

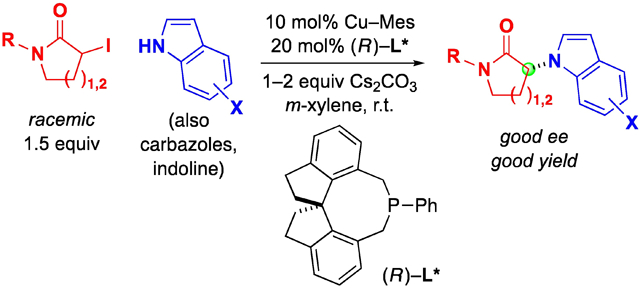
Abstract
Transition-metal catalysis has the potential to address shortcomings in the classic SN2 reaction of an amine with an alkyl electrophile, both with respect to reactivity and to enantioselectivity. In this study, we describe the development of a user-friendly method (reaction at room temperature, with commercially available catalyst components) for the enantioconvergent nucleophilic substitution of racemic secondary alkyl halides (α-iodolactams) by indoles. Mechanistic studies are consistent with the formation of a copper(I)–indolyl complex that reacts at different rates with the two enantiomers of the electrophile, which interconvert under the reaction conditions (dynamic kinetic resolution). This investigation complements earlier work on photoinduced enantioconvergent N-alkylation, supporting the premise that this important challenge can be addressed by a range of strategies.
Graphical Abstract
INTRODUCTION
Because a wide array of organic molecules (including pharmaceuticals, agrochemicals, and polymers) contain carbon–nitrogen bonds, the development of methods for the construction of C–N bonds is of great interest.1 The nucleophilic substitution of an electrophile by an amine is a particularly straightforward strategy for the generation of C–N bonds; unfortunately, however, thermal/uncatalyzed approaches do not satisfactorily address a variety of challenges with regard to scope and/or stereoselectivity (Figure 1). Consequently, efforts have turned to catalysis of this transformation by transition metals. In the case of substitution reactions of aryl electrophiles by amines, the Ullmann reaction (copper catalysis)2 and the Buchwald–Hartwig reaction (palladium catalysis)3 can achieve this important objective with a much broader spectrum of electrophiles than is possible using uncatalyzed approaches, such as nucleophilic aromatic substitution (left side of Figure 1).4
Figure 1.

Nucleophilic substitution of electrophiles by amines: Qualitative overview.
The uncatalyzed substitution reaction of alkyl electrophiles by amines is a widely applied approach to C–N bond construction; for example, in one analysis of reactions used by the pharmaceutical industry, the coupling of an alkyl electrophile with a nitrogen nucleophile was the most frequently employed method for achieving N-substitution, exceeding approaches such as reductive amination and Ullmann/Buchwald–Hartwig.5 Nevertheless, uncatalyzed SN2 reactions of alkyl electrophiles with amines have substantial limitations: with less-reactive electrophiles, elimination to form an olefin can occur in preference to substitution, whereas with more-reactive electrophiles, over-alkylation can be a significant side reaction. Transition-metal catalysis has the potential to address this challenge of controlling reactivity, and to thereby expand the scope of substitution reactions of alkyl electrophiles by amines; however, progress to date has been rather limited (right side of Figure 1).6,7
In the case of substitution reactions of alkyl electrophiles, there can be a challenge not only of achieving the desired C–N bond construction, but also of controlling stereochemistry in the event that a chiral electrophile is used (Figure 2a). Because an array of bioactive molecules include chiral amines wherein nitrogen is attached to a stereogenic carbon (Figure 2b),8,9 the development of catalysts that can simultaneously address the dual challenges of reactivity and stereoselectivity is a worthwhile goal.6,10,11
Figure 2.
