

Abstract
Our investigations on the reaction mechanism to account for regioselectivity on the addition of indoles to unsymmetrical silyloxyallyl cations are reported. Using both experimental and computational methods, we confirmed the significance of steric effects from the silyl ether group toward directing the inward approach of indoles, leading to nucleophilic attack at the less substituted electrophilic α′-carbon. The role of residual water toward accelerating the rate of reaction is established through stabilization of the participating silyloxyallyl cation.
Graphical Abstract

INTRODUCTION
Oxyallyl cations are useful reactive intermediates in organic synthesis. Commonly generated via Nazarov electrocyclization or Favorskii ionization,1 these species readily participate in carbon–carbon bond forming reactions via a variety of cycloaddition as well as nucleophilic addition processes.2,3 In recent years, studies on the exploitation of oxyallyl cations toward direct functionalization of ketones at the α-carbon via their capture with heteroatom-centered and π-carbon nucleophiles have emerged in the literature.4 A noteworthy innovation in these new reports was highlighted by the production of oxyallyl cations via base-induced ionization of ketones bearing a leaving group at the α-carbon (Scheme 1). While they are powerful, these methods nonetheless suffered from the lack of regioselectivity especially in substrates that partook in an intermediacy of α-branched unsymmetrical oxyallyl cations, viz. 1.4a,b
Scheme 1.

Regioselective Addition of Indoles to Unsymmetrical Silyloxyallyl Cations
Our group has recently contributed feasible solutions to this critical issue.5 We discovered that ionization of isomeric hydroxy silylenol ethers 2 and 3 with catalytic pyridinium triflate produced putative silyloxyallyl cations 4 that could be readily captured by substituted indoles exclusively at the less substituted α′-carbon to afford highly functionalized enol ethers 5 in high yields.5a,c In this article, we report our findings on the underlying mechanistic factors that implicated the observed regioselectivity using both experimental and computational methods.
RESULTS AND DISCUSSION
As depicted in Scheme 2, our studies commenced with the use of five-membered α′-hydroxy silylenol ether 6 as a model substrate. While our originally reported conditions identified the role of 4 Å molecular sieves in preventing protodesilylation by excluding any residual water molecules from the reaction mixture,5a our subsequent studies on related six-membered variations revealed that the presence of the water byproduct from ionization might be, in fact, beneficial toward improving the rate of reaction.5c To examine this effect on compound 6, we devised an improved ionization protocol (condition A) and found that the presence of trace water indeed substantially accelerated the rate of reaction. As evidenced by the preparative scale synthesis using various substituted indoles, this new condition afforded α′-indolyl silylenol ether 7a–7f in just a few hours with comparable yields and regioselectivity, as opposed to days using the original reaction condition B.
Scheme 2.

Investigations on the Effect of Trace Watera
aProducts were isolated with >20:1 regioselectivity.
The exquisite regioselectivity observed in our methodology was believed to have been governed by the tert-butyldimethylsilyl (TBS) ether based on our observation on the effects of the protecting group on the oxyallyl cation moiety from a series of hydroxy silylenol ethers 8 (Table 1). As shown in entries 1–3, exposure of starting materials 8a–8c, which were, respectively, elaborated with TBS, triethylsilane (TES), and tert-butyldiphenylsilyl (TBDPS) groups, to the new reaction conditions afforded their corresponding α′-indoyl silylenol ethers 9 as a single regioisomer. Unsurprisingly, an erosion of product yield was observed with the TES substrate presumably because of its relative instability under the Brønsted acidic conditions. It is also anticipated to note that the sterically imposing TBDPS group dramatically impeded the rate of reaction. As our study continued with O-alkyl substituents (entries 4–5), we noticed that both methyl and benzyl containing substrates 8d and 8e rendered the indole addition essentially non-regioselective as indoyl adducts 9 and 10 were produced as a mixture.
Table 1.
Effects of Protecting Groups
 | |||||
|---|---|---|---|---|---|
| entry | Substrate | R | time (h) | yield (%)a | 9/10b |
| 1 | 8a | −TBS | 4 | 85 | 20:1 |
| 2 | 8b | −TES | 8 | 61 | 20:1 |
| 3 | 8c | −TBDPS | 168 | 72 | 20:1 |
| 4 | 8d | −Bn | 5 | 73c | 2:1 |
| 5 | 8e | −Me | 4 | 80c | 1:1 |
Combined yield of 9 and 10 as these products were inseparable by flash column chromatography.
The ratio of products 9 and 10 was determined by 1H NMR.
Hydrolysis products were isolated upon aqueous workup.
To examine the underlying force that dictates this contrasting level of regioselectivity as well as to determine the mechanistic pathway including the rate determining step, electronic structure calculations at the density functional theory (DFT) level were carried out. The details of the computational methods used can be found in the Supporting Information.
Regioselectivity Calculations.
Electronic structure calculations on the participating intermediates and transition states for two model substrates 8a and 8e were carried out to determine the electronic energies, Gibbs free energies, and the Boltzmann distributions. Charge analysis was utilized to determine whether the regioselectivity is charge controlled or dominated by steric effects. Molecular orbitals were also investigated to assess whether oxyallylic π-bonding resonance by one of the oxygen’s lone pairs invoked the secondary orbital interaction with the indole ring. Symmetry-adapted perturbation theory (SAPT) calculations were performed in order to quantitatively determine the effect of steric hindrance.
Our findings are depicted in Figure 1 and Table 2. Based on a notion that our reaction proceeded via an SN1 mechanism (discussed in greater detail in the proceeding sub-section), we subjected putative silyloxyallyl and methyloxyallyl cations 11a and 11e to react with indoles. Depending on the relative facial orientation, the approach of indoles to these oxyallyl cations could hypothetically afford two pairs of diastereomeric indolium ion adducts, that is, 12 and 13 with nucleophilic addition occurring at the α-carbon, and 14 and 15 with the α′-carbon being the electrophilic center. To evaluate the energetics involved in this regiodetermining step, the associated transition states that led to the formation of each intermediate 12–15 were computed. Upon structural optimization, we identified two distinct modes of indole addition to oxyallyl cations 11a and 11e. The first mode involves an outward approach as indoles reacted with the putative oxyallyl cation from its periphery. Conversely, the second mode proceeds via an inward approach where the planes of the approaching indole and oxyallyl cation are stacked on top of each other as the bond-forming event took place.
Figure 1.

