

Abstract
Mutant IDH1/2 gliomas represent a more indolent form of cancer. However, how this group of tumors progress, in a microenvironment-dependent manner, is still a pending question. Here we describe that the expression of Tau, a gene classically associated with neurodegenerative diseases, is epigenetically controlled by the balance between wild-type and mutant IDH1/2 in gliomas. Moreover, Tau is almost absent from tumors with EGFR mutations, whereas its expression is inversely correlated with overall survival in gliomas carrying wild-type or amplified EGFR. We demonstrate that the overexpression of Tau, through the stabilization of microtubules, impairs the mesenchymal/pericyte transformation of EGFRamp/wt glioma cells through the blockade of the EGFR-NFκB-TAZ axis. However, mutant EGFR induces a constitutive activation of this pathway, which is no longer sensitive to Tau. By inhibiting the phenotypic plasticity of EGFRamp/wt glioma cells, Tau protein inhibits angiogenesis and favors vascular normalization, decreasing tumor aggressiveness.
Keywords: Glioma, Tau (MAPT), IDH1/2 mutations, EGFR, microtubule stabilizers, TAZ, NF-kB, Cancer-Stem-Cell (CSC) plasticity, epithelial-to-mesenchymal (EMT) transition, tumor-derived-pericytes, vascular normalization, tumor-microenvironment (TME)
Introduction
Diffuse gliomas are classified and graded according to histological criteria. They include low and intermediate-grade gliomas (herein called Lower-Grade Gliomas, LGG), which encompass World Health Organization (WHO) grades 2 and 3, and the highly aggressive WHO grade 4 glioblastomas (GB), with 5-year survival rates of 5%. They are categorized based on the increments in cellular atypia, mitotic activity and vessel density. On top of that, GB are characterized by the specific presence of areas of necrosis and robust neoangiogenesis, being considered one of the most vascularized cancers. LGG are more indolent tumors although many of them progress into secondary GB, albeit at highly variable intervals, with survival rates that goes from 1 to 15 years1. Unfortunately, little is known about the factors that drive this transition from LGG to GB.
A big effort has been made in the last two decades in order to characterize the genetic modifications associated with gliomas. Some of them have been incorporated into the novel WHO classification. Particularly, the identification of mutations in the IDH1/2 (Isocitrate dehydrogenase 1/2) genes, which are associated with a more favorable prognosis in gliomas2, is common now in the routine clinical practice. IDH1/2 mutated proteins induce the accumulation of the oncometabolite, 2-D-hydroxyglutarate (2-HG), which competes with the α-ketoglutarate (α-KG) produced by the wild-type IDH enzymes and blocks TET (Ten Eleven Translocation)-mediated DNA demethylation. This process generates a CpG island methylator phenotype (G-CIMP), which is associated with a general suppression of gene expression3. Moreover, 2-HG inhibits histone demethylases, which further contribute to this phenotype4. By contrast, wild-type IDH1 promotes the metabolic adaptation of GB cells to support aggressive growth5. Therefore, it has been proposed that the balance between the wild-type and the mutant IDH1/2 function determines the clinical outcome of gliomas, including their sensitivity to radiation and chemotherapy6.
Within the new subclasses of high grade gliomas (proneural (PN), classic (CL) or mesenchymal (MES)), IDH1/2 mutations are accumulated in the first group, which is enriched in secondary GB and includes tumors with a better clinical prognosis7. Mutations in EGFR (Epidermal growth factor receptor), by contrast, accumulate in the CL and MES subtypes. This gene is mutated and/or amplified in a large percentage of diffuse gliomas and it has been associated with proliferation and survival, as well as with the invasive properties of glioma cells7, 8.
Several cytoskeletal proteins have been involved in tumor progression. Tau, encoded by the gene MAPT (Microtubule-associated protein tau), is well-known for its relevance in Alzheimer’s disease (AD) although it is also expressed in healthy brains, where it controls neural development and synaptic transmission9. In addition, Tau and other microtubule-stabilizing agents like taxanes modulate protein and organelle trafficking10, 11, which could be relevant for cancer cells. Interestingly, a possible comorbidity of dementias and GB had been suggested12, which led us to perform a bioinformatic analysis. We found that Tau (MAPT), among other genes related to neurodegeneration, is expressed in gliomas, where it seems to correlate negatively with tumor progression (Gargini et al., Front. Aging Neurosci., in press). Based on these data, we decided to conduct a more comprehensive characterization of Tau in this deadly pathology. Here we show that the expression of this protein depends on the genetic status of IDH1/2, being enriched in LGG and PN gliomas, where it hinders the progression of the tumors. Mechanistically, we have found that Tau inhibits the EGFR-NF-kB-TAZ signaling pathway, provided that no EGFR mutations are present. By blocking this signaling pathway, it impedes the plasticity of the tumor cells and their capacity to generate mesenchymal/pericyte-like cells. As a consequence, Tau impairs the processes of angiogenesis and neo-vascularization, favoring normalization of the gliomas vasculature and hampering tumor progression. Therefore, we propose a novel role for Tau, acting downstream of IDH mutations in gliomas, to orchestrate the vascular phenotype and the aggressiviness of these tumors.
Results
High levels of Tau (MAPT) correlate inversely with glioma aggressiveness
The in silico analysis of the glioma TCGA data set showed that the level of Tau (MAPT) decreases as the tumor grade increases, at least in astrocytomas (Fig. 1a) (Suppl. Fig. 1a,b). In agreement with this, a higher expression of the Tau (MAPT) gene was associated with an increased overall survival of glioma patients (Fig. 1b,c) (Suppl. Fig. 1c–f). These results confirmed our previous data (Gargini et al., Front. Aging Neurosci., in press) and prompted us to perform an immunohistochemical (IHC) staining on glioma samples, which showed that Tau protein is clearly expressed in the cytoplasm of tumor cells, with a very different pattern to that observed in the normal tissue (NT) (Fig. 1d) (Suppl. Fig. 1g). Moreover, we found high levels of Tau in a subset of the gliomas analyzed by Western Blot (WB) (Fig. 1e). The quantification of the IHC staining (Fig. 1f) and the WB (Fig. 1g) confirmed that Tau protein is clearly enriched in LGG compared to GB. As this accumulation could explain on its own the survival data (Fig. 1b,c), we decided to dissect out the effect of Tau expression in the LGG and the GB separated TCGA cohorts.
Figure 1: Tau is expressed in gliomas and it is enriched in lower-grade tumors.
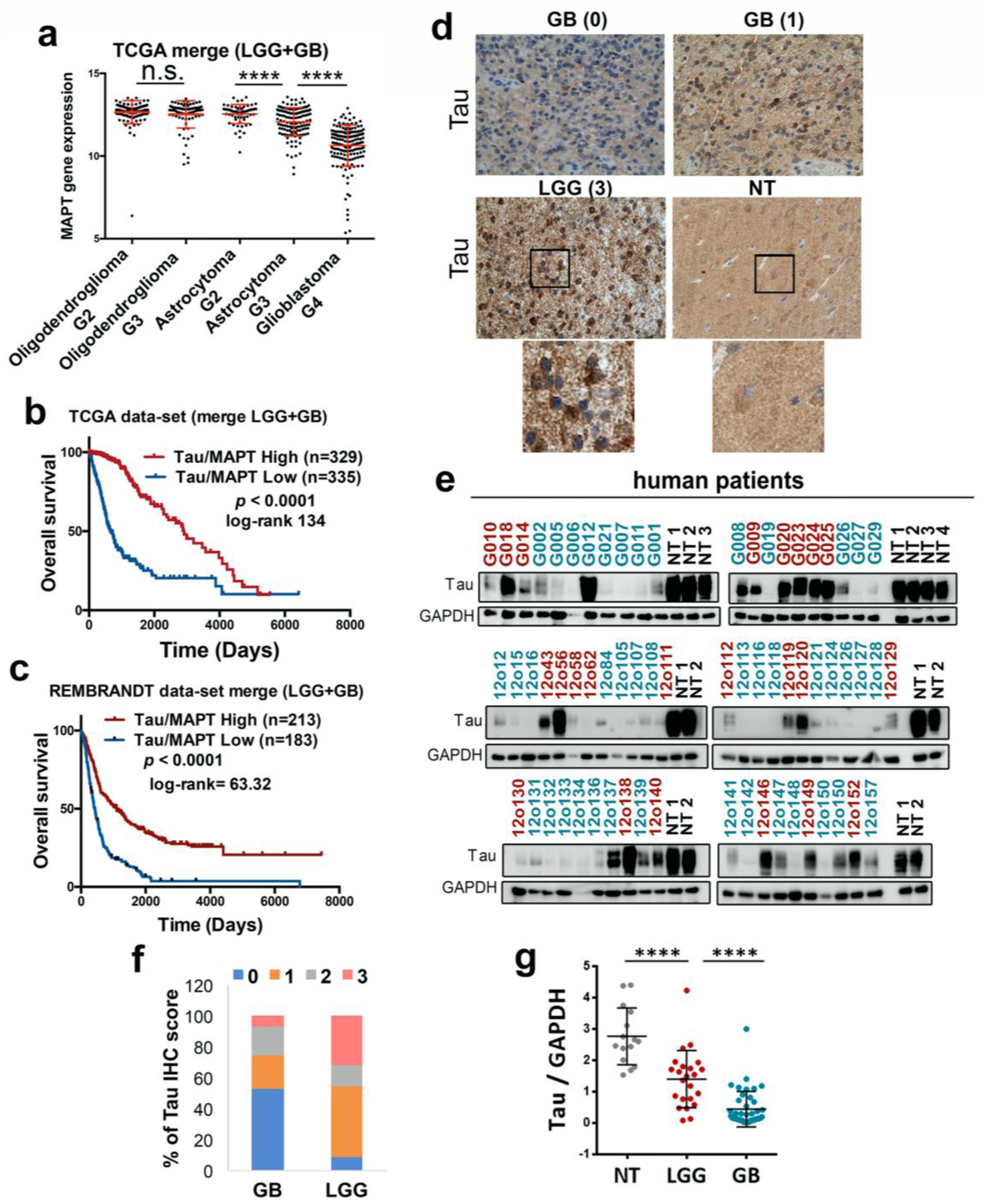
a, Analysis of Tau (MAPT) mRNA expression by RNAseq in gliomas (TCGA cohort) grouped according to the WHO classification (histological type and grade) (n=692) b,c, Kaplan-Meier overall survival curves of patients from the TCGA (LGG + GB) (n=664) and the Rembrandt (LGG + GB) (n=396) cohorts. Patients in each cohort were stratified into 2 groups based on high and low Tau (MAPT) expression values. d, Representative pictures of the IHC Tau staining of several gliomas and normal tissue (NT). The Tau IHC score is represented between brackets and an amplified section of the last two images is shown on the bottom. e, WB analysis of Tau expression in tumor tissue extracts from patients diagnosed with glioma. LGG samples are represented in red and GB samples in blue. NT was used as a control of Tau expression and GAPDH level as a loading control. f, Percentage of tumors (GB (n=55) and LGG (n=22)) with different Tau IHC score. g, Quantification of the relative amount of Tau in the WB in e. ****, P ≤0.0001. n.s. non significant.
Fig. 2a,b shows that the transcript level of Tau correlates with an increased overall suvival in both sets of patients. Similar results were obtained when we meassured Tau by RT-PCR analysis in our own GB cohort (Fig. 2c). Collectively, these results support the idea that Tau expression is associated with a less aggressive behavior of gliomas, independently of the tumor grade. Furthermore, we found a marked decrease in the expression of Tau in disease free vs progressed tumors (Fig. 2d). To confirm these observations we performed a longitudinal IHC quantification of Tau in primary LGG and their recurrent paired samples. Remarkably, we found a consistent decrease in Tau expression in those tumors that had progress into a more aggressive phenotype (Fig. 2e), which was not observed when there was no change in the histological classification of the recidives (Fig. 2f). These results suggest that Tau downregulation must be important for the tumors to relapse after surgical resection, which is a critical step in the mortality related to this pathology.
