in transduced cells reduce the responsiveness of CD19–28ζ CAR-T cells to stimulus of their native receptors (C). Values represent mean±SEM of EMD scores, n=3.
E) TIM3 and PD1 exhaustion marker levels in T cells expressing a CD19–28ζ, GD2–28ζ (huk666) or GD2–28ζ (14G2A) CAR. The gating for TIM3/PD1 are shown in Supplementary Figure S11, bars represent mean + SEM for n = 18 for non-transduced and n = 4 for each CAR.
We were interested in the strength of response downstream from a 28ζ CAR compared to that provided by stimulus of the native CD3 and CD28 receptors. We hypothesized that tonic CAR signaling would blunt T cell response because of the increased basal phospho-protein expression. Such effects have previously been demonstrated in fresh cells with different memory phenotypes—Krishnaswamy et al (18) showed that following T cell stimulation, there was lower co-dependency (lower DREMI) in signaling edges in mouse effector memory compared to naïve T cells, in part due to effector memory cells having higher basal phospho-protein expression.
Counter to expectation, the EMD scores for pERK and pMAPKAPK2 following CAR cross-linking were significantly higher than those generated by CD3+CD28 cross-linking in donor-matched untransduced expanded cells (Fig. 2D). This data supports the notion that CD3 and CD28 signaling from a CAR is stronger than from native receptors and suggests that tonic signaling is associated with the very effective signal transduction associated with CARs.
A recent study found CAR tonic signaling to be in part mediated by clustering, attributable to scFv framework interactions(12). One proposed sequela of tonic signaling is T-cell exhaustion, a factor which has been linked to poorer clinical efficacy in CAR-T cell products, and is associated with the expression of markers such as PD-1 and TIM3. Consistent with the recognized effects of continuous stimulus, we found that T-cells expressing any of three second-generation (28ζ) CARs were significantly more likely than untransduced cells to display an exhaustion phenotype of high PD1 and TIM3 expression (Fig. 2E). However, despite containing different scFvs targeting either GD2 (huk666(24) or 14G2A(25) scFvs) or CD19 (FMC63 scFv), there were no significant differences in the exhaustion profiles of the different CAR-T populations, suggesting that the exhausting potential of 28ζ CARs is independent of scFv identity. Given that the CARs in these experiments target antigens not expressed on T-cells, it is highly unlikely that the signaling is due to unexpected CAR ligation.
Sustained non-physiological stimulus hardwires T-cell receptor signaling networks
T cells are exposed to a variety of signaling events as a result of CAR-T engineering; expansion usually involves stimulation using CD3 and CD28 antibodies in the presence of IL-2, and CARs can produce unwanted tonic signals. We investigated whether ex vivo manipulation and CAR expression alter the signaling network itself in the process of human T-cell engineering.
To evaluate network state, which can be visualized using a network map as shown in fig. S3, we examined the variance in DREMI score across multiple stimuli. Since T cells from a single donor are all treated under the same conditions, for each stimulus that is applied, DREMI scores can be generated for edges of interest in each population (CD4+ or CD8+ for instance). The addition of stimulus affects DREMI scores to varying degrees, depending on the baseline state of the signaling edge and its susceptibility to change. For a defined cell population from one donor, variance in DREMI for a particular edge across a panel of stimuli indicates how much the signaling relationship can be influenced by stimulus in general—in other words, the plasticity of the network. Edges with low DREMI variance are not as susceptible to stimulus-induced change as edges with high variance, irrespective of the overall mean DREMI score.
Using a panel of stimulus conditions and unstimulated controls, we observed two broadly different behaviors categorized by signaling edge and T-cell subset. CD4+ cells tended to undergo network state changes in two steps, first upon expansion and again after transduction. In contrast, CD8+ cell networks mainly changed following expansion, with little further change following transduction, a factor particularly noticeable in the early TCR signaling edge CD3→pSLP-76. Fig. 3A illustrates this across all stimuli provided, and Fig. 3B shows representative DREVI plots for one donor’s CD8+ T-cells stimulated by αCD3 cross-linking for 360s. When examining the behavior of all of the measured signaling edges in αβT cells, two distinct patterns of network behavior were observed. Edges in the canonical TCR signaling pathway increased mean DREMI whilst retaining a low variance or even reducing variance following stimulation or transduction (Fig. 3C & E & fig. S4). Edges that signaled via pAKT showed a different behavior, increasing in mean and variance following expansion and transduction (Fig. 3C–D & fig. S4). The differences between CD4+ and CD8+ αβT cells described in Fig. 3A–B remained consistent for other signaling edges, though were most pronounced in the canonical TCR pathway (Fig. 3H, fig. S5 & Supplementary Tables 1–3). The increase in DREMI following CAR transduction but in the absence of further stimulus also generalizes to three second-generation CARs (Fig. 3I & fig. S6A–B). Expression of the exhaustion marker Tim-3 was higher in expanded CD8+ cells than in expanded CD4+ cells (fig. S7A), but these differences were lost when cells were transduced with second generation CARs (fig. S7B).
Figure 3. CAR expression reduces network plasticity in the canonical T-cell signaling pathway.
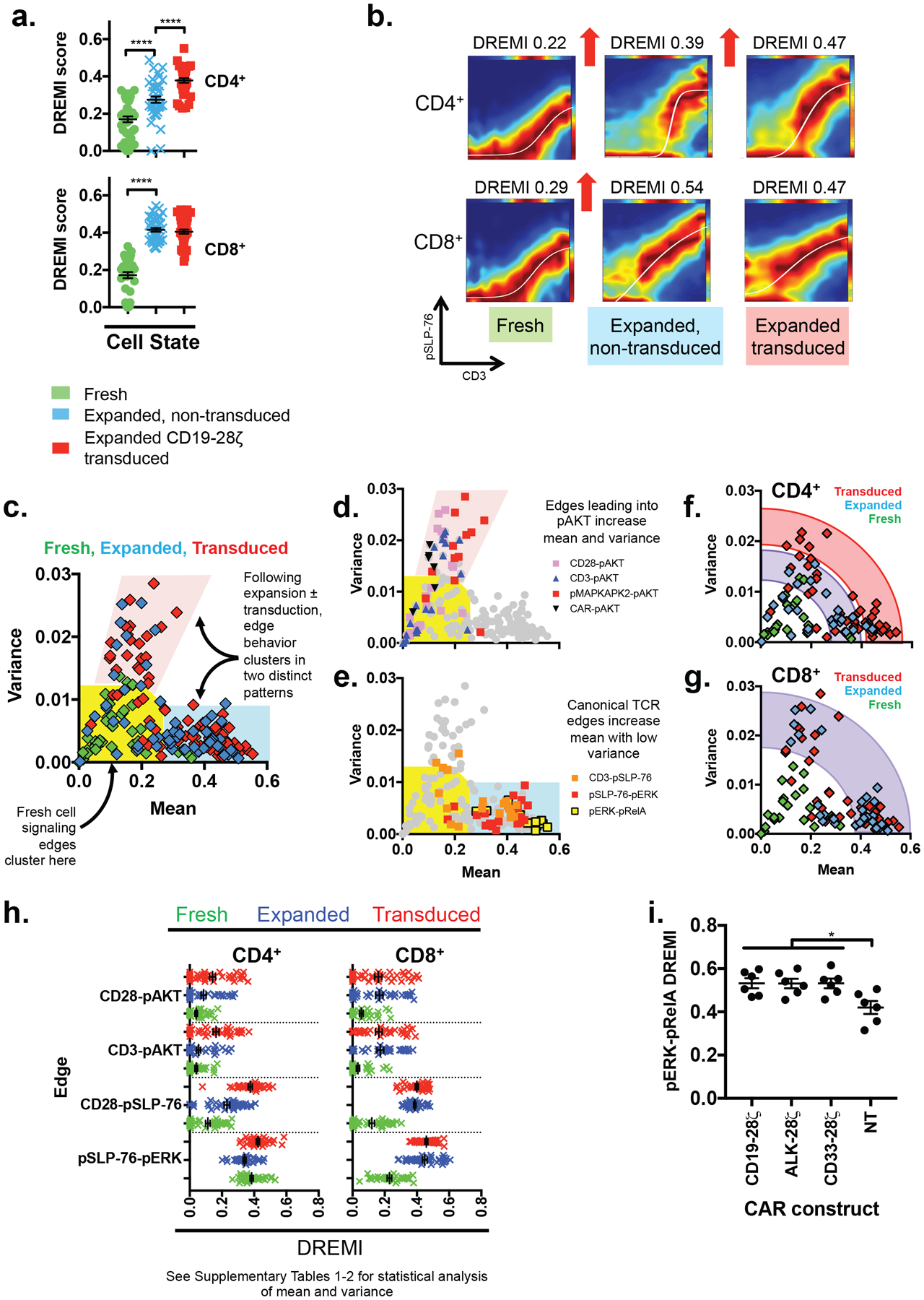
A) DREMI scores for canonical TCR signaling edge CD3→pSLP-76 in T-cells that are fresh, expanded and untransduced, or expanded and transduced with CD19–28ζ across a range of stimuli (αCD3, αCD28, αCD3+αCD28 for 60s, 180s or 360s), or that remain unstimulated for 0 s, 60 s, 180 s or 360 s. n = 3 donors. Each point represents a DREMI score for a single donor, stimulus condition and timepoint. **** indicates p<0.0001 using 1-way ANOVA with Sidak’s correction.
B) DREVI plots and DREMI scores for representative CD4+ and CD8+ T-cells stimulated for 360s with αCD3, corresponding to data in (A).
C) DREMI mean and variance calculated across the full panel of stimuli in (A). Each point represents an edge in one donor, and all measured known edges are shown. Edges from fresh cells (n = 3 donors) cluster near the axis origin (yellow area) whereas edges from expanded or transduced cells (n = 3 donors each) fall into two different clusters (pink and blue areas).
