

Abstract
Hierarchically assembled nanomaterials can find a variety of applications in medicine, energy, and electronics. Here, an automatically controlled dip-pulling method is developed and optimized to generate an unprecedented ordered nano-to-micro hierarchical nanoridge-in-microridge (NiM) structure from a bacteria-specific human-safe virus, the filamentous phage with or without genetically displaying a foreign peptide. The NiM structure is pictured as a window blind with each lath (the microridge) made of parallel phage bundles (the nanoridges). It is independent of the substrate materials supporting it. Surprisingly, it can induce the bidirectional differentiation of stem cells into neurons and astrocytes within a short timeframe (only 8 d) not seen before, which is highly desired because both neurons and astrocytes are needed simultaneously in treating neurodegenerative diseases. Since phages can direct tissue regeneration, template materials formation, sense molecules, and build electrodes, the NiM structures displaying different peptides and on varying materials hold promise in many technologically important fields.
Keywords: hierarchical structures, nanomaterials, stem cells, viruses
Hierarchical structures, either natural or artificial, can find potential applications in medicine, energy, and electronics.[1] Nature has produced biological materials with nano-to-micro hierarchical structures such as diatoms and bone.[1] However, it is still a daunting challenge to fabricate nano-to-micro hierarchical structures in the laboratory. Hence, we aimed to develop an unprecedented hierarchical structure and demonstrated one of its applications in serving as a matrix to induce the bidirectional neural differentiation of stem cells. The bidirectional neural differentiation of stem cells into neurons and astrocytes is desired because both neurons and astrocytes are the building blocks of the human brain and thus are needed simultaneously in treating neurodegenerative diseases (NDDs) such as Parkinson’s disease (PD) and Alzheimer’s disease (AD).
Hence, there is a pressing need to simultaneously regenerate both neurons and astrocytes to advance the NDD treatment[2] and to generate a coculture model for studying the interactions between both cells.[3] So far, there have been very limited studies on the use of nanomaterials to generate a neuron-astrocyte coculture model.[4] Moreover, these studies used rodent cells to generate the neuron-astrocyte coculture. Thus, generating neurons and astrocytes from the same human cell source will facilitate the development of NDD treatment strategies. Stem cells such as neural stem cells (NSCs) or neural progenitor cells (NPCs) are the direct source for neural differentiation, but the only tissue source for NSCs or NPCs is human fetal brain tissue. Thus using the tissue source to generate neurons and astrocytes presented ethical issue.[5] Human induced pluripotent stem cells (hiPSCs) or NPCs derived from them are the new cell sources for tissue regeneration.[6] A material can serve as a matrix for providing a microenvironment to modulate their differentiation. However, so far, a material has mainly been used to generate either neurons[7] or astrocytes[8] from a single cell source. To the best of our knowledge, almost no study has been dedicated to the use of a material to achieve the regeneration of both neurons and astrocytes from a human cell source such as NPCs derived from hiPSCs although both cells are needed for treating NDDs.[9,10]
Hence, we proposed to develop a nano-to-micro hierarchical structure (termed nanoridge-in-microridge, NiM, Scheme 1b–d) from filamentous M13 phage, by a novel method termed dip-pulling (Scheme 1a), and then employed the NiM structures to induce the bidirectional differentiation of hiPSC-derived NPCs into both neurons and astrocytes (Scheme 1). It should be noted that hiPSCs did not adhere to the phage films (probably due to the formation of suspended cell clusters called embryonic bodies during their differentiation into NPCs). However, the NPCs derived from them did not form the suspended clusters but adhered well to the phage films. Thus, the NPCs were first derived from hiPSCs (Figure S1, Supporting Information) and then cultured on the NiM structures to achieve the bidirectional differentiation of NPCs in the absence of additional differentiation inducers, because the NiM structures presented unique nanotopography. Moreover, we found that the NiM structures could be formed virtually on any type of material.
Scheme 1.
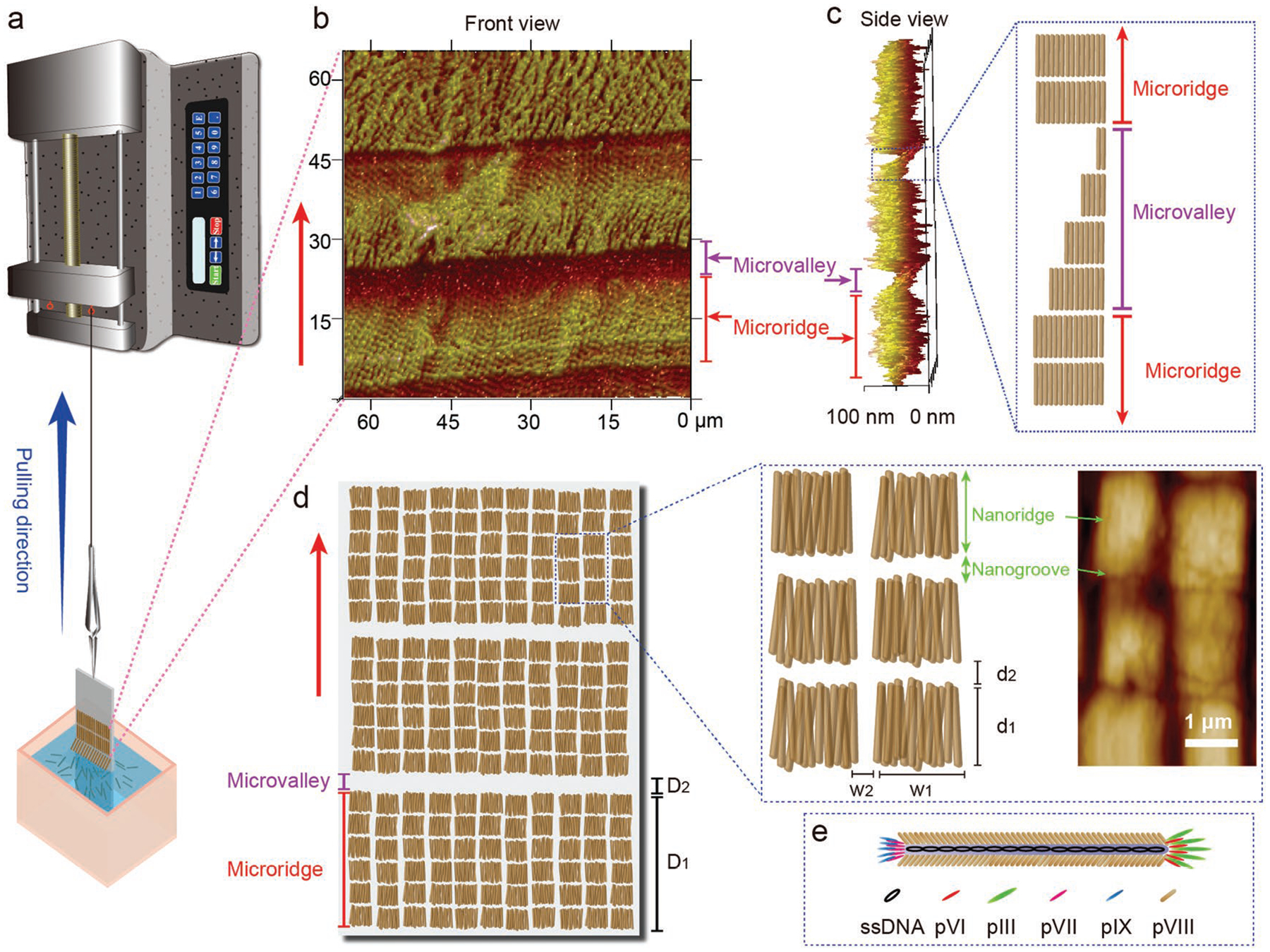
Self-assembly of phages into hierarchical nanoridge-in-microridge (NiM) structures by a dip-pulling method. a) Schematic illustration of the dip-pulling method. In this method, a substrate (e.g., glass slide) coated with positively charged polylysine was dipped into a phage solution and then vertically pulled out of the solution at a certain speed by a syringe pump. Under certain conditions (phage solution concentrations, pulling speeds, evaporation rates, pH values, and salt concentrations), this method generated a phage film with a nanoridge-in-microridge (NiM) structure. In the NiM, phages were parallel to the pulling force direction and became ordered into the parallel-aligned nanoridges that were further hierarchically assembled into the parallel microridges. The nanoridges (with a length of d1) were separated by nanogrooves (with a width of d2) and as wide as the length of phages (≈1.2 μm for wild-type phage and ≈550 nm for engineered phage). b) Front view and c) side view of the NiM phage film structure by AFM imaging. The side view (c) of the NiM shows the presence of a microvalley in between microridges with the microvalley made of phage layers of decreasing thickness. d) Schematic of the NiM structures by dip-pulling method and high magnification of the nanoridge and nanogroove structure by AFM image. D1 and D2 are the width of microridges and microvalleys, respectively. d1 and w1 are the length and width of the nanoridges, respectively. d2 and w2 are the size of the nanogrooves between the nanoridges along and perpendicular to the pulling direction, respectively. The nanoridges were organized into microridges separated by the microvalleys. Each nanoridge is just a bundle formed by parallel-aligned phages, and thus d1 should be theoretically the length of a phage nanofiber. e) schematic of a single phage. Red arrows denoted the pulling force direction. The AFM images show that the NiM film was assembled from wild-type phages with a concentration of 1 × 1014 pfu mL−1 by the dip-pulling method at pulling speeds of 1.5 μm s−1 (c) and 0.5 μm s−1 (b,d).