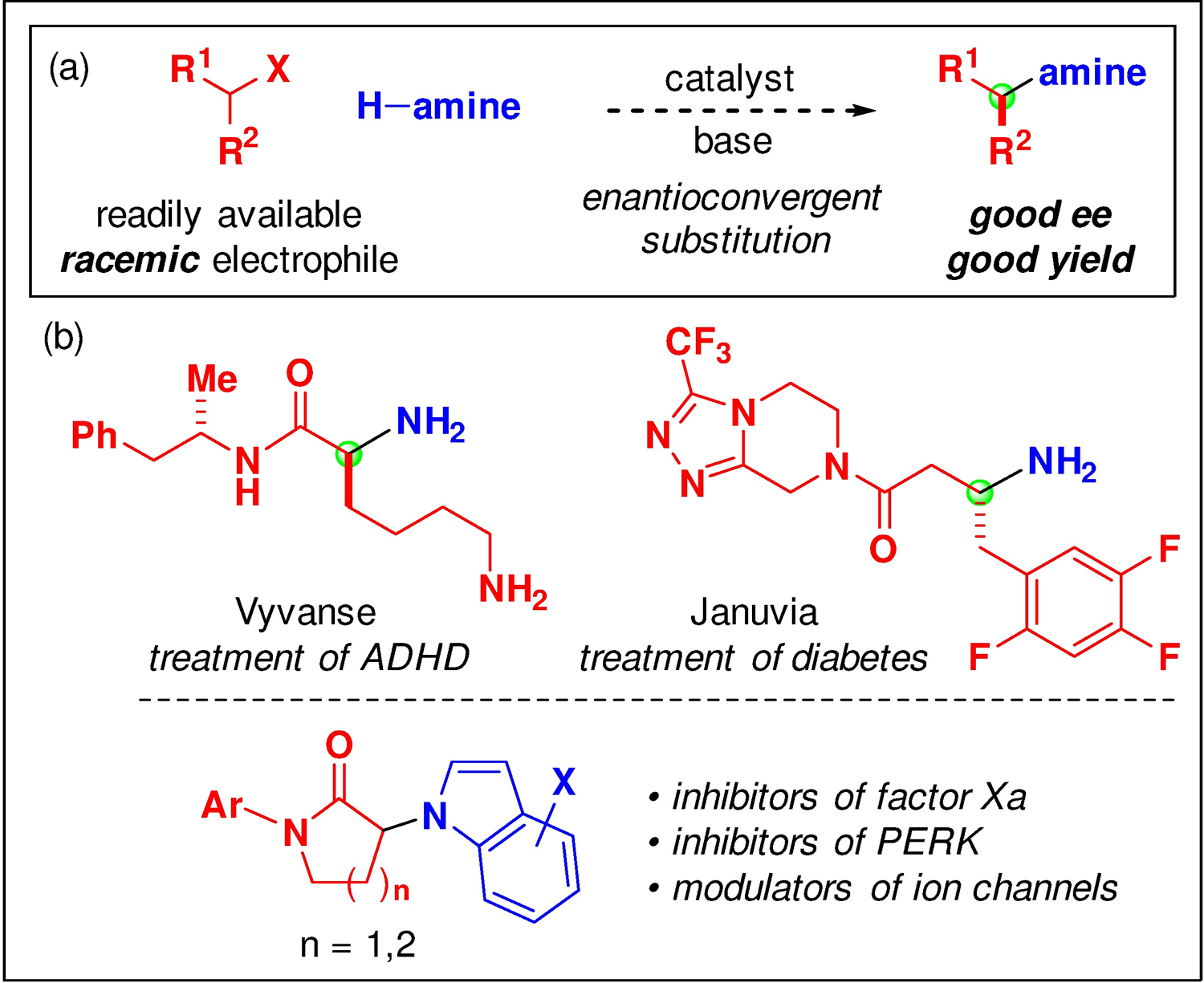
Chiral amines wherein nitrogen is attached to a stereogenic carbon: (a) Synthesis via metal-catalyzed enantioconvergent alkylation; (b) Examples of bioactive compounds.
We have recently demonstrated the feasibility of this objective, specifically, that enantioconvergent N-alkylations by racemic tertiary alkyl chlorides can be achieved in the presence of light and a chiral copper catalyst (Figure 3).11 In this report, we describe complementary reactivity, wherein, in the absence of light, a chiral copper catalyst effects enantioconvergent N-alkylations by racemic secondary alkyl iodides (Figure 3).
Figure 3.

Complementary metal-catalyzed enantioconvergent substitution reactions of alkyl electrophiles by amines.
RESULTS AND DISCUSSION
3-Indolyl-lactams exhibit a wide range of interesting bioactivity (Figure 2b).8,12 When the photoinduced, copper-catalyzed method that we had developed for enantioconvergent alkylations by racemic tertiary alkyl chlorides (top of Figure 3)11 was applied to the alkylation of 3-methylindole by secondary alkyl iodide A (Table 1), the results were unsatisfactory (<1% ee, 24% yield). Upon exploring a variety of conditions, we have determined that the desired enantioconvergent coupling can be achieved in the absence of light at room temperature (entry 1: 87% ee, 84% yield; both Cu–Mes and L* are commercially available). Control reactions have established that, if Cu–Mes and L* are both omitted from the reaction mixture, only a small amount of a background reaction is observed (entry 2: 8% yield), whereas, if only L* is omitted, considerable N-alkylation occurs (entry 3: 55% yield). The use of a different chiral monodentate phosphine, phosphepine L1, results in modest ee and yield (entry 4). With half of the standard catalyst loading, the ee and the yield decrease somewhat (entry 6), and with less phosphine L*, a significant loss in ee is observed (87→68% ee; entry 7). This method for the catalytic enantioconvergent synthesis of 3-indolyl-lactams is not highly air- or moisture-sensitive (entries 7 and 8).
Table 1.
Enantioconvergent N-alkylation: Effect of reaction parameters.
 | |||
|---|---|---|---|
| entry | change from the “standard conditions” | ee (%) | yield (%)a |
| 1 | none | 87 | 84 |
| 2 | no Cu–Mes, no L* | – | 8 |
| 3 | no L* | – | 55 |
| 4 | L1, instead of L* | 32 | 39 |
| 5 | 5 mol% Cu–Mes, 10 mol% L* | 82 | 64 |
| 6 | 14, instead of 20, mol% L* | 68 | 76 |
| 7 | under air in a capped vial | 83 | 67 |
| 8 | 0.1 equiv of water added | 85 | 74 |
All data represent the average of two experiments.