D) Signaling edges in the PI3K pathway and CD3→pAKT highlighted on the plot from (C), showing that both the DREMI mean and variance increase following prolonged stimulus.
E) Canonical TCR signaling edges highlighted on the plot from (C) showing that the DREMI mean increases whilst variance remains low or falls.
F) DREMI mean and variance for signaling edges in CD4+ cells. A full list of measured edges are shown in fig. S1.
G) DREMI mean and variance of CD8+ T-cells. A full list of measured edges are shown in fig. S1
H) DREMI scores for key PI3K and TCR signaling edges in fresh, expanded and transduced CD4+ and CD8+ T-cells from 3 independent donors. Each point represents a replicate of a particular stimulus condition. See Supplementary Tables 1–2 for statistical analysis of these changes.
I) DREMI scores for the pERK→pRelA edge in untransduced T-cells and T-cells expressing three 2nd generation CAR constructs with no further stimulus. CD4+ and CD8+ cells from 3 indpendent donors are plotted as separate points on the same graphs. (p = 0.011–0.012 by 2-way ANOVA with Sidak’s multiple comparison correction)
Taken together, our results show that second-generation CAR expression is associated with increased basal signaling in the canonical TCR pathway, leading to increased ERK and NFκB phosphorylation, greater exhaustion marker expression and higher DREMI scores. The attendant reduction of DREMI variance in canonical TCR pathway edges can be understood as a reduced ability of stimuli to alter information flow or DREMI scores, i.e., a loss of plasticity, or greater “hardwiring” of the network. The higher basal signaling closely resembles that which can be induced in CD8+ cells though prolonged expansion using CD3+CD28, but it affects both CD4+ and CD8+ T-cell subsets equally following CAR expression. High levels of tonic signaling and network hardwiring are associated with a more “exhausted” T-cell phenotype, known to be correlated with poor in vivo efficacy. We hypothesize that CAR technologies that lack tonic signaling would restore network plasticity and relieve T-cell exhaustion.
Boolean logic approach to remove tonic TCR pathway activity
Whilst αβTCRs interact with specific peptide fragments presented on the major histocompatibility complex, γδT cells expressing Vγ9Vδ2 TCR (Vδ2+ cells) recognize tumor cells in an MHC-independent manner. The Vγ9Vδ2 TCR interacts with stress markers that are present on malignant or infected cells but are absent on their untransformed/uninfected counterparts(26–30); it signals in a similar manner to conventional αβTCRs, via associated CD3ζ molecules. We have previously shown that an alternative chimeric costimulatory receptor (CCR) which provides only costimulation and relies on the γδTCR to provide CD3ζ signals, avoids reactivity against cells which do not engage the γδTCR(37). We therefore reasoned that this CCR design would provide beneficial costimulation without causing tonic CD3ζ signaling. Moreover, because T-cells typically require a TCR/CD3ζ signal and a costimulatory signal for full activation, removal of CD3ζ from the synthetic receptor should introduce two requirements for full activation—γδTCR and CCR ligation—forming a Boolean “AND” logic gate which would elicit less on-target off-tumor toxicity (Fig. 4A).
Figure 4. CCRs expressed in Vδ2 γδT cells avoid tonic signaling by acting as an AND gate for tumor antigens.

A) A schematic highlighting that CARs provide all activation signals from one receptor, whereas the CCR requires a CD3ζ signal to be provided from another source, in this case the engagement of target-cell butyrophilin (BTN3A1) by the Vδ2 γδTCR. Tumor cells with a high phosphoantigen burden (due to accumulated isopentenyl pyrophosphate, IPP) have the correct BTN3A1 confirmation for γδTCR engagement, whereas healthy cells do not.
B) Phospho-protein abundance measured using flow cytometry in Vδ2+ cells expressing a CD33-CD28 CCR and stimulated for 60s, 300s or 600s with either αCD3, αCCR or αCD3+αCCR. Values represent mean ± SEM., n = 3 independent donors.
C) TNFα production assessed by intracellular cytokine staining of Vδ2+ γδT cells expressing a CD33-CD28 CCR and co-cultured overnight in the presence of beads coated in αCD3, αCAR or αCD3+αCCR antibody. Error bars, SEM, n = 3 independent donors.
D) Specific cytotoxicity detected using a 51Cr release assay, for Vδ2 cells transduced with GD2-CD28 CCR and co-cultured for 4h with GD2+ neuroblastoma (LAN1) target cells at a range of effector:target ratios. Controls are untransduced Vδ2+ cells from the same donor. Values represent mean ± s.e.m., n = 4 independent donors.
E) EMD scores for phospho-protein expression in expanded Vδ2+ γδT cells following stimulation for 60s, 180s or 360s by cross-linking αCD3, αNKG2D or αCD3+αNKG2D. Values represent mean ± s.e.m., n = 4 independent donors.
F) EMD scores for phospho-protein expression in Vδ2+ cells expanded for the same duration as in (C) but also transduced to express a CD33-DAP10 CCR. Cells were stimulated for 60s, 180s or 360s by cross-linking αCD3, αCCR or αCD3+αCCR. Values represent mean ± SEM., n = 3 independent donors. **** denotes p<0.0001, 2-way ANOVA with Tukey’s correction.
To determine whether a CAR lacking CD3ζ could signal and induce important T-cell effector functions such as TH1 cytokine production and cytotoxicity, we created three CCRs (GD2-CD28, CD33-CD28 and ErbB2-CD28) and expressed them in Vδ2+ cells. Stimulation of both CD3 and CD33-CCR significantly enhanced phosphorylation of signaling proteins compared to CD3 stimulus alone, with ErbB2-CD28 and GD2-CD28 showing similar results (Fig. 4B & fig. S8A). Stimulation of CD33-CD28 CCR did not significantly increase expression of either pAKT or pERK. Addition of CCR stimulus to CD3 stimulus significantly increased production of the TH1 cytokine TNFα, a marker of functional immune activity (Fig. 4C) and the anti-GD2-CD28 CCR provided modest enhancement to killing of GD2+ LAN1 neuroblastoma compared to untransduced controls (Fig. 4D). There was little evidence of tonic signaling from CD28-CCRs—effectively none for pAKT and pRelA—although slight tonic signaling in pZAP70 and pERK was observed (fig. S8B). Next, we proceeded to test whether a CCR format comprising signaling motifs from the NKG2D receptor complex would provide a stronger stimulus to γδT cells.
NKG2D and DAP10-CCR signaling in Vδ2+ γδT cells
NKG2D is an innate immune receptor expressed on a range of effector lymphocytes including Vδ2+ γδT cells, which responds to danger-associated molecular markers such as MICA, MICB and ULBP1–6(31). Its role in γδT cell activation remains a matter of debate; some studies suggest it acts as a costimulatory molecule(32, 33) and others indicate that NKG2D alone is sufficient to activate γδT cells(31, 34). NKG2D does not contain any signaling endodomains, instead forming a complex with the adaptor protein DAP10(35). Engagement of DAP10 reportedly leads to increased PI3K pathway activity(31, 36) and SLP-76 phosphorylation(33). We previously showed that a CCR incorporating DAP10 enodomain provides effective Boolean AND gating in Vδ2+ γδT cells (37) and so wished to determine whether this approach could be extended to the antigen CD33 to avoid off target toxicity in the context of targeting acute myeloid leukemia, and to evaluate the signaling downstream from a DAP10 CCR.
We observed that Vδ2+ γδT cells retain responsiveness to CD3 and NKG2D stimulation following 10 days of expansion using zoledronate and IL-2. As expected, CD3 cross-linking led to increased phosphorylation of pZAP70, pSLP-76, pAKT, pMAPKAPK2, pERK and pS6 whereas NKG2D cross-linking only increased AKT phosphorylation, consistent with PI3K pathway activity (Fig. 4E). NKG2D stimulus provided no further increases in signal strength when combined with CD3 stimulus.
Cross-linking of CD33-DAP10-CCR had minimal effect on the proximal mediators of T-cell receptor signaling, ZAP70 and SLP-76, but induced strong responses in pMAPKAPK2, pAKT and pERK (Fig. 4F). DAP10-CCR induced pAKT more robustly than NKG2D stimulus, led to faster pMAPKAPK2 response than CD3 stimulus alone, and produced significantly larger effects on pMAPKAPK2 and pERK when combined with CD3 than either stimulus alone.
DAP10 CCR does not reduce network plasticity and synergizes with CD3 when both are stimulated
CD33-DAP10+ γδT cells upregulated TNFα production in response to CCR stimulus, though interestingly little response was seen to CD3 stimulus, perhaps suggesting that different innate receptors are responsible for the production of different TH1 cytokines (fig. S9). Comparison of EMD scores between untransduced Vδ2 and CD33-DAP10 or CD33–28ζ transduced Vδ2 in the absence of further stimulus showed significantly higher EMD in pSLP-76 and pERK in CD33–28ζ cells, consistent with tonic signaling arising from CD3ζ (Fig. 5A).
Figure 5. DAP10-CCR does not reduce network plasticity and synergizes with CD3 when stimulated.