Filamentous M13 phage is a human-safe bacteria-specific viral nanofiber (Scheme 1e) that is made of a sheath of coat proteins encapsulating a circular ssDNA.[11] The side wall of the nanofiber is assembled from ≈3000 copies of a major coat protein (termed p8). We used two types of M13 phage in this work, one is the wild-type phage (termed WT-phage) and another is the engineered phage (termed RGD-phage) with the side wall displaying Arg-Gly-Asp (RGD) peptide (through fusion of RGD to N-terminal of p8 by genetic means reported by us previously).[11] Both WT-phage and RGD-phage are nanofibers (≈7 nm wide) with a negatively charged protein shell.
To establish and optimize the dip-pulling method, we employed WT-phage simply because WT-phage could be amplified by infecting bacteria more efficiently than RGD-phage, and both phages do not show difference in phage assembly in the dip-pulling. It should be noted that WT-phage (≈1.2 μm long) is longer than RGD-phage (≈550 nm long). It is known that displaying a peptide on the side wall of M13 phage shortens the phage.[12] Our dip-pulling method was designed as a one-step operation (one dipping and one pulling) under automatic control to avoid the irreproducibility commonly seen in manual or multistep operation. Namely, a glass slide precoated with positively charged polylysine was dipped into a monodisperse phage solution, and then vertically pulled out of the solution at a stable speed controlled by a syringe pump (and thus avoiding manual operation).
During the dip-pulling process, the positively charged polylysine coated glass slide attracted negatively charged phages to its surface and the phages were parallelly aligned into a unique hierarchical NiM structure along the pulling direction (Scheme 1b–d). Such a NiM structure has never been reported before and is significantly different from reported alignment of bionanofibers.[13,14] The nanoridged pattern in the hierarchical structure looked like a window blind with each lath (the microridge) made of nanoridges (assembled from parallel-aligned phages). The NiM showed a periodic pattern at both nano- and microstructure. At the microscale, microridges were parallel to each other with microvalleys in between the neighboring ones. The microvalleys were made of phage bundles with decreasing thickness along the pulling direction (Scheme 1c). At the nanoscale, each microridge is made of many parallel nanoridges separated by nanogrooves (Scheme 1d). Each nanoridge is assembled from parallel phages and is as wide as the length of an individual phage. In such a structure, phage nanofibers are always aligned along the pulling force direction, probably because they have the smallest hydrodynamic interactions with fluid in the shear force direction.[15] We found that the NiM structure was only formed under certain conditions after we carried out a series of experiments optimizing these conditions, including the pulling speeds, evaporation rates, phage concentrations, salt concentrations, and pH values. Although we have not fully understood the NiM formation mechanism, we believe that it is related to the solvent evaporation at the meniscus of air–fluid–solid interface moving along the pulling force. It is very likely that the phage solution reaches a concentration higher than the critical concentration (4 × 1015 pfu mL−1) for forming a smectic liquid crystal phase at the meniscus.[16]
NiM structures were consistently formed at different pulling speeds (10, 4, 1.5, and 0.5 μm s−1, with 1 × 1014 pfu mL−1 phage solution), as confirmed by atomic force microscopy (AFM) (Figure 1). However, some size parameters of the nanoridges/microridges and the nanogrooves/microvalleys are different. To better understand the effect of pulling speeds on these size parameters, we define six size parameters as shown in Scheme 1, which were determined by a combination of three AFM images for each speed. D1 and D2 are the width of the microridges and microvalleys, respectively. d1 and w1 are the length and width of the nanoridges, respectively. d2 and w2 are the size of the nanogrooves between the nanoridges along and perpendicular to the pulling direction, respectively (Scheme 1d). Because each nanoridge is actually just a bundle formed by lateral aggregation of parallel-aligned phages, d1 should be the length of a phage nanofiber. This is confirmed by our finding that d1 was nearly constant and independent from the pulling speed change (Figure 1i). However, with the increase in the pulling speed, D1 and d2 were increasing whereas D2, w1, and w2 were decreasing (Figure 1h,i). The effect of periodic pulling speeds (10 and 0.5 μm s−1) on the phage film formation was also studied (Figure S2, Supporting Information). We found that d1, w1, and w2 were not significantly affected by the periodic speed change. However, D1 and d2 still followed our predicted trend in Figure 2 under both speeds; their values on the higher speed side are larger than those on the lower speed side (Figure S2, Supporting Information). On the speed boundary line, we could see a sudden change in the nanostructures when we changed the speed (Figure S2c, Supporting Information). On the fast speed side (10 μm s−1), the nanoridges tended to become bent along the pulling direction, while on the slow speed side (0.5 μm s−1), the nanoridges appeared to be more straight.