Determined through 1H NMR spectroscopy, with the aid an Internal standard.

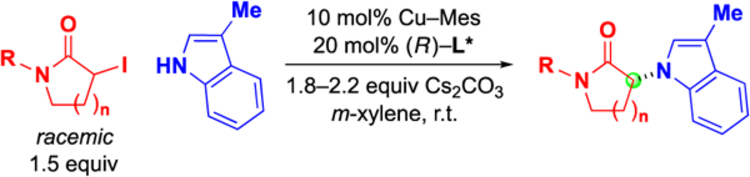
With suitable reaction conditions in hand, we explored the scope of this new enantioconvergent N-alkylation with respect to the electrophile (Table 2). The substituent on the nitrogen of the lactam can be an electron-rich or an electron-poor aromatic ring (entries 2 and 3), or it can be an alkyl group (entries 4 and 5).13 Furthermore, six-membered α-iodolactams can serve as electrophiles (entries 6 and 7). Finally, in the case of an electrophile that bears a second stereogenic center, the catalyst can predominantly control the stereochemistry of the N-alkylation product (Figure 4).
Table 2.
Enantioconvergent N-alkylation: Scope with respect to the electrophile.
 | ||||
|---|---|---|---|---|
| entry | R | n | ee (%) | yield (%)a |
| 1 | Ph | 1 | 88 | 73 |
| 2 | 4-(OMe)C6H4 | 1 | 91 | 78 |
| 3 | 4-(CF3)C6H4 | 1 | 90 | 73 |
| 4 | Bn | 1 | 90 | 72 |
| 5 | Ph(CH2)3 | 1 | 82 | 80 |
| 6 | Ph | 2 | 80 | 88 |
| 7 | 4-(OMe)C6H4 | 2 | 88 | 74 |
All data represent the average of two experiments.
Yield of purified product.
Figure 4.

The stereochemistry of the catalyst, rather than of the substrate, predominantly controls the stereochemistry of an N-alkylation.
The standard conditions that we developed for copper-catalyzed N-alkylations by a racemic alkyl iodide can also be applied to an alkyl bromide (eq 1).14 A significantly higher yield is observed in the presence of a larger excess of the electrophile.
 |
(1) |
Next, we examined the scope of this copper-catalyzed stereoconvergent N-alkylation with respect to the nucleophile (Table 3). When indoles are used as the nucleophile (entries 1–11), substituents can be present in a range of positions, including the 2 position. Furthermore, a variety of functional groups are compatible with the coupling, such as an alkene, a cyano group, an ether, an aryl bromide, and a secondary amide. The method can be applied without further optimization to the enantioconvergent alkylation of carbazoles (entries 12 and 13) and an indoline (entry 14), with good enantioselectivity. On a gram scale, the stereoconvergent N-alkylation depicted in entry 12 proceeds in 92% ee and 85% yield (1.06 g of product).15
Table 3.
Enantioconvergent N-alkylation: Scope with respect to the nucleophile.
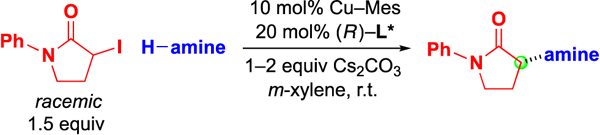 | |||
|---|---|---|---|
| entry | H-amine | ee (%) | yield (%)a |
| 1 |  |
96 | 66 |
| 2 | 98 | 69 | |
| 3 |  |
83 | 52 |
| 4 | 90 | 84 | |
| 5 | 87 | 94 | |
| 6 | 87 | 54 | |
| 7 | 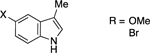 |
90 | 84 |
| 8 | 88 | 70 | |
| 9 | 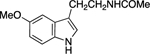 |
84 | 56 |
| 10 | 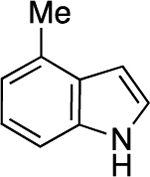 |
91 | 58 |
| 11 |  |
84 | 53 |
| 12 |  |
96 | 82 |
| 13 |  |
94 | 80 |
| 14 |  |
88 | 64 |
All data represent the average of two experiments.
Yield of purified product.
Taken together, the ee and the yield establish that the copper-catalyzed substitution of alkyl iodide A by 3-methylindole (Table 1: 87% ee, 84% yield) is an enantioconvergent process (not a simple kinetic resolution), wherein both enantiomers of the electrophile are being transformed by the chiral catalyst into a particular enantiomer of the product. To gain insight into the mechanism, we monitored the ee of the unreacted electrophile as a function of time (Figure 5). We hypothesize that the initial increase in ee is due to the chiral copper catalyst reacting more rapidly with one enantiomer of the electrophile than the other. With each N-alkylation, iodide anion is generated, which serves as a nucleophile that racemizes the unreacted electrophile via an SN2 reaction, thereby enabling a dynamic kinetic resolution, as well as leading to a decrease in the ee of the electrophile during the later stages of the reaction.