Figure 2: The expression of Tau in gliomas correlates inversely with tumor aggressiveness.
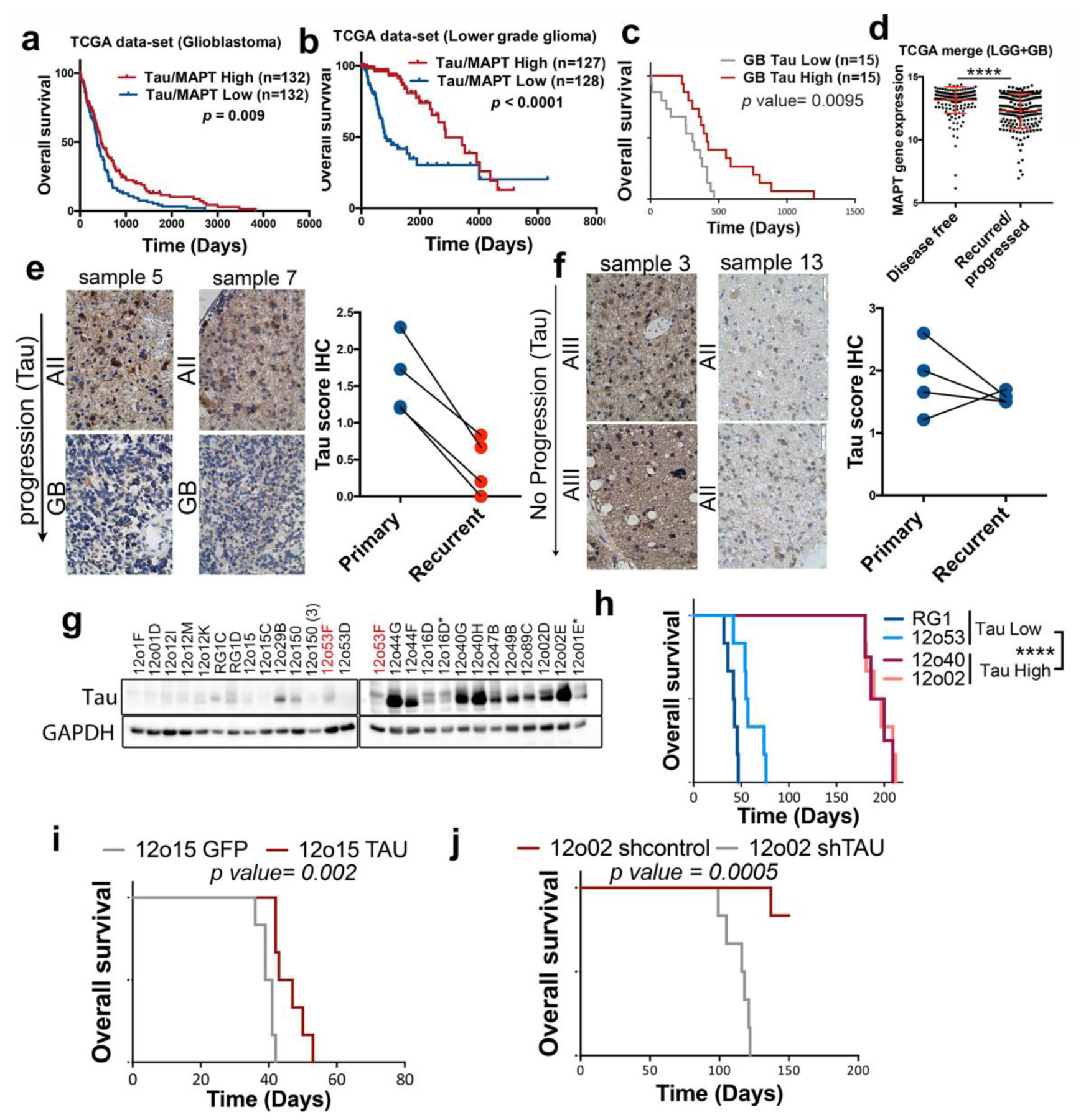
a,b, Kaplan-Meier overall survival curves of patients from the TCGA, the GB cohort (n=264) (A) or the LGG cohort (n= 155). Patients in each cohort were stratified into 2 groups based on high and low Tau (MAPT) expression values. c, Kaplan-Meier overall survival curves of patients from our own GB cohort (n=30). Patients in each cohort were stratified into 2 groups based on high and low Tau (MAPT) expression values (measured by RT-PCR using HPRT levels for normalization). d, Analysis of Tau (MAPT) mRNA expression by RNAseq in gliomas (TCGA cohort) grouped according to the clinical evolution of the tumors (n=386). e,f, Representative pictures of the IHC Tau staining of paired glioma samples (primary and recurrent tumor). Average Tau IHC score of the paired samples in shown on the right. g, WB analysis of Tau expression in tumor tissue extracts from subcutaneous patient-derived xenografts (PDXs). GAPDH was used as a loading control. h, Kaplan-Meier overall survival curves of mice that were orthotopically injected with different primary GB cell lines. i, Kaplan-Meier overall survival curves of mice that were orthotopically injected with 12o15 cells overexpressing GFP or Tau (n=6). j, Kaplan-Meier overall survival curves of mice that were orthotopically injected with 12o02 control or 12o02-shTau cells (n=6). The statistical analysis is shown on the bottom. ****, P ≤0.0001.
To further asses the participation of Tau in glioma aggressiveness we measured its expression in a panel of patient-derived-xenografts (PDX), obtained from GB surgeries. As expected, the amount of Tau protein expressed by the PDX was variable (Fig. 2g). However, those primary cell lines with higher levels of Tau grew slowlier when they were implanted in the brain of immunodeficient mice (Fig. 2h). In agreement with this observation, the overexpression of this gene in a Tau-deficient glioma cell line (12o15) delayed tumor formation (Fig. 2i), whereas its downregulation in Tau-enriched cells (12o02) clearly increased their aggressive behavior (Fig. 2j). Altogether, our results confirm that Tau is expressed in the tumor cells of several gliomas, especially in the less aggressive ones. Additionally, they suggest that Tau could be playing an active role as an inhibitor of tumor progression.
The expression of Tau (MAPT) in gliomas is regulated by IDH function
We analyzed the genetic background of gliomas in relation to the levels of Tau (MAPT) and we found that IDH1 mutations accumulate in high-Tau gliomas (Fig. 3a). This correlation was validated using a Volcano Plot analysis (Fig. 3b). Furthermore, Tau protein was detected in the majority of tumor cells that express the most common IDH1 mutation (R132H) (Supplementary Fig. 2a,b). This observation confirmed that the protein is truly expressed in the tumor cells and not only in the trapped neurons. We then quantified the amount of Tau IHC staining in wild-type and mutant IDH gliomas and we found a clear enrichment of high and medium stained sections in the second group (Fig. 3c). These results suggest that there is a strong correlation between the presence of IDH mutations and the expression of Tau. Remarkably, this seems to be independent of the tumor grade as we found higher levels of Tau in IDH mutant compared to IDH wild-type tumors in the GB (Supplementary Fig. 2c), as well as in the LGG (Supplementary Fig. 2d) TCGA cohorts. In order to confirm the relationship between Tau and IDH mut, we analyzed a recently published glioma model, in which IDH1 wt or IDH R132H had been expressed in an ATRX mutant background13. As expected, there was a clear delay in the tumor growth after the expression of mutant IDH1 (Fig. 3d). Moreover, when we dissected out the intracranial tumors we measured a clear increase in Tau protein levels in the mutant compared to the wild-type allografts (Fig. 3e). These results suggest that the expression of Tau in gliomas is induced by mutant IDH proteins.
Figure 3: Tau expression is regulated by IDH1/2 function.
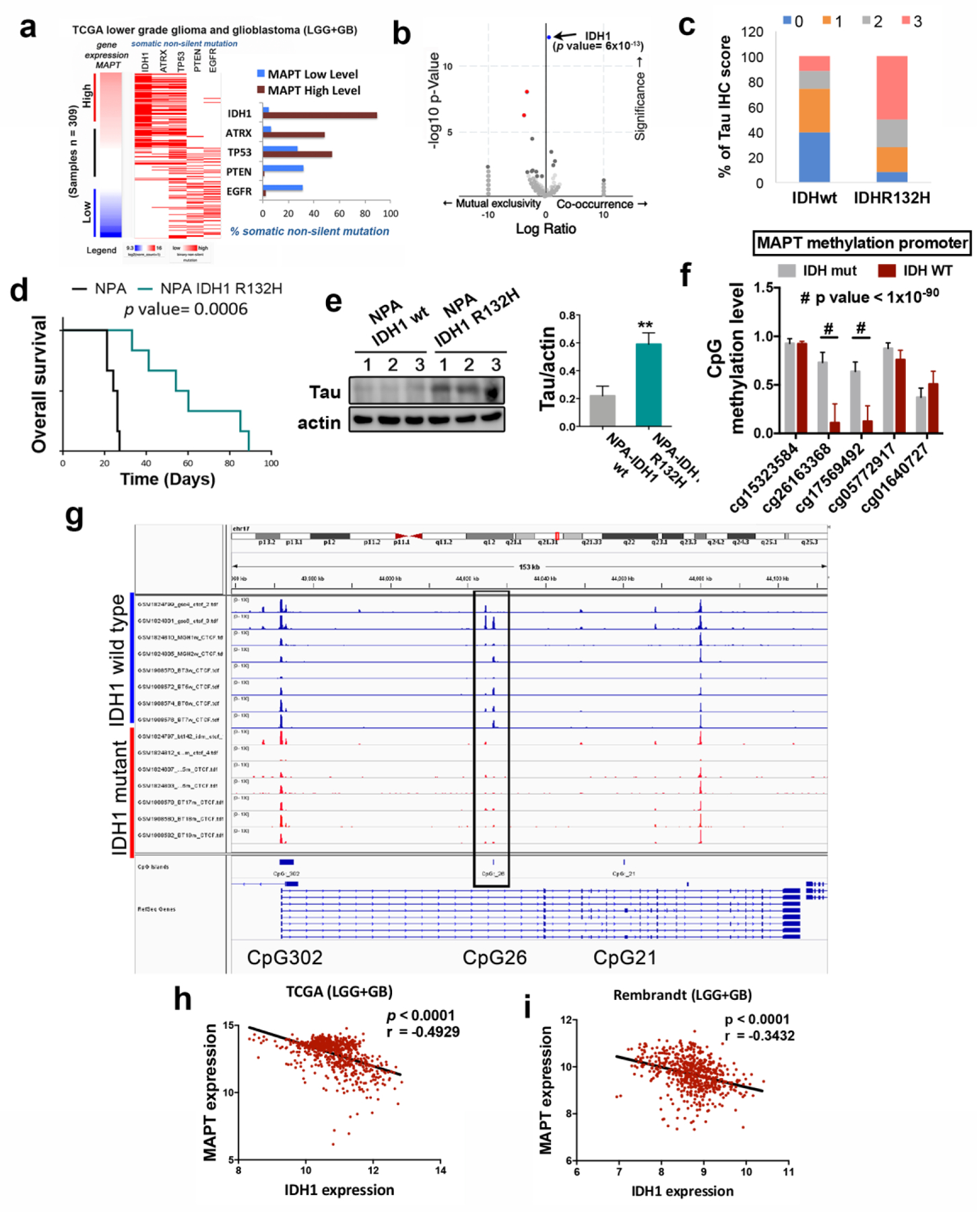
a, Analysis of non-silent somatic mutations in genes commonly modified in diffuse glioma grouped based on high or low expression of Tau (MAPT). b, Volcano plots showing mutated genes with differential distribution in glioma comparing tumors with high and low level of Tau. The arrow points to IDH1 mutations. c, Percentage of tumors with different Tau IHC score in wt (n= 35) and mutant (n=36) IDH1 gliomas. d, Kaplan-Meier overall survival curves of mice that were orthotopically injected with NPA IDH1 wt or NPA-IDH1 R32H cells (n=6). e, WB analysis and quantification of Tau expression in intracranial tumors from (d). Actin was used as loading control. f, Quantification of the methylation of CpG26 using 5 different probes and comparing IDHwt vs IDHmut gliomas. g, CTCF-binding profiles for the Tau (MAPT) CpG26 locus in IDH-mutant and IDH wild-type tumors, normalized by average signal. h,i, Correlation of the expression of Tau (MAPT) with that of wild-type IDH1 using the TCGA-merge (LGG+GB) (n=) (h) and the Rembrandt (LGG+GB) (n=580) (i) cohorts . **, P ≤0.01; ****, P ≤0.0001; #, P ≤1×10−90
The G-CIMP phenotype, which is associated with the presence of IDH mutations, is supposed to block the transcription of many genes. However, it also activates certain others, especially those involved in the tumorigenesis of LGG (like the PDGFRA (Platelet Derived Growth Factor Receptor Alpha) oncogene) through the disruption of the binding site of the CTCF insulator protein14. Interestingly, we observed an increased expression of Tau in the G-CIMP GB subtype (Supplementary Fig. 3a), as well as a significant correlation between the amount of Tau (MAPT) and PDGFRA mRNAs in the TCGA samples (Supplementary Fig. 3b). These results support a model in which Tau promoter might be controlled by the epigenetic changes induced by IDH mutations. To test this theory, we first analyzed the presence of CpG islands in the Tau (MAPT) promoter region by using the xena genome browser. We identified three of them in the 5′region of transcription initiation site (Supplementary Fig. 3c), which correlate with the sites previously described15. Using epigenetic data from the TCGA (LGG cohort) we compared the methylation of the whole promoter region in mutant vs wild-type IDH tumors. We observed an increased methylation in CpG:26 (Fig. 3f; Supplementary Fig. 3d,e) and in some parts of CpG:302 (Supplementary Fig. 3d,e). However, when we analyzed the ChIP-seq data of the CTCF binding to the different clusters, we observed that only the binding to the CpG:26 site was lost in the IDHmut tumors (Fig. 3g). In order to obtain an independent confirmation of the epigenetic upregulation of Tau (MAPT) in response to mutant IDH, we treated primary GB cells with 2-HG and we observed a dose-dependent accumulation of its mRNA (Supplementary Fig. 3f), an effect that was reversed in the presence of azacytidine (AZA) (Supplementary Fig. 3g). In summary, these results strongly suggest that the increase in the methylation of the CpG:26 region, induced by IDH mutant proteins, changes the chromosomal insulator topology and the binding of CTCF to the Tau promoter, activating its transcription.
