

Abstract
Background
Riluzole is approved for the treatment of amyotrophic lateral sclerosis in most countries. Questions persist about its clinical utility because of high cost and modest efficacy.
Objectives
To examine the efficacy of riluzole in prolonging survival and in delaying the use of surrogates (tracheostomy and mechanical ventilation) to sustain survival, and to assess the effect of riluzole upon functional health.
Search methods
We searched the Cochrane Neuromuscular Disease Group Specialized Register (20 April 2011), the Cochrane Central Register of Controlled Trials (CENTRAL) (2011, Issue 2), MEDLINE (1966 to April 2011), EMBASE (1980 to May 2011) and made enquiries of authors of trials, Aventis (manufacturer of riluzole) and other experts in the field.
Selection criteria
Types of studies: randomized controlled trials Types of participants: adults with a diagnosis of amyotrophic lateral sclerosis Types of interventions: treatment with riluzole or placebo Types of outcome measures: Primary: pooled hazard ratio of tracheostomy‐free survival over all time points with riluzole 100 mg. Secondary: per cent mortality with riluzole 50 mg, 100 mg and 200 mg; neurologic function, muscle strength and adverse events.
Data collection and analysis
One author performed data extraction and two other authors checked them. One author checked the data and entered them into the computer. The other authors verified the data entry. We obtained missing data from the trial authors whenever possible.
Main results
The four trials examining tracheostomy‐free survival included a total of 974 riluzole‐treated patients and 503 placebo‐treated patients. No new randomized controlled trials were found when we updated the searches for this update in 2011. The methodological quality was acceptable and three trials were easily comparable, although one trial (169 participants) included older patients in more advanced stages of amyotrophic lateral sclerosis and one (195 participants) had multiple primary endpoints. Riluzole 100 mg per day provided a benefit for the homogeneous group of patients in the first two trials (hazard ratio (HR) 0.80, 95% confidence internal (CI) 0.64 to 0.99, P= 0.042) and there was no evidence of heterogeneity (P = 0.33). When the third trial (which included older and more seriously affected patients) was added, there was evidence of heterogeneity (P < 0.0001) and the overall treatment effect was reduced but still significant (HR 0.84, 95% CI 0.698 to 0.997, P= 0.046). This represented a 9% gain in the probability of surviving one year (49% in the placebo and 58% in the riluzole group), and increased median survival from 11.8 to 14.8 months. There was a small beneficial effect on both bulbar and limb function, but not on muscle strength. A three‐fold increase in serum alanine transferase was more frequent in riluzole‐treated patients than controls (mean difference 2.62, 95% CI 1.59 to 4.31).
Authors' conclusions
Riluzole 100 mg daily is reasonably safe and probably prolongs median survival by about two to three months in patients with amyotrophic lateral sclerosis.
Plain language summary
Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND)
Amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) is a fatal neurological disease which produces paralysis of the limb, swallowing and breathing muscles. There is no available treatment to stop or reverse its progressive course. In this review, we examine the evidence from four randomized clinical trials involving 1477 people with ALS. The methodological quality of the trials was acceptable and three of the trials were easily comparable (although one of them included older patients with more advanced ALS). The searches for this review were last updated in 2011, when we found no new randomized controlled trials. The results indicate that riluzole 100 mg probably prolongs median survival in people with ALS by two to three months and the safety of the drug is not a major concern. The evidence from randomized controlled trials indicates that participants taking riluzole probably survive longer than participants taking placebo. The beneficial effects are very modest and the drug is expensive. There was a small beneficial effect on both bulbar and limb function, but not on muscle strength. Adverse effects from riluzole are relatively minor and for the most part reversible after stopping the drug.
Summary of findings
for the main comparison.
| Riluzole compared with placebo for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) | ||||||
|
Patient or population: patients with ALS/MND Settings: large European and US ALS centers Intervention: riluzole Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Riluzole | |||||
| Mortality or tracheostomy at 12 months (pooled hazard ratio)(riluzole 100 mg) | 510 per 1000 | 419 per 1000 (367 to 475) | HR 0.83 (0.70 to 1.00) |
1282 (3 studies) |
++++ high | When the results of the third trial of older and more seriously affected patients were included, the results were still significant (P = 0.046). This relative effect represents a three month increase in median survival from 11.8 to 14.8 months |
| Per cent mortality at 12 months (riluzole 100 mg) | 440 per 1000 | 343 per 1000 (286 to 405) | RR 0.78 (0.65 to 0.92) | 799 (3 studies) | ++++ high | |
|
Rate of decline of Norris Scale ‐ Norris Limb
(riluzole 100 mg) Follow up: 12 months |
23.1 per year weighted mean rate of decline | The mean rate of decline of Norris Scale ‐ Norris Limb in the intervention groups was 3.94 slower (7.25 to 0.64 slower) | 731 (3 studies) | ++++ high | Although the change in function was not significant in the individual studies, the pooled data show a slower decline of limb function in the treated group | |
|
Rate of decline of Norris Scale ‐ Norris Bulbar
(riluzole 100 mg) Follow up: 12 months |
11.1 per year weighted mean rate of decline | The mean rate of decline of Norris Scale ‐ Norris Bulbar in the intervention groups was 2.06 slower (3.86 to 0.27 slower) | 742 (3 studies) | ++++ high | Although the change in function was not significant in the individual studies, the pooled data show a slower decline of bulbar function in the treated group | |
| Adverse event: nausea | 91 per 1000 | 142 per 1000 (96 to 207) | RR 1.55 (1.06 to 2.28) | 801 3 studies |
++++ high | |
| Adverse event: asthenia | 116 per 1000 | 175 per 1000 (124 to 246) | RR 1.5 (1.07 to 2.12) | 801 3 studies |
++++ high | |
| Adverse event: 3‐fold increase in alanine transferase | 49 per 1000 | 129 per 1000 (78 to 211) | RR 2.62 (1.59 to 4.31) | 801 3 studies |
++++ high | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Background
Amyotrophic lateral sclerosis (ALS) is a progressive degenerative motor neuron disease characterized by weakness in limb and bulbar muscles with atrophy, spasticity, weight loss and ultimately respiratory failure. The incidence is approximately 2 per 100,000 per annum, and it is estimated that there are about 25,000 prevalent patients in North America (McGuire 1996). The disease is virtually always fatal and approximately half of patients die within three to four years after the onset of symptomatic weakness. There is a combination of upper motor neuron and lower motor neuron abnormalities, and relentless and nearly linear progression of impaired function in almost all patients.
The burden of disease upon patients, family members and caregivers is substantial with increasing cost associated with increasing disability and the need for assisted medical care. At present in ALS, disease‐specific therapy can at best only slow disease progression and does not stabilize or improve the underlying disorder.
There have been many controlled clinical trials of disease‐specific therapy for ALS. Until the mid 1990s, all were negative. Emerging evidence that chronic glutamate excitotoxicity may accumulate to toxic levels and contribute to neuronal death in ALS provided a rational basis for undertaking a clinical trial with riluzole, a drug with complex effects, but which appears to block the presynaptic release of glutamate (Rothstein 1996). The first randomized controlled trial (RCT) demonstrated a modest increase in survival in treated participants compared to placebo controls (Bensimon 1994). However, many questions were raised by this study, especially in view of the disproportionate benefit observed in participants with onset of disease in bulbar (oropharyngeal) as opposed to limb muscles (Rowland 1994).
To address these concerns, a much larger dose‐ranging study was carried out and again there was a small but statistically significant prolongation of survival in participants receiving the intermediate and high dose of riluzole (Lacomblez 1996). A third trial was also carried out in France and Belgium involving people with more advanced ALS who did not qualify for the large trial (Bensimon 2002). In this study, there was no significant survival advantage from riluzole. A fourth trial was carried out in Japan with multiple outcome measures that differed from the other three trials (Yanagisawa 1997). This study, which involved small numbers of participants and different end points, was negative. The results from this trial are included in this review although the difference in outcomes and the lack of survival‐specific data prevented us from including it in the meta‐analyses.
Subsequently, the drug was approved in the USA and just recently in Australia and Canada and in a many European countries.
A number of concerns about the therapeutic effect persist: the lack of benefit observed for some secondary measures of efficacy, the modest prolongation of survival (on average a few months), and the relatively high cost of the drug (approximately $10,000 per year in the US, and approximately £4056 per year in the UK). A Practice Advisory was issued in 1997 by the Quality Standards Subcommittee of the American Academy of Neurology recommending that the drug should be offered to patients, but with some restrictions (Neurology 1997). Some published reviews have favored the use of riluzole, but their conclusions were not based on a systematic review of the evidence (Hugon 1996a; Hugon 1996b; Meininger 1997; Miller 1996; Wokke 1996). The Trent Institute report on purchasing did not recommend riluzole, expressing concern about cost effectiveness (Chilcott 1997). A report from Wessex reached a similar conclusion (Booth‐Clibborn 1997). The Committee for Proprietary Medicinal Products (CPMP) of the European Agency for the evaluation of additional products reported that riluzole had demonstrated a modest prolongation of survival (CPMP 1999). Their report indicated that there was adequate evidence of efficacy of riluzole and a satisfactory benefit profile to recommend marketing authorization. The National Institute for Clinical Effectiveness recommended riluzole use in the UK, based upon the systematic review from the Midlands Group as well as input from experts and user groups (HTA 2001; NICE 2001). Recently an evidence‐based practice parameter issued by the American Academy of Neurology, which updated the Practice Advisory of 1997, recommended riluzole to slow disease progression for patients with ALS (Miller 2009). The present review was initially published in the Cochrane Library including only the first two clinical trials (Bensimon 1994; Lacomblez 1996) and subsequently we fully included a third trial in the analysis with partial data from a fourth. We identified no new trials for this update.