Calculated reaction pathways for the intermediate step; the addition of indoles to the various cations. Energies are given in kcal/mol and are with respect to the reactant of each pathway. Gibbs free energies are given first, followed by potential energies in parenthesis. The free energies marked in red indicate cases where the product is higher in energy than the corresponding reactant (12a-I, 13a-I, 13e-O, and 13e-I), or the transition state is lower in energy than the corresponding reactant (15a-O). Analogous calculations were carried out for the most stable motifs for the methyl and TBS cases with water as the implicit solvent, and the same trends were observed (see Supporting Information).
Table 2.
Comparison of the Activation Free Energy, ΔG‡, for the Various Transition States as Well as the Difference in Free Energy of the Reactants/Transition States/Products with Respect to the Most Stable Structure, ΔΔG, for Each Casea
| oxyallyl cation | reactive site | structure | ΔG‡[b] (kcal/mol) | structure | ΔΔG[c] (kcal/mol) | structure | ΔΔG[c] (kcal/mol) | structure | ΔΔG[c] (kcal/mol) |
|---|---|---|---|---|---|---|---|---|---|
 |
α | TS-12a-0 | 6.0 | pre-12a-0 | 5.4 | TS-12a-0 | 6.8 | 12a-0 | 7.7 |
| α | TS-13a-0 | 4.1 | pre-13a-0 | 6.1 | TS-13a-0 | 5.6 | 13a-0 | 6.1 | |
| α’ | TS-14a-0 | 3.7 | pre-14a-0 | 4.8 | TS-14a-0 | 3.9 | 14a-0 | 3.9 | |
| α’ | TS-15a-0 | −0.2 | pre-15a-0 | 7.5 | TS-15a-0 | 2.7 | 15,1–0 | 5.0 | |
| α | TS-12a-l | 9.4 | pre-12a-l | 2.2 | TS-12a-l | 6.9 | 12a-l | 6.2 | |
| α | TS-13a-l | 7.3 | pre-13a-l | 1.8 | TS-13a-l | 4.5 | 13a-l | 6.3 | |
| α’ | TS-14a-l | 4.8 | pre-14a-l | 2.7 | TS-14a-l | 2.8 | 14a-l | 4.3 | |
| α’ | TS-15a-l | 4.6 | pre-15a-l | 0.0 | TS-15a-l | 0.0 | 15a-l | 0.0 | |
 |
α | TS-12e-0 | 6.2 | pre-12e-0 | 2.2 | TS-12e-0 | 2.1 | 12e-0 | 3.1 |
| α | TS-13e-0 | 6.4 | pre-13e-0 | 1.1 | TS-13e-0 | 1.2 | 13e-0 | 2.3 | |
| α’ | TS-14e-0 | 3.6 | pre-14e-0 | 4.3 | TS-14e-0 | 1.7 | 14e-0 | 0.0 | |
| α’ | TS-15e-0 | 2.8 | pre-15e-0 | 4.2 | TS-15e-0 | 0.7 | 15e-0 | 1.2 | |
| α | TS-12e-l | 6.7 | pre-12e-l | 1.5 | TS-12e-l | 2.0 | 12e-l | 1.0 | |
| α | TS-13e-l | 6.7 | pre-13e-l | 0.1 | TS-13e-l | 0.6 | 13e-l | 2.2 | |
| α’ | TS-14e-l | 5.3 | pre-14e-l | 1.7 | TS-14e-l | 0.8 | I4e-I | 0.5 | |
| α’ | TS-15e-l | 6.3 | pre-15e-l | 0.0 | TS-15e-l | 0.0 | 1Se-l | 0.6 |
Values are given for both the TBS and methyl model systems.
Free energy barriers (difference between each transition state and its corresponding reactant).
Relative free energy of each reactant/product/transition state with respect to the most stable reactant/product/transition states for the Me and TBS cases.
As presented in Table 2, the reactants, products, and transition states in the outward approach for the silyloxyallyl case were found to be consistently higher in Gibbs free energy (and potential energy) than those corresponding to the inward approach. Considering that the outward reactant structures for the silyloxyallyl system (pre-12a-O to pre-15a-O) are approximately 3 kcal/mol or higher than the corresponding inward structures (pre-12a-I to pre-15a-I), our discussions on this case will be restricted only to the more productive pathways that involved the inward orientation of indoles.
Among the inward structures, intermediate reactant pre-15a-I, intermediate transition TS-15a-I, and intermediate product 15a-I were substantially more stable than the other inward motifs including those that are involved in addition at the α-carbon, that is, 12a-I and 13a-I (Table 2). Furthermore, the Gibbs free energy profiles in Figure 1 show that the intermediate products in 12a-I and 13a-I are higher in energy than the reactant, resulting in the reverse step possessing a lower activation energy than the forward step. From Tables 1 and S1 (Supporting Information), the products formed due to addition at the α′-carbon (motifs 14a-I and 15a-I) are more thermodynamically favorable than those leading to the indole addition at the α-carbon (motifs 12a-I and 13a-I). In addition, from Figure 1, it is clear that the energy difference (both potential energy and free energy) between the transition states and the corresponding reactants is lower for motifs 14a-I and 15a-I and, therefore, kinetically favored. Thus, addition to the α′-carbon is both thermodynamically and kinetically favored. Boltzmann distributions were calculated at 300 K to further assess the regioselectivity that was observed experimentally, and the results are detailed in the Table S1 (Supporting Information). From these distributions, the obvious preference for indole addition is at the α′-carbon. While there is some small probability of the existence of pre-12a-I at 300 K, the ratio of TS-15a-I to TS-12a-I is nearly zero.
Applications of analogous assessment on the indole approaches to the methyloxyallyl variant 11e revealed interesting and yet subtle differences between the participating transition states when compared to those of silyloxyallyl 11a. For instance, while the inward TS-12e-I–TS-15e-I structures generally possess higher activation energies than those of the outward counterparts, there is a lack of meaningful energetic differences between these transition states. The lack of regioselectivity observed with the methyloxyallyl system could be attributed to two highly competitive pathways. One pathway traversing through pre-15e-I (the most stable of the intermediate reactants within the methyloxyallyl motif) and TS-15e-I (the most thermodynamically favored transition state). The other pathway, toward the formation of 13e-I, exhibits very similar free energies for both the intermediate reactants with a nonsignificant energy difference of only 0.1 kcal/mol and the transition states with a barrier of 6.7 kcal/mol versus 6.3 kcal/mol. The final thermodynamic stability of the resulting indolium ion is also very similar.
Unlike the silyloxyallyl systems which favorably produced the inward α′-(S,R) 15a-I as the most stable adduct, there was no significant thermodynamic differences between 12e-O–15e-I in the methyloxyallyl systems. Gibbs free energy calculations follow the same trends as the electronic energies calculated, however intermediate structures 13e-I and 13e-O were both found to be slightly less stable than their reactant. The Boltzmann distributions (Table S1) for the 11e variants also show that both inward and outward orientations, as well as bonding at the α and α′-carbon will occur, further supporting the lack of regioselectivity seen in the experiments. We believed that the two competitive pathways for the addition of indoles to methyloxyallyl cation 11e most likely proceeded via 13e-I and 15e-I, which gave rise to the α and α′-adducts, respectively. Our theory was supported by calculations on the energetic requirement to ionize substrate 8e to the corresponding methyloxyallyl cation in the form of its eight possible reactant structures pre-12e-O to pre-15e-I. As summarized in Figure 2, formation of reactant structures pre-13e-I and pre-15e-I produced the largest energy barrier for the reverse reaction (reincorporation of the leaving group, i.e., water) when compared to those of pre-12e-I, pre-14e-I, pre-12e-O and pre-13e-O, pre-14e-O, and pre-15e-O. Such subtle differences in the reverse activation energy enjoyed by pre-13e-I and pre-15e-I rendered the regeneration of substrate 8e (to be exact, pre-ionization organization structure pre-11e) to be less competitive, thus enabling for the indole addition to occur. In contrast, the reverse reaction of pre-14e-O and pre-15e-O is essentially barrier-less. Given the unimpeded opportunity for these structures to return to the pre-11e form, the likelihood for the forward carbon–carbon bond forming reaction, that is, indole addition, becomes inconsequential.
Figure 2.