Figure 5.

Copper-catalyzed enantioconvergent N-alkylation of an indole by a racemic alkyl iodide via a dynamic kinetic resolution: ee of the electrophile as a function of time.
We have synthesized a possible intermediate in this copper-catalyzed enantioconvergent N-alkylation. Treatment of Cu–Mes with 3-methylindole in the presence of 2 equivalents of chiral phosphine L* provided a 65% yield of L*2Cu(2-methylindolyl) (B; eq 2), which we have crystallographically characterized (see the Supporting Information). When complex B (10%) is used in place of Cu–Mes/L* for the N-alkylation illustrated in Table 1, the enantioconvergent coupling proceeds in similar ee and yield (78% ee and 96% yield). Furthermore, in a stoichiometric reaction, copper complex B couples with electrophile A within 15 minutes at room temperature to afford the N-alkylation product with high ee and high yield, thereby establishing its competence as a possible intermediate (eq 3; 87% ee and 94% yield).16 An investigation of the impact of the ee of L* on the ee of the product revealed a small positive non-linear effect, consistent with the presence of a copper complex that contains more than one L*.
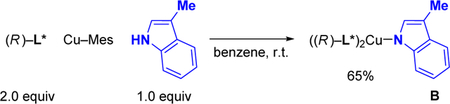 |
(2) |
 |
(3) |
We have also examined the mechanism of the copper-catalyzed enantioselective N-alkylation of a racemic alkyl bromide (eq 1), and we have observed a noteworthy difference versus the corresponding iodide. In the case of the bromide, a time-course experiment revealed that kinetic resolution of the electrophile occurs without significant racemization (Figure 6), in contrast to our data for the iodide (Figure 5). Given that racemization of the alkyl iodide is not extremely rapid under the reaction conditions (Figure 5), the lack of significant racemization of the alkyl bromide is understandable on the basis of its lower propensity to undergo an SN2 reaction, as well as consistent with our observation that the use of 2.0, rather than 1.5, equivalents of the alkyl bromide is necessary to obtain a high yield in the asymmetric substitution reaction (eq 1).
Figure 6.

Copper-catalyzed asymmetric N-alkylation of an indole by a racemic alkyl bromide via a simple kinetic resolution: ee of the electrophile as a function of time.