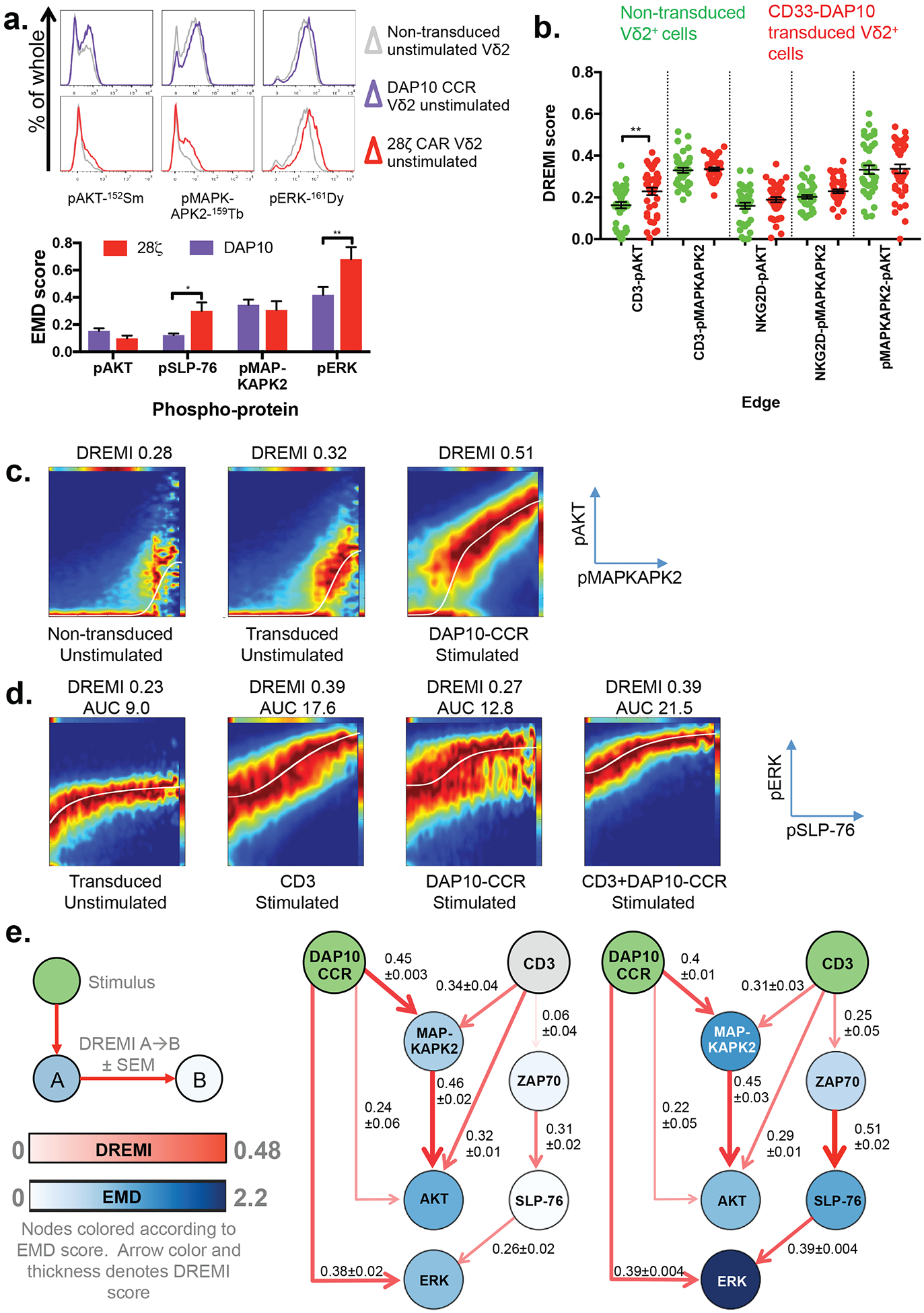
A) Basal phospho-protein expression in expanded, untransduced Vδ2+ cells and Vδ2+ cells expanded and transduced to express a CD33-DAP10 CCR or CD33–28ζ CAR. Representative histograms of one of three independent donors showing signaling markers known be activated through the DAP10 CCR. EMD scores between expanded non-transduced and expanded, transduced Vδ2 expressing either CD33-DAP10 (purple) or CD33–28ζ (n = 3 independent donors) are also shown.
B) DREMI scores for pAKT and pMAPKAPK2 signaling edges in expanded, CD33-DAP10-CCR transduced Vδ2+ cells and expanded untransduced controls, across a range of stimuli applied for 60s, 180s or 360s. Untransduced cells were stimulated with αCD3, αNKG2D or αCD3+αNKG2D whereas CD33-DAP10 transduced cells received αCCR in place of αNKG2D stimulus. Each point represents DREMI score for a single stimulus condition and time point from one of 3 donors. Comparison between transduced and untransduced was done using one-way ANOVA with Sidak’s correction for multiple comparisons.
C) Representative DREVI plots for DAP10-CCR transduced and untransduced Vδ2+ cells for the pMAKPAPK2→pAKT signaling edge.
D) Representative DREVI plots for the pSLP-76→pERK signaling edge in DAP10-CCR transduced Vδ2+ cells.
E) Network representation of signaling in DAP10-CCR transduced Vδ2+ cells stimulated for 360s with either DAP10-CCR cross-linking or DAP10-CCR+CD3 cross-linking. Green, stimulus inputs. Grey, non-stimulated receptors. Other nodes are colored according to EMD score (compared to time and donor matched unstimulated controls, n=3). Connector color and thickness is determined by the mean DREMI score across 3 independent donors.
To look for more subtle evidence of tonic activity, we used DREMI analysis of a CD33-DAP10 CAR and evaluated all pAKT and pMAPKAPK2 signaling edges, as these were strongly influenced by DAP10-CCR cross-linking. Using DREMI we were also able to examine any simultaneous rises in pAKT and pMAPKAPK2. No edges apart from CD3→pAKT demonstrated changes in mean DREMI across a range of donor and time-matched stimulus conditions (Fig. 5B & fig. S10). In contrast, stimulation of the CCR led to a substantial rise in pMAPKAPK2-pAKT DREMI score (Fig. 5C). Hence, whilst free of tonic signaling, the DAP10-CCR, when activated, is capable of rapidly (Fig. 4F) triggering a downstream signaling response.
To detect the influence of more than one signaling molecule on the activation of a downstream target, it is possible to analyze the area under a curve (AUC) fitted to the points of maximum density of a DREVI plot. This may be more useful than DREMI scores at uncovering additive or synergistic effects, since AUC accounts for different absolute expression levels represented in the maximum density curve (i.e., higher Y-axis intercept) that can result from independent signaling inputs. For example, although DAP10 does not influence pSLP-76 (Fig. 4F) and pSLP-76-pERK DREMI score remains low following DAP10 CCR stimulus, the pSLP-76-pERK AUC rises compared to unstimulated as a result of pSLP-76 independent ERK phosphorylation (Fig. 5D). When both CD3 and the DAP10 CCR are engaged, the AUC reaches maximal levels as both signals contribute to pERK expression.
Considering these combinatorial signals across the network rapidly becomes complex and difficult to visualize without a means of showing codependence and changes in phosphoprotein expression simultaneously. To gain a network-level understanding of the combinatorial effects of DAP10-CCR and CD3 stimuli, we built upon the “network DREMI” visualization(18). By using the post-stimulation EMD score to color nodes in the network and DREMI to define the color and thickness of connectors, we were able to intuitively visualize and summarize changes in codependency in addition to the resulting signals (Fig. 5E).
A range of different CCRs enhance effector function without on-target off-tumor toxicity
We were interested in whether the AND gate demonstrated for the CD33-DAP10 CCR generalizes to other CCRs, to strengthen the aim of overcoming conventional CAR exhaustion and off-tumor toxicity effects. In Vδ2+ γδT cells, expression of a second-generation CD19 CAR, but not a CD19-DAP10 CCR, was associated with significant increase in exhaustion markers compared with untransduced controls (Fig. 6A). The avoidance of exhaustion was not specific to the CD19 scFv and DAP10 endodomain; the same pattern was observed with a GD2 scFv and CD28 CCR with longer-term culture (Fig. 6B) and with a CD33-DAP10 CAR (Fig. 6C).
Figure 6. CCR engineering provides a general strategy for avoiding on-target off-tumor toxicity.