Figure 1.
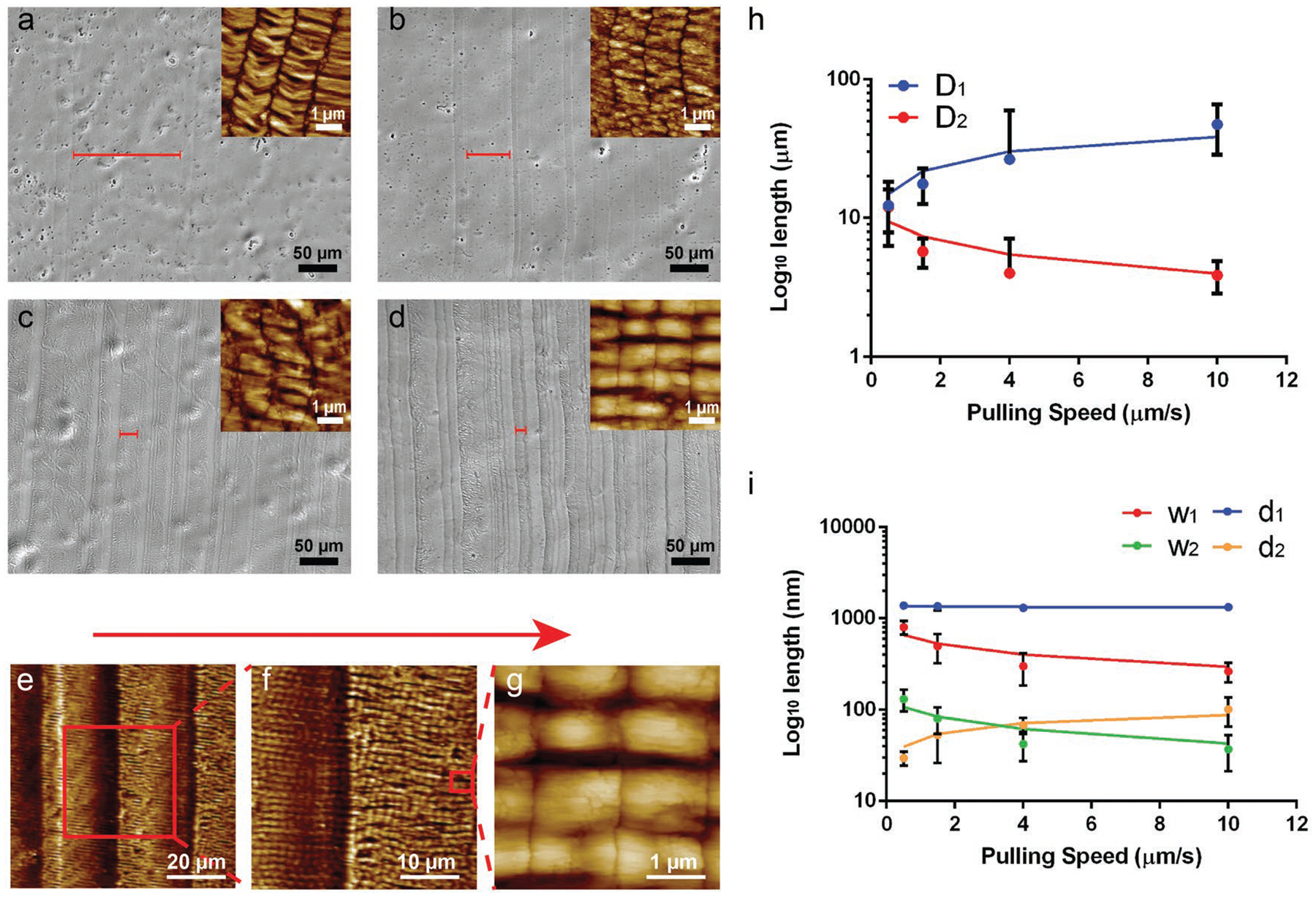
Surface morphologies of ordered phage films assembled from a phage solution (1 × 1014 pfu mL−1) at different pulling speeds by the dip-pulling method. The surface morphologies were observed by a bright field optical microscopy imaging at a low magnification and by AFM imaging at a high magnification (shown as insets). a–d) The optical imaging of the film structures at different pulling speeds: a) 10 μm s−1; b) 4 μm s−1; c) 1.5 μm s−1; d) 0.5 μm s−1). The red bar on the images indicated the average width (D1) of the microridges at different speeds. As the speed decreased, the width (D1) of the microridges became smaller. e–g) AFM images of a NiM structure at different magnifications showing the microridges/microvalleys and nanoridges/nanogrooves (schematically shown in Scheme 1d). The red arrow denotes the pulling force direction. h,i), Relationship between the pulling speed and the six size parameters of the NiM (D1, D2, w1, w2, d1, d2). The length of d1 didn’t change with the speeds because it reflects the phage length, while the other five factors showed an increase (D1, d2) or a decrease (D2, w1, w2).
Figure 2.