We have examined the reaction of enantiopure copper complex B with each enantiomer of 3-bromo-1-phenylpyrrolidin-2-one (Table 4). In each case, the product is generated with complete inversion at the carbon undergoing substitution.17 Chiral copper complex B reacts with the S enantiomer of the electrophile more than 10 times faster than with the R enantiomer.
Table 4.
Stoichiometric substitution reactions of an enantiopure electrophile by enantiopure copper complex B: Inversion of stereochemistry.
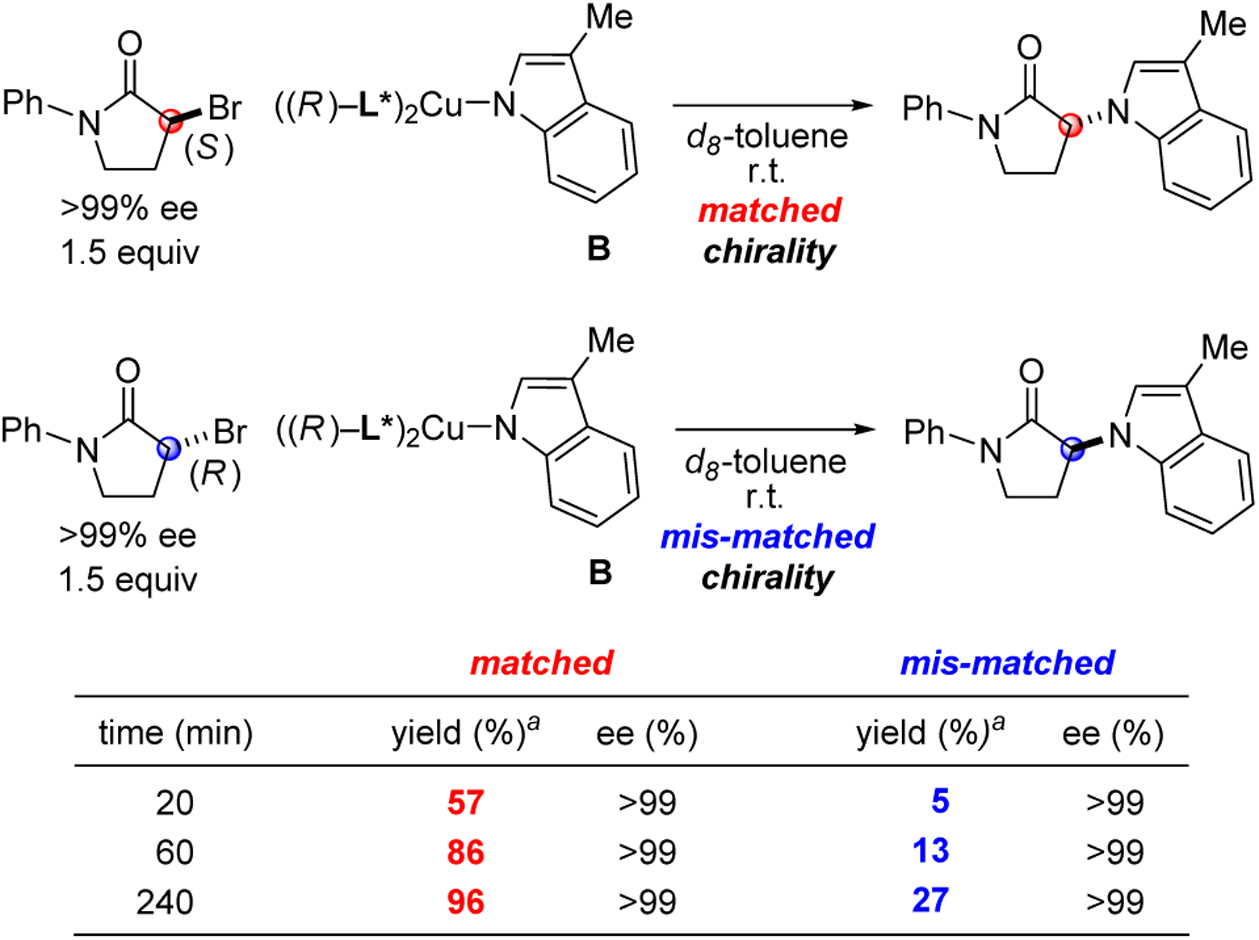 |
Determined through 1H NMR spectroscopy, with the aid of an internal standard.
Two catalytic cycles that are consistent with the above mechanistic observations are provided in Figure 7.18 In one pathway (Figure 7a), copper(I)–nucleophile complex C (complex B, above) undergoes oxidative addition of the electrophile via a two-step SN2 mechanism to afford organocopper(III) intermediate D. Reductive elimination of D then furnishes N-alkylation product E and copper(I)–halide complex F, which engages in ligand exchange to regenerate complex C.
Figure 7.
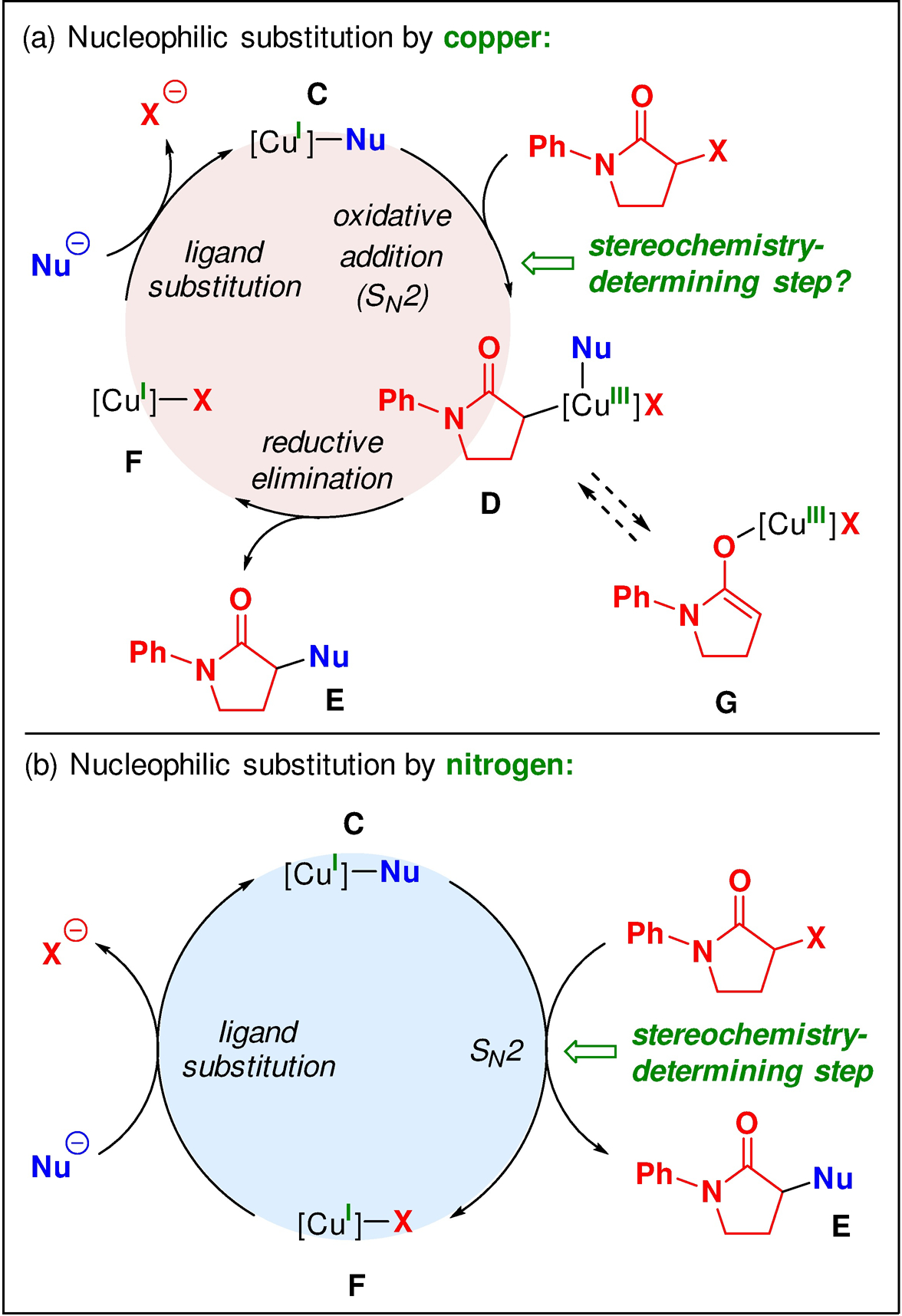
Outline of two possible mechanisms for copper-catalyzed enantioselective N-alkylations of indoles by racemic secondary alkyl electrophiles. (a) Copper as the nucleophile: Oxidative addition via an SN2 reaction to form a copper(III) intermediate. (b) Nitrogen as the nucleophile: Direct SN2 reaction.
Alternatively, copper complex C may simply serve as a chiral nitrogen-based nucleophile, reacting directly with the electrophile via an SN2 reaction to generate N-alkylation product E and copper–halide complex F (Figure 7b). Complex F then undergoes ligand exchange with the nucleophile to regenerate copper–nucleophile complex C.
The time-course experiments for these enantioselective N-alkylations indicate that the electrophile undergoes racemization under the reaction conditions in the case of the alkyl iodide, but not of the bromide (Figures 5 and 6; Figure 8). Thus, in the case of the iodide, a dynamic kinetic resolution is occurring, whereas, in the case of the bromide, a simple (not a dynamic) kinetic resolution is operative. For the mechanism outlined in Figure 7a, our observation that, for both enantiomers of the alkyl bromide, substitution by enantiopure copper complex B occurs with essentially complete inversion of stereochemistry (Table 4), suggests that copper enolate G is likely not accessible under the coupling conditions.19