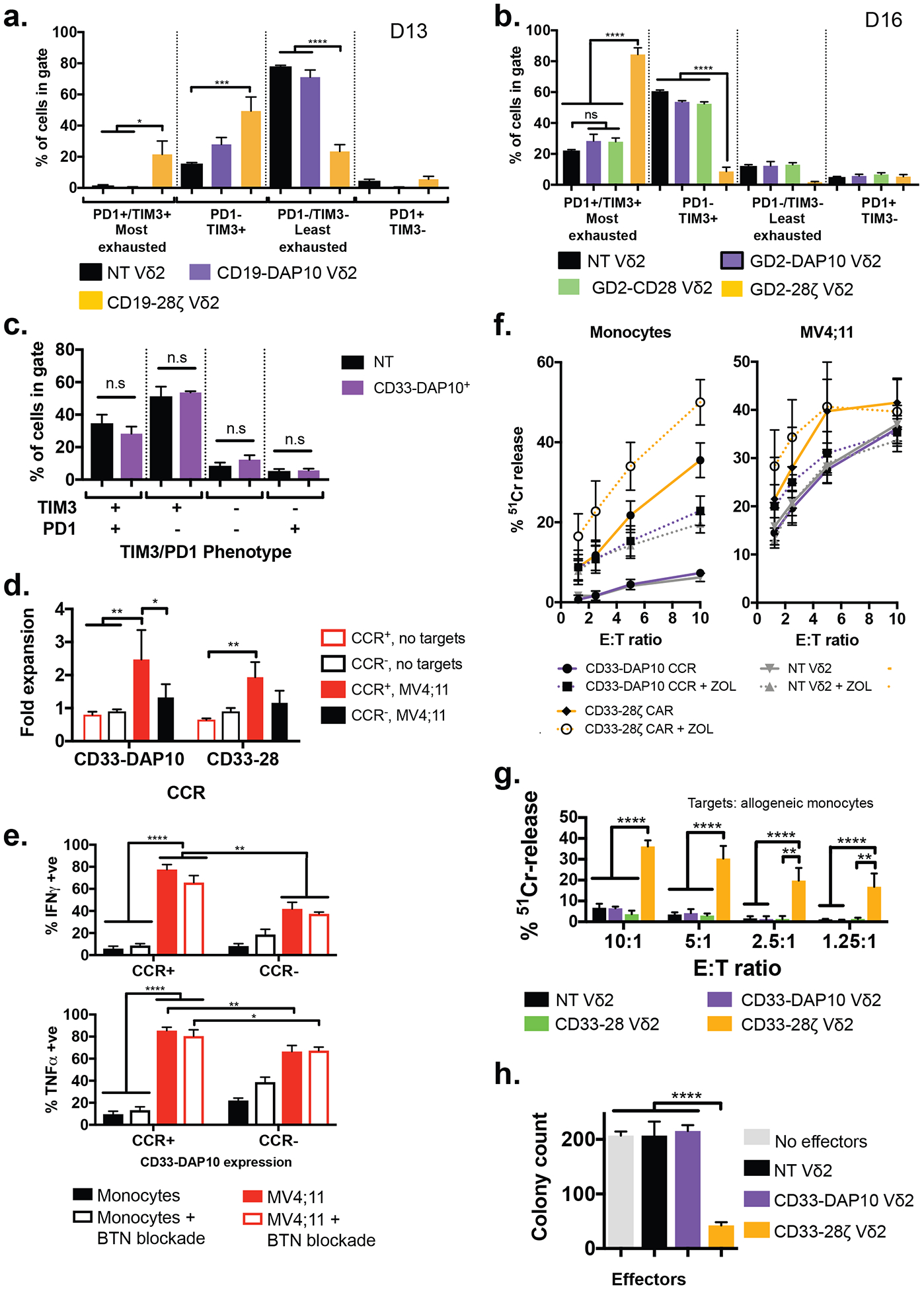
A) Exhaustion status as defined by TIM3 and PD1 expression of Vδ2+ cells expressing a CD19–28ζ CAR or a CD19-DAP10 CCR (identical aside from the endodomain configuration), compared to donor-matched untransduced cells (n = 3 donors, values mean ± SEM). Cells were analyzed 13 days after initial stimulation, 8 days post transduction. * p <0.033, ** p <0.0021, *** p<0.0002, **** p<0.0001 by one-way ANOVA with Sidak’s correction. Example gating is shown in fig. S15.
B) Exhaustion status defined as in (A) for Vδ2+ cells expressing either the GD2-CD28-CCR (n = 3), GD2-DAP10-CCR (n = 3, data from Fisher et al(37)) or GD2–28ζ CAR (n = 3), compared to untransduced controls (n = 18) cultured for 16 days following initial stimulation (11 days post transduction). * p <0.033, ** p <0.0021, *** p<0.0002, **** p<0.0001 by one-way ANOVA with Sidak’s correction.
C) Exhaustion status for CD33-DAP10 Vδ2+ cells as in (A) shows no significant difference to that of untransduced Vδ2+ cells after 16 days of culture (11 days post transduction, n = 3, values represent mean ± SEM compared by one-way ANOVA with Sidak’s correction)
D) Fold expansion of Vδ2+ cells ± CD33-DAP10 or CD33-CD28 CCRs in the presence of irradiated MV4;11 AML cells. MV4;11 induced significant proliferation only in CCR+ cells (p = 0.0025 and p = 0.0097, respectively). CD33-DAP10 expression confers significant (p = 0.016) AML-induced proliferative benefit over untransduced controls. (2-way ANOVA with Tukey’s multiple comparisons correction, bars show mean ±SEM of 3 independent donors).
E) Production of IFNγ and TNFα by DAP10-CCR+/− Vδ2+ γδT cells co-cultured overnight with either MV4;11 or allogeneic monocytes at a 1:1 effector:target ratio, in the presence or absence of butyrophillin 3A1 blockade. When in contact with MV4;11, CCR+ Vδ2 express significantly more of both cytokines (p = 0.008 for TNFα and 0.002 for IFNγ). Cytokine production by DAP10-CCR+ cells is significantly higher when in contact with MV4;11 than it is when in contact with monocytes (p<0.0001). Blockade of butyrophillin did not affect cytokine production. Bars are mean ± SEM of 3 independent donors and significance determined using 2-way ANOVA with Sidak’s correction.
F) 4-hour 51Cr release assay detecting killing of allogeneic monocytes or MV4;11 by non-transduced Vδ2 γδT cells or those expressing either CD33-DAP10 CCR or CD33–28ζ CAR, in the presence or absence of zoledronic acid. Zoledronic acid treatment enhanced killing of monocytes by NT Vδ2 (p = 0.01) or CD33-DAP10 Vδ2 (p=0.0005) but not of MV4;11. CD33-DAP10 did not enhance cytotoxicity against either target, and CD33–28ζ significantly enhanced killing of monocytes (p<0.0001) but not MV4;11. Data shows mean ± SEM of 3–10 independent donors, analysis by 2-way ANOVA with Sidak’s correction.
G) CD33-DAP10 (n=6) or CD33-CD28 (n=3) Vδ2+ cells do not kill healthy allogeneic monocytes and more than untransduced Vδ2 (n = 9), unlike CD33–28ζ CAR-transduced Vδ2+ cells (n=6) (comparisions by 2-way ANOVA with Tukey’s multiple comparison correction). Data is from 4h 51Cr release assays at the range of effector:target (E:T) ratios shown.
H) Myeloid colony formation assay for Vδ2+ cells expressing either CD33–28ζ or CD33-DAP10 and co-cultured overnight with healthy bone marrow. Only CD33–28ζ led to significant reduction in myeloid colony formation, demonstrating the lack of CD33-DAP10 Vδ2+ cell toxicity against healthy myeloid progenitors. Results for 3 independent γδT cell donors, mean ± SEM is shown, compared by one-way ANOVA with Sidak’s correction, ****p<0.0001.
We previously published evidence suggesting that a GD2-DAP10 CCR could enhance antigen- and tumor-specific immune responses(37). Because of the particular challenge of on-target off-tumor toxicity in targeting myeloid malignancies, we investigated whether this approach was more broadly applicable by studying key effector functions such as proliferation, TH1 cytokine production and cytotoxicity in the context of anti-CD33 CCRs. Using a multiplexed assay of protein tyrosine kinase activity, we confirmed that the AML cell line (MV4;11) produced activation of the TCR signaling pathway in Vδ2+ γδT cells. Upon comparing Vδ2+ cells co-cultured with MV4;11 with target-free Vδ2, the most specifically upregulated kinases were Syk, Abl and ZAP70, all canonical markers of TCR signaling(38) (fig. S11). We found that the presence of irradiated CD33+ MV4;11 AML cells induced significant proliferation in both CD33-DAP10 and CD33-CD28 CCR Vδ2 T-cells compared to target-free controls. Presence of the DAP10-CCR enhanced MV;411 induced proliferation above that of untransduced controls (Fig. 6D). Upon overnight co-culture with MV4;11 AML cells, CD33-DAP10 CCR+ Vδ2 were significantly more likely to stain positively for TNFα and IFNγ than CD33-DAP10-CCR− cells or than either group of γδT cells co-cultured with allogeneic monocytes (Fig. 6E & fig. S12A). The enhanced effector function against MV4;11 was not blocked by the presence of the butyrophillin antibody 103.2, suggesting it is independent of TCR engagement (26) (see Fig. 4A) and whilst a detailed analysis of the interaction between γδT cells and AML is beyond the scope of this work, experiments using a panel of blocking antibodies suggest that both the γδTCR, NKG2D and DNAM-1 are involved (fig. S12B), results consistent with previously published studies on the interaction between γδT cells and AML(39). CD33-DAP10 Vδ2 showed minimal cytotoxicity against allogeneic monocytes or myeloid progenitors whereas CD33–28ζ Vδ2 were highly cytotoxic to myeloid cells in both short and long term co-culture (Fig. 6F–H), and killing was increased by pre-treating monocytes with zoledronic acid (Fig. 6F). Interestingly, the baseline cytotoxicity of Vδ2 cells against the AML cell line MV4;11 was high but resistant to further enhancement using either CD33–28ζ, zoledronic acid treatment or CD33-DAP10 (Fig. 6F). Given the multiplem pathways involved in γδT cell recognition of AML alluded to in fig. S11B, this perhaps implies a maximum level of cytotoxicity has already been reached in these short term assays, whilst other effector functions such as proliferation and cytokine production can still be enhanced. For example, the pre-existance of NKG2D ligands on the surface of AML cells may already be producing efficient NKG2D signaling which is not further enhanced by the CCR. However, the inability of CCRs to enhance cytotoxicity has been previously described in studies of costimulation in trans (40, 41) and does not necessarily limit their utility.
Of significance for the cellular therapy field, even in the allogeneic setting, there is minimal off-tumor toxicity. This approach therefore both addresses the problem of on-target off-tumor toxicity and brings the field closer to developing a cellular therapy with limited allo-reactivity. Taken together, an empirical approach to signaling based on single cell analysis has been used to identify the signal aberrations related to tonic signaling and has led directly to fine tuning of CAR design.
Discussion
The remarkable success of second generation CAR-T cells in the treatment of lymphoid malignancies(1–5) has been frustrated in other settings. Targeting of myeloid malignancies has been complicated by a lack of AML-specific antigens(42), such that patients receiving CAR-T cell therapy for AML typically require rescue by hematopoietic stem cell transplantation. In solid tumors, the CAR-T experience has been punctuated by failure of T-cell expansion and significant off-tumor toxicity(8, 10).