The goal of the present review is to examine systematically all evidence from RCTs relating to the effects of riluzole in ALS, in order to supply the best evidence currently available on which to base clinical decision making and future research.
Objectives
To examine the efficacy of riluzole in prolonging survival and in delaying the use of surrogates (tracheostomy and mechanical ventilation) to sustain survival, as well as assessing the effect of riluzole upon functional health.
Methods
Criteria for considering studies for this review
Types of studies
RCTs involving riluzole treatment of ALS.
Types of participants
Adults with a clinical diagnosis of ALS.
Types of interventions
Treatment with oral riluzole or placebo.
Types of outcome measures
Primary outcomes
Pooled hazard ratio (HR) based on per cent mortality (or tracheostomy) for 100 mg riluzole versus placebo over all time points.
Secondary outcomes
Risk ratios (RRs) based on per cent mortality at 12 and 18 months for 100 mg riluzole versus placebo.
RRs based on per cent mortality as a function of time at 12 months ‐ all doses of riluzole versus placebo.
Muscle strength assessed by manual muscle testing.
Functional scales.
Quality of life of patients and caregivers.
Adverse effects from riluzole.
Search methods for identification of studies
We searched the Cochrane Neuromuscular Disease Group Specialized Register (11 April 2011) with 'amyotrophic lateral sclerosis' OR 'motor neuron disease' OR 'motor neurone' OR 'motoneurone disease' as the search terms. We also searched the Cochrane Central Register of Controlled Trials (CENTRAL) (2011, Issue 2 in the Cochrane Library), MEDLINE (January 1966 to April 2011) and EMBASE (January 1980 to April 2011). For the original version of the review we contacted the company (Aventis) and the authors of trials identified to find additional published or unpublished data and to clarify issues concerning trial design and loss of patients to follow up. We also obtained the reviews of the Food and Drug Administration, the Trent Institute (UK) (Chilcott 1997), the European Agency for the Evaluation of Medicinal Products (UK) (CPMP 1999), Booth‐Clibborn 1997, Chilcott 1997, and HTA 2001 and checked their references.
Electronic search strategies
See Appendix 1 (CENTRAL), Appendix 2 (MEDLINE) and Appendix 3 (EMBASE).
Data collection and analysis
Selection of studies
Two review authors checked titles and abstracts identified from the searches. The review authors obtained full texts of all potentially relevant studies for independent assessment by all authors. The authors decided which trials fitted the inclusion criteria and graded their methodological quality. The review authors contacted the study authors for clarification of data where necessary. The authors resolved disagreements about inclusion criteria and methodological quality by discussion.
Data extraction and management
One review author performed data extraction and two other authors performed checks. One author entered data into the Cochrane Collaboration statistical software Review Manager 2011. The other authors verified the data entry. We obtained missing data from the trial authors whenever possible.
Assessment of risk of bias in included studies
Two review authors independently assessed the risk of bias in included studies according to methods in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008). The studies were graded in the following domains: adequate sequence generation, allocation concealment, blinding, incomplete outcome data addressed, free of selective reporting and free of other bias. For 'other bias' we took into account, for example, baseline comparison of experimental groups and explicit diagnostic criteria. Studies were graded 'Low risk' when they were at low risk of bias, 'High risk' when the risk of bias was high and 'Unclear' when the criterion did not apply or when the risk of bias was unknown, for example when there was too little information to make a judgment.
Data synthesis
For the primary outcome variable, we calculated an overall measure of treatment efficacy which combined survival results at different time points. This measure is based on estimating a pooled HR (i.e. risk of death for treated divided by risk of death for controls) for each study and then calculating a weighted average of the pooled HR for each study. The weights are inversely proportional to the variances of the natural logs of the pooled HRs. The methods are described in Parmar 1998. We used life table methods to estimate survival at three‐month intervals for each study and each dose of riluzole. We pooled data from different studies (and different doses) and applied life table methods to the pooled data to obtain summary survival curves for combined treated and for combined control participants across different studies and doses. We estimated median survival for treated and for control participants by interpolation from the survival curves. The only dose of riluzole that was included in all trials was 100 mg, therefore we chose this dose for the intervention comparisons as the primary outcome measure. We also evaluated, as a secondary outcome measure chosen for this review, survival at 12 months, because it was the longest time point common to all studies. In addition, we created separate tables for each time point.
Tracheostomy was a surrogate endpoint for survival but there were no criteria which stipulated when tracheostomy should be performed. Timing of tracheostomy is a variable of patient care that may influence study outcome. Since a very small number of participants received tracheostomy, it appears unlikely that this influenced the results. The number of participants who had tracheostomy or intubation was low and balanced (riluzole versus placebo: 17 versus 16) when results from the two main studies were pooled (Bensimon 1994; Lacomblez 1996). The mortality, on the other hand, was clearly better in the riluzole group (124 deaths in the riluzole group and 156 deaths in the placebo group). Pulmonary function tests were measured infrequently (six month intervals) and were not well standardized.
Manual muscle testing was carried out using the Medical Research Council system (Lacomblez 1989). Limb function and bulbar function were evaluated every two months with modified Norris Scales (Lacomblez 1989). Quality of life was not measured but health status was assessed using a classification of five discrete health states which reflect increasing impairment in activities of daily living (Riviere 1998).
For secondary outcomes, including survival at individual time points, we calculated a weighted treatment effect (using a fixed‐effect analysis) across trials using Review Manager 2011. We expressed results as RRs and 95% confidence intervals (CIs) for dichotomous outcomes, and mean difference (MD) and 95% CI for continuous outcomes. We performed analysis on both short‐term outcomes (adverse effects of riluzole, quality of life, strength and functional scale ratings) and long‐term outcomes survival.
We tested for heterogeneity of the results across studies based on predicting the number of deaths at different time points from a pooled hazard compared with predictions based on estimating separate hazards for each study.
We examined the effects of known prognostic factors on survival (Stambler 1998) including age, gender, anatomical site of onset, disease severity (forced vital capacity) and disease duration (defined as being from the onset of weakness to randomization).
For this (2011) update we included a 'Summary of findings' table for the following outcomes:
pooled HR based on per cent mortality or tracheostomy (riluzole 100 mg);
per cent mortality at 12 months (riluzole 100 mg);
rate of decline of Norris Scale ‐ Norris Limb (riluzole 100 mg);
rate of decline of Norris Scale ‐ Norris Bulbar (riluzole 100 mg);
adverse events (riluzole 100 mg): nausea, asthenia, raised alanine transferase.
Results
Description of studies
The number of papers found by the new, current strategies were: MEDLINE = 259 (52 new papers), EMBASE = 128 (29 new papers), Cochrane Neuromuscular Disease Group Specialized Register = 36 (1 new paper), CENTRAL = 40.
Four studies fulfilled the selection criteria. Of these, two were included in the original version of the review (Bensimon 1994; Lacomblez 1996) and two were added at a subsequent update (Bensimon 2002; Yanagisawa 1997). The review authors considered, but excluded, a total of twelve studies for possible eligibility, including one at this update (Meininger 2004).
Only three of the four included trials contained full data on tracheostomy‐free survival, these included results on a total of 876 riluzole‐treated participants and 406 placebo‐treated participants. The first was a smaller trial which compared riluzole 100 mg to placebo in 155 severely affected patients (Bensimon 1994). The second, a larger trial, was a dose ranging study comparing 50 mg, 100 mg and 200 mg riluzole with placebo in 959 participants (Lacomblez 1996). The third trial involved participants with more advanced disease (age > 75, duration of illness > 5 years, forced vital capacity (FVC) < 60%) and compared riluzole 100 mg with placebo in 168 participants (Bensimon 2002). The fourth trial involved 195 participants in Japan (Yanagisawa 1997), with inclusion criteria that were comparable to the first two trials. Unfortunately, full data on tracheostomy‐free survival are not available from this trial. Despite repeated attempts, we have been unable to obtain comparable data on survival. Thus the trial was not included in the meta‐analyses. The primary outcome measure in the Japanese trial was disease progression utilizing multiple measures including walking, arm function, tracheostomy, ventilation and tube feeding.
The reasons for exclusion were: seven studies were not RCTs (Arriada‐Mendicoa 1999; Couratier 2000; Desiato 1999; Kalra 1998; Pongratz 1999; Riviere 1998; Sojka 1997); two did not compare riluzole to placebo (Graf 2005; Palma 2000); one study was an audit of outcomes in patients taking riluzole versus those not taking the drug (Mitchell 2006); one was a population‐based comparison (Zoccolella 2007); and for one study (Meininger 2004) we were unable to obtain the key data for main outcomes. We included Meininger 2004 as a 'study awaiting classification' in the previous version of this review because the report did not provide complete data in terms of death at different time points for the placebo or riluzole groups. We requested from the authors more complete data about death and vital capacity changes over time, but as we were unable to obtain these data, we excluded the study in this 2011 update. However, the summary data are of interest and are briefly reviewed in the Discussion.
