

Supplemental Digital Content is available in the text.
Keywords: calcium signaling; endothelium; hypertension; muscle, smooth, vascular; obesity
Abstract
Obesity-related hypertension is one of the world’s leading causes of death and yet little is understood as to how it develops. As a result, effective targeted therapies are lacking and pharmacological treatment is unfocused. To investigate underlying microvascular mechanisms, we studied small artery dysfunction in a high fat–fed mouse model of obesity. Pressure-induced constriction and responses to endothelial and vascular smooth muscle agonists were studied using myography; the corresponding intracellular Ca2+ signaling pathways were examined using confocal microscopy. Principally, we observed that the enhanced basal tone of mesenteric resistance arteries was due to failure of intraluminal pressure-induced Ca2+ spark activation of the large conductance Ca2+ activated K+ potassium channel (BK) within vascular smooth muscle cells. Specifically, the uncoupling site of this mechanotransduction pathway was at the sarcoplasmic reticulum, distal to intraluminal pressure-induced oxidation of Protein Kinase G. In contrast, the vasodilatory function of the endothelium and the underlying endothelial IP-3 and TRPV4 (vanilloid 4 transient receptor potential ion channel) Ca2+ signaling pathways were not affected by the high-fat diet or the elevated blood pressure. There were no structural alterations of the arterial wall. Our work emphasizes the importance of the intricate cellular pathway by which intraluminal pressure maintains Ca2+ spark vasoregulation in the origin of obesity-related hypertension and suggests previously unsuspected avenues for pharmacological intervention.
Obesity-related cardiovascular disease is set to become one of the defining epidemics of the 21st century. In the United Kingdom, 30% of all children are either overweight or obese by the time they leave primary school aged 11 years old1: This phenotype is then maintained into adulthood. The metabolic consequences of obesity have profound effects on the cardiovascular system at almost every level. One of the most prominent of these obesity-related complications is hypertension.2 Thus, obese people are more than twice as likely to develop hypertension than normal weight people.3,4
The pathophysiology underlying the rise in blood pressure accompanying weight gain is undoubtedly multifactorial, but much attention has focused on the abnormal function of small resistance arteries in obesity. This microvascular dysfunction is important principally as the increased collective constriction of resistance arteries drives peripheral resistance and thus elevates the central blood pressure.
The defining characteristic of small arteries is their capacity to constrict in response to intraluminal pressure. This pressure-induced constriction is known as myogenic autoregulation and is regulated by the vascular smooth muscle cell (VSMC) layer of the artery. The basal tone maintained by the pressure-induced constriction of VSMC is rapidly and reactively modulated by the vascular endothelial cells (ECs) through mechanisms to hyperpolarize and thus vasodilate the adjacent VSMC. These vasoregulatory processes of both VSMC and EC are controlled by cellular coupling processes whereby tightly regulated, spatiotemporally brief changes in local Ca2+ concentrations activate adjacent Ca2+-activated K+ channels on the plasma membrane. The resultant efflux of K+ from the cell hyperpolarizes the membrane, vasodilating the artery. Within VSMC, the Ca2+ release events from the sarcoplasmic reticulum are Ca2+ sparks, each of which activates around 30 neighboring large conductance Ca2+ activated K+ channels (BK).5 The hyperpolarisation of the VSMC membrane by BK channel activation partially hyperpolarises the cell and blunts the inherent pressure induced constriction. Within ECs, a broadly similar process occurs whereby Ca2+ is released from the endoplasmic reticulum in response to IP-3 (Inositol Trisphosphate; Ca2+ pulsars) and these activate adjacent K+ channels6 (the intermediate conductance Ca2+ activated K+ channels [IK]). In addition, muscarinic activation triggers additional localized Ca2+ entry into the EC through TRPV4 (vanilloid 4 transient receptor potential ion channel) channels (Ca2+ sparklets), which also trigger IK activation and thus vasodilation.7 Both the TRPV4 and IP-3 Receptor Ca2+ events within the EC occur within a unique mast-like projection of the EC which extends through the internal elastic lamina of the artery to plant directly onto the VSMC. This structure is called the myoendothelial projection.