Functional assays dominate the CAR-T cell literature, and while many studies assess whether similar CARs will target new antigens, very few have dissected the mechanism of existing designs. Quantitative, reproducible and detailed signaling analysis is needed to inform future CAR designs. This will help address mechanistic problems such as target-autonomous CAR signaling, which have been linked to T-cell exhaustion and poor efficacy(12, 43). Our analysis of CAR-T signaling allowed the first detailed description of a CAR-T signaling response within a population based on single-cell analysis. We also examined the effects of changing the cellular “chassis” within which CARs are expressed—in γδT cells, CCRs eliminated the tonic signaling associated with conventional costimulatory domains by creating an AND gate that is only capable of full signal response upon TCR engagement. This example of signaling-data-driven design to avoid off-tumor toxicity may prove a paradigm in CAR construction.
CAR-T engineering produces a number of artificial signaling events. Expansion using CD3+CD28 antibodies gives sustained stimulus, and introduction of a 28ζ CAR at day 3 provides continuous and autonomous CD3ζ signals, allowing dissection of brief, sustained and tonic stimulation. The introduction of DREMI as a meaure of codependence between phosphoproteins adds an additional dimension to the characterization of cellular response, beyond changes in phosphoprotein abundance.
DREMI is a surrogate for information flow between signaling nodes, and in fresh cells, brief stimulus elicits increases in phosphoprotein expression (quantified by EMD) as well as in DREMI score. Expanded cells, having already received a sustained stimulus, had higher baseline DREMI scores in the canonical TCR pathway than fresh cells, and further stimulus with CD3, or CD3 and CD28, was less able to alter DREMI, resulting in reduced DREMI variance, in other words reduced plasticity of established signaling relationships. 28ζ CAR-T cells had the highest DREMI scores, indicating a large amount of signal flow that resists augmentation using further stimulus, as indicated by the low DREMI variance. PI3K pathway edges did not show this reduction in plasticity, retaining the potential to be altered by further stimulation (Fig. 3D) and consistent with tonic signaling being restricted to the CD3ζ endodomain (Fig. 2C). These findings support a model whereby continuous signal flow “erodes” highly co-dependent but rigid connections between signaling nodes. Thus, signaling edges in the canonical TCR pathway increase mean DREMI and reduce DREMI variance as stimulus progresses from brief to sustained to tonic, and restimulation of expanded or transduced cells cannot further increase DREMI (aka transmit information), despite eliciting protein phosphorylation.
We observed additional differences in network hardwiring in another context. Expanded CD8+ cells, which exhibit less plastic canonical TCR edges than expanded CD4+ cells, also express TIM-3 at higher levels. Whether a direct link exists between T-cell functional exhaustion and the “hardening” of network connections (as observed by reduced DREMI variance) remains to be seen. In support of this possibility, 28ζ CAR-expressing cells have the highest DREMI means with lowest variance in canonical TCR pathway edges, and are also much more likely to have an “exhausted” TIM3+/PD1+ phenotype than expanded untransduced controls (Fig. 2E). Due to tonic signaling, 28ζ CAR transduced cells have higher baseline phosphoprotein expression in the TCR signaling pathway (pSLP-26, pERK and pRelA, Fig. 2C) with better-connected nodes (higher DREMI) at baseline. Chronically higher MEK/ERK pathway activity and chronic IKK activation (with resultant NFκB p65 RelA phosphorylation) are both associated with T-cell exhaustion, to the extent that MEK inhibitors will reverse tumor-induced CD8+ T-cell dysfunction(44, 45). Together, these observations suggest that a rigid signaling network, with reduced capacity to remodel in response to stimulus, may lead to reduced phenotypic responses and a functionally “exhausted” phenotype. The functional in vivo correlates of this network rigidity are impractical to determine due to the large number of cells required for CyTOF analysis, and short-term cytotoxicity assays are known to be poor indicators of long-term CAR-T cell health. However, there is substantial pre-existing evidence that tonic signaling, now demonstrated to be associated with reduced network plasticity and increased exhaustion marker expression, leads to reduced longevity and proliferative capacity of CAR-T cells(12, 46). The emerging constellation of characteristics displayed by tonically signaling CAR-T cells: increased exhaustion marker expression, reduced in vivo longevity and now loss of network plasticity is tantalizing, and hints at network plasticity being a marker of cellular health after expansion and transduction. A broader screen of other CAR constructs and endodomain combinations would provide more information to validate this association.
Our analysis of tonic signaling from CAR-T cells with conventional signaling endodomains led us to examine alternative CAR designs. Excess and redundant CD3ζ signals from 28ζ CARs are an established phenomenon; removal of 1/3 or 2/3 CD3ζ ITAMs leads to enhanced in vivo persistence of CAR-T cells without reducing efficacy(46). Our alternative approach of removing the synthetic CD3ζ signaling motif was made possible by using γδT as a cellular chassis. Whilst often overlooked, tumor-infiltrating γδT cells are the strongest predictor of positive outcome in solid tumors(47). These cells readily accept both conventional CARs and CCRs(37, 48–51) without loss of innate function or tumor tropism. The MHC-unrestricted promiscuity of the γδTCR gives them a key advantage over αβT cells, insomuch as the native γδTCR can be used to provide a CD3ζ signal, removing the requirement to include it in a synthetic sonstruct. By not relying on a CAR to deliver a CD3ζ signal, we abrogate tonic signaling; at the same time, we harness the ability of γδT cells to detect cancer-associated danger signals for perceiving whether a target cell is “healthy” or “tumor”. Our DAP10-CCR design was informed by the important role of NKG2D in γδT cell activation(33) and its susceptibility to tumor immune escape mechanisms(52, 53). Perhaps due to differences in intracellular packing or expression levels between NKG2D/DAP10 complexes and DAP10-CCRs, stimulation of the synthetic receptor generates more potent signals and activates p38 MAPK and MEK pathways which were not activated by NKG2D stimulus. Importantly, this activation is only observed when the CCR is stimulated (Fig. 5), preserving the plasticity of signaling and having no effect on γδT cell exhaustion profiles (Fig. 6A–C).
Cancer immunotherapeutic strategies that redirect the immune system against a tumor-associated antigen must strike a balance between efficacy and toxicity. In CAR-T trials targeting lymphoid malignancies, depletion of healthy CD19+ cells is a recognized but tolerable side effect. Ablation of healthy myeloid cells by T cells expressing CD33–28ζ CARs (Figs. 6F–H) could be profoundly toxic if the CAR-T cells persisted in a patient. Inter-species differences between mouse and human γδT cell biology and immunological niche preclude modeling this in murine model without introducing problematic confounders such as weekly zoledronic acid treatments (54, 55) which sensitize healthy myeloid cells to γδT cell cytotoxicity. Whilst a full analysis of efficacy is beyond the scope of this paper, we have demonstrated that CD33-DAP10 enhances the function of Vδ2+ γδT cells, conferring significant proliferative advantage over untransduced cells upon encountering AML (Fig. 6D). CD33-DAP10 Vδ2 cells are also capable of killing AML cells without demonstrating toxicity against allogeneic monocytes or myeloid progenitors (Figs. 6F–H).
In summary, we have shown that mass cytometry of native and synthetic T-cell signaling in combination with DREMI and EMD analysis provides a rapid means of assessing the mechanisms of novel CAR or CCR constructs. This analysis allowed us to reveal important differences between fresh and expanded T-cells and isolate the source of tonic signaling to inform improved CAR designs. CCR-expressing γδT cells may provide a means of improving the safety of cellular immunotherapy by avoiding on-target off-tumor toxicity. Furthermore, they may allow targeting of antigens which to date have been off-limits due to toxicity concerns. Clearly, more work is needed to determine whether these cells are able to be used as a viable cellular therapy chassis, but studies using unmodified γδT cell infusions have been promising(56, 57), and we look forward to seeing early phase clinical studies of this technology.
Materials and methods
Isolation and pre-stimulation handling of fresh PBMC
20 ml of whole blood were diluted with 10 ml PBS + 500 μl 100 mM EDTA and layered on 20 ml Percoll. Interface PBMCs (20 min, 300xg, RT) were washed in PBS and re-suspended in 25 ml T-cell medium (X-VIVO 15 (Lonza BioWhittaker, Maryland, USA) supplemented with penicillin/streptomycin (100 IU/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich, Missouri, USA)) and cultured overnight before use.
T-cell expansion
PBMC were isolated as described above. Cells were cultured in RPMI-1640 medium supplemented with L-glutamine (2 mM, Sigma-Aldrich), penicillin/streptomycin (100 IU/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich)) and 10% FCS (v/v, (Gibco, Massachusetts, USA)). T-cell expansion was induced by addition of anti-CD3 (OKT3) and anti-CD28 (clone CD28.2). 100 IU/ml IL-2 (Aldesleukin, Novartis, Frimley, UK) was added to PBMC suspension after PBMC isolation (day 1) and was replenished every 2–3 days by removing half of the media from the well and replacing with fresh media containing 200 IU/ml IL-2.
Vδ2+ T-cell expansion
For specific Vδ2+ γδ T-cell expansion, PBMC were isolated as described above. They were cultured in RPMI-1640 medium supplemented with L-glutamine (2 mM, Sigma-Aldrich), penicillin/streptomycin (100 IU/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich)) and 10% FCS (v/v, (Gibco, Massachusetts, USA)). Vδ2+ γδT cell expansion was stimulated using 5 μM zoledronic acid (Actavis, New Jersey, USA) and 100 IU/ml IL-2 (Aldesleukin, Novartis, Frimley, UK), which was added to PBMC suspension after PBMC isolation (day 1). IL-2 was replenished every 2–3 days by removing half of the media from the well and replacing with fresh media containing 200 IU/ml IL-2.