IDH1/2 mutations define a distinct subset of gliomas with a better outcome. By contrast the expression of IDH wild-type in LGG and GB defines a subgroup with poorer prognosis (Supplementary Fig. 3h). In fact, we found an inverse correlation between the expression of wild-type IDH1 and Tau on the TCGA (Fig. 3h) and the Rembrandt (Fig. 3i) cohorts. These results allow us to propose that the epigenetic induction of Tau transcription might depend on the equilibrium between wild-type and mutant IDH expression.
Tau opposes EGFR signaling in gliomas
The data presented here indicate that Tau is highly expressed in a subset of gliomas, especially in IDH mutant tumors, where it seems to impair tumor aggressiveness. To gain insight into the mechanism behind this behavior, we performed a DAVID gene analysis of the pathways co-upregulated with Tau in gliomas. As expected, we found a positive association with microtubule and neurogenic-related processes. However, the other Tau-linked pathways (vesicle-transport, MAPK/GTPase regulation and Pleckstrin-related processes) were somehow related to receptor tyrosine kinases signaling (Supplementary Fig. 4a). In addition, our in silico analysis indicated that IDH mutations, as well as higher levels of Tau are mutually exclusive with EGFR and PTEN mutations (Fig. 3a, Fig. 4a and Supplementary Fig. 4b), which was confirmed in a Volcano Plot analysis (Fig. 4b). To interrogate if Tau could be modulating this signaling pathway, we expressed IDH wt or IDH R132H in a EGFR amplified cell line (RG1). As we have previously observed in the mouse glioma model (Fig. 3d), IDH mut impaired tumor burden (Fig. 4c). However, we also confirmed that the overexpression of IDHwt increases the aggressiveness of the glioma cells (Fig. 4c). These effects were paralleled by changes in Tau expression, which augmented in IDH mut and decreased in IDH wt tumors, compared to the control ones (Fig. 4d,e). Interestingly, the phosphorylation of EGFR showed a striking inverse correlation with Tau levels (Fig. 4d,f). In agreement with this observation, the in silico analysis showed that higher levels of phosphorylated EGFR are observed in tumors with lower Tau expression (Supplementary Fig. 4c,d). More importantly, the expression of Tau correlated with overall survival in the group of EGFRamp/wt gliomas (Fig. 4g) but it had no clinical relevance in the presence of additional mutations in EGFR (Fig. 4h). Taken together, these results suggest that there could be a negative effect of IDH/Tau on the pathway activated by this receptor, but only in the group of EGFRamp/wt gliomas.
Figure 4: Tau opposes EGFR in gliomas.
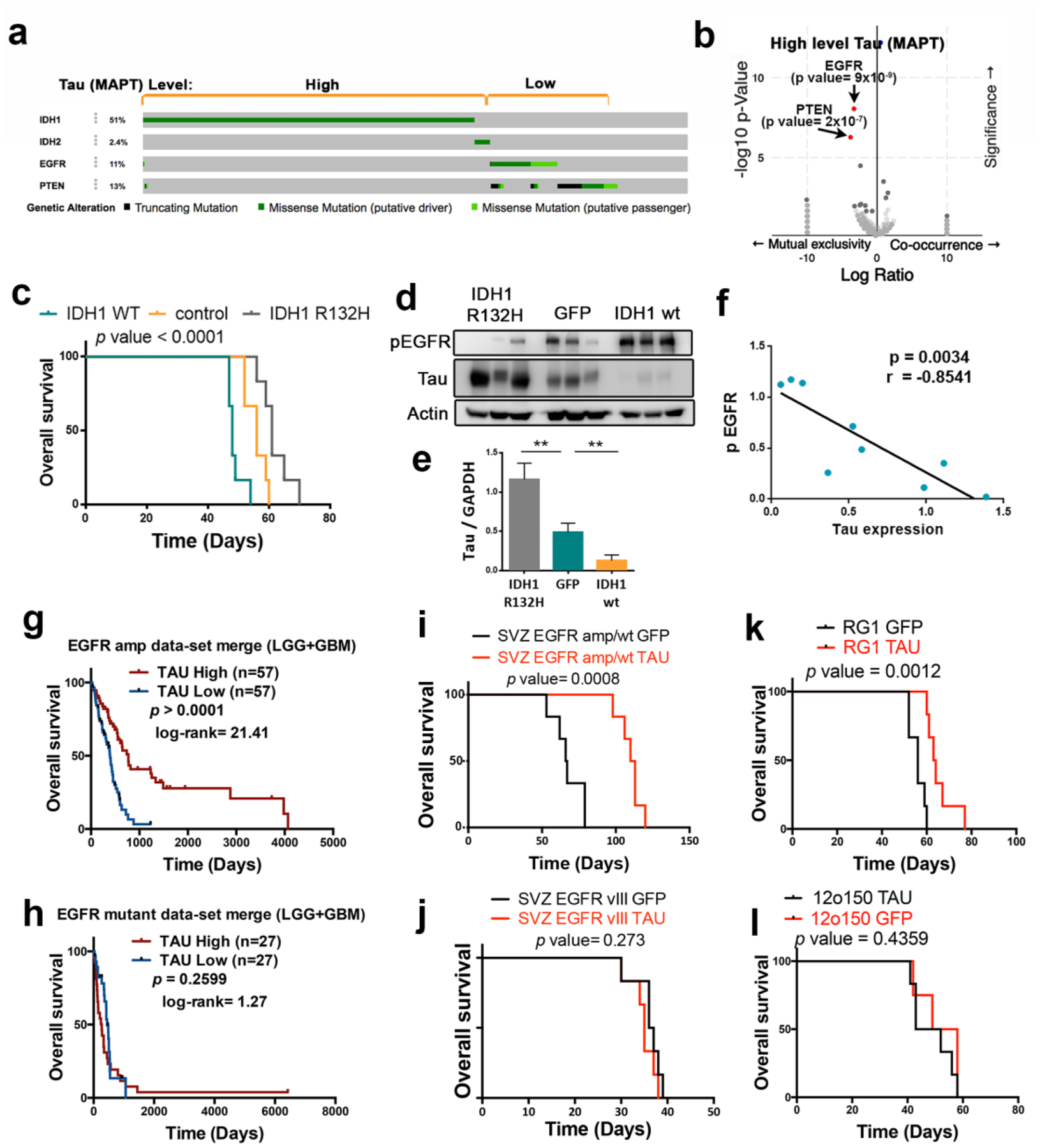
a, Distribution of somatic non-silent mutations in IDH1/2, EGFR and PTEN in a glioma cohort (TCGA, n=812). b, Volcano plots showing mutated genes with differential distribution in gliomas, comparing tumors with high and low level of Tau. The arrows point to PTEN and EGFR mutations. c, Kaplan-Meier overall survival curves of mice that were orthotopically injected with RG1, RG1 IDH1 wt or RG1 IDH1 R32H cells (n=6). d, WB analysis of phosphorylated EGFR (pEGFR) and Tau in intracranial tumors from (c). Actin was used as a loading control. e, Quantification of the amount of Tau in (d). f, Correlation between the levels of Tau and phospho-EGFR in RG1 tumors. g,h, Kaplan-Meier overall survival curves of patients from the GB+LGG TCGA cohort. Patients were separated based on the EGFR status: tumors without mutations (amplified or wild type) (n=114) (g) and amplified tumors with mutations (n=54) (h). Then, they were stratified into 2 groups based on Tau (MAPT) expression value. i-l, Kaplan-Meier overall survival curves of mice that were orthotopically injected with SVZ-EGFRamp/wt (i), SVZ-EGFRvIII (j), RG1 (EGFRamp) (k) or 12o150 (EGFRmut) (l) cells, overexpressing either GFP or Tau (n=6). **, P ≤0.01.
To evaluate the effect of Tau on EGFR signaling, we overexpressed this protein in two mouse glioma models: SVZ-EGFRamp/wt and SVZ-EGFRvIII, and we analized their capacity to grow as intracranial allografts. These models were generated by transforming subventricular zone (SVZ) progenitors from p16/p19 ko mice with retrovirus expressing either the wild-type or the variant III (vIII) isoform or the receptor. Both type of cells depend on EGFR signaling in vitro and in vivo and they generate gliomas with a high penetrance (Segura, Gargini et al., manuscript in preparation). As expected, overexpression of Tau clearly inhibited the growth of SVZ-EGFRamp/wt mouse gliomas (Fig. 4i) but it had no effect on SVZ-EGFRvIII tumors (Fig. 4j). Similar results were obtained when we overexpressed Tau in two human primary cell lines, RG1 (EGFRamp) and 12o150 (EGFR amplified and mutated, EGFRmut). Tau expression impaired the growth of the EGFRamp cell line (Fig. 4k) but it had no effect on EGFRmut cells (Fig. 4l). Remarkably, Tau did not inhibit the survival or the self-renewal capacity of the mouse (Supplementary Fig. 4e,f) or the human (Supplementary Fig. 4g,h) glioma cells in vitro, suggesting that the consequence of Tau overexpression on EGFR signaling pathway must be only relevant in the context of the tumor microenvironment (TME).