The intricate and interconnected ionic processes within EC and VSMC which regulate small artery tonic activity have all been implicated in the pathogenesis of obesity-related cardiovascular disease. However, to date, studies have often been incomplete, either in the breadth of the imaging of the individual Ca2+ events or in the assessment of the effect of changes to ion channel activity on basal pressure induced constriction of the arteries. The picture is also clouded due to conflicting data: for example, endothelial function is variably reported as equivalent8 or damaged,9–11 dependent on mode of study of the arteries (eg, wire10,11 versus pressure8,9 myography) or the model of obesity used (genetic versus diet-induced). In this study, we have used one of the commonest approaches; chronic high-fat feeding and in addition have studied all the predominant Ca2+ signaling events controlling the pressure-induced tone of small resistance arteries. We also studied the structure of the small arteries to assess remodeling of the arterial wall. Using this approach, we find normal endothelial function but have identified the key importance of an intraluminal pressure-sensitive Ca2+ signaling pathway within the vascular smooth muscle of the artery, which is associated with the development of hypertension due to obesity.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request. Third-order mesenteric arteries from high-fat diet (HFD) and low-fat diet (LFD) fed mice were studied using pressure myography, high-speed confocal microscopy of pressurized arteries (VSMC Ca2+ sparks) and an en-face slit open artery preparation (endothelial Ca2+ IP-3 pulsars and TRPV4 sparklets) and Western Blot approaches. For the pressurized artery protocols, arteries were mounted on borosilicate glass pipettes in an arteriography chamber. The arteries were tied in place with nylon string and connected to a servo-controlled pressure-regulator (Living Systems Instrumentation). For the diameter studies, arteries were studied once pressure-induced constriction (myogenic constriction) had developed at 80 mm Hg intraluminal pressure and in bicarbonate-buffered PSS (119 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgCl2, 2 mmol/L CaCl2, 7 mmol/L glucose, 24 mmol/L NaHCO3, and 2.3 mmol/L EDTA) heated to 37°C and gassed with biological gas (95% air and 5% CO2). Pressure-induced constriction was calculated as the difference between the intraluminal diameter of the vessel in Ca2+-containing and Ca2+-free solutions, expressed as a percentage of the diameter in Ca2+-free solution. Internal lumen diameter was recorded using videomicroscopy and edge detection software (Living Systems Instrumentation and IonOptix). For complete methods section, see Expanded materials and methods in the online-only Data Supplement. Student t-test or multiple 1-way ANOVA were used for statistical analysis testing. P≤0.05 was used as a threshold to determine statistical significance. n values are shown for number of animals and number of arteries studied.
Results
Obesity-Related Hypertension Is Associated With Preservation of Vasodilatory Endothelial Calcium Signaling
Mice on a high-fat diet developed obesity, hypertension, and glucose intolerance, in a similar pattern to previous studies,12 which have used this dietary approach before (Figure S1 in the online-only Data Supplement). We studied the effect of high-fat feeding on vascular vasodilatory Ca2+ signals, which are generated by activation of the TRPV4 channel (Ca2+ sparklets). Activation of TRPV4 channels following muscarinic stimulation of the endothelium occurs almost exclusively within the myoendothelial projection (MEP).7 Within the mouse mesenteric MEP, the anchoring protein AKAP-150 underpins a vasodilatory microdomain whereby TRPV4 channels exist within 4 channel clusters. It is therefore possible to observe 1 to 4 channel patterns of activation based on established fluorescence levels for individual TRPV4 channel opening events.7 Furthermore, previous studies in obesity have suggested that changes to MEP structure and function account for alterations in the mechanisms by which the predominant endothelial hyperpolarization pathways are configured.13 We used the en-face preparation to study the TRPV4 signals, both at baseline and in response to increasing concentrations of Carbachol. Representative images from the endothelial preparation are shown in Figure 1A and 1B. Figure 1C and 1D are representative traces from the regions of interest indicated on the en-face preparations. The dashed lines indicate previously described quantal levels for TRPV4 activity based on the fluorescence generated by Ca2+ entry through between 1 and 4 channels, consistent with previous publications.7,14 To suppress IP-3-mediated Ca2+ signals, which would obscure the TRPV4 channel opening Ca2+ entry events, the preparations were incubated with the SERCA inhibitor, cyclopiazonic acid. A low concentration of GSK1016970A (a TRPV4 activator, hereafter referred to as GSK101) was added as per previous imaging protocols to boost signal acquisition. Figure 1E and 1F indicate no difference in relative TRPV4 activity per field or the open probability of the channel based on the Ca2+ entry events between the low fat–fed and high fat–fed groups. Furthermore, consistent with previous reports of TRPV4 activity in the endothelium, all the Carbachol-induced TRPV4 activity was observed at the MEP sites (Figure 1E). Amplitude histograms were created to illustrate quantal changes in fluorescence based upon the number of channels open in response to the pharmacological stimuli (Figure S2). Increasing concentrations of Carbachol sequentially increased the amplitude histogram event counts in both high- and low-fat groups equivalently.