Risk of bias in included studies
In all four trials, participants were randomly assigned to receive riluzole or an identical appearing placebo and we considered the allocation concealment adequate. Participant as well as evaluator blinding was intended in both trials, but no information was provided to assess the effectiveness of patient or evaluator blinding.
Each trial used internationally accepted diagnostic criteria. All trials examined baseline demographic and clinical features, and there were no marked differences between placebo‐treated and riluzole‐treated participants at entry. In all trials, the study authors carried out a full intention‐to‐treat analysis, and all randomized participants were accounted for as dead or alive. In Bensimon 1994, 24 participants were included although they did not entirely meet inclusion criteria (details not available). The participants were distributed evenly between groups (11 riluzole, 13 placebo) and probably had little impact on the results. No protocol violations were reported in the other two trials. In the study by Lacomblez 1996, although the balance of clinical features was not different at baseline between the study groups, participants from France and Belgium were more severely affected at the start of the study than those from other regions. When these differences were adjusted for in the survival analysis, a beneficial treatment effect was still present. In Bensimon 2002, disease severity in participants (% FVC, duration of illness, age and weight) was more marked compared to the other two trials.
The risk of bias assessment took into account random sequence generation, allocation concealment, patient blinding, observer blinding, incomplete outcome data, selective reporting and other sources of bias such as explicit diagnostic criteria, explicit outcome criteria and how studies dealt with baseline differences of the groups. All studies explicitly stated their diagnostic criteria as being those of the World Federation of Neurology (Brooks 1994), thus we considered all three trials at low risk of bias for this measure. With the exceptions of sequence generation in the Lacomblez trial, which we graded 'Unclear risk' and incomplete outcome data in Yanagisawa 1997, which we assessed at 'High risk' of bias, we graded the studies at low risk of bias for all criteria. Figure 1 summarises the review authors' risk of bias assessments.
1.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
See: Table 1
Primary outcome measure: pooled hazard ratio based on per cent mortality (or tracheostomy) for patients on riluzole 100 mg versus placebo from three trials over all time points
Using methods for combining survival results from different studies (Parmar 1998), we found riluzole 100 mg per day provided a slight benefit for the homogeneous group of patients in the first two trials (P = 0.042, HR 0.80, CI 0.64 to 0.99) and there was no evidence of heterogeneity (P = 0.33). When the 100 mg results of the two trials were pooled, the median survival was 15.5 months for treated participants and 13.2 months for placebo, a 2.3 month difference. When the third trial (which included older and more seriously affected patients) was added, there was evidence of heterogeneity (P < 0.0001) and the overall treatment effect was reduced but still significant (HR 0.84, 95% CI 0.698 to 0.997, P value = 0.046). This represented a 9% gain in the probability of surviving one year (49% in the placebo and 58% in the riluzole group), and increased median survival from 11.8 to 14.8 months. The calculations are shown in additional Table 11. The graph at Figure 2 shows the pooled analyses for survival. The pooled HR for the three trials decreased from zero to six months and then increased from six to 18 months (data not shown). An overall assessment, based on the HRs for the three trials at all time points, indicated a 16% reduction in the HR for those taking 100 mg riluzole, which was not quite statistically significant (P = 0.056). This represents a 10% absolute increase in the probability of surviving for one year (56% in the placebo group and 66% in the riluzole group).
1. Parmar analysis calculations for three studies at 100 mg dose.
| Time (months) | Pooled Drug | Pooled placebo | ||||||||
| Alive | Deaths | Lost | Ri | Alive | Deaths | Lost | Ri | ln(HRi) | var(lnRHi) | |
| 0 | 385 | 32 | 1 | 378.5 | 406 | 41 | 0 | 386 | ‐0.230 | 0.050 |
| 3 | 362 | 41 | 0 | 341.5 | 365 | 48 | 1 | 341 | ‐0.161 | 0.039 |
| 6 | 321 | 23 | 0 | 309.5 | 316 | 45 | 0 | 294 | ‐0.724 | 0.059 |
| 9 | 298 | 38 | 0 | 279 | 271 | 44 | 0 | 249 | ‐0.260 | 0.041 |
| 12 | 260 | 27 | 17 | 238 | 227 | 25 | 11 | 209 | ‐0.053 | 0.068 |
| 15 | 216 | 40 | 82 | 155 | 191 | 26 | 84 | 136 | 0.300 | 0.050 |
| 18 | 94 | 0 | 0 | 94 | 81 | 4 | 0 | 79 | ‐ | ‐ |
| ln(HR) | ‐0.181 | |||||||||
| se(ln(HR)) | 0.091 | |||||||||
| z‐statistic | ‐1.991 | |||||||||
| 2‐sided P | 0.046 | |||||||||
| Pooled Hazard Ratio | 0.835 | |||||||||
| 95% Conf Int | (0.698,0.997) | |||||||||
2.
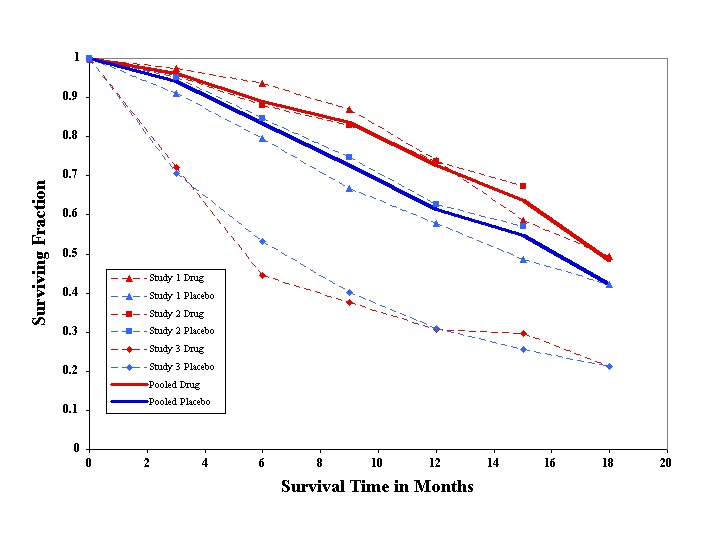
Pooled survival time in months. Solid lines show pooled results from the two trials that were homogeneous.
Results for combined doses
When the data from all three doses in Lacomblez 1996 were combined with those for Bensimon 1994 (only 100 mg), the estimated HR from these two homogeneous studies was 0.80 (95% CI 0.66 to 0.95, P = 0.013). Median survival was estimated to be 15.5 months in placebo and 17.2 months in treated participants, a difference of 1.7 months.
Secondary outcome measures
Risk ratio based on per cent mortality at 12 months for riluzole 100 mg versus placebo
In the earlier trial by Bensimon 1994, there was significantly lower per cent mortality in riluzole‐treated participants than in placebo‐treated participants at 12 months (RR 0.61, 95% CI 0.39 to 0.97). In Lacomblez 1996 there was lower per cent mortality in riluzole‐treated participants at 12 months (HR 0.71, 95% CI 0.54 to 0.92). In Bensimon 2002, there was no significant difference between riluzole‐treated participants and placebo‐treated participants (RR 0.99, 95% CI 0.79 to 1.25). From a combined analysis of all three trials, there was a survival advantage (P = 0.004) with riluzole at 12 months with RR of 0.78 (95% CI 0.65 to 0.92) (see Analysis 1.1).
1.1. Analysis.
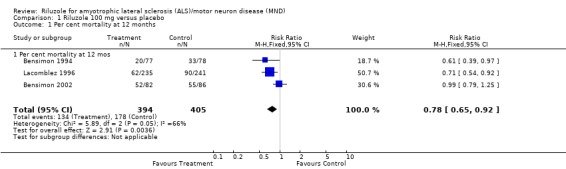
Comparison 1 Riluzole 100 mg versus placebo, Outcome 1 Per cent mortality at 12 months.
There was evidence of heterogeneity in the results (worse survival in the third trial) attributable to the inclusion of participants with more advanced ALS in the later trial by Bensimon et al. (Bensimon 2002. However, the combined results in terms of RR from all three trials were nearly the same as those based on the first two published trials. The HR for the combined data from all three studies was 0.78 (95% CI 0.65 to 0.92) at 12 months, compared to the RR for the combined data from the studies by Bensimon 1994 and Lacomblez 1996 (excluding the later trial Bensimon 2002) which was 0.68 (95% CI 0.54 to 0.86) at 12 months. Although the survival data show heterogeneity, there was virtually no impact of combining the studies on the overall RR results because of the relatively small size of the trial by Bensimon 2002. Bensimon 2002 did not show a beneficial effect, but because of the small size of the trial, this result should not be interpreted as proving that there is no effect in patients with advanced ALS.
Per cent mortality at 12 and 18 months ‐ riluzole 100 mg
In Bensimon 1994 there was significantly lower per cent mortality in riluzole‐treated participants than placebo‐treated participants at six, nine and twelve months but the differences were not significant at 3, 15 or 18 months. In Lacomblez 1996 there was a lower per cent mortality in riluzole‐treated participants at 9, 12 and 15 months but it was not significantly lower at 3, 6 or 18 months. In Bensimon 2002, there were no significant differences in mortality at any time point. From the combined analysis of all three trials there was a survival advantage with riluzole at 12 months but not at 18 months (see Analysis 1.1 and Analysis 2.1, Table 11 and Figure 2).