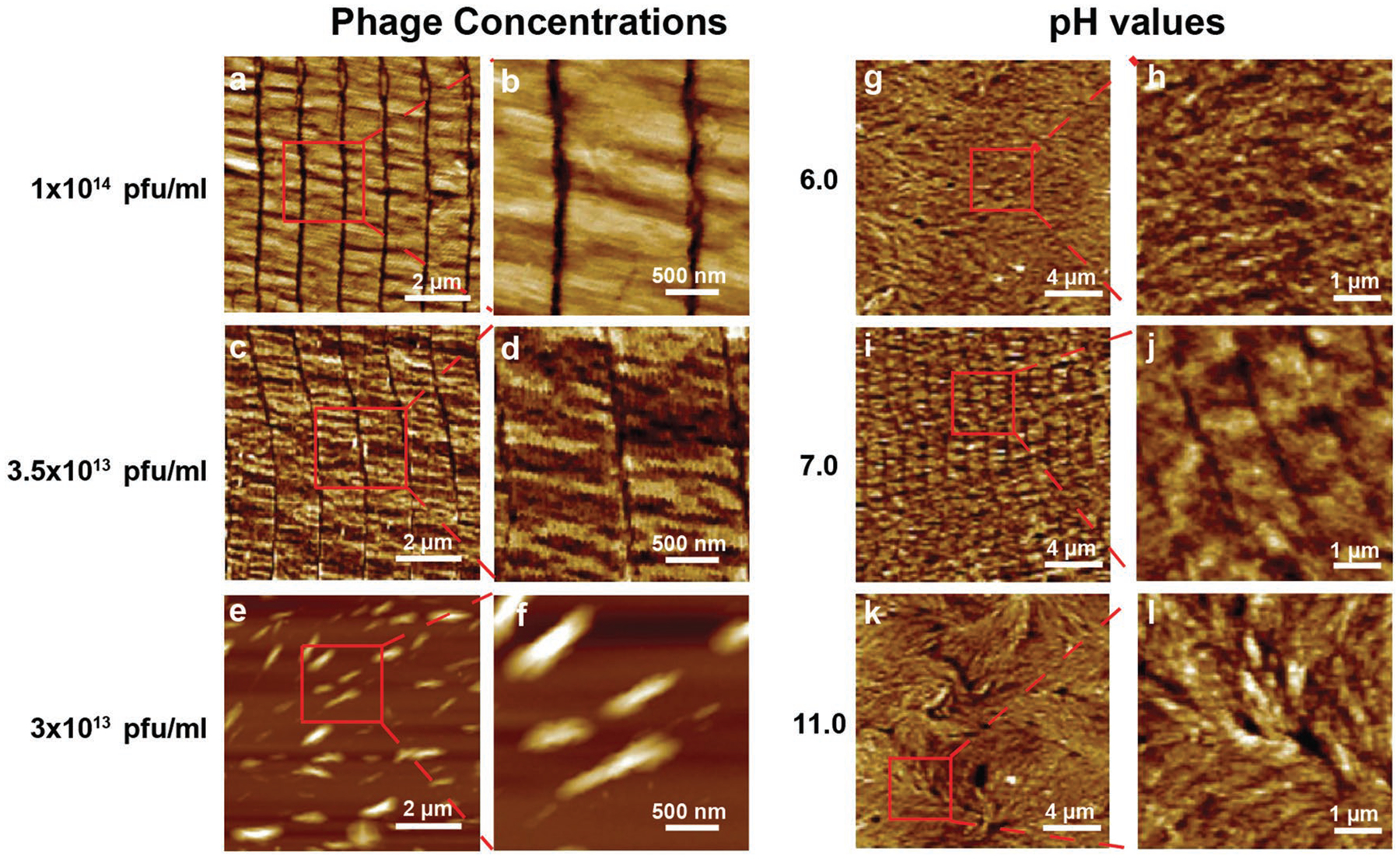
Surface morphologies of wild-type phage films assembled form wild-type phages at different phage concentrations and pH values by the dip-pulling method. a–f) Phage concentration manipulates the phage film formation. The phages were assembled into similar ordered nanoridged patterns when their concentration was higher than 3.5 × 1013 pfu mL−1, below which such patterns were not formed. Pulling speed, 1.5 μm s−1. g–l) pH value of the phage solution manipulates the phage film formation. When the pH was between 6 and 11, the parallel-aligned nanoridged pattern could be seen under the AFM. When the pH was out of this range, such pattern was not formed. (Phage concentration, 5 × 1013 pfu mL−1; pulling speed, 1.5 μm s−1).
We then investigated the effect of evaporation rates on the phage film formation. We found that the best evaporation condition for NiM structure formation was the normal room condition at room temperature. When the dip-pulling was operated inside a chemical fume hood to accelerate the evaporation, we found that the NiM structure was disturbed at the microscales (Figure S3, Supporting Information). In comparison to the normal room evaporation, the distribution of the microridges and microgrooves was not as uniform under the fast evaporation. Moreover, under the fast evaporation, microridge width D1 significantly decreased. However, the nanoridge structures stayed the same under different evaporation conditions.
We then studied the effect of the phage concentrations on the NiM structure formation. Different concentrations of phage solutions, ranging from 1 × 1013 to 2 × 1014 pfu mL−1, were proved to affect the phage film structures at a pulling speed of 1.5 μm s−1. The concentration changes of the phage solutions did not disturb the NiM structure and the six sizes parameters (D1, D2, w1, w2, d1, d2) when their concentration was higher than 3.5 × 1013 pfu mL−1 (Figure 2a–d). When the phage concentrations were below 3.5 × 1013 pfu mL−1, the NiM structure could not be formed (Figure 2e,f). These data suggest that the formation of NiM only occurred when the phage concentration was above a critical concentration, further indicating that the NiM structure was probably driven by the aforementioned liquid crystal phase formation at the moving meniscus of the air–fluid–solid interface when the glass slide was being pulled.
To understand the effect of ionic strengths on the formation of NiM structures, we studied the NiM structure formation under different NaCl concentrations in the phage solution with a certain phage concentration (7 × 1013 pfu mL−1) and pulling speed (1.5 μm s−1). We found that the NaCl concentrations significantly affected the NiM structure formation and generally lower NaCl concentrations benefited the formation of the NiM structures (Figure S4, Supporting Information). When NaCl concentrations were higher than 0.01 m, phages could not be assembled into NiM structures, but only formed nonparallel, crossed patterns. When the NaCl concentrations were increased from 0.02 to 0.05 m, the crossed patterns became more disordered. When the NaCl concentrations reached 0.1 m, the phages were assembled into a completely disordered structure. Thus, overall a higher NaCl concentration tended to result in a more disordered phage film.