Microtubule stabilizators like Tau and EpoD favor the degradation of wild-type EGFR
When we analyzed the dissected tumors after Tau overexpression we observed that the levels of phospho-EGFR were attenuated in SVZ-EGFRamp/wt tumors (Fig. 5a) as well as in EGFRamp xenografts (Fig. 5b), whereas there was no change in SVZ-EGFRvIII gliomas (Fig. 5c) or in EGFRmut xenografts (Fig. 5d) after Tau upregulation. These data reinforce the notion that Tau opposes wild-type but not mutant EGFR signaling.
Figure 5: Tau and microtubule-stabilizing compounds impair wild-type EGFR stability and signaling in gliomas.
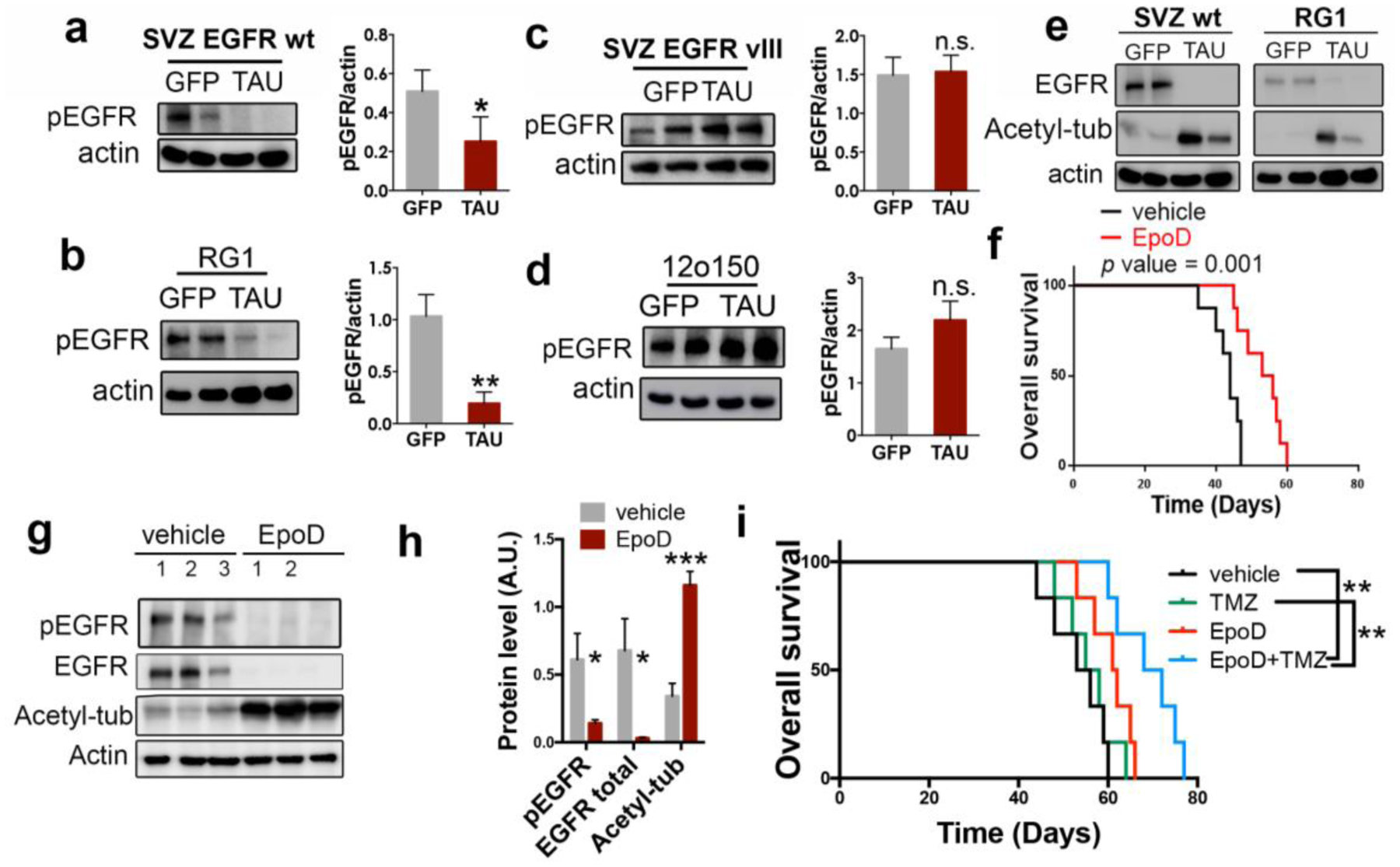
a–d, WB analysis and quantification of phosphorylated EGFR (pEGFR) in SVZ EGFRwt/amp (a), RG1 (b), SVZ EGFR vIII (c) and 12o150 (d) tumors after the overexpression of GFP or Tau. Actin was used as a loading control. e, WB analysis of total EGFR and Acetylated tubulin (Acetyl-tub) in SVZ EGFR wt/amp and RG1 tumors after the overexpression of GFP or Tau. Acting was used as a loading control. f, Kaplan-Meier overall survival curves of mice that were orthotopically injected with RG1 cells and subsequently treated with intraperitoneal injections (2/week) of EpoD (1 mg/Kg) (n=8). g,h WB analysis and quantification of pEGFR, total EGFR and Acetyl-tub in the tumors (f). Actin was used as loading control. i, Kaplan-Meier overall survival curves of mice that were orthotopically injected with RG1 cells and subsequently treated with intraperitoneal injections (2/week) of EpoD (1mg/Kg) and/or TMZ (5mg/Kg/day) (n=6). *, P ≤0.05, **, P ≤0.01, ***, P ≤0.001. n.s. non significant.
Previous results from our group have demonstrated that, as a result of Tau function, stabilized microtubules become heavily acetylated through the inhibition of HDAC6 (histone deacetylase 6). In fact, the increase in tubulin acetylation can serve as a readout of Tau experssion16. Moreover, it has been proposed that this acetylation promotes the subsequent degradation of EGFR due to changes in the microtubule-dependent endocytic machinery17. In agreement with these published data, we observed high levels of acetylated tubulin in Tau overexpressing gliomas (mouse or human), which were paralled by a strong decrease in the amount of EGFR protein (Fig. 5e). Collectively, these results suggest that Tau impairs EGFR signaling through the stabilization of the microtubules and the subsequent alteration of the receptor trafficking, which leads to its degradation. To validate this hypothesis we treated RG1(EGFRamp)-injected mice with a Taxol derivative, Epothilone D (EpoD). This kind of components can stabilize the microtubules as well. In fact, they compete with tau for the same tubulin-binding site. EpoD in particular has been proved to reach the brain and revert some of the axonal defects of a Tau loss-of-function model18. Systemic treatment with EpoD significantly delayed RG1 tumor formation (Fig. 5f) and reduced the amount of phospho-EGFR in the tumors (Fig. 5g,h). This was accompanied by a strong downregulation of the levels of EGFR and a clear increase in the amount of acetylated tubuline (Fig. 5g,h). Altogether, our data support the notion that the microtubule-related function of Tau is responsible for its effect on EGFR signaling and tumor growth in gliomas. Moreover, they suggest that taxol derivatives could reduce the aggressiviness of gliomas. In fact, we observed that EpoD treatment made RG1 tumors more sensitive to chemotherapy (Fig. 5i) so there could be a therapeutic opportunity for these compounds to be used in glioma patients.
Tau blocks the mesenchymalization of EGFRamp/wt glioma cells.
The bioinformatic analysis showed that the levels of Tau (MAPT) correlate positively with the PN signature while they exhibit a strong inverse correlation with the MES expression profile of gliomas (Fig. 6a, Supplementary Fig. 5a–f). In addition, we observed that Tau expression inversely correlates with the NF-κB (Nuclear factor kappa-light-chain-enhancer of activated B cells) and the inflammatory pathways, but not with other signatures, like those associated with hypoxia or PDGFRα (Fig. 6a,b). These in silico observations were confirmed by the WB analysis of RG1 (EGFRamp) xenografts, which showed that overexpression of Tau induces a clear inhibition of the NF-κB subunit phosphorylation, as well as a strong reduction in the protein level of TAZ, a master regulator of the MES phenotype19 (Fig. 6c,d). Similar results were obtained in the WB analysis of 12o15 xenografts (Fig. 6e,f), a primary cell line that overexpresses the receptor in the absence of gene amplification20. However, Tau did not change the expression of other MES genes (Fig. 6g) and it had no effect on TAZ expression or NF-κB activation in EGFRmut tumors, where there was no inhibition of the receptor upon Tau induction (Supplementary Fig. 5g). These data suggest that EGFR signaling induces the MES features in gliomas, which can be impaired by Tau, provided that no additional mutations in the receptor are present.
Figure 6: Tau blocks the mesenchymal features of EGFRamp/wt cells through the inhibition of the EGFR/NF-κB/TAZ axis.