2.1. Analysis.
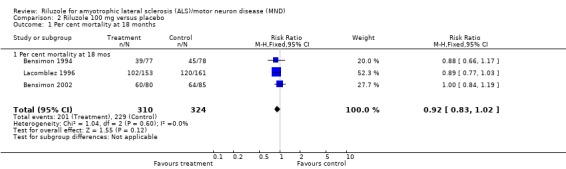
Comparison 2 Riluzole 100 mg versus placebo, Outcome 1 Per cent mortality at 18 months.
Per cent mortality ‐ all doses of riluzole
Pooled data from the 50 mg, 100 mg and 200 mg dose groups across all three trials showed no significant difference in mortality with riluzole compared to placebo at 12 months (see Analysis 3.1).
3.1. Analysis.
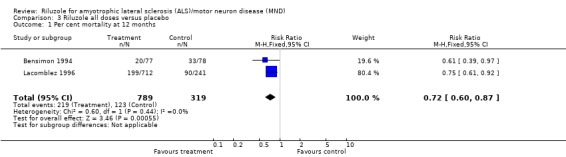
Comparison 3 Riluzole all doses versus placebo, Outcome 1 Per cent mortality at 12 months.
Muscle strength assessed by manual muscle testing
In Bensimon 1994 there was a beneficial effect upon strength (Medical Research Council Scale) in participants treated with riluzole compared to placebo (MD ‐11.50, 95% CI ‐21.69 to ‐1.36). However, in Lacomblez 1996 and in Bensimon 2002, no beneficial effect was seen (MD 0.40, 95% CI ‐4.18 to 4.98 for Lacomblez 1996; MD ‐3.90, 95% CI ‐15.00 to 7.20 for Bensimon 2002). When the data were combined, there was no positive effect from riluzole (MD ‐1.88, 95% CI ‐5.79 to 2.03) (see Analysis 4.1).
4.1. Analysis.
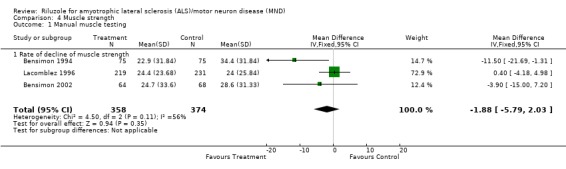
Comparison 4 Muscle strength, Outcome 1 Manual muscle testing.
Functional scales
Bulbar function
Although there was no beneficial effect of riluzole on bulbar function in any of the three trials, there was a beneficial effect in the combined data (MD ‐2.06, 95% CI ‐3.86 to ‐0.27) (see Analysis 5.1).
5.1. Analysis.
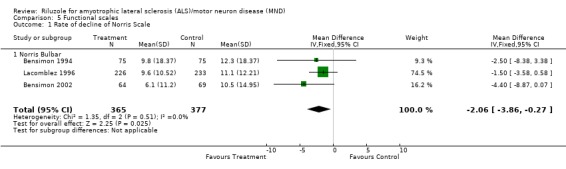
Comparison 5 Functional scales, Outcome 1 Rate of decline of Norris Scale.
Limb function
There was a small positive benefit on limb function in Lacomblez 1996 (MD ‐4.00, 95% CI ‐7.89 to ‐0.11) and a positive effect on the combined limb data (MD ‐3.94, 95% CI ‐7.25 to ‐0.64) (see Analysis 6.1).
6.1. Analysis.
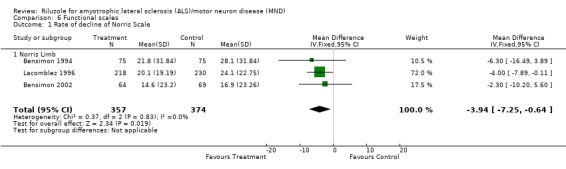
Comparison 6 Functional scales, Outcome 1 Rate of decline of Norris Scale.
Quality of life of patients and caregivers
There are no data which directly measured quality of life from the published trials, but participants treated with riluzole in Lacomblez 1996 remained in a more moderately affected health state significantly longer than placebo‐treated participants (MD 35.5 days, 95% CI 5.9 to 65.0). There was no significant prolongation of the mild, severe or terminal health states. When the mild and moderate health states were combined, participants receiving riluzole remained in these states longer than participants receiving placebo. There was no significant prolongation of the combined severe and terminal states.
Adverse effects from riluzole
Nausea
In Lacomblez 1996, nausea was more frequent in riluzole‐treated participants than with placebo. Similar results were found when the data from the three studies (Bensimon 1994; Bensimon 2002; Lacomblez 1996) were combined (RR 1.55, 95% CI 1.06 to 2.28) (see Analysis 7.1).
7.1. Analysis.
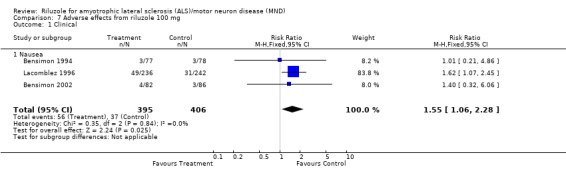
Comparison 7 Adverse effects from riluzole 100 mg, Outcome 1 Clinical.
Asthenia
There was a trend toward more asthenia among the treated participants in each trial, and this became statistically significant when the data from the three trials (Bensimon 1994; Bensimon 2002; Lacomblez 1996) were combined (RR 1.50, 95% CI 1.07 to 2.12) (see Analysis 8.1).
8.1. Analysis.
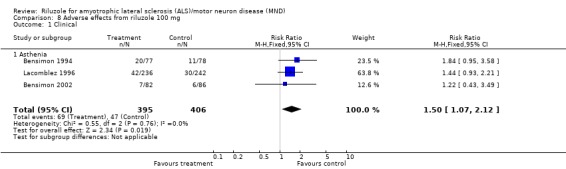
Comparison 8 Adverse effects from riluzole 100 mg, Outcome 1 Clinical.
Other clinical adverse effects
Vomiting, diarrhoea, anorexia and dizziness were somewhat more frequent in treated participants compared to controls, but differences did not reach statistical significance. Five riluzole‐treated participants reported circumoral paresthesias in Lacomblez 1996 but this symptom was not reported by any controls (MD 7.71, 95% CI 1.33 to 44.84).
Increased alanine transferase (more than three times the upper limit of normal)
More treated participants developed a threefold or greater elevation of serum alanine transferase compared to controls in Lacomblez 1996, Bensimon 2002 and in the combined data (RR 2.62, 95% CI 1.59 to 4.31) (see Analysis 9.1).
9.1. Analysis.
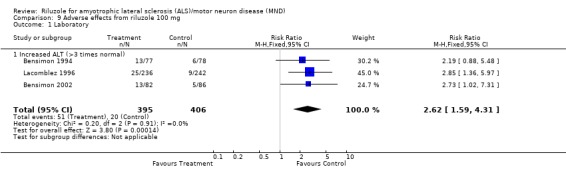
Comparison 9 Adverse effects from riluzole 100 mg, Outcome 1 Laboratory.
Low hemoglobin
There was a trend to low hemoglobin in treated participants in Lacomblez 1996, but this was not significant (MD 4.36, 95% CI 0.98 to 19.37).
Subgroup analysis
In Bensimon 1994 and Lacomblez 1996 there was a significant association of survival and three prognostic variables: age, disease severity (forced vital capacity) and disease duration. However, when each of these variables was incorporated into the Cox model, there was no impact of any variable upon the drug treatment effect.
Bulbar score was significantly correlated with survival in Bensimon 1994 but not in Lacomblez 1996 nor in Bensimon 2002. Gender as a prognostic variable was not reported.
Discussion
Four reports of randomized trials of riluzole in a total of 1477 people with ALS were available for this review. The fourth report from Japan was not sufficiently detailed to include in the meta‐analysis. The methodological quality of these trials was judged as adequate by the reviewers. The therapeutic effects of riluzole at a 100 mg dose on survival were significant when the homogeneous group of participants in the first two trials were considered (P = 0.039). However, when all three trials are analyzed, there was heterogeneity (P < 0.0001) due to the addition of more seriously affected and older patients, and the combined treatment effect fell just short of significance (P = 0.056). Thus, the difference in survival pooled over all participants at all time periods was not quite statistically significant, and the increase in median survival for the riluzole group was very modest (two to three months). Although the authors described a dose response in Lacomblez 1996, we agree with the Health Technology Assessment (HTA) that there is no statistically significant evidence for a dose response, and that the claim in Lacomblez 1996 is based on faulty statistics. The detailed statistical issues involved in the dose response analysis are discussed in the HTA report (HTA 2001). Also, there was modest impact on functional measures. The studies were stratified to balance the number of participants with bulbar onset and limb onset in each treatment arm because of the important prognostic significance of this variable, with shorter survival on average in patients with bulbar onset. In the earlier study by Bensimon 1994, the therapeutic effect was most prominent in participants with bulbar onset. In the trial by Lacomblez et al., there was no significant difference in therapeutic response between the bulbar‐ and limb‐onset groups. In Bensimon 2002, participants with bulbar onset had a worsening of mortality from riluzole. Overall, there was no correlation between site of onset and benefit from riluzole. In Bensimon 1994, 24 patients were enrolled who did not meet inclusion criteria, constituting protocol violations. When these patients were dropped in a separate post hoc analysis, the therapeutic effect of riluzole was not statistically significant, possibly due to reduced power. In Lacomblez 1996, protocol violations were found in 35 participants (details not provided) and these participants were included in the intent‐to‐treat analysis. Data on protocol violations were not available for Bensimon 2002 or Yanagisawa 1997.