Figure 8.
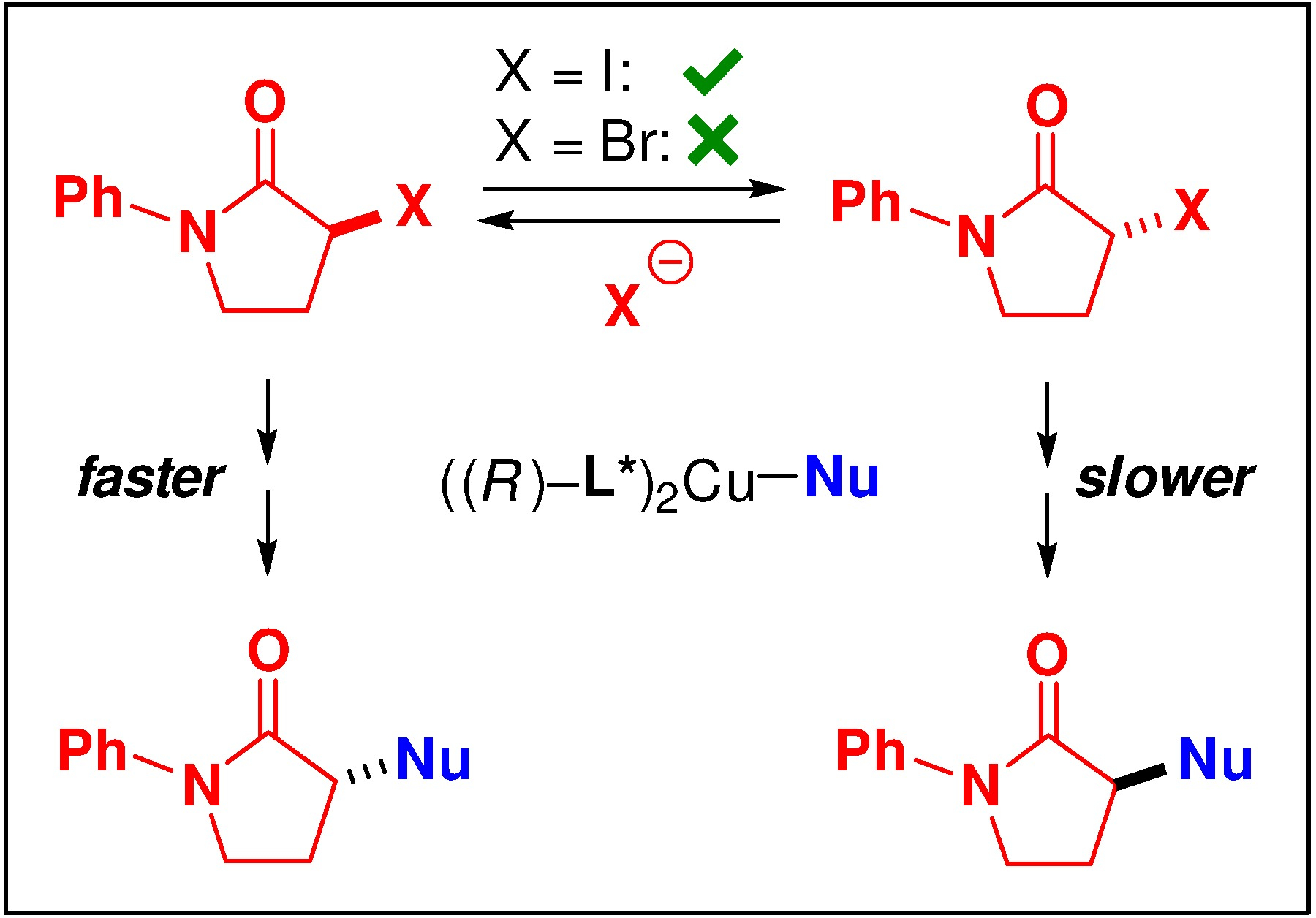
Divergence between copper-catalyzed enantioselective N-alkylations by an alkyl iodide (dynamic kinetic resolution) versus an alkyl bromide (simple kinetic resolution).
CONCLUSIONS
We have developed a straightforward method for the enantioconvergent alkylation of amines by racemic alkyl electrophiles (primarily, couplings of indoles with secondary α-iodolactams) that proceeds in the absence of light; this complements our earlier investigation, which focused on photoinduced, enantioconvergent reactions of carbazoles (and some indoles) with racemic tertiary alkyl chlorides. We have isolated and structurally characterized a proposed intermediate (a copper(I)–indolyl complex) and demonstrated that it is competent in C–N bond formation when treated with an α-iodolactam. Furthermore, we have established that the starting alkyl iodide racemizes under the reaction conditions, which enables a dynamic kinetic resolution. Further investigations of copper-catalyzed enantioconvergent N-alkylations, both photoinduced and non-photoinduced, are underway.
Supplementary Material
ACKNOWLEDGMENTS
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences; grant R01-GM109194), the Bengt Lundqvist Memorial Foundation of the Swedish Chemical Society (fellowship for A.B.), Boehringer-Ingelheim Pharmaceuticals (grant to G.C.F.), and the Dow Next Generation Educator Fund (grant to Caltech). We thank Prof. Jonas C. Peters, Dr. Quirin M. Kainz, Dr. J. M. Ahn, Lawrence M. Henling, Dr. Nathan D. Schley, Dr. Mona Shahgholi, Dr. Michael K. Takase, Dr. Scott C. Virgil, and Dr. Susan L. Zultanski for assistance and helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Procedures and characterization data (PDF).