Figure 1.
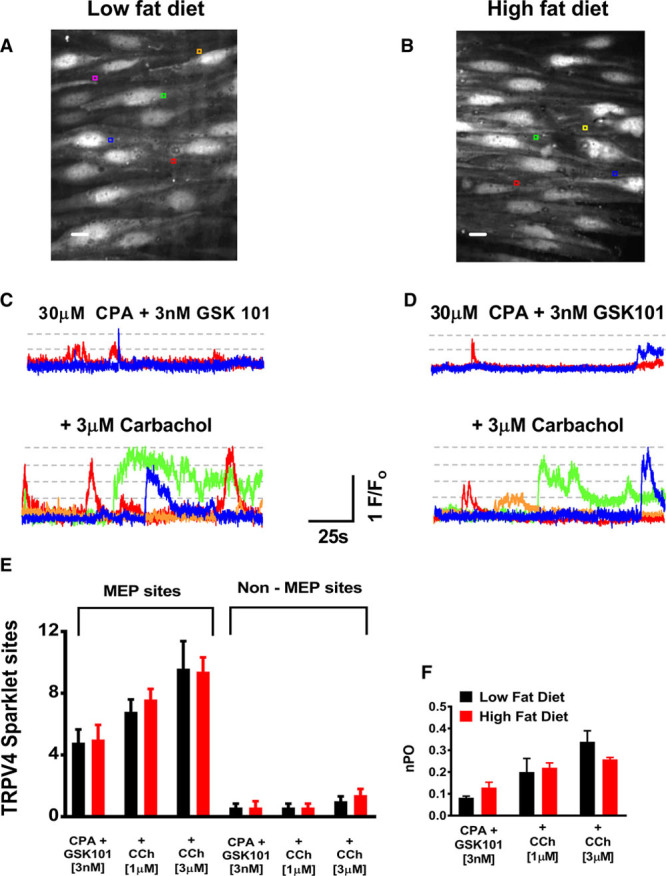
Endothelial TRPV4 Ca2+ signal quantification. A and B, Representative en-face images of the mesenteric endothelium from low-fat diet (LFD) and high-fat diet (HFD) fed mice. Scale bars=10 µm. C and D, Representative changes in F/Fo at region of interest positioned on A and B. Dashed lines indicate published levels of TRPV4 fluorescence based on quantal opening of 1 to 4 channels. Traces shown under baseline conditions (CPA and 3 nM GSK101) and subsequently with Carbachol 3 µM. Scale bars=10 µm. E, Number of TRPV4 sparklet sites per field studied indicating activity at MEP rather than non-MEP locations. F, NPo per TRPV4 site studied between LFD (black) and HFD (red) arteries (LFD: n=5 arteries from 4 mice. HFD: n=5 arteries from 3 mice). CPA indicates cyclopiazonic acid; F/Fo, fluorescence intensities relative to baseline; MEP, myoendothelial projection; and TRPV4, vanilloid 4 transient receptor potential ion channel.
We also studied IP-3-induced Ca2+ signals from the endoplasmic reticulum within the MEPs (Ca2+ pulsars) using the endothelial en-face approach (Figure S3). There were no differences in frequency (Figure S3C), amplitude (Figure S3D) of these Ca2+ events seen between the mesenteric arteries from the low fat–fed and high fat–fed mice, although the duration of the IP-3-mediated Ca2+ signals was higher in the high fat fed mice (Figure S3E).