Free-energy diagram on the ionization of α′-hydroxy methylenol ether 8e. All energies are in kcal/mol.
Rate-Determining Step.
Considering that isomeric starting materials 2 and 3 produced an identical α′-indolyl product,5a and that unsubstituted and strongly electron-deficient substrates failed to react,5e we believed that our methodology proceeded via an SN1 mechanism through an intermediary unsymmetrical silyloxyallyl cation 4 that was produced upon activation of the substrates with the Brønsted acid catalyst.6 To test this hypothesis, we modeled a possible reaction mechanism in the formation of silyloxyallyl cation 11a, revealing four important results:7
The ionization involved simultaneous proton transfer from the pyridinium ion and departure of the water molecule from the substrate. The calculated electronic energy found that this step required 15.9 kcal/mol (13.9 kcal/mol from Gibbs free energy calculations) of activation barrier and is thereby rate-determining. Interestingly, the indole appeared to yield additional stabilization at this step. Analogous calculations in the absence of indoles resulted in a higher activation energy barrier (18.0 kcal/mol electronic energy, 14.6 kcal/mol Gibbs free energy) and reduced stability of the resulting oxyallyl cations (17.1 kcal/mol electronic energy, 11.7 kcal/mol Gibbs free energy). Without the interaction between the cation and indole, not only is the barrier higher, but the stability of the intermediate reactant is also significantly reduced as the barrier for the reverse reaction is only 2.5 kcal/mol (free energy) putting the second step, despite its “low” barrier, in competition with the reverse reaction. This extra stability appears to originate from the stabilizing π–π interaction between the highest occupied molecular orbital (HOMO) of indoles and lowest occupied molecular orbital (LUMO) of the emerging cationic intermediate. Exemplified by methyloxyallyl cation 11e (leading to pre-12e-I) as shown in Figure 3, this π–π interaction with indoles interestingly involves both α and α′-carbons, thus affecting the stacking of the two reaction components. Overall, in addition to a primary interaction that ultimately yields the forward carbon–carbon bond formation between indoles and the cationic species, we have identified a secondary interaction, which explains the stability observed at the intermediate reactants in the inward position (pre-12e-I to pre-15e-I).
The alternative SN2 mechanism, in which addition of indoles occurred simultaneously as the water-leaving group departed, was not probable. As shown through ab initio metadynamics simulations (see Supporting Information for details), an attempt to model this pathway did not lead to productive results.
Indeed, the observed role of water in expediting the rate of reaction is confirmed through stabilization of the resulting carbocation. To quantify the stabilizing effect water has on the reaction, the binding energy of the carbocation and water was calculated. We found that the binding energy of pre-15a-I cation with water is −7.7 kcal/mol, TS-15a-I is −9.8 kcal/mol, and 15a-I is −10.5 kcal/mol.
The regioselectivity involving the TBS substrate seemed to be controlled by steric effects imposed by the bulky silyl ether. SAPT calculations were performed to quantify the steric hindrance, as the exchange term from SAPT calculations is indicative of this property. While TS-15e-I and TS-13e-I had similar exchange contributions (~84.5 kcal/mol), the exchange term for TS-13a-I is 26.7 kcal/mol higher than TS-15a-I. The notion that regioselectivity is due to the sterics was reinforced by the charge analysis on relevant silyloxyallyl and methyloxyallyl cations, which revealed that the charge distribution at the α versus α′-carbon centers did not change significantly, regardless the nature of the protecting groups. The s–p hybridization of the oxygen atom in silyloxyallyl and methyloxyallyl cations 11a and 11e was investigated to determine any possibilities of secondary orbital interactions between oxyallylic π-bonding invoked from one of the oxygen’s lone pairs and the indole ring. The results, as seen in the Supporting Information, show a strong p character in 11a, thus signifying that the oxygen atom is sp3 hybridized in the silyloxyallyl system. With the methyloxyallyl system 11e, the analogous oxygen atom was found to be sp2 hybridized because of its weaker p character. This analysis also indicates that one of the lone pairs resides in the p orbital, and it readily participates in the oxyallylic π-bonding resonance. By visualizing the molecular orbitals of carbocations 11a and 11e (see Supporting Information), there was no evidence for such secondary orbital interactions in both systems, despite the contrasting hybridization of their respective oxygen atoms.
Figure 3.