Finally, we investigated the effect of pH values of the phage solutions on the NiM structure formation when the salt concentration was 0.01 m, the phage concentration was 5 × 1013 pfu mL−1 and the pulling speed was 1.5 μm s−1. The isoelectric point (pI) of M13 phage is 4.5.[17] The pH of phage solutions was 7–8. Phages could stay in pH 3–11 for 20 min without losing their infectivity.[18] Thus, we studied the effect of pH values when the pH values of the phage solutions were buffered from pH 3 to 11 (Table S1, Supporting Information). When the pH value was between 6 and 11, the NiM structures were still formed. When the pH value was higher than 11 or lower than 6, the NiM structures could not be formed (Figure 2g–l).
Our method can be extended to the formation of ordered phage structures on a variety of substrates including amorphous materials (e.g., glass and silicon oxide on a silicon wafer), crystalline inorganic materials (e.g., gold, titanium, stainless steel), and organic materials (e.g., polycarbonate plastics) (Figure 3). The key is that these substrates should be pretreated by being dipped into a polylysine solution to form a polylysine coating. We found that phages could be assembled into NiM structures once these substrates were pretreated with polylysine. However, such structures were usually not formed on the nontreated substrates (Figure 3). For example, less organized phage structures were formed on the nontreated glass and gold substrates, and phages could not be assembled on the noncoated substrates of titanium, silicon, plastic, and stainless steel. Namely, only after the polylysine pretreatment, could phages be assembled into highly ordered NiM structures on all of the substrates with inherently different compositions and crystallinities. It should be noted that surface treatment by only polylysine did not lead to the formation of NiM structures (Figure S5, Supporting Information).
Figure 3.

Phage structures on a variety of substrates before and after polylysine treatment. Before polylysine treatment, M13 phages are assembled into nanoridges and nanogrooves on glass and gold substrate with less organized structure, whereas no phage patterns were found on titanium, silicon, plastic, and stainless steel substrate. After the polylysine pretreatment, phages formed highly ordered NiM structures on all of substrates. Red arrow indicated the pulling direction.
We used the optimized conditions (phage concentration, 1 × 1014 pfu mL−1; NaCl concentration, 0 m; pH, 7; pulling speed, 1.5 μm s−1) to prepare phage films with the NiM structures of WT-phage or RGD-phage. Thus, we had three substrate groups, WT-phage, RGD-phage (Figure S6, Supporting Information) and control (the polylysine-coated glass slide without phage), in testing the bidirectional differentiation of iPSC-derived NPCs into neurons and astrocytes in the absence of differentiation inducers in the culture medium. hiPSC-derived NPCs were seeded on the three substrates in a nondifferentiation medium. We observed obvious differences in cell morphology on different substrates. Starting from Day 2 after cell seeding, the cells on the phage films showed an elongated morphology while those on the control substrate were not elongated (Figure S7a–c, Supporting Information). The elongated cells on the WT-phage film tended to form clusters without preferential orientation, whereas those on the RGD-phage film, either isolated or clustered, were preferentially aligned along the length direction of microridges (Figure S7c,d, Supporting Information). Surprisingly, we found that the NiM structures of both WT-phage and RGD-phage could induce the bidirectional differentiation of NPCs into neurons and astrocytes (Figure S7d,e, Supporting Information).
In order to verify that the phage films induced the intended bidirectional differentiation, we tested the possible neural differentiation of NPCs into neurons, astrocytes or oligodendrocytes by detecting their corresponding cellular markers (Figure 4; Figure S7d,e, Supporting Information). The mRNA and protein expression levels of different markers were tested by qRT-PCR and immunofluorescence, respectively, on the cells cultured on the phage groups at two time points (Day 4 and 8) (Figure 4; Figure S8, Supporting Information). It should be noted that Nestin is an NPC marker, βIII-Tubulin (also known as TBB3) and MAP2 are early and late stage neuron markers, GFAP is an astrocyte marker, and Olig2 is an oligodendrocyte marker. The results from the phage film groups were normalized to those of the control group. We could detect NPC markers for all groups. However, compared to the control group, we could detect a significantly high level of the markers for both neurons and astrocytes, but could not detect the markers for oligodendrocytes, indicating that the phage films could only induce hiPSC-derived NPCs to differentiate into both neurons and astrocytes, but not oligodendrocytes. Moreover, at the two time points, the cells on the WT-phage films showed higher expression levels of the early stage (TBB3) and late stage (MAP2) neuron markers than those on the RGD-phage films at the both mRNA and protein levels (Figure 4a–d). On the other hand, at both time points, the cells on the RGD-phage films presented a higher expression level of astrocyte marker (GFAP) than those on the WT-phage films. Therefore, although both WT-phage and RGD-phage films induced the bidirectional differentiation of NPCs into both neurons and astrocytes, the former and latter more favored the differentiation into neurons and astrocytes, respectively (Figure 4).
Figure 4.