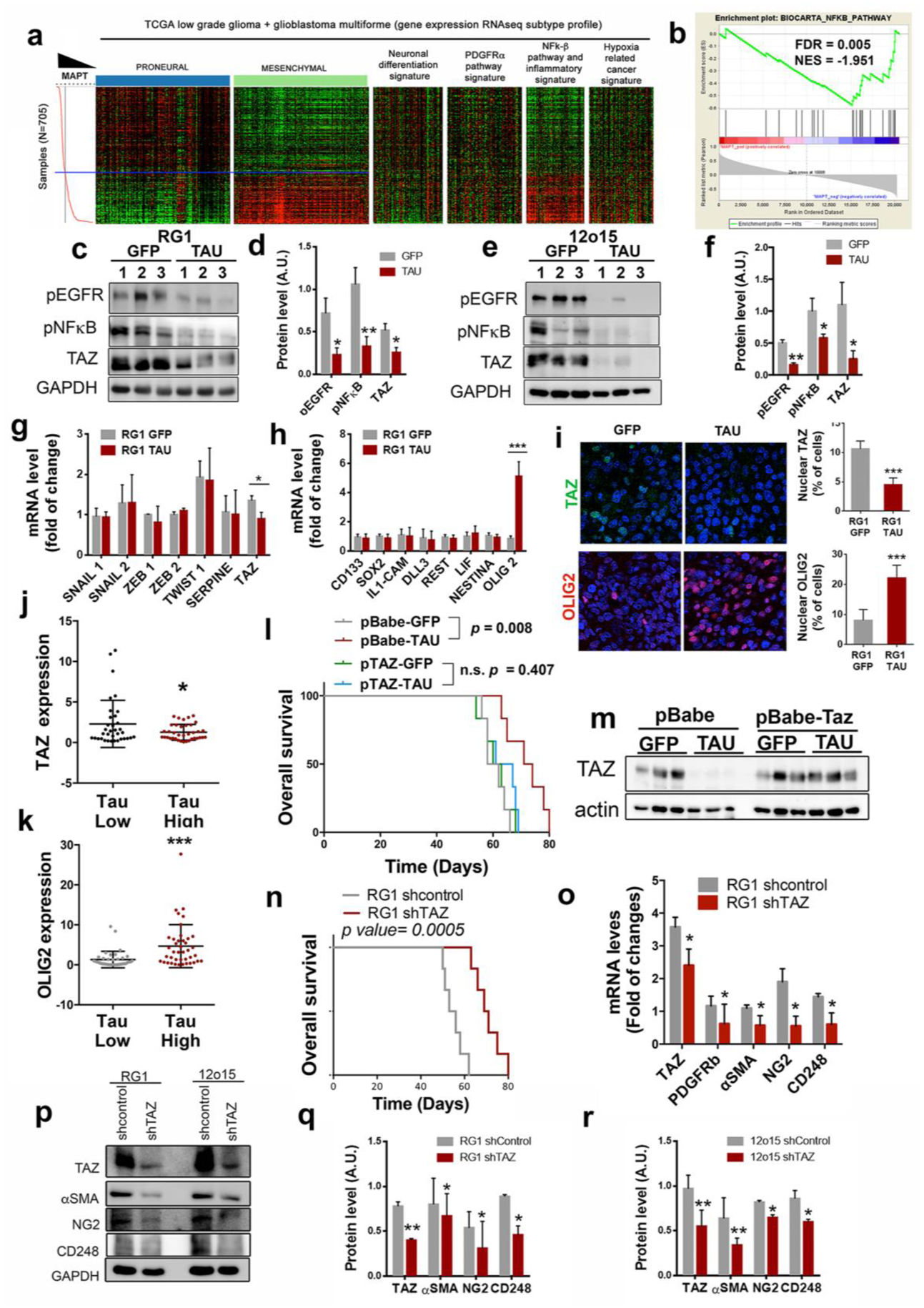
a, Heatmap of PN, MES, neuronal differentiation, PDGFRα pathway, NF-κB/inflammation, and hypoxia-related cancer expression genes depending on Tau (MAPT) expression levels. b, GSEA enrichment plot analysis using Tau gene expression values as template and the NF-κB pathway geneset from the Biocarta pathways database. c–f, WB analysis (c,e) and quantification (d,f) of phosphorylated EGFR (pEGFR), phosphorylated pNF-κB (p65) and TAZ in RG1 (c,d) and 12o15 (e,f) xenografts expressing either GFP or Tau. GADPH was used as a loading control. g,h, qRT-PCR analysis of MES (g) and PN-subtype (h) related genes in RG1 xenografts expressing GFP or Tau. HPRT was used for normalization. i, Representative images of TAZ (top) or OLIG2 (bottom) IF staining of sections from GFP or Tau overexpressing RG1 gliomas. Quantification is shown on the right. j,k, qRT-PCR analysis of TAZ (j) and OLIG2 (k) in gliomas. Tumors were classified in two groups based on the expression of Tau (MAPT). (n=72). HPRT expression was used for normalization. l, Kaplan-Meier overall survival curves of mice that were orthotopically injected with RG1 cells overexpressing GFP, Tau or Tau plus TAZ (n=6). m,WB analysis of TAZ in the tumors (l). Actin was using as loading control. n, Kaplan-Meier overall survival curves of mice that were orthotopically injected with RG1 shControl or RG1 shTAZ cells (n=6). o, qRT-PCR analysis of pericytic–related genes in RG1 shControl and RG1 shTAZ xenografts. p–r, WB analysis (p) and quantification (q,r) of TAZ, αSMA, NG2 and CD248 in RG1 (p,q) or 12O15 (p,r) cells after TAZ downregulation. GADPH levels were used for normalization *, P ≤0.05; ***, P ≤0.001.
Tau overexpression also induced the expression of OLIG2 (Fig. 6h), a transcription factor that is frequently used as a bona fide PN marker21. Moreover, the IF analysis of RG1 gliomas confirmed that Tau represses the nuclear expression of TAZ, whereas it induces the nuclear expression of OLIG2 in the tumor cells (Fig. 6i). These data were confirmed by RT-PCR analysis of human samples (Fig. 6j,k), and by in silico studies (Supplementary Fig. 5h), which showed that Tau correlates inversely with TAZ and positively with OLIG2 expression. Altogether, our results indicate that Tau induces a change in the glioma phenotype, repressing MES regulators and inducing PN promoters in the tumor cells through the regulation of the EGFR/TAZ/NF-kB pathway. This transition is responsible for the growth inhibition induced by Tau as TAZ overexpression was able to rescue the effect of Tau in RG1 tumors (Fig. 6l,m).
Previous results from our group and others have show that, in comparison with PN cancer stem cells (CSCs), MES CSCs are more capable of glioma initiation, probably associated with their increased angiogenic capacity22, 23. In agreement with this observation, the overexpression of Tau in RG1 cells impaired their capacity to form subcutaneous xenografts (Supplementary Fig. 5i,j). Furthermore, we found that most of the genes upregulated with TAZ in gliomas are associated with angiogenesis and tumor vasculature (Supplementary Fig. 6a–d). Particularly, we noticed that markers of pericytes, which are considered as perivascular MES stem cells24, were among the genes which correlated better with TAZ in gliomas (Supplementary Fig. 6e,f). In order to examine if this transcription factor could modulate the function and/or the number of mural cells we induced the expression of shTAZ in RG1 cells and we performed an ortothopic in vivo experiment. As expected, downregulation of this protein clearly reduced the aggressiviness of the tumors (Fig. 6n). Moreover, we observed a significant inhibition of the expression of several pericyte markers in RG1 shTAZ compared to control tumor samples (Fig. 6o). It is worth mentioning that TAZ seems to regulate peryctic differentiation in a cell-autonomous way, as we also observed a reduction of these markers after the in vitro expression of shTAZ in RG1 and 12o15 cells (Fig. 6p–r). These results are in accordance with some recent publications showing that most of the pericytic functions in GB are performed by the highly plastic CSCs, which undergo a transdifferentiation process, acquiring mesenchymal and mural cell features25–27.
Tau reduces the amount of tumor-derived-pericytes and normalizes the vasculature of EGFRamp gliomas
Our results indicate that Tau impairs the mesenchymalization of glioma cells by blocking the expression of TAZ, which is induced in response to EGFR activation. Moreover, we could hypothesize that, as a result of this inhibition, there is an important blockade of the capacity of the tumor cells to transdifferentiate into pericytes. Therefore, we decided to analyze the vascular component of the tumors after the overexpression of Tau in RG1 (EGFRamp) cells. We observed a marked decrease in the number of αSMA and CD248 positive cells (Fig. 7a), together with a significant inhibition of the expression of human pericytic markers, in Tau-expressing tumors (Fig. 7b). A similar result was observed in 12o15 (EGFRwt) (Supplementary Fig. 7a), but not in 12o150 (EGFRmut) xenografts (Supplementary Fig. 7b). Remarkably, Tau did not impaired the expression of mouse pericityc genes (Fig. 7c), reinforcing the idea that this protein inhibits the glioma-to-pericyte transdifferentiation but it does not influence the amount and/or the differentiation capacity of host pericytes.
Figure 7: Tau overexpression blocks the appearance of tumor-derived-pericytes in EGFRwt/amp gliomas, inhibiting angiogenesis and normalizing the tumor vasculature.
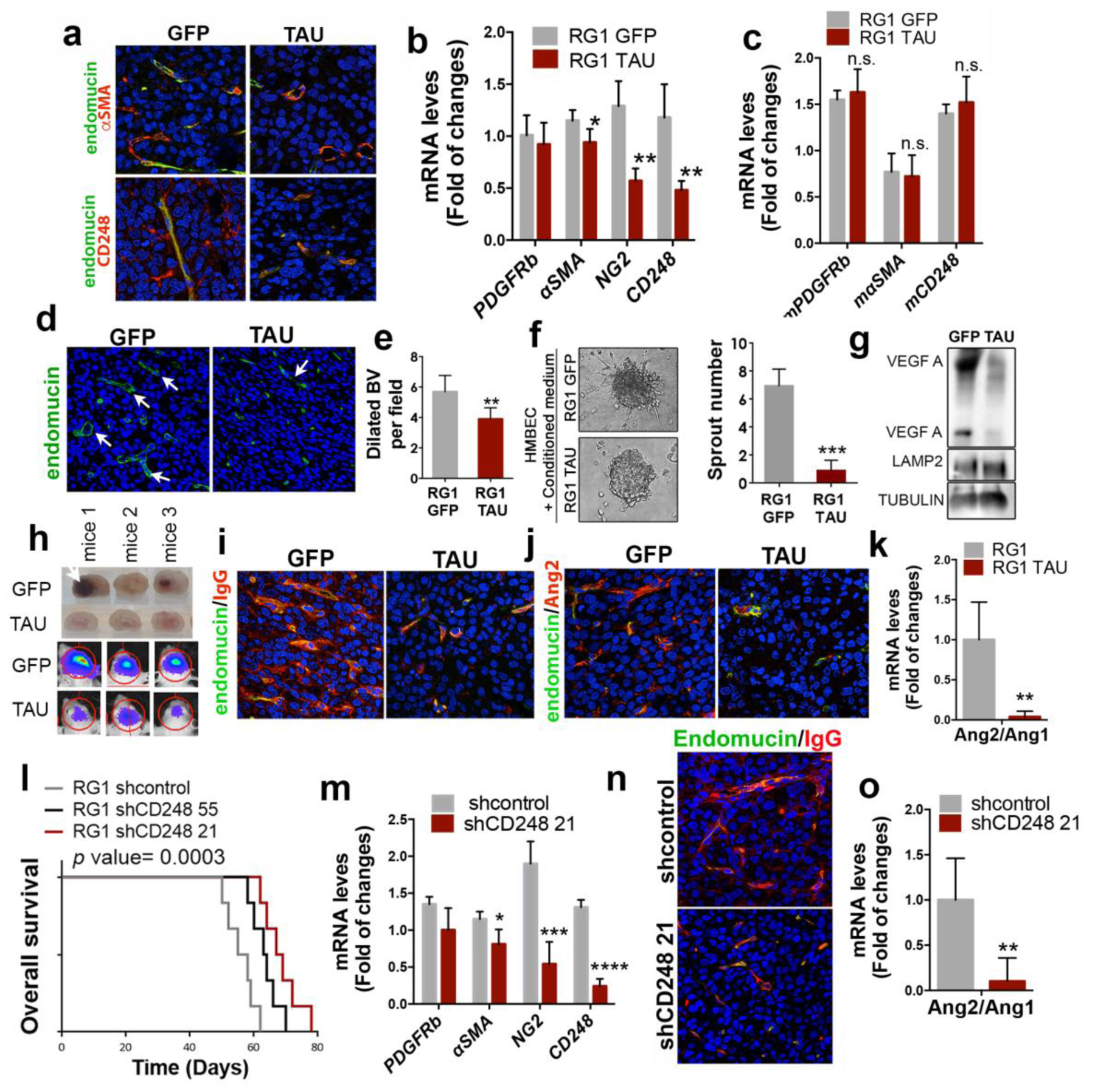
a, Representative images of endomucin and αSMA (top) or endomucin and CD248 (bottom) IF co-staining of sections from RG1 xenografts expressing GFP or Tau. b, qRTPCR analysis of pericytic–related genes (using human-specific primers) in RG1 GFP and Tau xenografts. HPRT was used for normalization. c, qRT-PCR analysis of pericytic–related genes (using mouse-specific primers) in RG1 GFP and Tau xenografts. Actin was used for normalization. d, Representative images of endomucin IF staining of sections from RG1 xenografts expressing GFP or Tau. e, Quantification of dilated blood vessels in (d) (indicated by arrows). f, Representative contrast phase images of HMBEC cells cultured in the presence of conditioned media from RG1 GFP or RG1 Tau cells. The number of sprouts is shown on the right. g, WB of VEGF in the supernatant of RG1 cells after overexpression of GFP or Tau. LAMP2 and TUBULIN were used as loading controls. h, Representative images of sectioned mouse brains (top) and pseudo-color images of Fluc bioluminescence (bottom), after the injection of GFP or Tau-expressing RG1 cells. i,j, Representative images of IF co-staining of endomucin and IgG (i) or endomucin and Ang2 (j) on RG1 xenografts expressing GFP or Tau. k, Ratio of Ang2/Ang1 expression levels measured by qRT-PCR in RG1 gliomas after Tau overexpression. Actin was used for normalization. l, Kaplan-Meier overall survival curves of mice that were orthotopically injected with RG1 cells expressing shcontrol, shCD248 55 or shCD248 21 (n=6). m, qRT-PCR analysis of pericytic-related genes in RG1 tumors expressing shcontrol or shCD248 21. n, Representative images of IF co-staining of endomucin and IgG from RG1 xenografts expressing shControl or shCD248 21. o, Ratio of Ang2/Ang1 expression levels measured by qRT-PCR in RG1 gliomas after CD248 downregulation. Actin was used for normalization. *, P ≤0.05; **, P ≤0.01; ***, P ≤0.001; ****, P ≤0.0001. n.s., nonsignificant.