There was a significant beneficial effect of riluzole in two of four studies, although not at all time points. The survival data for the 100 mg dose of riluzole, the only dosage common to all studies, did not show significance in the early and late time periods, perhaps related to the diminished numbers of events and power in those time frames. When data from all doses of riluzole from three trials are combined, no comparison was significant, perhaps related to the very modest and non‐significant beneficial effect of the 50 mg dose. The absolute risk reduction with the 100 mg dose at 12 months was 9%. Therefore the number‐needed‐to‐treat to delay one death until after 12 months is 11.
In the 2006 revision of this review, we reported that riluzole 100 mg appears to prolong survival in patients with ALS by about two to three months which is somewhat shorter than that suggested in earlier versions of this review. This reduction in estimated survival prolongation occurred as a result of the inclusion of a study (Bensimon 2002), not included in earlier versions, which enrolled older patients with more advanced disease. Inclusion of such a study might a priori be expected to weaken the evidence of efficacy in terms of survival prolongation. Conversely, recent studies using large databases spanning five to 10 years have suggested that treatment with riluzole might be associated with a median survival prolongation of six months (Meininger 2000), 10 months (Mitchell 2006) 14 months (Brooks 2001), 12 months (Traynor 2001), or even 21 months (Turner 2001). A recent population‐based study in Italy found a six‐month overall survival benefit, that was significant in bulbar‐onset and in elderly patients, but not in limb‐onset patients (Zoccolella 2007). It is not clear to what extent the greater reported efficacy of riluzole in these uncontrolled studies was influenced by other factors, such as riluzole users having less advanced or more slowly progressive disease than non‐users, or differential use of interventions such as gastrostomy and non‐invasive respiratory support. There is a trend for patients with more severe and rapidly progressive disease to avoid RCT, but the differences between trial and registry patients deserve further study (Logroscino 2007). These studies had the advantage of longer term follow‐up than the RCTs (Bensimon 1994; Bensimon 2002; Lacomblez 1996) and included participants treated earlier in the course of ALS, which may approximate routine clinical practice more closely, but the effects of uncontrolled potential confounders on survival could have biased the survival results. Although the therapeutic effects of riluzole on survival were consistent in two of the three studies with comparable outcomes, the impact on functional measures varied among the studies. There was no positive effect on muscle strength when the data were combined. Small beneficial effects on patient function were found in the limb and bulbar scale at the 100 mg dose. The beneficial effect of drug on health status was derived from post hoc analysis of blinded data from the study by Lacomblez et al. (Riviere 1998). Participants treated with riluzole remained longer in a more moderately affected health state compared with placebo‐treated participants. These results should be interpreted with caution, however, since no validation study of remaining in a specific health state has been carried out in ALS. Although we were unable to obtain tracheostomy‐free survival for Yanagisawa 1997, an addendum to the HTA report indicated that some data were made available by Aventis. The HTA analysis including the Yanagisawa 1997 data shifts the pooled HR result from 0.83 (95% CI 0.69 to 0.99) to 0.89 (95% CI 0.75 to 1.05) (HTA 2001). The authors of the HTA report concluded that "the differences between these results are of no practical importance". They also stated that the impression of heterogeneity is strengthened. Thus the results from all four trials do not differ significantly from our previous review of the first three trials. Future versions of this review should focus on survival at 12 and 18 months and also upon a pooled HR of all doses of riluzole upon survival. Future riluzole trials should include health‐related quality of life measures as an outcome measure. Cost effectiveness calculations should also be included in the trial design since this is an expensive drug (approximately US $10,000 per year, although cheaper in the UK at approximately £4056 per year). Moreover, in future trials where survival is a primary outcome measure, the standards of care must be carefully delineated in the protocol because percutaneous endoscopic gastrostomy and non‐invasive mechanical ventilation appear to extend or prolong survival to a significant degree (Bourke 2006; Miller 1999; Miller 2009). Future trials should focus more carefully on gathering pulmonary function data because of the critical role of respiratory function in prognosis. The ALSFRS‐R should be utilized to assess functionality. Older and more advanced patients should also be studied to determine whether or not they receive the same benefit as younger, less advanced patients.
There were no serious adverse effects from riluzole in any study. Nausea and asthenia were the most frequently documented adverse events from riluzole treatment. Elevated liver function tests were also seen in patients treated with riluzole and support the clinical recommendations: (1) to undertake monthly liver function tests for the first three months and then at three month intervals thereafter, and (2) to avoid riluzole in patients with significant hepatic impairment.
Two recent phase III studies of xaliproden included a placebo arm in one study, and an arm of participants taking riluzole plus placebo in another (both reported in Meininger 2004). There was no randomization as such between riluzole plus placebo and placebo. In one of the studies, xaliproden versus placebo was compared in an RCT where no participants took riluzole. In the other study, all participants were taking riluzole; and xaliproden plus riluzole was compared with placebo plus riluzole. Thus it was possible to compare one cohort treated with placebo alone in the first study, and another treated with riluzole plus placebo in the second study, all in the context of an identical protocol. These participants were all studied in the context of a placebo‐controlled trial even though the main objective was evaluating xaliproden, not comparing riluzole with placebo. The enrolment criteria were the same as those used in the original riluzole trial and the two primary endpoints were: (1) time to death, tracheostomy, or permanent assisted ventilation and; (2) time to vital capacity less than 50%, or permanent assisted ventilation, whichever occurred first. The participant characteristics were similar between the two studies except that participants taking riluzole had a longer disease duration (mean 25.7 months) compared to the placebo participants (mean 19.2 months).
A total of 692 participants in Meininger 2004 received no xaliproden: 286 participants received only placebo and 406 participants received riluzole alone. One hundred and thirty two participants reached the primary endpoint of death, tracheostomy or permanent ventilation in the placebo group (46.2%) compared with 153 participants in the riluzole group (37.7%) (P = 0.03 based on number of events, ignoring time to event. Survival analysis results, which take into account follow‐up time, are not available in the published report). The longer disease duration in the group receiving riluzole, which may be due to more slowly progressive disease, may partially explain the trend to a lower mortality rate in the riluzole‐treated participants, or it may have been a riluzole effect, or possibly due to both factors.
The percentage of participants in Meininger 2004 who reached vital capacity less than 50% was very similar in the riluzole group (45.8%) compared to the placebo group (46.6%), (P = 0.9 based on number of events, ignoring time to event and initial FVC values. More detailed data not available in the published report). The combination of time to failure of vital capacity less than 50% or death, tracheostomy, or mechanical ventilation dependency was also similar (57.9%) in the riluzole‐treated group, compared with (62.1%) in the placebo group (P = 0.29 based on number of events). With both endpoints, there was a trend toward a benefit from riluzole but it was not convincing nor were sufficient data available to conduct more rigorous tests.
Similar results for riluzole can be obtained by comparing 1 and 2 mg doses of xaliproden in the arms with and without riluzole. For example, 119 of 293 (40.6%) of those taking 1 mg xaliproden without riluzole compared to 141 of 394 (35.8%) of those taking 1 mg xaliproden with riluzole reached the primary endpoint (P = 0.2 based on number of events). At the 2 mg xaliproden dose the comparison was 114/288 (39.6%) without riluzole versus 160/410 (39.0%) with riluzole (P = 0.88, again based on number of events rather than survival analysis).
In terms of safety, there was no significant increase in adverse events in the riluzole‐treated participants compared with placebo. A number of reports of adverse events have accumulated as case reports, presenting as an acute hypersensitivity reaction. Individual cases of pancreatitis (Rodrigo 2001), hepatitis (Remy 1999), pneumonitis (Cassiman 2003; Borderias‐Clau 2006), neutropenia (Weber 2004) and one systemic inflammatory reaction (Sorenson 2008) have been reported as likely secondary to riluzole, with improvement after stopping the drug.
Two trials involved add‐on therapy to riluzole. A double‐blind placebo‐controlled randomized parallel group clinical trial was conducted to determine whether vitamin E, 5000 mg/day, added to riluzole and compared with riluzole plus placebo might slow disease progression (Graf 2005). No difference between participants treated with vitamin E and placebo were detected and there were no significant safety concerns with megadose vitamin E during the study.
A randomized open study has been carried out to evaluate the efficacy of riluzole alone versus riluzole plus gabapentin in 50 people (23 males, 27 females) affected by ALS (Palma 2000). There was no significant benefit observed with gabapentin. Most ALS clinical trials now include riluzole treatment as standard care.
Authors' conclusions
Implications for practice.
Riluzole 100 mg daily probably prolongs median survival by two to three months in patients with probable and definite amyotrophic lateral sclerosis with symptoms less than five years, forced vital capacity greater than 60% and aged less than 75 years. More studies are needed, especially to determine whether patients treated earlier or older, more advanced patients with longstanding disease derive the same benefit. Benefits are not apparent to individual patients.
The most frequent side effects are nausea and asthenia. Liver function becomes altered and requires monitoring.
Implications for research.
Future trials should examine the effect on quality of life, functionality (ALSFRS‐R), and in different subgroups (for example, more severely affected and older compared with mildly affected and younger patients). Data from all clinical trials should be made available to the scientific community. Genotyping in future trials might be useful to analyze the heterogeneity of responses to therapy.