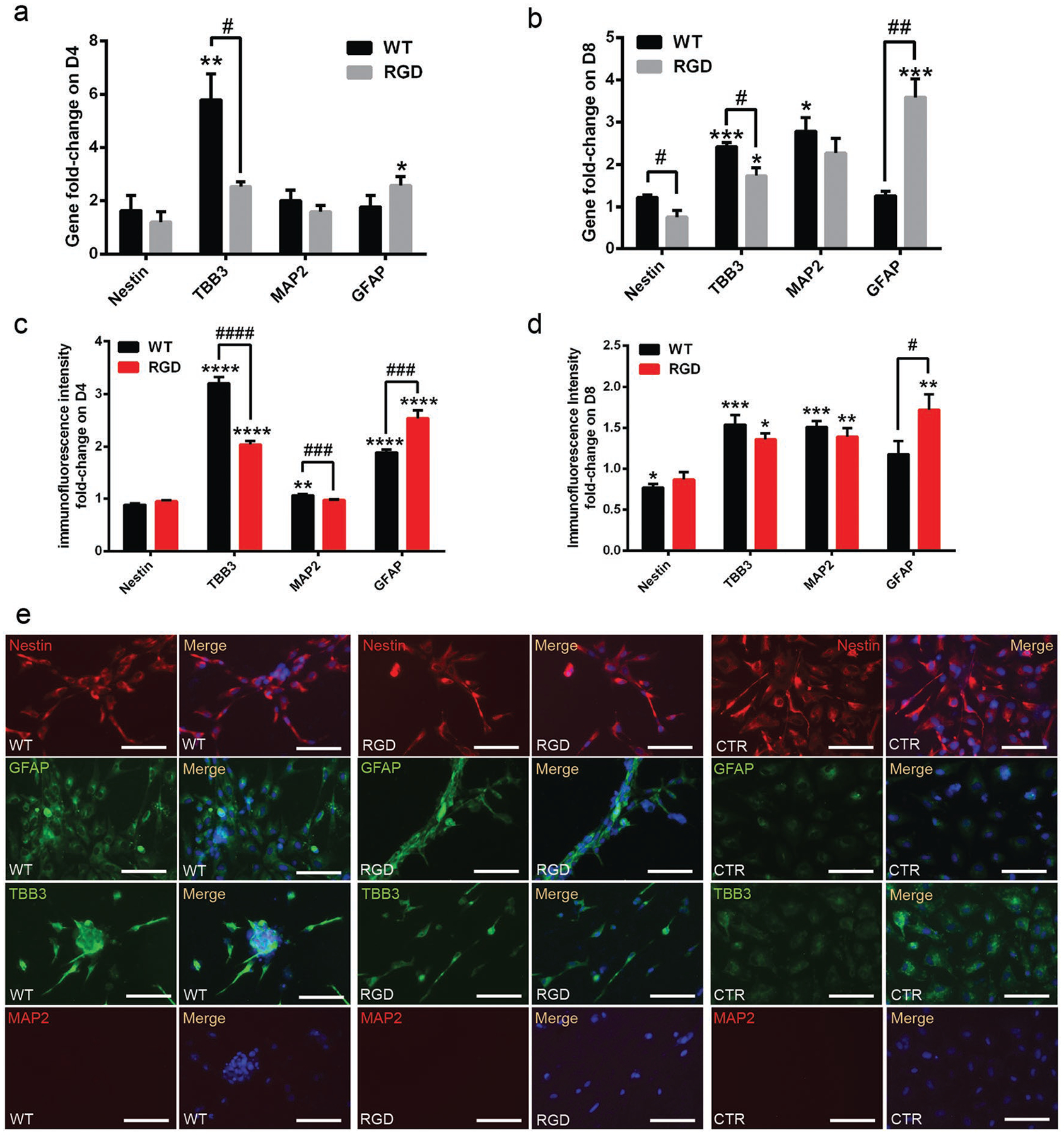
Neural differentiation of NPCs on phage films prepared by the dip-pulling method under the same conditions (Phage concentration, 1 × 1014 pfu mL−1; pulling speed, 1.5 μm s−1.). a–d) Neural differentiation of the NPCs verified by: a,b) qRT-PCR and c,d) immunofluorescent intensity fold change of the markers for NPCs, neurons and astrocytes on: a,c) Day 4 and b,d) Day 8. c,d) Generated by analysis of multiple immunofluorescence images for each marker using image J. The immuno-fluorescent intensity level of each marker in CTR (nonphage control group) were set to 1. In (c) and (d), the black columns indicate WT/CTR fluorescent intensity ratio and the red columns indicate RGD/CTR fluorescent intensity ratio. Nestin is an NPC marker. TBB3 and MAP2 are early and late stage neuron markers, respectively. GFAP is an astrocyte marker. The NPCs on the WT-phage films showed higher expression levels of TBB3 and MAP2 marker, while the cells on the RGD-phage films showed a higher expression level of GFAP. e) Immunofluorescence imaging results of the markers for the neural differentiation of the NPCs on Day 4. Cells on the WT-phage films had a higher expression level of TBB3 marker, while those on the RGD-phage films showed a higher expression level of GFAP marker. Scale bar: 100 μm. These results indicate that WT-phage and RGD-phage films more efficiently induced the differentiation of NPCs into neurons and astrocytes, respectively. (* or #, P < 0.05; ** or ##, P < 0.01; *** or ###, P < 0.001; **** or ####, P < 0.0001. * indicates the significant difference between WT/RGD and CTR, # indicates the significant difference between WT and RGD).
On Day 4, the expression level of the NPC marker (Nestin) was similar in each group, but the expression levels of TBB3 and GFAP were significantly increased in both WT-phage and RGD-phage film groups compared to the control group. Compared to the control group, the cells on the WT-phage film expressed a 3.2-fold and 1.9-fold higher fluorescent intensity of TBB3 and GFAP, respectively, while those on the RGD-phage film showed a 2-fold and 2.5-fold fluorescent intensity increase in TBB3 and GFAP, respectively (Figure 4c,d). On Day 4, the cells on the WT-phage films showed 5-fold higher mRNA expression of TBB3 than the control group (Figure 4a). These results collectively indicated that the unique phage film structure could induce bidirectional differentiation of hiPSC-derived NPCs within 4 d.
From Day 4 to Day 8, the level of TBB3 expression dropped, but the level of MAP2 expression increased, indicating that the neurons were mature within 8 d (Figure S6a–d, Supporting Information). It usually took more than two weeks for NPCs to become mature neurons.[19] However, our phage films achieved the differentiation of NPCs into mature neurons in 8 d, suggesting that our phage films could guide and accelerate the neuron maturation of the hiPSC-derived NPCs due to their unique topographies.
Synthetic topography cues greatly influenced the stem cell fate.[20] Anisotropic patterns (such as gratings and grooves) and isotropic patterns (such as pillars and wells) of the substrates were reported to favor neuronal and glial differentiation, respectively.[20] Grating structures were considered to provide stem cells with a stronger support in connecting adjacent cells and promote the neurite growth and neuronal cell polarization.[21] In comparison to the nonpatterned group, gratings were reported to enhance bidirectional differentiation in stem cell cultures.[20] The grating size proved to induce neural differentiation ranged from hundreds of nanometers to a few micrometers.[20,22] Thus, we believe that microridges and microgrooves would have a stronger effect on neural differentiation of the hiPSC-derived NPCs than the nanoridges and nanogrooves in our NiM structures. On the other hand, nanofibers were reported to induce astrocyte differentiation[23] and manipulate astrocyte activation.[24] Nanofibrous polymers are one of the most used materials to induce the bidirectional differentiation.[9,10,25] They can be arranged into nanogratings by nanoimprinting[25] or form nanofibers by electrospinning.[10] Our phage films exhibited the topographical features of both anisotropic grating and nanofibers. Hence, such unique topographies induced the bidirectional differentiation of NPCs into neurons and astrocytes without neural differentiation inducers in the culture medium. On the basis of the unique morphology of phages, the peptides displayed on them are the secondary factor that can fine tune the bidirectional differentiation. For instance, in this work we found that both WT-phage and RGD-phage could induce the differentiation of stem cells into both neurons and astrocytes but with a different target cell ratio. Therefore, this work, along with our earlier studies where phages were only assembled into nanoridges, collectively showed that the unique morphology of filamentous phages endowed them to bear the ability to direct stem cell differentiation, although their different self-assembly patterns led to different target cells.[14,26]
Indeed, we found that in a very short duration (within 8 d) the WT-phage films highly induced the hiPSC-derived NPCs to express neuron markers, whereas the RGD-phage films had a higher induction in astrocyte formation. The generation of neurons through neuronal differentiation from NPCs using neuronal induction medium usually need more than two weeks.[19,27] The generation of astrocytes from NPCs or NSCs by using astrocyte induction medium usually needs more than 30 d.[28] Thus, the unique NiM structure of the phage films largely shortened the neural differentiation duration and provided us a way to study the relationship between neurons and astrocytes in tissue regeneration in the future.