(a) HOMO–LUMO interaction of methyloxyallyl cation 11e and indoles, leading to pre-12e-I (pyridine excluded). (b) Schematic of the molecular orbitals for protected oxyallyl cations (R = TBS or Me) and indoles that result in inward orientation.
Lastly, we evaluated the conversion of an indolium ion in the silyloxyallyl system to the final product. Using the most stable α′-(S,R) 15a-I intermediate as a starting point and pyridine as the base, re-aromatization (see Figure 4) to the observed α′-indolyl silylenol ether enjoyed an overall exothermic stabilization by 19.4 kcal/mol in electronic energy (14.9 kcal/mol Gibbs free energy) with a barrier of 5.2 kcal/mol in electronic energy (3.6 kcal/mol Gibbs free energy). With these data in hand, we were able to put together a complete schematic for the mechanism of our reaction. As depicted in Figure 5, the energy barrier for the reverse reaction for the formation of each intermediate in the mechanism is unambiguously higher than the corresponding forward step, essentially rendering the regioselective indole addition at the less-substituted α′-carbon of unsymmetrical silyloxyallyl cation 11a an irreversible process. For comparison, the overall profile for the methyloxyallyl cation 11e is presented in the Supporting Information (Figure S4).
Figure 4.
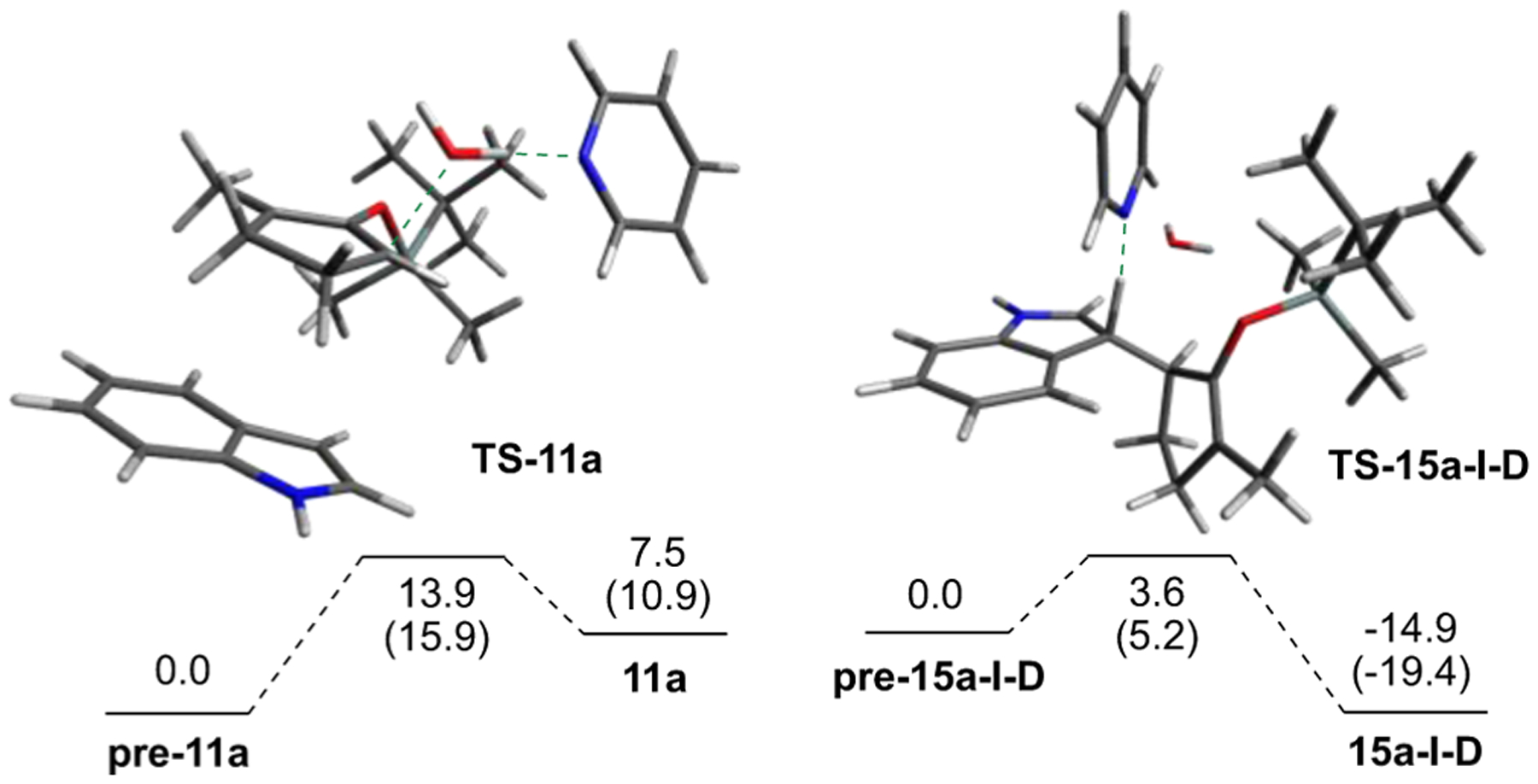
Identification of the rate-determining step and rear-omization of indolium ions. Gibbs free energies are given first, followed by potential energies in parenthesis. All energies are in kcal/mol.
Figure 5.

Overall free energy profile for regioselective indole addition at the α′-carbon of silyloxyallyl cation 11a. All energies are in kcal/mol.
CONCLUSIONS
In conclusion, the underlying role in the origin of regioselectivity in the addition of silyloxyallyl cations has been investigated. Using DFT-based electronic structure calculations, we identified the significance of the TBS ether in unsymmetrical silyloxyallyl cations toward biasing indole addition at the α′-carbon. The origin of this regiocontrol can be traced to its steric effects, which directs the approach of indole addition, providing crucial stabilization of the indolium ion intermediates. Furthermore, we confirmed that this methodology proceeded via an SN1 mechanism by which the formation of the unsymmetrical silyloxyallyl cations is rate-determining. This mechanistic understanding will aid in the further development of relevant synthetic reactions based on this cationic species, which will be reported in due course.
EXPERIMENTAL SECTION
General Information.
All materials, unless otherwise stated, were purchased from commercial sources and utilized without further purification. Anhydrous reactions were conducted in an oven-dried glassware, which was then cooled under vacuum and purged with nitrogen gas. Anhydrous solvents (dichloromethane, toluene, acetonitrile, diethyl ether, and tetrahydrofuran) were filtered through activated 3 Å molecular sieves under nitrogen in a solvent purification system. Reactions were either monitored by analytical thin layer chromatography (TLC silica gel 60 F254, glass plates) and analyzed using 254 nm UV light and anisaldehyde–sulfuric acid or potassium permanganate stains or via gas chromatography–mass spectrometry (GC–MS). The column for the GC–MS system was TG-SQC (15 m × 0.25 mm × 0.25 μm). Low and high mass readings were set to 60 to 400 amu, respectively. Oven, inlet, and detector temperatures were set to 250 °C, and helium was used as the inert carrier gas. Column chromatography was completed using silica gel or neutral alumina. Unless otherwise noted, all 1H and 13C NMR spectra were recorded in CDCl3 using spectrometers operating at either 400 MHz for 1H and 100 MHz for 13C or 500 MHz for 1H and 125 MHz for 13C. Chemical shifts (δ) are reported in ppm relative to residual CHCl3 as an internal reference (1H: 7.26 ppm, 13C: 77.00 ppm). Coupling constants (J) are reported in hertz. Peak multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), x (septet), h (heptet), b (broad), and m (multiplet). Fourier transform infrared spectra were recorded using thin film, and absorption frequencies were reported in reciprocal centimeters. High-resolution mass spectrometry (HRMS) analyses were performed using the electron spray ionization-time of flight (ESI-TOF) method.
3-(2-((tert-Butyldimethylsilyl)oxy)-3-methylcyclopent-2-en-1-yl)-1H-indole (7a).5a