What's new
| Date | Event | Description |
|---|---|---|
| 19 December 2011 | New citation required but conclusions have not changed | Searches were updated to 20 April 2011. |
| 1 August 2011 | New search has been performed | No new randomized trials found. 'Risk of bias' tables and 'Summary of findings' table added. Mary Lyons withdrew from authorship. |
History
Protocol first published: Issue 1, 1999 Review first published: Issue 1, 2000
| Date | Event | Description |
|---|---|---|
| 12 August 2008 | New search has been performed | A Search of the Cochrane Neuromuscular Disease Group Specialized Register in January 2008, MEDLINE (January 1966 to January 13 2008) and EMBASE (January 1966 to January 20 2008) identified two potentially relevant new studies. Neither was a randomised controlled trial. One was a population‐based comparison and the other was an audit of outcomes in a large center. |
| 11 August 2008 | Amended | Converted to new review format. |
| 25 August 2006 | New citation required and conclusions have changed | A search of the Cochrane Neuromuscular Disease Group Register was last undertaken in October 2006, MEDLINE (January 1966 to August 25th 2006) and EMBASE (January 1980 to September 30th 2006). These searches identified one potentially relevant new trial for which we are attempting to obtain additional data. Two randomized trials looking at add‐on therapy to riluzole have been added to the 'Discussion' section. |
Notes
The authors consider that further research is very unlikely to change the conclusions of this review; therefore, the next planned update will be four years from the current date of search rather than the usual two years. If new evidence emerges contrary to this, an earlier update will be scheduled.
Acknowledgements
The authors are grateful to Ms Kate Jewitt and Ms Chow Saephanh for help and support in preparing this review. The authors wish to acknowledge the untimely death of our co‐author Prof Douglas Mitchell on February 13, 2011. He was a very active contributor to the Cochrane Neuromuscular Disease Group and a productive clinical investigator in the field of ALS/MND. He will be greatly missed.
The Cochrane Neuromuscular Disease Group is funded by the Motor Neuron Disease Association and the MRC Centre for Neuromuscular Diseases.
Appendices
Appendix 1. CENTRAL search strategy
#1MeSH descriptor Motor Neuron Disease explode all trees #2(moto* neuron* disease* or moto?neuron* disease) #3"Amyotrophic Lateral Sclerosis" #4("Lou Gehrig*" and (disease* or syndrome*)) #5(#1 OR #2 OR #3 OR #4) #6riluzole or rilutek #7(#5 AND #6)
Appendix 2. MEDLINE (OvidSP) search strategy
1 randomized controlled trial.pt. 2 controlled clinical trial.pt. 3 randomized.ab. 4 placebo.ab. 5 drug therapy.fs. 6 randomly.ab. 7 trial.ab. 8 groups.ab. 9 or/1‐8 10 exp animals/ not humans.sh. 11 9 not 10 12 exp Motor Neuron Disease/ 13 (moto$1 neuron$1 disease$1 or moto?neuron$1 disease).mp. 14 ((Lou Gehrig$1 adj5 syndrome$1) or (Lou Gehrig$1 adj5 disease)).mp. 15 charcot disease.tw. 16 Amyotrophic Lateral Sclerosis.mp. 17 or/12‐16 18 riluzole.tw. or RILUZOLE/ or rilutek.tw. 19 11 and 17 and 18
Appendix 3. EMBASE (OvidSP) search strategy
1 crossover‐procedure/ 2 double‐blind procedure/ 3 randomized controlled trial/ 4 single‐blind procedure/ 5 (random$ or factorial$ or crossover$ or cross over$ or cross‐over$ or placebo$ or (doubl$ adj blind$) or (singl$ adj blind$) or assign$ or allocat$ or volunteer$).tw. 6 or/1‐5 7 human/ 8 6 and 7 9 nonhuman/ or human/ 10 6 not 9 11 8 or 10 12 Motor Neuron Disease/ or amyotrophic lateral sclerosis/ 13 (moto$1 neuron$1 disease$1 or moto?neuron$1 disease$1).mp. 14 ((lou gehrig$1 adj5 disease$1) or (lou gehrig$1 adj5 syndrome$1)).mp. 15 charcot disease.tw. 16 Amyotrophic Lateral Sclerosis.tw. 17 or/12‐16 18 riluzole.tw. or RILUZOLE/ or rilutek.tw. 19 11 and 17 and 18 20 remove duplicates from 19 21 limit 20 to embase
Data and analyses
Comparison 1. Riluzole 100 mg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Per cent mortality at 12 months | 3 | 799 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.65, 0.92] |
| 1.1 Per cent mortality at 12 mos | 3 | 799 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.65, 0.92] |
Comparison 2. Riluzole 100 mg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Per cent mortality at 18 months | 3 | 634 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.83, 1.02] |
| 1.1 Per cent mortality at 18 mos | 3 | 634 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.83, 1.02] |
Comparison 3. Riluzole all doses versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Per cent mortality at 12 months | 2 | 1108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.60, 0.87] |
Comparison 4. Muscle strength.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Manual muscle testing | 3 | 732 | Mean Difference (IV, Fixed, 95% CI) | ‐1.88 [‐5.79, 2.03] |
| 1.1 Rate of decline of muscle strength | 3 | 732 | Mean Difference (IV, Fixed, 95% CI) | ‐1.88 [‐5.79, 2.03] |
Comparison 5. Functional scales.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Rate of decline of Norris Scale | 3 | 742 | Mean Difference (IV, Fixed, 95% CI) | ‐2.06 [‐3.86, ‐0.27] |
| 1.1 Norris Bulbar | 3 | 742 | Mean Difference (IV, Fixed, 95% CI) | ‐2.06 [‐3.86, ‐0.27] |
Comparison 6. Functional scales.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Rate of decline of Norris Scale | 3 | 731 | Mean Difference (IV, Fixed, 95% CI) | ‐3.94 [‐7.25, ‐0.64] |
| 1.1 Norris Limb | 3 | 731 | Mean Difference (IV, Fixed, 95% CI) | ‐3.94 [‐7.25, ‐0.64] |
Comparison 7. Adverse effects from riluzole 100 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical | 3 | 801 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [1.06, 2.28] |
| 1.1 Nausea | 3 | 801 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [1.06, 2.28] |
Comparison 8. Adverse effects from riluzole 100 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical | 3 | 801 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [1.07, 2.12] |
| 1.1 Asthenia | 3 | 801 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [1.07, 2.12] |
Comparison 9. Adverse effects from riluzole 100 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Laboratory | 3 | 801 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.62 [1.59, 4.31] |
| 1.1 Increased ALT (>3 times normal) | 3 | 801 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.62 [1.59, 4.31] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bensimon 1994.
| Methods | Double blind, parallel group, randomized, placebo‐controlled trial | |
| Participants | 155 adult patients with ALS | |
| Interventions | Oral placebo twice a day or riluzole 50 mg twice a day | |
| Outcomes | Primary: Per cent mortality, without tracheostomy or endotracheal intubation, and functional scales; tracheostomy‐free survival (time to death or tracheostomy); change in functional status after 12 months. Secondary: muscle strength scores, respiratory function, clinical global impression of change scale, patients's subjective evaluations | |
| Notes | International (France, Belgium), multicenter | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization codes established by blocking |
| Allocation concealment (selection bias) | Low risk | The allocation results were concealed from all evaluators and participants |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No loss to follow‐up for survival, primary endpoint |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting |
| Other bias | Low risk | None observed |
Bensimon 2002.
| Methods | Double blind, parallel group, placebo‐controlled, randomized trial | |
| Participants | 168 adult patients with ALS, not qualifying for Lacomblez 1996 (age >75, FVC < 60%, > five years duration | |
| Interventions | Oral placebo twice a day or riluzole 50 mg twice a day | |
| Outcomes | Primary: Per cent survival without tracheostomy or endotracheal intubation Secondary: functional scales, muscle strength, respiratory function | |
| Notes | International (France and Belgium), multicenter | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization codes established by blocking |
| Allocation concealment (selection bias) | Low risk | The allocation results were concealed from all evaluators and participants |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No loss to follow‐up for survival, primary endpoint |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting |
| Other bias | Low risk | None observed |
Lacomblez 1996.
| Methods | Double blind, parallel group, placebo‐controlled, randomized, dose‐ranging trial | |
| Participants | 959 adult patients with ALS fulfilling WFN criteria (Brooks 1994) | |
| Interventions | Placebo, riluzole 50 mg, riluzole 100 mg or riluzole 200 mg per day | |
| Outcomes | Primary: per cent mortality without tracheostomy or endotracheal intubation, by intention‐to‐treat analysis. Secondary: muscle strength, functional status, respiratory function, clinician's global impression scale and patient subjective assessments of fasciculations, cramps, tiredness, and stiffness. Respiratory function measured only at six month intervals | |
| Notes | International (Europe and North America), multicenter. Protocol violations (35 patients) included in the intention‐to‐treat analysis. Patients were evenly distributed among groups. Interim analysis (October 1994) did not meet conditions for stopping trial. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomization stratified by center and site of onset, but allocation sequence method not described |
| Allocation concealment (selection bias) | Low risk | Allocation was concealed from all but central coordinator |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Assignments were concealed to evaluators and patients and placebo and drug were identical |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patient mortality data obtained and reported to FDA later |
| Selective reporting (reporting bias) | Low risk | All patient mortality reported |
| Other bias | Low risk | None observed |
Yanagisawa 1997.