The IF analysis of the tissues also revealed that there is an important inhibition of cellular proliferation (Supplementary Fig. 7c,d) after Tau overexpression, accompanied by a significant decrease in the number of dilated blood vessels (Fig. 7d,e), typical of malignant gliomas28. The angiogenic signals that are reduced upon Tau induction must be derived, at least in part, from the tumor cells, as the supernatant of RG1 cells overexpressing Tau has a reduced capacity to induce HMBEC (Human Brain Microvascular Endothelial Cells) sprouting (Fig. 7f) and contains less VEGF (Vascular Endothelial Growth Factor) (Fig. 7g).
We had previously noticed the lack of hemorrhages in EGFRamp tumors overexpressing Tau, an event that was frequent in control tumors (Fig. 7h). In parallel, we observed a reduction in the extravasation of IgG after Tau overexpression (Fig. 7i). These observations suggest that Tau induces a less aberrant tumor vasculature, with an increase in the intregrity of the blood brain barrier (BBB). In agreement with this hypothesis, tumors overexpressing Tau had a clear reduction in the levels of Ang2 (Angiopoietin 2) (Fig. 7j), a key regulator of angiogenesis that has been associated with vascular abnormalities in glioma, acting as an antagonist of Ang129. In fact, we observed a significant decrease in the Ang2/Ang1 ratio after Tau overexpression (Fig. 7k) in the EGFRamp model, but not in the EGFRmut xenografts (Supplementary Fig. 7e). These results suggest that the transdifferentiation of glioma cells into mesenchymal/pericyte-like cells, induced by EGFR signaling, favors the secretion of angiogenic signals and the formation of an aberrant vasculature. Interestingly, this process can be impaired by Tau in EGFRwt/amp but not in EGFRmut gliomas.
In order to confirm the relevance of the tumor-derived-pericytes in the growth of EGFRamp gliomas, we downregulated CD248 expression in RG1 cells. CD248, also called endosialin, aids in supporting tumor microvasculature and it is expressed in pericytes, but only within malignant solid tumors, including high-grade gliomas26, 30. We picked the most effective shRNA sequences (Supplementary Fig. 7f) and we injected the interfered cells into the brains of immunodeficient mice, in the presence of normal host pericytes. We observed a significant delay in tumor growth after CD248 downregulation (Fig. 7l). Moreover, the inhibition of CD248 in the tumor cells reduced the expression of other pericytic markers in vitro (Supplementary Fig. 7g) and in vivo (Fig. 7m). These results suggest that CD248 is a key player in the transdifferentiation of tumor cells into pericytes. On top of that, tumors formed after CD248 downregulation showed a reduction in the extravasation of IgG (Fig. 7n) as well as a decreased Ang2/Ang1 ratio (Fig. 7o), wich further supports the hypothesis that the depletion of tumor-derived-pericytes (at least in EGFRwt/amp tumors) normalizes the glioma vasculature and reduces the aggressiviness of the tumors.
Tau expression is a surrogate marker of the less aggressive vascular behavior of gliomas
The data presented so far place Tau at the boundary between the most common genetic alterations of gliomas (those that affect IDH1/2 and EGFR) and the regulation of the tumor microenvironment, in particular the vascular phenotype. In order to translate the results obtained in mouse models into the clinical settings, we used our own cohort of patient’s samples (RT-PCR analysis) and the data from the TCGA (in silico analysis). We divided the tumors into IDH1 mut (those with higher transcript levels of Tau) and IDH1 wt, and we subclasified the second ones into High or Low Tau gliomas (Fig. 8a,b). We then analyzed the expression of CD248 and we observed a significant inverse correlation between the transcript levels of this pericyte-related gene and the expression of Tau (Fig. 8c,d). This correlation was confirmed by the IHC analysis of the tumors. The representative images in Figure 8e evidence the gradual normalization of the vasculature in parallel with the increase in Tau levels (Fig. 8e). Furthermore, the quantification of the CD248 score (Fig. 8f), the amount of CD34 (Fig. 8g) and the amount of dilated vessels (Fig. 8h) confirmed the visual differences and reinforced our model, in which Tau expression could function as a surrogate marker of the less aggressive vascular behavior in gliomas.
Fig 8: Analysis of Tau and vascular molecules in human samples.
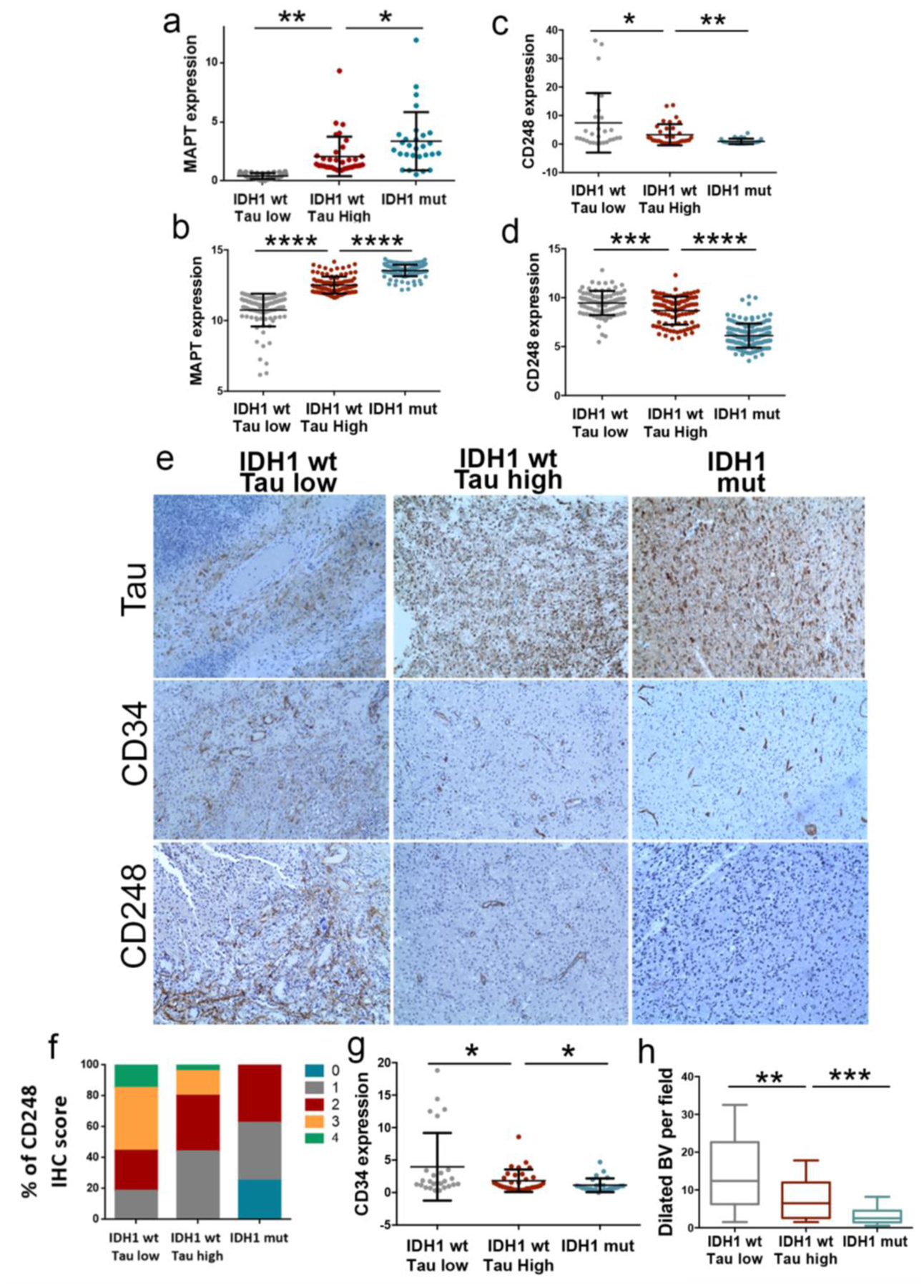
a–d, Tau (MAPT) (a,b), and CD248 (c,d) expression levels were determined by qRT-PCR analysis (Glioma cohort) (n=87) (a,c) or RNAseq analysis (TCGA-LGG+GB cohort) (n=319) (b,d). Tumors were classified in three groups: IDH1 wt (Tau Low), IDH1 wt (Tau High) and IDH1 mut. HPRT expression was used for normalization. e, Representative pictures of the IHC staining of Tau, CD34 and CD48 in three tumors, one for each of the groups. f, Percentage of tumors with different CD248 IHC score in e (n=68). g, CD34 levels were determined by qRT-PCR analysis (GB cohort) (n=87). h, Quantification of the dilated blood vessels in e (n=68). *, P ≤0.05; **, P ≤0.01; ***, P ≤0.001; ****, P ≤0.0001. n.s., non-significant.
Discussion
IDH1/2 mutations identify a genetically and clinically distinct glioma entity. Certainly, patients with such tumors have a much better prognosis, independently of the histological classification, and they show improved responses to chemotherapy and/or irradiation31, 32. Indeed, the re-expression of IDH1 R132H in GB can reduce tumor growth (Fig. 2a)13, 33. However, the molecular basis for the tumor-suppressor functions of IDH mutations are unclear. Here, we have identified Tau, a known microtubule stabilizer, as a new epigenetic target of IDHmut in gliomas. Based on our results we can propose that these mutant enzymes, acting through the increase in Tau expression, favor the normalization of the vasculature and impede the progression of the disease (Supplementary Fig. 8). In fact, Tau downregulation induces a dramatic increase in tumor burden in the orthotopic xenografts. Moreover, the longitudinal analysis of a set of paired samples showed that Tau expression decreases as LGG evolve into higher grade lesions. Interestingly, IDH1/2 status seems to be consistent during glioma progression34, 35, suggesting that some other mechanism is inhibiting Tau transcription in the recurrent tumors. One possible explanation would be that an increase in the levels of wild-type IDH protein in the recidives could impair Tau expression, as it happens in the mouse xenografts (Fig. 4d). Indeed, it has been recently shown that the non-mutated IDH1 is upregulated in primary GB in comparison with secondary GB or LGG, where it promotes aggressive growth and therapy resistance5.
From the moment that IDH1/2 mutations were identified in gliomas, it seemed clear that they represent early events in the process of tumorigenesis36. A decade later we know that these proteins induce important metabolic and epigenomic changes, which could explain their oncogenic properties13, 14, 37–39. Consequently, a plethora of inhibitors have been developed to block IDHmut function in different cancers. Indeed, some of them have shown anti-tumor acitivty in mouse glioma models40 and they have been approved to be tested in IDHmut patients41. However, the tumor-suppressor functions of this protein in gliomas, including the vascular normalization, could represent a severe pitfall for these approaches. In fact, the treatment with IDHmut inhibitors has been associated with a radioprotective effect42 and a decreased sensitivity to cisplatin43. Regarding IDHwt gliomas, our results suggest that microtubule stabilizers could imitate Tau function and reduce the aggressiveness of the tumors, which could render them sensitive to conventional chemotherapy (Fig. 5i). The same compounds could be tested in combination with IDHmut inhibitors in LGG, in an attempt to impair tumor growth and, at the same time, block the transformation into a more aggressive tumors.