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Amino Group Chemistry: From Synthesis to the Life Sciences; Ricci A, Ed.; Wiley–VCH: Weinheim, Germany, 2008. [Google Scholar]; (b) Lawrence SA Amines: Synthesis, Properties, and Applications; Cambridge University Press: Cambridge, U.K., 2004. [Google Scholar]; (c) Marvin CC Synthesis of Amines and Ammonium Salts In Comprehensive Organic Synthesis; Knochel P, Molander GA, Eds.; Elsevier: Amsterdam, Netherlands, 2015; Vol. 6, pp 34–99. [Google Scholar]
- (2).Copper-Mediated Cross-Coupling Reactions; Evano G, Blanchard N, Eds.; John Wiley & Sons: Hoboken, NJ, 2013; Chapters 1 and 2. [Google Scholar]
- (3).Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Terrier F Modern Nucleophilic Aromatic Substitution; Wiley–VCH: Weinheim, Germany, 2013. [Google Scholar]
- (5).Table 11 in:; Carey JS; Laffan D; Thomson C; Williams MT Analysis of the Reactions Used for the Preparation of Drug Candidate Molecules. Org. Biomol. Chem 2006, 4, 2337–2347. [DOI] [PubMed] [Google Scholar]
- (6).For examples of metal-catalyzed substitutions of allylic electrophiles by nitrogen nucleophiles, see:; Grange RL; Clizbe EA; Evans PA Recent Developments in Asymmetric Allylic Amination Reactions. Synthesis 2016, 48, 2911–2968. [Google Scholar]
- (7).For some examples of metal-catalyzed substitutions of unactivated secondary and tertiary alkyl electrophiles by nitrogen nucleophiles, see:; (a) Bissember AC; Lundgren RJ; Creutz SE; Peters JC; Fu GC Transition-Metal-Catalyzed Alkylations of Amines with Alkyl Halides: Photoinduced, Copper-Catalyzed Couplings of Carbazoles. Angew. Chem. Int. Ed 2013, 52, 5129–5133. [DOI] [PubMed] [Google Scholar]; (b) Do H-Q; Bachman S; Bissember AC; Peters JC; Fu GC Photoinduced, Copper-Catalyzed Alkylation of Amides with Unactivated Secondary Alkyl Halides at Room Temperature. J. Am. Chem. Soc 2014, 136, 2162–2167. [DOI] [PubMed] [Google Scholar]; (c) Peacock DM; Roos CB; Hartwig JF Palladium-Catalyzed Cross Coupling of Secondary and Tertiary Alkyl Bromides with a Nitrogen Nucleophile. ACS Cent. Sci 2016, 2, 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Jacobson IC; Quan ML; Wexler RR Fused Heterocyclic Inhibitors of Factor Xa. Patent WO 02/080853 A2, October 17, 2002.; (b) Axten JM; Medina JR Chemical Compounds Acting as PERK Inhibitors. WO 2015/136463 Al, September 17, 2015.; (c) Wilson D, et al. Heterocyclic Derivatives as Modulators of Ion Channels. Patent WO 2007/075895 A2, July 5, 2007.