Construction of retroviral constructs
The gammaretroviral vector used in all constructs was SFG (58), pseudotyped with an RD114 envelope. DNA fragments were amplified using the Phusion HT II polymerase according to the manufacturer’s instructions (Thermo Scientific, Massachusetts, USA). PCR was carried out in a PTC-200 DNA Engine (MJ Research, Massachusetts, USA). PCR products were extracted from 1% agarose gels using the Wizard SV Gel & PCR Clean-Up kit (Promega, Wisconsin, USA). Sample concentrations were determined using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Massachusetts, USA). The CAR ectodomain comprised one of a range of scFvs against a panel of targets including human GD2 (clone HUK666 or 14G2A), human CD33(clone 113), human ErbB2 (clone 4D5) or human CD19 (clone FMC63) and a spacer derived from the human IgG4 CH2-CH3 portion which has been described before in third generation format(24).
The endodomains were generated using oligo-assembly PCR based on a codon optimized sequence of the human CD3ζ, CD28 and DAP10 endodomains. In addition to the CAR construct, RQR8 which is a marker bearing a CD34 epitope (59) was included, separated from the CAR by a cleavable 2A peptide. This allows CAR expressing cells to be detected by flow cytometry by staining using the anti-CD34 antibody clone QBend10. The CAR and CCR constructs used in this study are shown in fig. S13.
Production of viral particles by transfection
1.5×106 293T cells/100 mm2 dish (Nucleon Delta Surface, Thermo Fisher) were plated at day 1 in 293T medium (D-MEM, 10 % FCS (v/v)). γ-retroviral particles were produced by co-transfection of 293T cells at day 2 using Gene Juice Transfection Reagent (Novagen/Millipore, Massachusetts, USA) in accordance with manufacturer’s directions. Viral particle-containing supernatants were harvested at day 4; medium was replenished, and harvested at day 5. Supernatants were pooled, filtered (0.45-μm filter, Millipore) and directly used for transductions or stored overnight at 4°C before use.
Transduction of T cells
Transduction of T cells was carried out in Retronectin (Takara Bio, Tokyo, Japan) coated 24-well plates, which were pre-loaded with viral supernatant. 0.5×106 T cells suspended in 0.5 ml T-cell medium + 400 IU IL-2 were combined with 1.5 ml viral supernatant and centrifuged for 40 min, 1000xg at RT. Typically, 12×106 T cells per donor were plated for transduction.
Timing of transduction differed between γδT cells and αβT cells due to expansion dynamics. γδT-cell expansion was stimulated with 5 μM zoledronic acid (Actavis, New Jersey, USA) and 100 IU/ml IL-2 (Aldesleukin,) and transduction performed at day 5. At day 8 of culture (day 3 after transduction) cells were pooled, washed and plated at 2×106 cells/ml in T-cell medium + 100 IU IL-2/ml (24-well plates, Nucleon Delta Surface, Thermo Scientific, Massachusetts, USA). Transduction efficiency was determined by flow cytometry at day 10 (day 5 after transduction).
In the case of αβ T cells, transduction was performed 72 h following stimulation with anti-CD3/CD28 antibodies in the presence of 100 IU/ml IL-2 (Aldesleukin), with re-plating 3 days after transduction. Signaling analysis and cytotoxicity assays were performed 6 days post transduction in both cases.
Stimulation of T-cells using receptor-antibody cross linking
Prior to stimulation, T-cells were cultured overnight in serum-free media (XVIVO-15, X-VIVO 15 (Lonza BioWhittaker, Maryland, USA) to reduce background phosphorylation levels. Single samples containing 0.5–1×106 cells were used for each stimulation condition
Primary antibodies targeting the receptors to be stimulated (ms αCD28, ms αCD3, ms αNKG2D, gt αhuFc) were loaded on ice for 10m and the cells were then washed in ice cold phosphate buffered saline at 3°C. Secondary antibodies specific to the species of the primary antibody to be cross-linked (donkey anti-ms or donkey anti-gt) were resuspended in XVIVO-15 and pre-warmed to 37°C. Primary antibody labelled T-cells were added to the pre-warmed tube containing secondary antibody and stimulated for 60, 180 or 360s. Stimulation was halted by the addition of paraformaldehyde to a final concentration of 4%.
Control samples were made for baseline (t = 0s) and for every time point (60, 180 and 360s). Control samples were treated identically to stimulated samples, but the primary and secondary antibodies were excluded. A diagram of the stimulation protocol is shown in fig. S14
Mass cytometry
All samples from a given donor and stimulation run were barcoded using the Cell-ID™ 20-plex Pd barcoding kit (Fluidigm, San Francisco, California, USA) in accordance with the manufacturer’s protocol. Up to 20 samples were therefore stained simultaneously in the same tube in a total volume of 300μl. A two step staining procedure was used whereby surface markers including CD3, CD45RA, CD45RO, CD27, CCR7, TIM-3, CD4, CD8, NKG2D, CD16 and TCRVδ2 were stained, followed by intracellular markers including pERK1/2, pZAP70, pMAPKAPK2, pRelA(p65), pS6, pSLP-76 and pAKT. Cell fixation, permeabilization and staining were performed as previously described (60). To ensure maximum comparability between samples, all data was acquired using internal metal isotope bead standards (EQ Beads, Fluidigm). A list of antibodies used and their conjugates is shown in Supplementary Table S4.
Mass cytometric analysis was performed using a Helios Mass Cytometer (Fluidigm). Approximately 100,000 events were acquired per sample – totaling 2×106 events for a full barcoded set.
CyTOF data post-processing
Individual time series were normalized to the internal bead standards using the method described in (61) and bead events were removed from the resulting FCS files. In addition, as described in (60), abundance values reported by the mass cytometer were transformed using a scaled arcsinh, with a scaling factor of 5. Prior to arcsinh transformation with a cofactor = 5, values of zero in the FCS files had gaussian noise with mean 0 and a standard deviation of 0.5 added to them to enhance visualization using conventional flow cytometry packages and to later enable easier computation of density estimates. Given the minimal SD of the noise compared to actual “positive” signal intensities and the in-built noise and outlier handling capacity of DREMI (Figure S6 of [18]), the addition of noise is not expected to significantly alter the DREMI scores. Furthermore, as the calculation of DREMI scores is done by binning across the dynamic range of values, addition of a small amount of noise to the bottom bins will not propagate across the rest of the distribution,
Debarcoded FCS files were post-processed using a combination of FlowJo Vx and bespoke Python scripts. Gating was performed in FlowJo VX and gated populations were exported prior to computational analysis.
Gating of cellular subsets
Cells were filtered through a series of gates to ensure compatibility between compared samples. Singlets were identified using DNA content, and live cells were detected by exclusion of Cell-ID Cisplatin (Fluidigm) which only stains dead cells. T-cell subsets were identified by expression of CD3 ± CD4, CD8 or TCRVδ2. CAR expression was detected using anti-human Fc, which stains the stalk portion of the CAR. Ane example of the gating strategy used is shown in fig. S15A.
DREMI and DREVI analysis
DREMI and DREVI are information theoretic based methods developed to quantify and visualize relationship between two molecular epitopes(18). Given two proteins epitopes X and Y, and assume that we are interested in assessing the influence of X on Y, then DREVI visualizes the conditional dependence of Y on X. Specifically, DREVI computes the conditional probability density of Y given X, , where the joint distribution is computed using a heat-diffusion based kernel density estimation procedure(62). Once the conditional density is computed, the result is visualized as a heatmap.
DREMI quantifies the strength of relationship between two protein epitopes, using mutual information based metric. While a traditional mutual information metric relies on the joint distribution, it is more likely to be biased by dense regions thereby missing out on interesting biology shown by extreme expression levels of proteins. To circumvent this, DREMI computes the mutual information on the conditional density function (as computed for the DREVI visualization) as opposed to on the joint density.
Thus .
Effectively DREMI reweighs the regular mutual information so that all observed range of expression contributes uniformally. In this manuscript, we utilized the simpledremi implementation of DREVI and DREMI for analysis (63).
Calculation of Earth Movers Distance
Earth movers distance (EMD) was computed between stimulated samples and time & donor matched unstimulated controls. EMD was calculated between T-cell populations that had undergone the same post-processing in terms of gating of specific T-cell populations. The python module wasserstein_distance, which is a component of scipy.stats was used to calculate EMD between samples.
Flow cytometry
Flow cytometric analysis was performed using an LSRII flow cytometer (BD, New Jersey, USA). Data was collected using BD FACSDiva™ V8.0.1 and was analyzed using FlowJo VX. Compensation was calculated based on OneComp eBeads (Thermo Fisher, Massachusetts, USA) stained with single color antibodies.
When flow cytometry was used as an alternative to mass cytometry for rapid stimulation experiments, cells stimulated and fixed as described above were stained in a 2-step protocol whereby surface markers (CD3, TCRVδ2, human-Fc) were stained first, after which the cells were permeabilized and intracellular markers (pZAP70, pERK, pRelA and pAKT) were subsequently stained. A list of antibodies used is shown in Supplementary Table S5.
An example of the gating strategy used when flow cytometry was used to determine TIM3/PD-1 expression of transduced and non-transduced cells is shown in fig. S15B.