Starting material 6 (145 mg, 0.635 mmol) was dissolved in toluene (3.2 mL). Indole (82 mg, 0.698 mmol) was then added, followed by pyridinium triflate (15 mg, 0.064 mmol). The reaction was stirred for 4 h when it reached completion as monitored by TLC. The mixture was then concentrated in vacuo to obtain the crude material, which was purified by flash column chromatography with 95:5 hexanes/EtOAc to give product 7a in 85% yield (177 mg, 0.540 mmol) as a clear oil. Rf: 0.70 (70:30 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.86 (s, 1H), 7.65 (d, J = 9.7 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.21 (t, J = 7.2 Hz, 1H), 7.14 (t, J = 7.1 Hz, 1H), 7.00 (s, 1H), 4.03 (m, 1H), 2.49–2.35 (m, 2H), 2.34–2.23 (m, 1H), 1.92 (m, 1H), 1.75 (s, 3H), 0.88 (s, 9H), 0.01 (s, 3H), −0.04 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) 147.9, 136.5, 127.1, 121.5, 119.4, 119.3, 118.9, 113.5, 110.9, 76.8, 42.5, 32.3, 29.8, 25.6, 18.1, 12.5, −4.2, −4.3.
3-(2-((tert-Butyldimethylsilyl)oxy)-3-methylcyclopent-2-en-1-yl)-1-methyl-1H-indole (7b).5a

Starting material 6 (175 mg, 0.327 mmol) was dissolved in toluene (1.6 mL). N-Methylindole (45 μL, 0.360 mmol) was then added, followed by pyridinium triflate (7 mg, 0.032 mmol). The reaction was stirred for 9 h when it reached completion as monitored by TLC. The mixture was then concentrated in vacuo to obtain the crude material, which was purified by flash column chromatography with 88:12 hexanes/CH2Cl2 to give product 7b in 56% yield (63 mg, 0.184 mmol) as a clear oil. Rf: 0.60 (80:20 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.58 (d, J = 7.9 Hz, 1H), 7.27 (d, J = 8.2 Hz, 1H), 7.20 (t, J = 7.1 Hz, 1H), 7.07 (t, J = 7.4 Hz, 1H), 6.85 (s, 1H), 3.99–3.94 (m, 1H), 3.73 (s, 3H), 2.42–2.30 (m, 2H), 2.28–2.21 (m, 1H), 1.88–1.80 (m, 1H), 1.69 (s, 3H), 0.82 (s, 9H), −0.05 (s, 3H), −0.11 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ (ppm) 148.0, 137.2, 127.6, 126.4, 121.1, 119.4, 118.3, 117.8, 113.4, 109.0, 42.4, 32.5, 32.2, 30.1, 25.7, 18.1, 12.5, −4.2, −4.2.
3-(2-((tert-Butyldimethylsilyl)oxy)-3-methylcyclopent-2-en-1-yl)-2-phenyl-1H-indole (7c).5a

Starting material 6 (111 mg, 0.489 mmol) was dissolved in toluene (2.4 mL). 2-Phenylindole (104 mg, 0.538 mmol) was then added, followed by pyridinium triflate (11 mg, 0.048 mmol). The reaction was stirred for 3 h when it reached completion as monitored by TLC. The mixture was then concentrated in vacuo to obtain the crude material, which was purified by flash column chromatography with 99:1 hexanes/EtOAc to give product 7c in 89% yield (176 mg, 0.432 mmol) as a brown oil. Rf: 0.68 (80:20 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.92 (s, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 7.0 Hz, 2H), 7.46 (t, J = 7.6 Hz, 2H), 7.37 (d, J = 7.4 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.17 (t, J = 8.1 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 4.18, 4.21–4.15 (m, 1H), 2.53–2.44 (m, 1H), 2.43–2.33 (m, 2H), 2.21–2.11 (m, 1H), 1.68 (s, 3H), 0.64 (s, 9H), −0.35 (s, 3H), −0.45 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ (ppm) 148.0, 136.2, 135.2, 133.3, 128.6, 128.6, 128.1, 127.6, 121.9, 120.8, 119.2, 115.5, 112.4, 42.3, 32.8, 29.0, 25.5, 18.0, 12.7, −4.5, −4.7.
5-Bromo-3-(2-((tert-butyldimethylsilyl)oxy)-3-methylcyclopent-2-en-1-yl)-1H-indole (7d).5a

Starting material 6 (112 mg, 0.490 mmol) was dissolved in toluene (2.5 mL). 5-Bromoindole (106 mg, 0.539 mmol) was then added, followed by pyridinium triflate (11 mg, 0.049 mmol). The reaction was stirred for 2 h when it reached completion as monitored by TLC. The mixture was then concentrated in vacuo to obtain the crude material, which was purified by flash column chromatography with 95:5 hexanes/EtOAc to give product 7d in 70% yield (138 mg, 0.339 mmol) as a clear oil. Rf: 0.79 (70:30 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.92 (s, 1H), 7.71 (s, 1H), 7.26–7.23 (m, 1H), 7.19 (d, J = 8.6 Hz, 1H), 6.99 (d, J = 3.1 Hz, 1H), 3.95–3.88 (m, 1H), 2.42–2.23 (m, 3H), 1.87–1.77 (m, 1H), 1.69 (s, 3H), 0.82 (s, 9H), −0.03 (s, 3H), −0.09 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) 147.4, 135.1, 128.9, 124.4, 122.7, 121.9, 119.2, 113.9, 112.3, 112.3, 42.4, 32.2, 29.7, 25.6, 25.3, 18.1, 12.4, −4.2, −4.3.
3-(2-((tert-Butyldimethylsilyl)oxy)-3-methylcyclopent-2-en-1-yl)-1H-indole-4-carbonitrile.5a

Starting material 6 (125 mg, 0.547 mmol) was dissolved in toluene (2.7 mL). 4-Cyanoindole (85 mg, 0.601 mmol) was then added, followed by pyridinium triflate (12 mg, 0.054 mmol). The reaction was stirred for 7 h when it reached completion as monitored by TLC. The mixture was then concentrated in vacuo to obtain the crude material, which was purified by flash column chromatography with 95:5 hexanes/EtOAc to give product 7e in 65% yield (63 mg, 0.184 mmol) as a yellow solid. Rf: 0.57 (80:20 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.30 (s, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.47 (d, J = 7.4 Hz, 1H), 7.22–7.17 (m, 1H), 7.16 (d, J = 2.4 Hz, 1H), 4.46–4.34 (m, 1H), 2.68–2.50 (m, 1H), 2.24 (t, J = 7.0 Hz, 2H), 1.77–1.70 (m, 1H), 1.69 (s, 3H), 0.84 (s, 9H), 0.06 (s, 3H), −0.10 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ (ppm) 146.7, 136.6, 126.9, 126.1, 124.9, 121.1, 119.6, 119.3, 115.8, 114.9, 101.9, 41.6, 31.7, 31.4, 25.6, 18.1, 12.3, −4.3, −4.3.
Methyl 3-(2-((tert-Butyldimethylsilyl)oxy)-3-methylcyclopent-2-en-1-yl)-1H-indole-5-carboxylate (7f).5a