- (9).Nugent TC Chiral Amine Synthesis; Wiley–VCH: Weinheim, Germany, 2010. [Google Scholar]
- (10).Zhang H; Kang H; Hong L; Dong W; Li G; Zheng X; Wang R Construction of the N1−C3 Linkage Stereogenic Centers by Catalytic Asymmetric Amination Reaction of 3‐Bromooxindoles with Indolines. Org. Lett 2014, 16, 2394–2397. [DOI] [PubMed] [Google Scholar]
- (11).Kainz QM; Matier CM; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C–N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For examples of discussions of indoles as privileged structures in bioactive molecules, see:; (a) de Sa Alves FR; Barreiro EJ; Fraga CAM From Nature to Drug Discovery: The Indole Scaffold as a ‘Privileged Structure’. Mini Rev. Med. Chem 2009, 9, 782–793. [DOI] [PubMed] [Google Scholar]; (b) Kumari A; Singh RK Medicinal Chemistry of Indole Derivatives: Current to Future Therapeutic Prospectives. Bioorg. Chem 2019, 89; 10.1016/j.bioorg.2019.103021. [DOI] [PubMed] [Google Scholar]; (c) Table 1 in:Welsch ME; Snyder SA; Stockwell BR Privileged Scaffolds for Library Design and Drug Discovery. Curr. Opin. Chem. Biol 2010, 14, 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For examples of the removal of N-benzyl and N-(p-anisyl) groups of tertiary lactams to generate secondary lactams, see:; (a) benzyl: Abe T; Takeda H; Takahashi Y; Miwa Y; Yamada K; Ishikura M Metal-Catalyzed Reactions between 2-Azabicyclo[2.2.1]hept-5-en-3-ones and Arylboronic Acids. Eur. J. Org. Chem 2010, 3281–3294. [Google Scholar]; (b) p-anisyl: Jette CI; Geibel I; Bachman S; Hayashi M; Sakurai S; Shimizu H; Morgan JB; Stoltz BM Palladium-Catalyzed Construction of Quaternary Stereocenters by Enantioselective Arylation of γ-Lactams with Aryl Chlorides and Bromides. Angew. Chem. Int. Ed 2019, 58, 4297–4301. [DOI] [PubMed] [Google Scholar]
- (14).Under the same conditions, the corresponding alkyl chloride does not react with 3-methylindole.
- (15). If desired, virtually all (94%) of the chiral phosphine, L*, can be recovered after oxidation to the corresponding phosphine oxide (see the Supporting Information).
- (16).In order that the coupling could be monitored by 1H NMR spectroscopy, it was conducted in d8-toluene. The standard reaction (Table 1) proceeeds with similar ee and yield in toluene and in m-xylene.
- (17).For examples of copper-catalyzed substitution reactions that proceed with inversion of stereochemistry when carbon nucleophiles are used, see:; (a) Terao J; Todo H; Begum SA; Kuniyasu H; Kambe N Copper-Catalyzed Cross-Coupling Reaction of Grignard Reagents with Primary-Alkyl Halides: Remarkable Effect of 1-Phenylpropyne. Angew. Chem. Int. Ed 2007, 46, 2086–2089. [DOI] [PubMed] [Google Scholar]; (b) Yang C-T; Zhang Z-Q; Liang J; Liu J-H; Lu X-Y; Chen H-H; Liu L Copper-Catalyzed Cross-Coupling of Nonactivated Secondary Alkyl Halides and Tosylates with Secondary Alkyl Grignard Reagents. J. Am. Chem. Soc 2012, 134, 11124–11127. [DOI] [PubMed] [Google Scholar]
- (18).These copper-catalyzed substitution reactions could of course proceed through different pathways for alkyl iodides versus alkyl bromides.
- (19).The Chemistry of Metal Enolates; Zabicky J, Ed.; Wiley: Chichester, U.K., 2009; Vol. 1 and 2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.