In this work, a simple pretreatment of different substrates by polylysine consistently led to the formation of same NiM structures. This suggests that our dip-pulling method is a universal one for the formation of NiM structures on a variety of different substrates. This also further confirms that the electrostatic interaction between phages and the polylysine is one of the driving forces for the assembly of phages into NiM structures on the substrates. Since phages can be used to direct tissue regeneration,[14] template functional materials formation,[29] sense molecules,[30] and build battery materials,[31] the capability of forming NiM phage structures on different materials holds promise in finding versatile applications in many technologically important fields such medicine, energy, and electronics.
Although we have not fully understood the mechanism of NiM structure formation, we hypothesize that different experimental parameters affect the structure formation by influencing the liquid crystal behavior of M13 phage at the liquid–air–solid interface. M13 phage becomes highly concentrated to favor liquid crystal formation on the newly exposed solid substrate area that just leaves the liquid phase at the interface during the dip-pulling. The concentration and speed changes directly affect the critical concentration for liquid crystal formation at the liquid–air–solid interface, and the liquid crystal phase change might occur at different critical concentrations.[16] A change in the salt concentration and pH values can alter the surface charge of single phages, which in turn changes the electrostatic interaction in the phage solution.[32] In addition, it is known that electrostatic interaction is the main factor for directing liquid crystal phase transitions.[33] These known influences on the liquid crystal formation by the parameters (concentrations, pulling speeds, salt concentration, and pH value) shed light into the possible mechanisms by which these parameters control the structure formation of phage films observed in our study and indicate an optimal combination of these parameters leads to the formation of NiM structures.
In summary, we developed a new dip-pulling method for the assembly of both WT-phage and RGD-phage into a unique and unprecedented NiM structure. This method could be extended to the other substrates by a simple polylysine pretreatment before the dip-pulling process. We also identified the best conditions for forming the NiM structure. The key conditions included the pulling speeds, the phage concentrations, the salt concentrations, evaporation rates, and the pH values. We also found that the resultant NiM structures could induce the bidirectional differentiation of hiPSC-derived NPCs into both neurons and astrocytes in the absence of any differentiation inducer in the culture medium. The NiM structures could induce the bidirectional differentiation within 8 d and thus are significantly faster than the reported methods (more than two weeks).[19,27,28] When the RGD peptide was displayed on the WT-phage to form RGD-phage, the resultant NiM structures could lead to the formation of more astrocytes in the bidirectional differentiation. Both neurons and astrocytes are building blocks in a healthy CNS. Hence, the neurons and astrocytes generated by our NiM structures can not only be used for treating NDDs but also can establish a neuron-astrocyte coculture model for studying their interactions to gain insights for treating NDDs.
Supplementary Material
Acknowledgements
The authors would like to thank the financial support from National Institutes of Health (EB021339 and GM116116) and the Institute for Biomedical Engineering, Science and Technology of the University of Oklahoma. The hiPSCs used in this study were obtained from ALSTEM, Inc.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adma.201905577.
Contributor Information
Ningyun Zhou, Department of Chemistry & Biochemistry, Stephenson Life Sciences Research Center, University of Oklahoma, 101 Stephenson Parkway, Norman, OK 73019, USA.
Yan Li, Department of Chemistry & Biochemistry, Stephenson Life Sciences Research Center, University of Oklahoma, 101 Stephenson Parkway, Norman, OK 73019, USA.
Christian H. Loveland, Department of Chemistry & Biochemistry, Stephenson Life Sciences Research Center, University of Oklahoma, 101 Stephenson Parkway, Norman, OK 73019, USA
Megan J. Wilson, Department of Chemistry & Biochemistry, Stephenson Life Sciences Research Center, University of Oklahoma, 101 Stephenson Parkway, Norman, OK 73019, USA
Binrui Cao, Department of Chemistry & Biochemistry, Stephenson Life Sciences Research Center, University of Oklahoma, 101 Stephenson Parkway, Norman, OK 73019, USA.
Penghe Qiu, Department of Chemistry & Biochemistry, Stephenson Life Sciences Research Center, University of Oklahoma, 101 Stephenson Parkway, Norman, OK 73019, USA.
Mingying Yang, Institute of Applied Bioresource Research, College of Animal Science, Zhejiang University, Hangzhou, Zhejiang 310058, P. R. China.
Chuanbin Mao, Department of Chemistry & Biochemistry, Stephenson Life Sciences Research Center, University of Oklahoma, 101 Stephenson Parkway, Norman, OK 73019, USA.