Starting material 6 (112 mg, 0.490 mmol) was dissolved in toluene (2.4 mL). Methyl indole-5-carboxylate (91 mg, 0.520 mmol) was then added, followed by pyridinium triflate (11 mg, 0.047 mmol). The reaction was stirred for 3 h when it reached completion as monitored by TLC. The mixture was then concentrated in vacuo to obtain the crude material, which was purified by flash column chromatography with 95:5 hexanes/EtOAc to give product 7f in 83% yield (151 mg, 0.392 mmol) as a clear oil. Rf: 0.69 (70:30 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.36 (s, 1H), 8.12 (s, 1H), 7.88 (d, J = 6.2, 1.6 Hz, 1H), 7.34 (d, J = 7.7 Hz, 1H), 7.05 (s, 1H), 4.04–3.97 (m, 1H), 3.93 (s, 3H), 2.48–2.21 (m, 3H), 1.81 (m, 1H), 1.69 (s, 3H), 0.81 (s, 9H), −0.03 (s, 3H), −0.11 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) 147.4, 139.1, 126.9, 123.1, 122.6, 122.4, 121.1, 121.0, 114.1, 110.6, 51.8, 42.3, 32.2, 30.0, 25.6, 18.1, 12.4, −4.2, −4.3.
3-Methyl-2-((triethylsilyl)oxy)cyclopent-2-en-1-ol (8b).

2-Hydroxy-3-methylcyclopent-2-enone (318 mg, 2.841 mmol) was dissolved in CH2Cl2 (2.8 mL). Subsequently, imidazole (289 mg, 4.261 mmol) followed by TESCl (0.5 mL, 3.168 mmol) were added. The reaction mixture was stirred at room temperature for 19 h when it reached completion as monitored by TLC. The reaction mixture was cooled to 0 °C and quenched with 2 M HCl (3 mL). Upon separation of the two layers, the aqueous layer was extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by flash column chromatography with 90:10 hexanes/EtOAc to give product S-2 in 96% yield (614 mg, 2.71 mmol) as a colorless oil. Rf: 0.56 (80:20 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 2.42–2.38 (m, 2H), 2.34–2.30 (m, 2H), 1.95 (s, 3H), 0.97 (t, J = 7.9 Hz, 9H), 0.72 (q, J = 8.0 Hz, 6H). 13C{1H} NMR (125 MHz, CDCl3): δ (ppm) 202.9, 151.2, 149.7, 32.1, 26.9, 14.7, 6.7, 5.6. IR (cm−1): 2954, 2911, 1707, 1641, 1459, 1395, 1343, 1215, 1115, 1008, 839, 729. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H23O2Si, 227.1461; found, 227.1464.

Starting material S-2 (387 mg, 1.72 mmol) was dissolved in dry CH2Cl2 (8.6 mL) and cooled to 0 °C. Diisobutylaluminium (DIBAL) (3.4 mL, 1 M in heptane) was then added dropwise and the mixture was allowed to warm to room temperature. The reaction was stirred for 2 h when it reached completion as monitored by TLC. The reaction mixture was cooled to 0 °C and quenched with 1 M HCl (10 mL). Upon separation of the two layers, the aqueous layer was extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by flash column chromatography with 95:5 hexanes/EtOAc to give product 8b in 60% yield (235 mg, 1.03 mmol) as a colorless oil. Rf: 0.50 (80:20 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3): δ (ppm) 4.52–4.45 (m, 1H), 2.36–2.26 (m, 1H), 2.26–2.16 (m, 1H), 2.13–2.04 (m, 1H), 1.69–1.63 (m, 1H), 1.60 (s, 3H), 1.00 (t, J = 7.9 Hz, 9H), 0.70 (q, J = 8.1 Hz, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ (ppm) 148.0, 117.0, 76.2, 30.5, 30.4, 12.2, 6.7, 5.4. IR (cm–1): 2414, 2995, 2910, 2878, 1711, 1458, 1412, 1237, 1192, 1071, 1007, 736. HRMS (ESI-TOF) m/z: [M − H2O]+ calcd for C12H22OSi, 210.1434; found, 210.1440.
(±)-2-(tert-Butyldiphenylsilyloxy)-3-methylcyclopent-2-enol (8c).

2-Hydroxy-3-methylcyclopent-2-enone (1.00 g, 8.93 mmol) was dissolved in dry CH2Cl2 (45 mL). Subsequently, imidazole (1.8 g, 26.73 mmol) followed by TBDPSCl (4.9 mL, 17.83 mmol) were added. The reaction mixture was stirred at room temperature for 72 h when it reached completion as monitored by TLC. The reaction mixture was cooled to 0 °C and quenched with 2 M HCl (15 mL). Upon separation of the two layers, the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo to give crude S-3.
Crude material S-3 (500 mg, 1.72 mmol) was dissolved in CH2Cl2 (7.5 mL) and cooled to 0 °C. DIBAL (2.3 mL, 1 M in toluene) was then added dropwise and the mixture was warmed to room temperature. The reaction was stirred for 1 h when it reached completion as monitored by TLC. The reaction mixture was cooled to 0 °C and quenched with 1 M HCl (10 mL). Upon separation of the two layers, the aqueous layer was extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by flash column chromatography with 95:5 hexanes/EtOAc to give product 8c in 70% yield (350 mg, 0.993 mmol) as a colorless oil. Rf: 0.63 (70:30 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.81–7.74 (m, 4H), 7.46–7.39 (m, 6H), 4.31–4.24 (m, 1H), 2.75 (br s, 1H), 2.32–2.27 (m, 1H), 2.07–1.97 (m, 2H), 1.59 (s, 3H), 1.58–1.54 (m, 1H), 1.15 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3): δ (ppm) 148.1, 135.6, 135.6, 135.5, 135.0, 134.0, 133.9, 130.2, 130.2, 129.7, 128.0, 127.8, 117.4, 75.5, 30.7, 29.8, 26.9, 26.7, 19.7, 12.6. IR (cm−1): 2930, 2855, 1687, 1427, 1250, 1110, 860, 740, 698. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H29O2Si, 353.1931; found, 353.1934.
2-(Benzyloxy)-3-methylcyclopent-2-enol (8d).

2-Hydroxy-3-methylcyclopent-2-enone (500 mg, 4.46 mmol) was dissolved in dry acetone (22 mL). K2CO3 (1.23 g, 8.92 mmol) and then benzyl bromide (0.82 mL, 6.69 mmol) were added. The reaction mixture was stirred at room temperature for 24 h when it reached completion, as monitored by TLC. After concentrating the reaction mixture in vacuo, the crude residue was partitioned in 1:1 EtOAc/H2O (100 mL). The aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic layers were then washed with brine, dried over Na2SO4, and concentrated in vacuo to give crude S-4 as yellow oil.