| Methods | Double blind, parallel group, randomized placebo‐controlled | |
| Participants | 195 adult patients with ALS | |
| Interventions | Oral placebo or riluzole, 50 mg twice a day | |
| Outcomes | Disease progression (walking, arm function, ventilation, tube feeding and nutrition, loss of upper extremity function, independent ambulation, time to death and tracheostomy); Overall survival. Secondary: muscle strength, Japanese Norris scales (limb and bulbar), grip, back extension, pinch, FVC, safety | |
| Notes | Japan, 48 centers | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Allocation done by study controller by biased coin method |
| Allocation concealment (selection bias) | Low risk | Key codes were sealed and kept by controller until opening after study completion |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Allocation concealed and placebo and study drug identical in appearance |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Efficacy evaluable population of 154 patients reflects incomplete follow‐up of 41 patients (intention‐to‐treat population of 195 patients) |
| Selective reporting (reporting bias) | Low risk | Non‐standard outcome measures |
| Other bias | Low risk | Small numbers of patients at many centers and non‐standard outcome measures prevented meta‐analysis |
WFN: World Federation of Neurology. FDA: US Food and Drug Administration.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arriada‐Mendicoa 1999 | Open‐label non‐randomized uncontrolled study of 50 Mexican patients. The study suggested that progression was slowed in patients taking riluzole but was excluded because it was not randomized and was uncontrolled |
| Couratier 2000 | An observational series of 340 patients with ALS at a single center, half of whom were treated with riluzole. Excluded because it was not randomized |
| Desiato 1999 | A small study of 31 patients employed transcranial magnetic stimulation in 31 patients with ALS receiving riluzole and 30 controls. Differences in motor evoked potential duration and silent period duration were noted between treated patients and controls. Excluded because it was not randomized |
| Graf 2005 | A randomized controlled trial of megadose vitamin E (5000 mg/day) versus placebo in patients who were all taking riluzole. Results of this study have been excluded and are included in the Discussion only, because riluzole was not compared with placebo |
| Kalra 1998 | Analyzed magnetic resonance spectroscopy (MRS) data in 11 patients and found that riluzole improved results. Results were quite variable. Excluded because it was not randomized |
| Meininger 2004 | A large phase III study of xaliproden comparing xaliproden to placebo in patients not taking riluzole and then a separate study comparing patients taking riluzole alone with riluzole plus xaliproden. Complete data were not provided about the riluzole and placebo groups in terms of death at different time points. Unable to obtain the key data for main outcomes |
| Mitchell 2006 | Audit of outcomes in motor neuron disease (MND) patients treated with riluzole |
| Palma 2000 | Randomized but open. Compared gabapentin plus riluzole to riluzole alone, not riluzole against placebo. This was only published in abstract form and is now included in the Discussion only as the study involved add‐on therapy to riluzole |
| Pongratz 1999 | Studied primarily the safety of riluzole in an open‐label German study involving 7916 patients with ALS. The major result was that serious adverse events associated with riluzole use occurred in only 1.7% of patients. This study was excluded because it was uncontrolled and not randomized |
| Riviere 1998 | A post hoc analysis of the health states of patients in the larger trial (Lacomblez 1996). This study was the only one with data that bear on quality of life and was discussed in our review. There was significant prolongation of the mild health state in patients taking riluzole |
| Sojka 1997 | Compared symptom progression during a lead‐in phase and a treatment phase in five patients with ALS taking riluzole. The results were highly variable. The study was excluded because it was non‐randomized and uncontrolled |
| Zoccolella 2007 | Population‐based study in Italy comparing patients taking riluzole with those not taking the drug |
Differences between protocol and review
The 'Risk of bias' methodology has been revised according to the 2008 Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008), and a 'Summary of findings' table included. We have removed 'RRs based on per cent mortality as a function of time at 18 months ‐ all doses of riluzole versus placebo' from the secondary outcomes.
Contributions of authors
The first version of the review was jointly written by all four authors at that time. The lead author (RGM) coordinated the review, abstracted data from the papers, requested additional data from authors, entered the data into Revman and wrote the review. The co‐reviewers (JDM, DHM) checked the data, appraised the quality of the studies (especially for allocation concealment) and offered revisions of the review. One co‐reviewer is a statistician (DHM). He offered help and advice to the lead author at all stages, and performed the additional statistical analysis of survival at multiple time points not provided by Revman. One co‐reviewer (ML) was a nurse and patient advocate and offered revisions of earlier versions of the review. This update was undertaken by RGM with statistical support from DHM. JDM provided substantive help with this update before his death in February 2011 but did not review all revisions.
Sources of support
Internal sources
None, Not specified.
External sources
None, Not specified.
Declarations of interest
Both Drs Miller and Mitchell were investigators in the second large trial of riluzole in ALS, but neither participated in data analysis or manuscript preparation. Dr Mitchell participated in other scientific activities (Consensus conferences, ALS CARE National database and ALS Practice Parameters) where financial support came from Aventis.
Dr Miller is a consultant for several pharmaceutical entities, but none are related to riluzole.
Dan H Moore, PhD received an honorarium for his participation in the ALS CARE program, supported by Aventis. He is a biostatistical consultant for several pharmaceutical entities, but none are related to riluzole.
For Dr Mitchell (deceased), declarations of interest are as published in the previous update of this review.
Deceased.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Bensimon 1994 {published and unpublished data}
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. The ALS/Riluzole Study Group. New England Journal of Medicine 1994;330(9):585‐91. [PUBMED: 8302340] [DOI] [PubMed] [Google Scholar]
Bensimon 2002 {published data only}
- Bensimon G, Lacomblez L, Delumeau JC, Bejuit R, Truffinet P, Meininger V. A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. Journal of Neurology 2002;249(5):609‐15. [PUBMED: 12021952] [DOI] [PubMed] [Google Scholar]
- Meininger V, Lacomblez L, Bensimon G. Unpublished report: controlled trial of riluzole in patients with advanced ALS. RP 54272‐302 1995.
Lacomblez 1996 {published and unpublished data}
- Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V, and the Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Dose‐ranging study of riluzole in amyotrophic lateral sclerosis. Lancet 1996;347(9013):1425‐31. [PUBMED: 8676624] [DOI] [PubMed] [Google Scholar]
Yanagisawa 1997 {published data only}
- Yanagisawa N, Tashiro K, Tohgi H, Mizuno Y, Kowa H, Kimuma J, et al. Efficacy and safety of riluzole in patients with amyotrophic lateral sclerosis: double‐blind placebo‐controlled study in Japan. Igakuno Ayumi 1997;182:851‐66. [Google Scholar]
References to studies excluded from this review
Arriada‐Mendicoa 1999 {published data only}
- Arriada‐Mendicoa N, Otero‐Siliceo E, Burbano G, Corona‐Vazquez T. Open label study of riluzole for the treatment of amyotrophic lateral sclerosis. Revista Ecuatoriana de Neurologia 1999;8:33‐6. [Google Scholar]
Couratier 2000 {published data only}
- Couratier P, Druet‐Cabanac M, Truong CT, Bernet‐Bernady P, Dumas M, Vallat JM, et al. Interest of a computerized ALS database in the diagnosis and follow‐up of patients with ALS [Interets d'une base de donnees informatisee dans le diagnostic et le suivi de patients atteints de la sclerose laterale amyotrophique]. Revue Neurologique 2000;156(4):357‐63. [PubMed] [Google Scholar]
Desiato 1999 {published data only}
- Desiato MT, Palmieri MG, Giacomini P, Scalise A, Arciprete F, Caramia MD. The effect of riluzole in amyotrophic lateral sclerosis: a study with cortical stimulation. Journal of Neurological Sciences 1999;169(1‐2):98‐107. [DOI] [PubMed] [Google Scholar]
Graf 2005 {published data only}
- Graf M, Ecker D, Horowski R, Kramer B, Riderer P, Gerlach M, et al. High dose vitamin E therapy in amyotrophic lateral sclerosis as add‐on therapy to riluzole: results of a placebo‐controlled double‐blind study. Journal of Neural Transmission 2005;112(5):649‐60. [DOI] [PubMed] [Google Scholar]
Kalra 1998 {published data only}
- Kalra S, Cashman NR, Genge A, Arnold DL. Recovery of N‐acetylaspartate in corticomotor neurons of patients with ALS after riluzole therapy. Neuroreport 1998;9(8):1757‐61. [DOI] [PubMed] [Google Scholar]
Meininger 2004 {published data only}
- Meininger V, Bensimon G, Bradley WG, Brooks BR, Douillet P, Eisen AA, et al. Efficacy and safety of xaliproden in amyotrophic lateral sclerosis: Results of two phase III trials. Amyotrophic Lateral Sclerosis & Other Motor Neuron Disorders 2004;5(2):107‐17. [DOI] [PubMed] [Google Scholar]
Mitchell 2006 {published data only}
- Mitchell JD, O'Brien MR, Joshi M. Audit of outcomes in motor neuron disease (MND) patients treated with riluzole. Amyotrophic lateral sclerosis and other motor neuron disorders 2006;7:67‐71. [DOI] [PubMed] [Google Scholar]
Palma 2000 {published data only}
- Palma V, Brescia Morra V, Polverino M, Santoro L, Caruso G. An open study of riluzole versus riluzole plus gabapentin in amyotrophic lateral sclerosis. Journal of the Peripheral Nervous System 2000;5(1):46. [Google Scholar]
Pongratz 1999 {published data only}
- Pongratz D, Neundorfer B. Open‐label trial of riluzole 50 mg b.i.d. in treatment of amyotrophic lateral sclerosis (ALS). Aktuelle Neurologie 1999;26(5):225‐9. [DOI] [PubMed] [Google Scholar]
Riviere 1998 {published data only}
- Riviere M, Meininger V, Zeisser P, Munsat T. An analysis of extended survival in patients with amyotrophic lateral sclerosis treated with riluzole. Archives of Neurology 1998;55(4):526‐8. [DOI] [PubMed] [Google Scholar]
Sojka 1997 {published data only}
- Sojka P, Anderson PM, Forsgren L. Effects of riluzole on symptom progression in amyotrophic lateral sclerosis. Lancet 1997;349(9046):176‐7. [DOI] [PubMed] [Google Scholar]
Zoccolella 2007 {published data only}
- Zoccolella S, Beghi E, Palagano G, Fraddosio A, et al. Riluzole and amyotrophic lateral sclerosis survival: a population based study in Southern Italy. European Journal of Neurology 2007;14(3):262‐8. [DOI] [PubMed] [Google Scholar]
Additional references
Booth‐Clibborn 1997
- Booth‐Clibborn N, Best L, Stein K. Riluzole for motor neurone disease. Southampton: Wessex Institute for Health Research & Development (DEC Report 73). Southampton, 1997.