Our results indicate that Tau is a key inhibitor of wild-type EGFR signaling in gliomas. Accordingly, drugs that interfere with microtubule function have been associated with EGFR inactivation in other cancers44, as they seem to alter endocytic trafficking11. Moreover, it has been shown that microtubule acetylation, which is promoted by Tau function16, promotes the degradation of EGFR through changes in the microtubule-dependent endocytic trafficking17. We propose that a similar mechanism could be operating in gliomas, but only in the absence of EGFR mutations. In that sense, there is plenty of literature that suggest that EGFR mutant proteins, especially the vIII isoform, can maintain the signal in the absence of the ligand45, possibly due to changes in the turnover of the mutant receptor46. Moreover, it has been proposed that some of the EGFR downstream targets, like NF-κB, become constitutively activated in GB after vIII expression47. The data presented here further supports that EGFR mutations induce a constitutive activation of NF-κB, which would be unsensitive to microtubule modulators.
Downstream of EGFR/NF-κB signaling, we have found that Tau overexpression severely reduced the amount of TAZ in GB. TAZ is a transcriptional coactivator that play a prominent role in gliomas48, controlling the MES signature19. Moreover, TAZ is induced by NF-κB activation in gliomas and promotes radio-resistance19. It has been recently proposed that TAZ promoter is a direct target of NF-κB49 so Tau could be inhibiting indirectly the transcription of the TAZ gene. However, we cannot discard a possible regulation of the protein stability downstream of EGFR/NF-κB signaling.
It is well known that IDH mutant gliomas are characterized by a lesser extent of contrast enhancement than wild-type tumors, even if we only consider GB50. However, the molecular explanation for this behavior was still missing. Our results show that Tau, acting downstream of IDHmut, could normalize the blood vessels and reduce the BBB leakage. These changes could cooperate or even precede other alterations observed in the TME of IDHmut gliomas, like the absence of microthrombi51 as well as the decrease in necrosis areas50 and hypoxia-induced angiogenesis52. Moreover, the immune component and its pro- or anti-tumoral properties could also be affected by the vascular phenotype of IDHmut gliomas53, explaining the decrease in the immune infiltrate observed in mouse models and human samples33. Future experiments are warranted in order to decipher the participation of Tau in these other phenotypes.
Although cancer and neurodegenerative diseases are considered as opposite phenomena, there could be a positive association between AD prevalence and GB incidence12. In AD and other tauopathies, there is an accumulation of Tau aggregates (gain of toxic function). However, this aggregation of Tau compromises its microtubule-stabilising functions (loss of physiological function), favoring the evolution of the pathology. In agreement with the loss-of-function model, we have reported behavioral changes and neurogenesis in aged Tau knockout mice54. Interestingly, EMT genes are enriched in the affected areas of AD brains55. Moreover, there is an important neurovascular dysfunction at early stages of AD, associated with BBB breakdown and inflammation56, which could be linked to pericyte loss57. Although there is still missing evidence of the relevance of Tau (especially the astrocytic Tau) in these phenotypes, it is tempting to speculate that the loss of function of this protein could represent a link between the progression of the two types of brain disease. If this hypothesis is correct, targeting pericytes or the mesenchymal transdifferentiation of the astrocytes could be an interesting therapeutic approach to be tested in several neurological disorders. Moreover, it would be worth studying if AD related drugs could be repurposed to treat brain tumors. In that sense, we have demonstrated that EpoD, a microtubule stabilizing agent that reduces AD pathology in mouse models18, slows down in vivo glioma growth without any apparent secondary effect. Another second-generation taxane, cabazitaxel, has demonstrated a good anti-glioma activity in preclinical tests58 and is being evaluated as a novel cytotoxic therapy (; ). However, we propose that lower doses of these compounds could imitate the effect of Tau overexpression, reducing the aggressiviness of gliomas with less secondary effects for the patients.
Collectively, our results provide and explanation for the better outcome of IDHmut gliomas, which could have important implications for several aspects of glioma research and clinical practice. Moreover, the understanding of how Tau governs the vascular component of the TME in gliomas could provide novel explanations for the neurovascular dysfunction observed in AD and other dementias.
Online Methods
Human samples
Glioma tissues embedded in paraffin were obtained after patient’s written consent and with the approval of the Ethical Committees of “Hospital 12 de Octubre” (CEI 14/023) and “Hospitales de Madrid” (14.10.632-GHM; 18.12.1337-GHM) (Supplementary Table 1,2).
Human and mouse glioma cells
RG1 cells were kindly donated by Rosella Galli (San Raffaele Scientific Institute). The rest of the human cells (Supplementary Table 3) were obtained by dissociation of surgical specimens from patients treated at the “Hospital 12 de Octubre” (Madrid, Spain). They were obtained after patient’s written consent and with the approval of the Ethical Committee (CEI 14/023). They belong to the Biobank of that Hospital. Cells were grown in Complete Media (CM): Neurobasal supplemented with B27 (1:50) and GlutaMAX (1:100) (Thermo-Fisher-Scientific); penicillin-streptomycin (1:100) (Lonza); 0.4% heparin (Sigma-Aldrich); and 40 ng/ml EGF and 20 ng/ml bFGF2 (Peprotech). Mouse SVZ models were obtained by retroviral expression of EGFRwt or EGFRvIII in primary neural stem cell (NSC) cultures from p16/p19 ko mice. The NSCs were obtained as previously described59 and they were grown in CM. After infection, the cells were injected into nude mice and the tumors that grew were dissociated and established as SVZ-EGFRamp/wt or SVZ-EGFRvIII models. Both models express GFP and luciferase as reporters. NPA (NRAS, shP53, shATRX) IDH1 wt and NPA (NRAS, shP53, shATRX) IDH1 R132H were provided by Maria G. Castro (University of Michigan)13 and cultured in CM.
DNA constructs and lentiviral/retroviral production
The lentivirus that encodes the longest wild type isoform of Tau in human brain harboring four repeats and two N-terminal inserts followed by GFP linked by an IRES was a gift from by Prof. Kenneth S. Kosik (UC Santa Barbara). As a control we used a lentivector encoding E-GFP, pRRLSIN.cPPT.PGK-GFP.WPRE (Addgene plasmid 12252). pLV-Hygro-Luciferase (VectorBuilder #VB150916–10098) was used as reporter. Lentiviral vector to express shRNAs were: shCD248 (Sigma #SHCLNG-NM_020404: TRCN0000053455, TRCN0000053457, TRCN00000443679, TRCN00000429396, TRCN0000043782) and shTAZ (TRCN0000370006, TRCN0000370007). Retroviral vectors used were pBabe-EGFR wt (#11011), MSCV-XZ066-GFP-EGFR vIII (#20737), pBabe-puro-Flag-IDH1 (#62923), pBabe-puro-Flag-IDH1-R132H (#62924) (Addgene) and pBabePuroTAZ-WT was a generous gift from Kun-Liang Guan. To obtain the virus, the 293T cells were transiently co-transfected with 5 μg of appropriate lentivector plasmid, 5 μg packaging plasmid pCMVdR8.74 (Addgene #Plasmid 22036) and 2 μg VSV-G envelope protein plasmid pMD2G (Addgene #Plasmid 12259) using Lipofectamine Plus reagent (Invitrogen). Retrovirus and lentivirus supernatant was prepared by transfection of 293T cells and collection of the supernatant 48 hr after.
Intracraneal tumor formation and treatment in vivo
Animal experiments were reviewed and approved by the Research Ethics and Animal Welfare Committee at “Instituto de Salud Carlos III” (PROEX 244/14 and 02/16), in agreement with the European Union and national directives. Intracranial transplantation to establish orthotopic xeno- and allo-grafts was performed injecting 100.000–300.000 cells (resuspended in 2 μl of culture stem cell medium) with a Hamilton syringe into athymic Nude-Foxn1nu brains (Harlan Iberica). The injections were made into the striatum (coordinates: A–P, −0.5 mm; M–L, +2 mm, D–V, −3 mm; related to Bregma) using a Stoelting Stereotaxic device. When applicable, tumor growth was monitored in an IVIS equipment (Perkin Elmer) after intraperitoneal injection of D-luciferin (75 mg/Kg) (PerkinElmer). The animals were sacrificed at the onset of symptoms. Mice were treated with Epothilone D (Abcam, ab143616) (1 mg/kg two days per week through intraperitoneal injection) and/or Temozolomide (Sigma Aldrich) (5mg/kg daily through intraperitoneal injection). Epothilone D was dissolved in 4% DMSO +10%Polysorbate. Temozolomide was dissolved in 1% BSA+PBS. Control animals were treated with these solvents.
Growth curve and sphere formation assay
Glioma cells were infected by the control lentivirus (LV-GFP) or lentivirus directing expression of TAU (LV-TAU-GFP). The tumor spheres were Accumax-dissociated to single cells, and 500/1000 cells of each condition were plated in a p24-well-plate in triplicate. Five days after plating, spheres and cell number were measured.
Endothelial cell sprouting assay
In order to generate endothelial cell spheroids, 1×106 cells of HMBEC were suspended in CM and seeded in nonadherent plates. These spheroids were harvest within 48 hours and embedded into matrigel in 96-well plates with conditioned medium generated for each cell line. After 24 h, sprouting was induced and the number of sprouts for each spheorid were quantified. For the generation of the conditioned media, RG1 (GFP or Tau) were grown during 48 hours in serum-free culture media (DMEM-F12 supplemented with FGF2 (50ng/ml) and penicillin-streptomycin). The medium was filtered with a 70-μm filter before use.
In vivo limiting dilution assay
Increasin numbers of RG1 (GFP or TAU) cells were re-suspended in CM and Matrigel (BD) (1:10) and injected subcutaneously injected in nude mice. Animals were sacrificed before tumors reached a 1.5 cm in diameter. The statistical significances were calculated using the Extreme Limiting Dilution Analysis software (http://bioinf.wehi.edu.au/software/limdil/index.html).
Inmunofluorescent (IF) and Inmunohistochemical (IHC) staining
Slides were heated at 60°C for 1 hour followed by deparaffinization and hydration, washed with water, placed into antigen retrieval solution (pressure cooking) in 10 mM sodium citrate pH 6.0. Paraffin sections were permeabilized with 1% Triton X-100 (Sigma-Aldrich) in PBS and blocked for 1 hour in PBS with 5% BSA (Sigma), 10% FBS (Sigma) and 0,1% Triton X-100 (Sigma). The following primary antibodies (Supplementary Table 4) were incubated O/N at 4°C. The second day, sections were washed with PBS three times prior to incubation with the appropriate secondary antibody (Supplementary Table 4) (1:200 dilution) for 2h at room temperature. Prior to coverslip application, nuclei were counterstained with DAPI and imaging was done with Leica SP-5 confocal microscope. Otherwise, IHC sections were incubated with biotinylated secondary antibodies (1:200 dilution). Target proteins were detected with the ABC Kit and the DAB kit (Vector Laboratories).
IHC quantification
The IHC score was judged from 0 (no staining) to 3 (Tau staining) or 4 (CD248 staining) on those samples with the strongest positive staining. For the longitudinal analysis of the primary and relapsed tumors we calculated the score of 10 high magnification pictures of each sample. The data depicted in Fig. 2e,f is the average of these 10 pictures. For the quantification of the vasculature, we counted the number of dilated vessels per high-magnification field.