Crude material S-4 (870 mg) was dissolved in dry CH2Cl2 (22 mL) and cooled to 0 °C. DIBAL (6.5 mL, 1 M in toluene) was then added dropwise. The reaction mixture was warmed to room temperature. The reaction was stirred for 1 h when it reached completion as monitored by TLC. The reaction was cooled to 0 °C and then quenched with H2O (20 mL). Upon separation of the two layers, the aqueous layer was extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by flash column chromatography with 80:20 hexanes/EtOAc to give product 8d in 71% yield (650 mg, 3.18 mmol) as a colorless oil. Rf: 0.32 (80:20 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.39–7.29 (m, 5H), 5.04 (d, J = 12.0 Hz, 1H), 4.89 (d, J = 12.0 Hz, 1H), 4.79–4.71 (m, 1H), 2.37–2.32 (m, 1H), 2.26–2.17 (m, 1H), 2.11–2.07 (m, 1H), 1.85 (br s, 1H), 1.72–1.69 (m, 1H), 1.66 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ (ppm) 151.4, 138.4, 128.6, 127.9, 127.6, 118.7, 73.7, 71.3, 31.1, 31.0, 12.4. IR (cm−1): 3368, 2935, 2849, 1691, 1441, 1333, 1217, 1041, 995, 633. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H16NaO2, 227.1043; found, 227.1044.
2-Methoxy-3-methylcyclopent-2-enol (8e).5b

2-Hydroxy-3-methylcyclopent-2-enone (4.00 g, 35.71 mmol) was dissolved in dry acetone (180 mL). K2CO3 (9.90 g, 71.43 mmol) and then methyl iodide (4.2 mL, 71.43 mmol) were added. The reaction mixture was stirred at room temperature for 48 h when it reached completion, as monitored by TLC. After concentrating the reaction mixture in vacuo, the crude residue was partitioned in 1:1 EtOAc/H2O (200 mL). The aqueous layer extracted with EtOAc (3 × 50 mL). The combined organic layers were then washed with brine, dried over Na2SO4, and concentrated in vacuo to give crude S-5 as yellow oil.

Crude ketone S-4 (4.50 g) was dissolved in dry CH2Cl2 (180 mL) and cooled to 0 °C. DIBAL (53 mL, 1 M in toluene) was then added dropwise. The reaction mixture was warmed to room temperature. The reaction was stirred for 1 h when it reached completion as monitored by TLC. The reaction was cooled to 0 °C and then quenched with H2O (100 mL). Upon separation of the two layers, the aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by flash column chromatography with 80:20 hexanes/EtOAc to give product 8e in 71% yield (3.5 g, 27.31 mmol) as a colorless oil. Rf: 0.29 (70:30 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 4.82–4.73 (m, 1H), 3.71 (s, 3H), 2.38–2.32 (m, 1H), 2.26–2.19 (m, 1H), 2.12–2.06 (m, 1H), 1.71–1.68 (m, 2H), 1.65 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ (ppm) 152.3, 116.9, 73.6, 57.4, 31.3, 31.0, 12.3.
3-(3-Methyl-2-((triethylsilyl)oxy)cyclopent-2-en-1-yl)-1H-indole (9b).

Starting material 8b (60 mg, 0.265 mmol) was dissolved in toluene (1.3 mL). Indole (34 mg, 0.291 mmol) was then added, followed by pyridinium triflate (6 mg, 0.026 mmol). The reaction was stirred for 7 h when it reached to completion as monitored by TLC. The mixture was then concentrated in vacuo to obtain the crude material, which was purified by flash column chromatography with 98:2 hexanes/EtOAc to give product 9b in 61% yield (53 mg, 0.162 mmol) as a clear oil. Rf: 0.72 (80:20 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.89 (s, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.17 (t, J = 8.0 Hz, 1H), 7.09 (t, J = 7.9 Hz, 1H), 6.99 (d, J = 2.2 Hz, 1H), 3.97 (s, 1H), 2.41–2.31 (m, 2H), 2.28–2.20 (m, 1H), 1.91–1.83 (m, 1H), 1.68 (s, 3H), 0.82 (t, J = 7.9 Hz, 9H), 0.49 (q, J = 7.9 Hz, 6H). 13C{1H} NMR (125 MHz, CDCl3): δ (ppm) 148.1, 136.6, 127.2, 121.6, 121.4, 119.3, 119.0, 113.6, 110.9, 42.7, 32.2, 29.8, 12.3, 6.6, 5.3. IR (cm−1): 3416, 2954, 2910, 2875, 1686, 1456, 1325, 1268, 1242, 1212, 861, 833, 738. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H30NOSi, 328.2091; found, 328.2085.
3-(2-((tert-Butyldiphenylsilyl)oxy)-3-methylcyclopent-2-en-1-yl)-1H-indole (9c).

Starting material 8c (150 mg, 0.425 mmol) was dissolved in toluene (2.1 mL). Indole (55 mg, 0.467 mmol) was then added, followed by pyridinium triflate (10 mg, 0.043 mmol). The reaction was stirred for 168 h when it reached to completion as monitored by TLC. The mixture was then concentrated in vacuo to obtain the crude material, which was purified by flash column chromatography with 98:2 hexanes/EtOAc to give product 9c in 64% yield (123 mg, 0.273 mmol) as a brown oil. Rf: 0.73 (70:30 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.72 (s, 1H), 7.63–7.60 (m, 2H), 7.41–7.34 (m, 4H), 7.27 (h, J = 7.7 Hz, 5H), 7.17–7.12 (m, 1H), 7.11–7.06 (m, 2H), 7.06–7.01 (m, 1H), 6.72 (s, 1H), 3.72–3.65 (m, 1H), 2.35–2.25 (m, 1H), 2.21–2.10 (m, 2H), 1.81–1.73 (m, 1H), 1.54 (s, 3H), 0.94 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) 147.7, 136.7, 135.5, 135.3, 134.1, 133.9, 129.5, 129.2, 127.3, 127.0, 121.7, 121.5, 119.5, 119.1, 118.8, 113.4, 110.7, 77.2, 42.4, 32.3, 29.4, 26.6, 19.5, 12.7. IR (cm–1): 3424, 3050, 2956, 2855, 1456, 1380, 1325, 1213, 1090, 1009, 868, 738, 612, 423. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C30H34NOSi, 452.2410; found, 452.2404.
2-(1H-Indol-3-yl)-5-methylcyclopentan-1-one (S-6) and 2-(1HIndol-3-yl)-2-methylcyclopentan-1-one (S-7).