Borderias‐Clau 2006
- Borderías‐Clau L, Garrapiz‐López J, Val‐Adán P, Tordesillas‐Lía C, Alcacera‐López A, Bru‐Martín JL. Strong suspicion of lung toxicity due to riluzole [Alta sospecha de toxicidad pulmonar por riluzol]. Archivos de Bronconeumologia 2006;42(1):42‐4. [DOI] [PubMed] [Google Scholar]
Bourke 2006
- Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non‐invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurology 2006;5:140‐7. [DOI] [PubMed] [Google Scholar]
Brooks 1994
- Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial "Clinical limits of amyotrophic lateral sclerosis" workshop contributors. Journal of Neurological Sciences 1994;124(Suppl):96‐107. [DOI] [PubMed] [Google Scholar]
Brooks 2001
- Brooks BR, Belden DS, Roelke K, Parnell J, Peper S, Houdek A, et al. Survival in non‐riluzole treated amyotrophic lateral sclerosis (ALS) ‐ Motor neuron disease (MND) patients with disease onset before and since 1996 is identical: a clinic‐based epidemiological study. ALS and other motor neuron diseases 2001;2:60‐1. [Google Scholar]
Cassiman 2003
- Cassiman D, Thomeer M, Verbeken E, Robberecht W. Hypersensitivity pneumonitis possibly caused by riluzole therapy in ALS. Neurology 2003;61(8):1150‐1. [DOI] [PubMed] [Google Scholar]
Chilcott 1997
- Chilcott J, Golightly P, Jefferson D, McCabe CJ, Walters S. The use of riluzole in the treatment of Amyotrophic Lateral Sclerosis (Motor Neurone Disease). Working Group on Acute Purchasing. Trent Institute for Health Services Research, Universities of Leicester, Nottingham and Sheffield 1997.
CPMP 1999
- Committee for Proprietary Medicinal Products. European Public Assessment Report (EPAR) Rilutek. The European Agency for the Evaluation of Medicinal Products 1997 (CPMP/290/96).
Higgins 2008
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors) editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Chichester (UK): John Wiley & Sons, 2008. [Google Scholar]
HTA 2001
- Stewart A, Sandercock J, Bryan S, Hyde C, Barton PM, Fry‐Smith A, et al. The clinical effectiveness and cost‐effectiveness of riluzole for motor neurone disease: a rapid and systematic review. Health Technology Assessment 2001;5(2):1‐97. [DOI] [PubMed] [Google Scholar]
Hugon 1996a
- Hugon J. Riluzole and ALS therapy. Weiner Medizinische Wochenschrift 1996;146(9‐10):185‐7. [PubMed] [Google Scholar]
Hugon 1996b
- Hugon J. ALS therapy: targets for the future. Neurology 1996;47:251‐4. [DOI] [PubMed] [Google Scholar]
Lacomblez 1989
- Lacomblez L, Bouche P, Bensimon G, Meininger V. A double‐blind, placebo‐controlled trial of high doses of gangliosides in amyotrophic lateral sclerosis. Neurology 1989;39(12):1635‐7. [DOI] [PubMed] [Google Scholar]
Logroscino 2007
- Logroscino G, Zoccolella S. Efficacy of riluzole: Who are the patients enrolled in the studies?. Amyotrophic Lateral Sclerosis 2007;8:124‐5. [DOI] [PubMed] [Google Scholar]
McGuire 1996
- McGuire D, Garrison L, Miller RG. Relationship of the Tufts Quantitative Neuromuscular Exam (TQNE) and the Sickness Impact Profile (SIP) in measuring progression of ALS. Neurology 1996;46(5):1442‐4. [DOI] [PubMed] [Google Scholar]
Meininger 1997
- Meininger V, Dib M, Aubin F, Jourdain G, Zeisser P. The Riluzole Early Access Programme: descriptive analysis of 844 patients in France. ALS/Riluzole Study Group III. Neurology 1997;244(Suppl 2):S22‐5. [DOI] [PubMed] [Google Scholar]
Meininger 2000
- Meininger V, Bensimon G, Bradley WG, Brooks BR, Douillet P, Eisen AA, et al. Efficacy and safety of xaliproden in amyotrophic lateral sclerosis: results of two phase III trials. Amyotrophic lateral sclerosis and other motor neuron disorders 2004;5(2):107‐17. [DOI] [PubMed] [Google Scholar]
Miller 1996
- Miller RG, Bouchard JP, Duquette P, Eisen A, Gelinas D, Harati Y, et al. Clinical trials of riluzole in patients with amyotrophic lateral sclerosis. Neurology 1996;47(4 Suppl 2):S86‐S92. [DOI] [PubMed] [Google Scholar]
Miller 1999
- Miller RG, Rosenberg JA, Gelinas DF, Mitsumoto H, Newman D, Sufit R, et al. Practice parameter: The care of the patient with amyotrophic lateral sclerosis (an evidence‐based review). Neurology 1999;52(7):1311‐23. [DOI] [PubMed] [Google Scholar]
Miller 2009
- Miller RG, Jackson CE, Kasarkis EJ, England JD, Forshew D, Johnston W, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence‐based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009;73(15):1218‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Neurology 1997
- Quality Standards Subcommittee of the American Academy of Neurology. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole. Neurology 1997;49:657‐9. [DOI] [PubMed] [Google Scholar]
NICE 2001
- National Institute for Clinical Excellence. Guidance on the use of riluzole (Rilutek) for the treatment of motor neurone disease. Technology Appraisal Guidance 2001; Vol. 20.
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Remy 1999
- Remy AJ, Camu W, Ramos J, Blanc P, Larrey D. Acute hepatitis after riluzole administration. Journal of Hepatology 1999;30(3):527‐30. [DOI] [PubMed] [Google Scholar]
Review Manager 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Rodrigo 2001
- Rodrigo L, Moreno M, Calleja S, Mateos V, Andrade RJ, Lucena MI. Riluzole‐induced acute pancreatitis. American Journal of Gastroenterology 2001;96(7):2268‐9. [DOI] [PubMed] [Google Scholar]
Rothstein 1996
- Rothstein JD. Therapeutic horizons for amyotrophic lateral sclerosis. Current Opinion in Neurobiology 1996;6:679‐87. [DOI] [PubMed] [Google Scholar]
Rowland 1994
- Rowland LP. Riluzole for the treatment of amyotrophic lateral sclerosis ‐ too soon to tell?. New England Journal of Medicine 1994;330(9):636‐7. [DOI] [PubMed] [Google Scholar]
Sorenson 2008
- Sorenson EJ. An acute, life‐threatening, hypersensitivity reaction to riluzole. Neurology 2006;67(12):2260‐1. [DOI] [PubMed] [Google Scholar]
Stambler 1998
- Stambler N, Charatan M, Cedarbaum J and the ALS CNTF Treatment Study Group. Prognostic indicators of survival in ALS. Neurology 1998;50(1):66‐72. [DOI] [PubMed] [Google Scholar]
Traynor 2001
- Traynor BJ, Alexander M, Corr B, Frost E, Mahon L, Hardiman O. Riluzole and prognosis in ALS: Findings of the Irish ALS register over a five year study period (1999‐2000). Amyotrophic lateral sclerosis and other motor neuron disorders 2001;2(Suppl 2):43‐4. [Google Scholar]
Turner 2001
- Turner MR, Bakker M, Sham P, Shaw CE, Leigh PN, Al‐Chalabi A. The Kings' Database 1999‐2000: an analysis of the effect on survival of interventions in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders 2001;2(Suppl 2):43. [DOI] [PubMed] [Google Scholar]
Weber 2004
- Weber G, Bitterman H. Riluzole‐induced neutropenia. Neurology 2004;62(9):1648. [DOI] [PubMed] [Google Scholar]
Wokke 1996
- Wokke J. Riluzole. Lancet 1996;348(9030):795‐9. [DOI] [PubMed] [Google Scholar]