Western Blot analysis
Protein content was quantified using BCA Protein Assay Kit (Thermo-Fisher-Scientific). Approximately 20 μg of proteins were resolved by 10% or 12% SDS-PAGE and they were then transferred to a nitro cellulose membrane (Hybond-ECL, Amersham Biosciences). The membranes were blocked for 1 h at room temperature in TBS-T (10 mM Tris-HCl [pH 7.5], 100 mM NaCl, and 0.1% Tween-20) with 5% skimmed milk, and then incubated overnight at 4°C with the corresponding primary antibody diluted in TBS-T. The primary antibodies and the dilutions are shown in Supplementary Table 4. After washing 3 times with TBS-T, the membranes were incubated for 2 h at room temperature with their corresponding secondary antibody (HRP-conjugated anti mouse or anti rabbit, DAKO) diluted in TBS-T. Proteins were visible by enhanced chemiluminiscence with ECL (Pierce) using the Amersham Imager 680.
qRT-PCR assay
RNA was extracted from the tissue using RNA isolation Kit (Roche). Total RNA (1μg) was reverse transcribed with PrimeScript RT Reagent Kit (Takara). Quantitative real-time PCR was performed using the Light Cycler 1.5 (Roche) with the SYBR Premix Ex Taq (Takara). The primers used for each reaction are indicated in Supplementary Table 5. Gene expression was quantified by the delta-delta Ct method.
EGFR Sequencing of primary GBMs
To identify point EGFR mutations, cDNAs from the different cell lines were sequenced using the primers indicated in Supplementary Table 6. The sequences were aligned and collated with the EGFR transcript (NM_005228.4) in the NIH GenBank database using the multiple sequence alignment tool Clustal Omega (www.ebi.ac.uk/Tools/msa/clustalo/) and the Sequencing Analysis Software v5.3.1 from Applied Biosystems. The identified mutations were analyzed at cBioPortal (www.cbioportal.org).
Gene expression and survival analyses
Tau (MAPT) gene expression and follow-up overall survival data from human glioma tumors corresponding to TCGA Glioblastoma (GB) and Brain lower grade Glioma (LGG) data sets were downloaded respectively from cBioPortal (http://www.cbioportal.org/) and TCGA databases (http://tcga-data.nci.nhi.gov/docs/publications/lgggbm_2015) using UCSC cancer browser. Kaplan-Meier survival curves were done within TCGA-GB, TCGA-LGG, TCGA-GB-LGG, Rembrandt, Gravendeel, Ducray, Freije and Nutt cohorts upon stratification based into low and high groups using expression values from Tau (MAPT) gene. Significance of differences in survival between groups was calculated using the log-rank test. These data were obtained from UCSC Xena-Browser (https://xenabrowser.net) and Gliovis (http://gliovis.bioinfo.cnio.es). Classification into classical, mesenchymal, neural and proneural subtypes was retrieved from the TCGA GB data set (https://www.ncbi.nlm.nih.gov/pubmed/24120142) together with Tau/MAPT expression values. Differences in Tau expression between mesenchymal and other groups were calculated using Student’s t-test. Correlation between gene expression values of MAPT versus other genes was done using Pearson analysis. Gene Set Enrichment Analysis (GSEA) was computed into the TCGA-GBMLGG cohort (RNAseq (IlluminaHiSeq)) using Tau gene expression as a continuous class label and genesets from the “CGP: chemical and genetic perturbations” and “CP:BIOCARTA: BioCarta gene sets” (n=) from the MSigDB genesets database.
Analysis of methylation of the MAPT gene and CTCF Chip-seq binding
DNA-methylation analysis in human glioma within the MAPT locus was done using the TCGA data (Illumina Infinium HumanMethylation450 platform). Methylation beta values from 56 probes within MAPT locus were retrieved from the Xena browser (https://xenabrowser.net/) together with MAPT gene expression values. Pearson correlation values between each methylation probe and gene expression values, calculated for all samples, was calculated and represented together with the CpG islands located at the MAPT locus (CpG302, CpG26 and CpG21). Both TCGA LGG-GB and LGG cohorts were individually analyzed. Correlation values were independently calculated for IDH1 mutant or wild-type samples in the TCGA LGG cohort. ChIP-seq analysis using anti-CTCF antibody was performed from profiling in IDH1 mutant and wild-type glioma patient specimens and culture models (GSE70991). Briefly tdf files from GEO repository from both IDH1 mutant and wild-type samples were downloaded and visualized using IGV browser. CTCF occupancy at the CpG islands located from MAPT loci was visualized.
Statistical analysis
For bar graphs, the level of significance was determined by a two-tailed un-paired Student’s t-test. The difference between experimental groups was assessed by Paired t-Test and one-way ANOVA. For Kaplan-Meier survival curves, the level of significance was determined by the two-tailed log-rank test. For correlation analysis between each gene, expression data were tested by Pearson’s correlation coefficient and Spearman’s correlation coefficient. All analyses were performed with the GraphPad Prism 5 software. P values < 0.05 were considered significant (*p < 0.05; **p < 0.01; *** p< 0.001; **** p< 0.0001; n.s., non-significant). All quantitative data presented are the mean ± SEM from at least three simples or experiments per data point. Precise experimental details (number of animals or cells and experimental replicates) are provided in the Fig. legends.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Rosella Galli for donating RG1, Rafael Hortigüela and the Confocal service personell, for their technical support. The graphical abstract was created with images adapted from Servier Medical Art by Servier. Original images are licensed under a Creative Commons Attribution 3.0 Unported License.
Funding
Work was supported by NIH/NINDS Grant R01-NS105556 to MGC; by Ministerio de Economía y Competitividad: (Acción Estratégica en Salud) grants: PI13/01258 to AHL, PI17/01489 and CP11/00147 to AAS, PI18/00263 to RGE, and PI16/01 278 to JS; by “Asociación Española contra el Cancer (AECC) grants: Investigador Junior to RG and GCTRA16015SEDA to JMS and JS; and by Ministerio de Economía y Competitividad: SAF-2014-53040-P to JA, RTC-2015-3771-1 to JS and SAF2015-65175-R/FEDER to PSG.
Footnotes
Competing interests
The authors declare no competing financial interests.
Reference List
- 1.Louis DN et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 131, 803–820 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Yan H et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 360, 765–773 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turcan S et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu C et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calvert AE et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 19, 1858–1873 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molenaar RJ, Maciejewski JP, Wilmink JW, & van Noorden CJF Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 37, 1949–1960 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhaak RG et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ichimura K, Narita Y, & Hawkins CE Diffusely infiltrating astrocytomas: pathology, molecular mechanisms and markers. Acta Neuropathol. 129, 789–808 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Avila J et al. Looking for novel functions of tau. Biochem. Soc. Trans 40, 653–655 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Barreda EG & Avila J Tau regulates the subcellular localization of calmodulin. Biochem. Biophys. Res. Commun 408, 500–504 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Li H et al. Effects of paclitaxel on EGFR endocytic trafficking revealed using quantum dot tracking in single cells. PLoS. One 7, e45465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehrer S Glioblastoma and dementia may share a common cause. Med. Hypotheses 75, 67–68 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Nunez FJ et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flavahan WA et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caillet-Boudin ML, Buee L, Sergeant N, & Lefebvre B Regulation of human MAPT gene expression. Mol. Neurodegener 10, 28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez M et al. Tau--an inhibitor of deacetylase HDAC6 function. J. Neurochem 109, 1756–1766 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Gao YS, Hubbert CC, & Yao TP The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J. Biol. Chem 285, 11219–11226 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci 32, 3601–3611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhat KP et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24, 331–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pozo N et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J Clin Invest 123, 2475–2487 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu F et al. Olig2-Dependent Reciprocal Shift in PDGF and EGF Receptor Signaling Regulates Tumor Phenotype and Mitotic Growth in Malignant Glioma. Cancer Cell 29, 669–683 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Romero N et al. Cancer stem cells from human glioblastoma resemble but do not mimic original tumors after in vitro passaging in serum-free media. Oncotarget. 7, 65888–65901 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bougnaud S et al. Molecular crosstalk between tumour and brain parenchyma instructs histopathological features in glioblastoma. Oncotarget. 7, 31955–31971 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergers G & Song S The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol 7, 452–464 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scully S et al. Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J. Neurosci 32, 12950–12960 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng L et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153, 139–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou W et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell 21, 591–603 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardee ME & Zagzag D Mechanisms of glioma-associated neovascularization. Am. J. Pathol 181, 1126–1141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park JS et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 30, 953–967 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Simonavicius N et al. Endosialin (CD248) is a marker of tumor-associated pericytes in high-grade glioma. Mod. Pathol 21, 308–315 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Buckner JC, Chakravarti A, & Curran WJ Jr. Radiation plus Chemotherapy in Low-Grade Glioma. N. Engl. J. Med 375, 490–491 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Cairncross JG et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J. Clin. Oncol 32, 783–790 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amankulor NM et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 31, 774–786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao Y et al. Mutation analysis of IDH1 in paired gliomas revealed IDH1 mutation was not associated with malignant progression but predicted longer survival. PLoS. One 8, e67421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson BE et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe T, Nobusawa S, Kleihues P, & Ohgaki H IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol 174, 1149–1153 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bardella C et al. Expression of Idh1(R132H) in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 30, 578–594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu KV et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22, 21–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Philip B et al. Mutant IDH1 Promotes Glioma Formation In Vivo. Cell Rep. 23, 1553–1564 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohle D et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golub D et al. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front Oncol. 9, 417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molenaar RJ et al. Radioprotection of IDH1-Mutated Cancer Cells by the IDH1-Mutant Inhibitor AGI-5198. Cancer Res. 75, 4790–4802 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Khurshed M et al. IDH1-mutant cancer cells are sensitive to cisplatin and an IDH1-mutant inhibitor counteracts this sensitivity. FASEB J.fj201800547R (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu X et al. Microtubule inhibition causes epidermal growth factor receptor inactivation in oesophageal cancer cells. Int. J. Oncol 42, 297–304 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Zahonero C & Sanchez-Gomez P EGFR-dependent mechanisms in glioblastoma: towards a better therapeutic strategy. Cell Mol. Life Sci(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grandal MV et al. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 28, 1408–1417 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Bonavia R et al. EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene 31, 4054–4066 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gargini R et al. WIP Drives Tumor Progression through YAP/TAZ-Dependent Autonomous Cell Growth. Cell Rep. 17, 1962–1977 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Ferraiuolo M et al. Agave negatively regulates YAP and TAZ transcriptionally and post-translationally in osteosarcoma cell lines. Cancer Lett. 433, 18–32 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Lai A et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol 29, 4482–4490 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Unruh D et al. Mutant IDH1 and thrombosis in gliomas. Acta Neuropathol. 132, 917–930 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kickingereder P et al. IDH mutation status is associated with a distinct hypoxia/angiogenesis transcriptome signature which is non-invasively predictable with rCBV imaging in human glioma. Sci. Rep 5, 16238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Missiaen R, Mazzone M, & Bergers G The reciprocal function and regulation of tumor vessels and immune cells offers new therapeutic opportunities in cancer. Semin. Cancer Biol 52, 107–116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pallas-Bazarra N et al. Novel function of Tau in regulating the effects of external stimuli on adult hippocampal neurogenesis. EMBO J. 35, 1417–1436 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Podtelezhnikov AA et al. Molecular insights into the pathogenesis of Alzheimer’s disease and its relationship to normal aging. PLoS. One 6, e29610 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kisler K, Nelson AR, Montagne A, & Zlokovic BV Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci 18, 419–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sagare AP et al. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun 4, 2932 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Semiond D, Sidhu SS, Bissery MC, & Vrignaud P Can taxanes provide benefit in patients with CNS tumors and in pediatric patients with tumors? An update on the preclinical development of cabazitaxel. Cancer Chemother. Pharmacol 72, 515–528 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Ferron SR et al. A combined ex/in vivo assay to detect effects of exogenously added factors in neural stem cells. Nat. Protoc 2, 849–859 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.