51Cr-release killing assay
To assess the killing capacity of T cells, 5,000 51Cr-labelled target cells were co-cultured with 50,000, 25,000, 12,500 or 6,250 effector cells in 200 μl T-cell medium + 100 IU IL-2/ml. Cells were incubated in 96-well V-bottomed plates (Greiner Bio-one, Kremsmünster, Austria) for 4 h. 50 μl of supernatant were transferred into Isoplate-96 HB plates (PerkinElmer, Massachusetts, USA). After addition of 150 μl scintillation cocktail (Optiphase Supermix, PerkinElmer) samples were incubated overnight at RT. Counts were acquired using a 1450 MicroBeta TriLux scintillation counter (PerkinElmer). Percentage killing was calculated using the following formula:
Activation of T cells using antibody-coated beads
Anti-Biotin MACS iBeads (Miltenyi Biotec) were labeled with 10 μg anti-CD3-biotin (OKT3, BioLegend), 10 μg anti-IgG (Fc)-biotin (Novex/Life Technologies), or 10 μg of anti-CD3-biotin and 10 μg anti-IgG (Fc)-biotin, respectively. After incubation (10 min, 4°C), beads were washed twice with PBS and re-suspended in T-cell medium. Bead suspensions (100 μl) were plated in 96-well U-bottom plates (Thermo Fisher, Massachusetts, USA). Effector T-cell preparations were added (0.2×106 cells/well in 100 μl T-cell medium) and incubated with beads for 24 h. No exogenous IL-2 was added to the medium in either case. After 23h cells were harvested for analysis of cytokine secretion by intracellular cytokine staining. The gating strategy was identical to that in fig. S15, with the exception that “% cytokine +ve” was used as the readout.
Colony formation assay
Cells were isolated from healthy donor bone marrow using Ficoll density gradient separation and co-cultured overnight with Vδ2+ γδT cells expressing either CD33–28ζ, CD33-DAP10 or untransduced controls at a 1:1 Effector: Target ratio. After overnight co-culture remaining cells were transferred to semi-solid medium (MethoCult™ H4534 Classic Without EPO, StemmCell Technologies, Vancouver, Canada) and incubated at standard tissue culture conditions for 7 days. After incubation all myeloid colonies were counted manually by 2 independent investigators blinded to the culture conditions.
Multiplex protein tyrosine kinase assay
Expanded Vδ2+ γδT cells were co-cultued with live MV4;11 which had been pre-coated in magnetic anti-CD33 microbeads. Adter 30 minutes of co-culture the MV4;11 were removed using magnetic column separation (Miltenyi, Bergisch Gladbach, Germany). The flow through, containing effector cells only, was lysed and protein tyrosine kinase activity in the lysate was assessed using the PamChip PTK assay using a PamStation 12 (PamGene, Wolvenhoek, Netherlands). Data was processed using BioNavigator6 (PamGene, Wolvenhoek, Netherlands)
Supplementary Material
S1 – Networks examined and antibodies used
S2 – Responses of αβT cells to stimulus
S3 – Network behavior of αβT cells following CD3+CD28 or CD19–28ζ CAR stimulus
S4 – Representative DREVI plots for expanded CD8+ T-cells
S5 – DREMI scores for expanded transduced and non-transduced T-cells
S6 – Representative DREVI plots for expanded transduced and non-transduced T-cells
S7 – TIM-3 expression in expanded transduced and non-transduced αβT cells
S8 – Phosphoprotein expression in stimulated an unstimulated Vδ2+ cells expressing CD28 CCRs
S9 – Production of TNFα in response to CD33-DAP10 CCR stimulus
S10 – DREMI scores in expanded non-transduced and CD33-DAP10-CCR transduced Vδ2+ cells
S11 – Most strongly activated tyrosine kinases in response to MV4;11
S12 – Cytokine production in response to monocytes or MV4;11 ± BTN blockade
S13 – Synthetic constructs used in this study
S14 – T-cell stimulation experiment setup
S15 – Flow cytometric and mass cytometric gating strategies
Supplementary Table 1 - Statistical analysis of data displayed in Figure 3H (CD4+)
Supplementary Table 2 - Statistical analysis of data displayed in Figure 3H (CD8+)
Supplementary Table 3 - Statistical analysis of data displayed in Supplementary Figure S5
Supplementary Table 4 – antibodies used in mass cytometry
Supplementary Table 5 – antibodies used in flow cytometry
Acknowledgments:
We would like to thank staff at the UCL Cancer Institute Cytometry Core Facility for their technical assistance. We would like to thank Dr Martin Pule (UCL Cancer Institute) for several of the constructs used in this study. We would also like to thank Dr Tal Nawy (MSKCC) for his assistance in the production of the manuscript.
Funding: Supported by a Wellcome Trust Fellowship (JF, 110022/Z/15/Z), and by by awards from Olivia Hodson Cancer Fund via GOSHCC (DWD), GOSHCC infrastructure award, NIHR GOSH Biomedical Research Centre (JA), GOSHCC leadership grant (JA), Research in Childhood Cancer (RICC, JA), Great Ormond Street Hospital Charity (JA leadership award and grants W1134, VS0118, W1029 and W1076) Action Medical Research GN2400 (MB), German Academic Exchange Service (PA), DP receives funding from the following grants: NIH (DP, NIH DP1-HD084071, NIH R01CA164729), Cancer Center Support (DP, P30 CA008748) and Gerry Center for Metastasis and Tumor Ecosystems (DP).
Footnotes
Competing Interests: JA holds company stock in Autolus Ltd and is paid consultant for TC Biopharm. JF has also undertaken paid consultancy work for TC Biopharm. JA and JF are both inventors on a patent pertaining to chimeric costimulatory receptors in γδT cells which has been licensed to TC Biopharm (WO/2016/174461). MB performed the work contributing to this paper whilst emplotyed at Universdity College London but is now employed by TC Biopharm.
Data Availability: Data will be made available via OpenScience Framework (OSF.io, https://osf.io/2s38h/).
Ethics: Informed consent was obtained from healthy donors prior to blood drawing for PBMC isolation.
References
- 1.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan D-AN, Lanier BJ, Morgan RA, Rosenberg SA, Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19, Blood 116, 4099–4102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA, Chimeric antigen receptor T cells for sustained remissions in leukemia, N. Engl. J. Med 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Teachey DT, Porter DL, Grupp SA, CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia, Blood 125, 4017–4023 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, Raubitschek A, Forman SJ, Greenberg PD, Riddell SR, Press OW, CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4–1BB domains: pilot clinical trial results, Blood 119, 3940–3950 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brentjens RJ, Brentjens RJ, Davila ML, Davila ML, Rivière I, Rivière I, Park J, Park J, Wang X, Wang X, Cowell LG, Cowell LG, Bartido S, Bartido S, Stefanski J, Stefanski J, Taylor C, Taylor C, Olszewska M, Olszewska M, Bórquez-Ojeda O, Borquez-Ojeda O, Qu J, Qu J, Wasielewska T, Wasielewska T, He Q, He Q, Bernal Y, Bernal Y, Rijo IV, Rijo IV, Hedvat C, Hedvat C, Kobos R, Kobos R, Curran K, Curran K, Steinherz P, Steinherz P, Jurcic J, Jurcic J, Rosenblat T, Rosenblat T, Maslak P, Maslak P, Frattini M, Frattini M, Sadelain M, Sadelain M, CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia, Sci Transl Med 5, 177ra38–177ra38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, Voytovich UJ, Lin C, Sommermeyer D, Liu L, Whiteaker JR, Gottardo R, Paulovich AG, Riddell SR, Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function, Sci. Signal 11, eaat6753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karlsson H, Svensson E, Gigg C, Jarvius M, Olsson-Strömberg U, Savoldo B, Dotti G, Loskog A, Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors, PLoS ONE 10, e0144787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA, Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2, Mol. Ther 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brentjens R, Yeh R, Bernal Y, Rivière I, Sadelain M, Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial, Mol. Ther 18, 666–668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E, Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience, Journal of Clinical Oncology 24, e20–2 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, Byatte AJ, Kirillova N, Valle JW, Sharma SK, Chester KA, Westwood NB, Halford SER, Nabarro S, Wan S, Austin E, Hawkins RE, The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity, Cancer Immunol Immunother 66, 1425–1436 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL, 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors, Nat Med 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gonen M, Sadelain M, Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection, Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quintarelli C, Orlando D, Boffa I, Guercio M, Polito VA, Petretto A, Lavarello C, Sinibaldi M, Weber G, Del Bufalo F, Giorda E, Scarsella M, Petrini S, Pagliara D, Locatelli F, De Angelis B, Caruana I, Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma, Oncoimmunology 7, e1433518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The key to unlocking CARs, Nat. Biotechnol 35, 889 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Orlova DY, Zimmerman N, Meehan S, Meehan C, Waters J, Ghosn EEB, Filatenkov A, Kolyagin GA, Gernez Y, Tsuda S, Moore W, Moss RB, Herzenberg LA, Walther G, Earth Mover’s Distance (EMD): A True Metric for Comparing Biomarker Expression Levels in Cell Populations, PLoS ONE 11, e0151859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levine JH, Simonds EF, Bendall SC, Davis KL, Amir E-AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, Finck R, Gedman AL, Radtke I, Downing JR, Pe’er D, Nolan GP, Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis, Cell 162, 184–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishnaswamy S, Spitzer MH, Mingueneau M, Bendall SC, Litvin O, Stone E, Pe’er D, Nolan GP, Systems biology. Conditional density-based analysis of T cell signaling in single-cell data, Science 346, 1250689 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarro MN, Cantrell DA, Serine-threonine kinases in TCR signaling, Nat. Immunol 15, 808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kingeter LM, Paul S, Maynard SK, Cartwright NG, Schaefer BC, Cutting Edge: TCR Ligation Triggers Digital Activation of NF-κB, J. Immunol, 1001051 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rane MJ, Coxon PY, Powell DW, Webster R, Klein JB, Pierce W, Ping P, McLeish KR, p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils, J. Biol. Chem 276, 3517–3523 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Lampariello F, On the use of the Kolmogorov-Smirnov statistical test for immunofluorescence histogram comparison, Cytometry 39, 179–188 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Filion GJ, The signed Kolmogorov-Smirnov test: why it should not be used, Gigascience 4, 9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas S, Straathof K, Himoudi N, Anderson J, Pule M, Karagiannis SN, Ed. An Optimized GD2-Targeting Retroviral Cassette for More Potent and Safer Cellular Therapy of Neuroblastoma and Other Cancers, PLoS ONE 11, e0152196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mujoo K, Kipps TJ, Yang HM, Cheresh DA, Wargalla U, Sander DJ, Reisfeld RA, Functional Properties and Effect on Growth Suppression of Human Neuroblastoma Tumors by Isotype Switch Variants of Monoclonal Antiganglioside GD2 Antibody 14.18, Cancer Research 49, 2857–2861 (1989). [PubMed] [Google Scholar]