Starting material 8d (111 mg, 0.547 mmol) was dissolved in toluene (2.7 mL). Indole (70 mg, 0.602 mmol) was then added, followed by pyridinium triflate (12 mg, 0.055 mmol). The reaction was stirred for 5 h when it reached completion as monitored by TLC. The mixture was quenched with 2 M HCl (1 mL) and stirred for 12 h. After which, the mixture was diluted with deionized (DI) H2O (2 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The crude material was then purified by flash column chromatography with 80:20 hexanes/EtOAc to give products S-6 and S-7 as a 2:1 inseparable mixture of regioisomers in 73% yield (85 mg, 0.399 mmol) as a colorless oil. Rf: 0.45 (80:20 hexanes/EtOAc). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.12 (s, 3H), 7.71 (d, J = 8.4 Hz, 2H), 7.58 (dd, J = 24.0, 7.9 Hz, 3H), 7.35–7.29 (m, 2H), 7.22–7.17 (m, 4H), 7.15–7.07 (m, 3H), 7.01–6.94 (m, 1H), 6.95–6.86 (m, 3H), 3.79 (t, J = 7.7 Hz, 1H), 3.59 (dd, J = 11.7, 8.9 Hz, 1H), 2.73–2.64 (m, 2H), 2.59–2.35 (m, 10H), 2.32–2.19 (m, 2H), 2.12–1.90 (m, 7H), 1.80–1.72 (m, 1H), 1.68–1.58 (m, 2H), 1.53 (s, 3H), 1.25 (d, J = 6.6 Hz, 3H), 1.23 (d, J = 7.2 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) 221.2, 221.1, 220.7, 137.1, 136.4, 126.7, 126.6, 125.3, 122.1, 122.0, 121.9, 121.8, 121.8, 121.6, 121.5, 121.5, 120.4, 119.4, 119.3, 119.2, 119.2, 117.1, 113.3, 111.5, 111.3, 111.3, 49.0, 46.9, 46.0, 44.4, 42.8, 37.8, 37.3, 30.2, 29.5, 29.3, 28.7, 23.0, 19.1,15.7, 14.7.
2-(1H-Indol-3-yl)-5-methylcyclopentan-1-one (S-6) and 2-(1H-Indol-3-yl)-2-methylcyclopentan-1-one (S-7).
Starting material 8e (163 mg, 1.27 mmol) was dissolved in toluene (6.4 mL). Indole (164 mg, 1.40 mmol) was then added, followed by pyridinium triflate (29 mg, 0.127 mmol). The reaction was stirred for 4 h when it reached completion as monitored by TLC. The mixture was quenched with 2 M HCl (1 mL) and stirred for 12 h. After which, the mixture was diluted with DI H2O (2 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic was dried over Na2SO4 and concentrated in vacuo. The crude material was then purified by flash column chromatography with 80:20 hexanes/EtOAc to give products S-6 and S-7 as a 1:1 inseparable mixture of regioisomers in 80% yield (231 mg, 1.01 mmol) as a colorless oil.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM127649. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Generous financial supports from Louisiana State University are gratefully acknowledged. A.H.C. thanks the Louisiana Board of Regents for the Graduate Fellowship (LEQSF(2015-20)-GF-02). R.K. and C.G.B. are grateful to LSU HPC and the Louisiana Optical Network Initiative for computer time. A.M. is grateful for computer time of the CIMENT infrastructure (ANR-10-EQPX-29-01) and to the labex ARCANE (ANR-11-LABX-0003-01) for funding. This material is based upon research (C.G.B.) supported by the Chateaubriand Fellowship of the Office for Science & Technology of the Embassy of France in the United States.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.9b00853.
1H and 13C NMR spectra and computational methods (PDF)
Computational data for all relevant structures (ZIP)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Frontier AJ; Collison C Tetrahedron 2005, 61, 7577–7606. [Google Scholar]; (b) Grant TN; Rieder CJ; West FG Chem. Commun 2009, 5676–5688. [DOI] [PubMed] [Google Scholar]; (c) Nakanishi W; West FG Curr. Opin. Drug Discovery Dev 2009, 12, 732–751. [PubMed] [Google Scholar]; (d) Shimada N; Stewart C; Tius MA Tetrahedron 2011, 67, 5851–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Vaidya T; Eisenberg R; Frontier AJ ChemCatChem 2011, 3, 1531–1548. [Google Scholar]
- (2).(a) Lohse AG; Hsung RP Chem.—Eur. J 2011, 17, 3812–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Harmata M Chem. Commun 2010, 46, 8904–8922. [DOI] [PubMed] [Google Scholar]; (c) Harmata M Chem. Commun 2010, 46, 8886–8903. [DOI] [PubMed] [Google Scholar]; (d) Foley DA; Maguire AR Tetrahedron 2010, 66, 1131–1175. [Google Scholar]; (e) Battiste MA; Pelphrey PM; Wright DL Chem.—Eur. J 2006, 12, 3438–3447. [DOI] [PubMed] [Google Scholar]; (f) Harmata M Acc. Chem. Res 2001, 34, 595–605. [DOI] [PubMed] [Google Scholar]
- (3).(a) Krenske EH; He S; Huang J; Du Y; Houk KN; Hsung RP J. Am. Chem. Soc 2013, 135, 5242–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li H; Hughes RP; Wu JJ Am. Chem. Soc 2014, 136, 6288–6296. [DOI] [PubMed] [Google Scholar]; (c) Li H; Wu J Synthesis 2015, 47, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Tang Q; Chen X; Tiwari B; Chi YR Org. Lett 2012, 14, 1922–1925. [DOI] [PubMed] [Google Scholar]; (b) Wal MNV; Dilger AK; MacMillan DW C. Chem. Sci 2013, 4, 3075–3079. [Google Scholar]; (c) Luo J; Zhou H; Hu J; Wang R; Tang Q RSC Adv. 2014, 4, 17370–17377. [Google Scholar]; (d) Luo J; Jiang Q; Chen H; Tang Q RSC Adv. 2015, 5, 67901–67908. [Google Scholar]; (e) Liu C; Oblak EZ; Vander Wal MN; Dilger AK; Almstead DK; MacMillan DW C. J. Am. Chem. Soc 2016, 138, 2134–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Ayala CE; Dange NS; Fronczek FR; Kartika R Angew. Chem., Int. Ed 2015, 54, 4641–4645. [DOI] [PubMed] [Google Scholar]; (b) Dange NS; Stepherson JR; Ayala CE; Fronczek FR; Kartika R Chem. Sci 2015, 6, 6312–6319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Malone JA; Cleveland AH; Fronczek FR; Kartika R Org. Lett 2016, 18, 4408–4411. [DOI] [PubMed] [Google Scholar]; (d) Kartika R; Stepherson J; Ayala C; Dange N Synlett 2016, 27, 320–330. [Google Scholar]; (e) Stepherson JR; Fronczek FR; Kartika R Chem. Commun 2016, 52, 2300–2303. [DOI] [PubMed] [Google Scholar]; (f) Malone JA; Van Houten JP; Ganiu MO; Nepal B; Kartika RJ Org. Chem 2017, 82, 10659–10664. [DOI] [PubMed] [Google Scholar]
- (6).Attempts to directly characterize silyloxyallyl cation 11a by in situ 1H NMR observation via treatment of substrate 8a with pyridinium triflate in deuterated toluene was unfruitful.
- (7). See Supporting Information for detailed calculations.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.