References
- [1].Yao HB, Fang HY, Wang XH, Yu SH, Chem. Soc. Rev 2011, 40, 3764. [DOI] [PubMed] [Google Scholar]
- [2].Maragakis NJ, Rothstein JD, Nat. Clin. Pract. Neurol 2006, 2, 679. [DOI] [PubMed] [Google Scholar]
- [3].a) Johnson MA, Weick JP, Pearce RA, Zhang SC, J. Neurosci 2007, 27, 3069; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Allen NJ, Eroglu C, Neuron 2017, 96, 697; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ, J. Neurosci 2010, 30, 3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Blumenthal NR, Hermanson O, Heimrich B, Shastri VP, Proc. Natl. Acad. Sci. USA 2014, 111, 16124; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chapman CAR, Chen H, Stamou M, Biener J, Biener MM, Lein PJ, Seker E, ACS Appl. Mater. Interfaces 2015, 7, 7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL, Proc. Natl. Acad. Sci. USA 2000, 97, 14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Yamanaka S, Cell Stem Cell 2012, 10, 678; [DOI] [PubMed] [Google Scholar]; b) Weissman IL, Science 2000, 287, 1442. [DOI] [PubMed] [Google Scholar]
- [7].Sun Y, Yong KM, Villa-Diaz LG, Zhang X, Chen W, Philson R, Weng S, Xu H, Krebsbach PH, Fu J, Nat. Mater 2014, 13, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Julia TCW, Wang MH, Pimenova AA, Bowles KR, Hartley BJ, Lacin E, Machlovi SI, Abdelaal R, Karch CM, Phatnani H, Slesinger PA, Zhang B, Goate AM, Brennand KJ, Stem Cell Rep. 2017, 9, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Que RA, Arulmoli J, Da Silva NA, Flanagan LA, Wang SW, J. Biomed. Mater. Res., Part A 2018, 106, 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lin CK, Liu C, Zhang LM, Huang Z, Zhao PP, Chen RQ, Pang M, Chen ZX, He LM, Luo CX, Rong LM, Liu B, Int. J. Mol. Med 2018, 41, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cao BR, Yang MY, Mao CB, Acc. Chem. Res 2016, 49, 1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mao C, Flynn CE, Hayhurst A, Sweeney R, Qi J, Georgiou G, Iverson B, Belcher AM, Proc. Natl. Acad. Sci. USA 2003, 100, 6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Rong J, Lee LA, Li K, Harp B, Mello CM, Niu Z, Wang Q, Chem. Commun 2008, 5185; [DOI] [PubMed] [Google Scholar]; b) Chung WJ, Oh JW, Kwak K, Lee BY, Meyer J, Wang E, Hexemer A, Lee SW, Nature 2011, 478, 364; [DOI] [PubMed] [Google Scholar]; c) Seuring C, Ayyer K, Filippaki E, Barthelmess M, Longchamp JN, Ringler P, Pardini T, Wojtas DH, Coleman MA, Dorner K, Fuglerud S, Hammarin G, Habenstein B, Langkilde AE, Loquet A, Meents A, Riek R, Stahlberg H, Boutet S, Hunter MS, Koglin J, Liang M, Ginn HM, Millane RP, Frank M, Barty A, Chapman HN, Nat. Commun 2018, 9, 1836; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang G, Li XL, He Y, Ma JK, Ni GL, Zhou SB, Prog. Polym. Sci 2018, 81, 80. [Google Scholar]
- [14].Wang J, Wang L, Yang M, Zhu Y, Tomsia A, Mao C, Nano Lett. 2014, 14, 6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].He C, Ye T, Teng W, Fang Z, Ruan WS, Liu G, Chen H, Sun J, Hui L, Sheng F, Pan D, Yang C, Zheng Y, Luo MB, Yao K, Wang B, ACS Nano 2019, 13, 1910. [DOI] [PubMed] [Google Scholar]
- [16].Dogic Z, Fraden S, Curr. Opin. Colloid Interface Sci. 2006, 11, 47. [Google Scholar]
- [17].Branston S, Stanley E, Keshavarz-Moore E, Ward J, Biotechnol. Prog 2012, 28, 129. [DOI] [PubMed] [Google Scholar]
- [18].Branston SD, Stanley EC, Ward JM, Keshavarz-Moore E, Biotechnol. Bioprocess Eng. 2013, 18, 560. [Google Scholar]
- [19].Gunhanlar N, Shpak G, Van Der Kroeg M, Gouty-Colomer L, Munshi ST, Lendemeijer B, Ghazvini M, Dupont C, Hoogendijk W, Gribnau J, Mol. Psychiatry 2017, 23, 1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Yang K, Jung K, Ko E, Kim J, Park KI, Kim J, Cho SW, ACS Appl. Mater. Interfaces 2013, 5, 10529; [DOI] [PubMed] [Google Scholar]; b) Ankam S, Suryana M, Chan LY, Moe AA, Teo BK, Law JB, Sheetz MP, Low HY, Yim EK, Acta Biomater. 2013, 9, 4535. [DOI] [PubMed] [Google Scholar]
- [21].Song L, Wang K, Li Y, Yang Y, Colloids Surf., B 2016, 148, 49. [DOI] [PubMed] [Google Scholar]
- [22].a) Pan F, Zhang M, Wu GM, Lai YK, Greber B, Scholer HR, Chi LF, Biomaterials 2013, 34, 8131; [DOI] [PubMed] [Google Scholar]; b) Chan LY, Birch WR, Yim EK, Choo AB, Biomaterials 2013, 34, 382; [DOI] [PubMed] [Google Scholar]; c) Lee MR, Kwon KW, Jung H, Kim HN, Suh KY, Kim K, Kim KS, Biomaterials 2010, 31, 4360. [DOI] [PubMed] [Google Scholar]
- [23].Patel BB, Sharifi F, Stroud DP, Montazami R, Hashemi NN, Sakaguchi DS, Macromol. Biosci 2019, 19, 1800236. [DOI] [PubMed] [Google Scholar]
- [24].Tiryaki VM, Ayres VM, Ahmed I, Shreiber DI, Nanomedicine 2015, 10, 529. [DOI] [PubMed] [Google Scholar]
- [25].Moe AA, Suryana M, Marcy G, Lim SK, Ankam S, Goh JZ, Jin J, Teo BK, Law JB, Low HY, Goh EL, Sheetz MP, Yim EK, Small 2012, 8, 3050. [DOI] [PubMed] [Google Scholar]
- [26].Wang J, Wang L, Li X, Mao C, Sci. Rep 2013, 3, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li XJ, Du ZW, Zarnowska ED, Pankratz M, Hansen LO, Pearce RA, Zhang SC, Nat. Biotechnol 2005, 23, 215. [DOI] [PubMed] [Google Scholar]
- [28].Shaltouki A, Peng J, Liu Q, Rao MS, Zeng X, Stem Cells 2013, 31, 941. [DOI] [PubMed] [Google Scholar]
- [29].Mao C, Solis DJ, Reiss BD, Kottmann ST, Sweeney RY, Hayhurst A, Georgiou G, Iverson B, Belcher AM, Science 2004, 303, 213. [DOI] [PubMed] [Google Scholar]
- [30].Wang Y, Ju Z, Cao B, Gao X, Zhu Y, Qiu P, Xu H, Pan P, Bao H, Wang L, Mao C, ACS Nano 2015, 9, 4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nam KT, Kim DW, Yoo PJ, Chiang CY, Meethong N, Hammond PT, Chiang YM, Belcher AM, Science 2006, 312, 885. [DOI] [PubMed] [Google Scholar]
- [32].Dogic Z, Fraden S, Phys. Rev. Lett 1997, 78, 2417. [Google Scholar]
- [33].Purdy KR, Fraden S, Phys. Rev. E 2007, 76, 011705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.