- 26.Harly C, Guillaume Y, Nedellec S, Peigné C-M, Mönkkönen H, Mönkkönen J, Li J, Kuball J, Adams EJ, Netzer S, Déchanet-Merville J, Léger A, Herrmann T, Breathnach R, Olive D, Bonneville M, Scotet E, Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset, Blood 120, 2269–2279 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karunakaran MM, Herrmann T, The Vγ9Vδ2 T Cell Antigen Receptor and Butyrophilin-3 A1: Models of Interaction, the Possibility of Co-Evolution, and the Case of Dendritic Epidermal T Cells, Front Immunol 5, 648 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asslan R, Pradines A, Pratx C, Allal C, Favre G, Le Gaillard F, Epidermal growth factor stimulates 3-hydroxy-3-methylglutaryl-coenzyme A reductase expression via the ErbB-2 pathway in human breast adenocarcinoma cells, Biochem. Biophys. Res. Commun 260, 699–706 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Gober HJ, Kistowska M, Angman L, Jeno P, Mori L, De Libero G, Human T Cell Receptor γδ Cells Recognize Endogenous Mevalonate Metabolites in Tumor Cells, Journal of Experimental Medicine 197, 163–168 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poupot M, Fournié J-J, Non-peptide antigens activating human Vgamma9/Vdelta2 T lymphocytes, Immunology Letters 95, 129–138 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Ribeiro ST, Ribot JC, Silva-Santos B, Five Layers of Receptor Signaling in γδ T-Cell Differentiation and Activation, Front Immunol 6, 15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ribot JC, DeBarros A, Silva-Santos B, Searching for “signal 2”: costimulation requirements of γδ T cells, Cell. Mol. Life Sci 68, 2345–2355 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nedellec S, Sabourin C, Bonneville M, Scotet E, NKG2D costimulates human V gamma 9V delta 2 T cell antitumor cytotoxicity through protein kinase C theta-dependent modulation of early TCR-induced calcium and transduction signals, The Journal of Immunology 185, 55–63 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Nitahara A, Shimura H, Ito A, Tomiyama K, Ito M, Kawai K, NKG2D ligation without T cell receptor engagement triggers both cytotoxicity and cytokine production in dendritic epidermal T cells, J Investig Dermatol 126, 1052–1058 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ, NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway, Nat. Immunol 4, 557–564 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Silva-Santos B, Serre K, Norell H, γδ T cells in cancer, Nat. Rev. Immunol 15, 683–691 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Fisher J, Abramowski P, Wisidagamage Don ND, Flutter B, Capsomidis A, Cheung GW-K, Gustafsson K, Anderson J, Avoidance of On-Target Off-Tumor Activation Using a Co-stimulation-Only Chimeric Antigen Receptor, Mol. Ther (2017), doi: 10.1016/j.ymthe.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu JJ, Ryu JR, Pendergast AM, Abl tyrosine kinases in T‐cell signaling, Immunol. Rev 228, 170–183 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gertner-Dardenne J, Castellano R, Mamessier E, Garbit S, Kochbati E, Etienne A, Charbonnier A, Collette Y, Vey N, Olive D, Human Vγ9Vδ2 T cells specifically recognize and kill acute myeloid leukemic blasts, The Journal of Immunology 188, 4701–4708 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M, Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells, Nat. Biotechnol 31, 71–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, Powell DJ, Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo, Cancer Immunol Res 1, 43–53 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tasian SK, Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: how far up the road have we traveled? Ther Adv Hematol 9, 135–148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, Cabral JMS, Rooney CM, Orange JS, Brenner MK, Mamonkin M, Tonic 4–1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent, Cell Rep 21, 17–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, Gould SE, Maecker H, Irving BA, Kim JM, Belvin M, Mellman I, MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade, Immunity 44, 609–621 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Krishna S, Xie D, Gorentla B, Shin J, Gao J, Zhong X-P, Chronic activation of the kinase IKKβ impairs T cell function and survival, The Journal of Immunology 189, 1209–1219 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feucht J, Sun J, Eyquem J, Ho Y-J, Zhao Z, Leibold J, Dobrin A, Cabriolu A, Hamieh M, Sadelain M, Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency, Nat Med 25, 82–88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, Diehn M, West RB, Plevritis SK, Alizadeh AA, The prognostic landscape of genes and infiltrating immune cells across human cancers, Nat Med 21, 938–945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deniger DC, Switzer K, Mi T, Maiti S, Hurton L, Singh H, Huls H, Olivares S, Lee DA, Champlin RE, Cooper LJ, Bispecific T-cells Expressing Polyclonal Repertoire of Endogenous γδ T-cell Receptors and Introduced CD19-specific Chimeric Antigen Receptor, Mol. Ther 21, 638–647 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rischer M, Pscherer S, Duwe S, Vormoor J, Jurgens H, Rossig C, Human gammadelta T cells as mediators of chimaeric-receptor redirected anti-tumour immunity, Br J Haematol 126, 583–592 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Fisher J, Anderson J, Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy, Front Immunol (2018), doi: 10.3389/fimmu.2018.01409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, Majani Y, gileadi T, Wallace R, Gustafsson K, Flutter B, Anderson J, Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation, Mol. Ther 26, 354–365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, Pende D, Steinle A, Ferrone S, Pistoia V, Downregulation and/or Release of NKG2D Ligands as Immune Evasion Strategy of Human Neuroblastoma, Neoplasia (New York, N.Y.) 6, 558 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Acheampong DO, Acute Myeloid Leukemic Cells Express NKG2D or Shed off NKG2D Ligand to Escape Immune-Surveillance, HTIJ 5, 1–2 (2017). [Google Scholar]
- 54.Santolaria T, Robard M, Léger A, Catros V, Bonneville M, Scotet E, Repeated systemic administrations of both aminobisphosphonates and human Vγ9Vδ2 T cells efficiently control tumor development in vivo, The Journal of Immunology 191, 1993–2000 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Fisher JPH, Flutter B, Wesemann F, Frosch J, Rossig C, Gustafsson K, Anderson J, Effective combination treatment of GD2-expressing neuroblastoma and Ewing’s sarcoma using anti-GD2 ch14.18/CHO antibody with Vγ9Vδ2+ γδT cells, Oncoimmunology 5, e1025194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fisher JPH, Heuijerjans J, Yan M, Gustafsson K, Anderson J, γδ T cells for cancer immunotherapy: A systematic review of clinical trials, Oncoimmunology 3, e27572 1–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fournié J-J, Sicard H, Poupot M, Bezombes C, Blanc A, Romagné F, Ysebaert L, Laurent G, What lessons can be learned from γδ T cell-based cancer immunotherapy trials? Cell. Mol. Immunol, 1–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rivière I, Brose K, Mulligan RC, Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells, Proc. Natl. Acad. Sci. U.S.A 92, 6733–6737 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, Marin V, Marafioti T, Chakraverty R, Linch D, Quezada SA, Peggs KS, Pule M, A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy, Blood 124, 1277–1287 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Bendall SC, Simonds EF, Qiu P, Amir E-AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP, Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum, Science 332, 687–696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, Pe’er D, Nolan GP, Bendall SC, Normalization of mass cytometry data with bead standards, Cytometry Part A 83, 483–494 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Botev ZI, Grotowski JF, Kroese DP, Kernel density estimation via diffusion, The Annals of Statistics 38, 2916–2957 (2010). [Google Scholar]
- 63.Dana Pe’er Lab GitHub. https://github.com/dpeerlab/DREMI, last accessed 28/01/19
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 – Networks examined and antibodies used
S2 – Responses of αβT cells to stimulus
S3 – Network behavior of αβT cells following CD3+CD28 or CD19–28ζ CAR stimulus
S4 – Representative DREVI plots for expanded CD8+ T-cells
S5 – DREMI scores for expanded transduced and non-transduced T-cells
S6 – Representative DREVI plots for expanded transduced and non-transduced T-cells
S7 – TIM-3 expression in expanded transduced and non-transduced αβT cells
S8 – Phosphoprotein expression in stimulated an unstimulated Vδ2+ cells expressing CD28 CCRs
S9 – Production of TNFα in response to CD33-DAP10 CCR stimulus
S10 – DREMI scores in expanded non-transduced and CD33-DAP10-CCR transduced Vδ2+ cells
S11 – Most strongly activated tyrosine kinases in response to MV4;11
S12 – Cytokine production in response to monocytes or MV4;11 ± BTN blockade
S13 – Synthetic constructs used in this study
S14 – T-cell stimulation experiment setup
S15 – Flow cytometric and mass cytometric gating strategies
Supplementary Table 1 - Statistical analysis of data displayed in Figure 3H (CD4+)
Supplementary Table 2 - Statistical analysis of data displayed in Figure 3H (CD8+)
Supplementary Table 3 - Statistical analysis of data displayed in Supplementary Figure S5
Supplementary Table 4 – antibodies used in mass cytometry
Supplementary Table 5 – antibodies used in flow cytometry