Obesity-Related Hypertension Is Associated With Preservation of Endothelial Vasodilation
We examined the small artery endothelial vasodilation in these 2 groups of animals (low-fat diet versus high-fat diet) using pressure myography. When small resistance mesenteric arteries are preconstricted by increased intraluminal pressure (pressure-induced constriction or myogenic constriction), the hyperpolarizing pathway comprising endothelial TRPV4 channel–mediated Ca2+ sparklet entry leading to IK channel activation accounts for at least 80% of the total vasodilation to muscarinic agonism.7 We therefore studied the vasodilatory responses to carbachol (muscarinic agonist, Figure 2A through 2C), GSK101 (Figure 2D through 2F) and NS309 which activates the IKCa and SKCa channels (Figure S4A through S4C). There were no differences in the vasodilatory responses to any of these agonists between the low-fat diet–fed mice and their high fat–fed counterparts.
Figure 2.
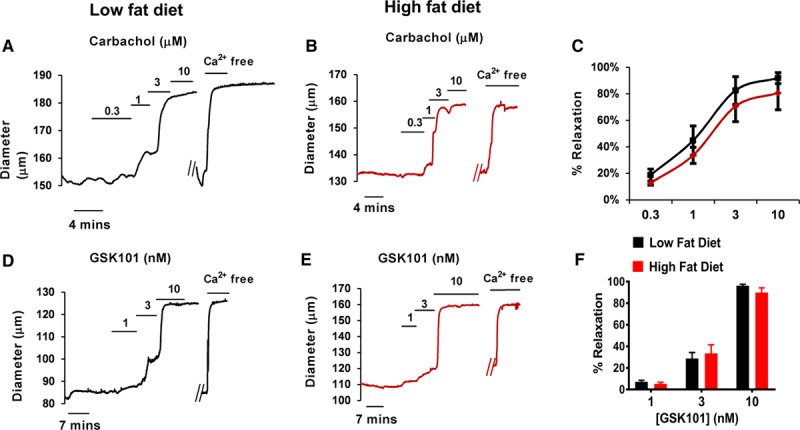
Endothelial-mediated vasodilation in obesity. Raw pressure myography traces are presented in this figure. The diameter of the artery (y axis) is shown in relation to time (x axis) and the vasodilatory responses of the arteries from the normal diet fed (black) and high fat–fed (red) mice are shown in response to various endothelial targeted vasodilatory agonists. Arterial diameter traces illustrate the concept of pressure-induced constriction whereby the artery maintains a stable internal diameter in response merely to the presence of 80 mmHg of intraluminal pressure. The artery is then exposed to a cumulative concentration range of endothelial agonists: Carbachol—a muscarinic agonist or GSK101—a TRPV4 (vanilloid 4 transient receptor potential ion channel) agonist. The raw data indicate that the pressure-constricted mesenteric arteries from low fat–fed (LFD) and high fat–fed (HFD) mice vasodilated equivalently to Carbachol (A–C, n=6 arteries from 5 mice for LFD, 12 arteries from 12 mice for HFD), GSK101 (D–F, n=3 arteries from 3 mice for LFD, 10 arteries from 10 mice HFD).
Increased Tone of Arteries in Obesity-Related Hypertension Is Associated With Impaired VSMC BK Channel Activation
Although the endothelial function of the small resistance arteries did not differ between the high fat–fed and low fat–fed groups, judged either using vasodilation to agonists or in the pattern of the vasodilatory Ca2+ signaling events, we noted enhanced steady state constriction in the mesenteric arteries from the high fat–fed mice compared with those from the low fat–fed mice (Figure 3A through 3C). This has been described previously and is attributed to dysfunction of the BK channel; more specifically due to aberrant trafficking of the β-subunit of the channel.12 Consistent with this, we showed that the increased pressure-induced constriction of the mesenteric small arteries from the high fat–fed mice was associated with a reduction in the constriction to the specific BK channel blocker Iberiotoxin, compared with the arteries from the low fat–fed mice (Figure 3D). However, when the degree of constriction was studied following incubation with Iberiotoxin, there was equivalence between the low fat–fed group and the high fat–fed group (Figure 3E). There was no appreciable change in diameter of the pressure constricted arteries in response to TRPV4 inhibition (HC067047 or HC) in either the high fat–fed group or the low fat–fed group (Figure 3D).
Figure 3.
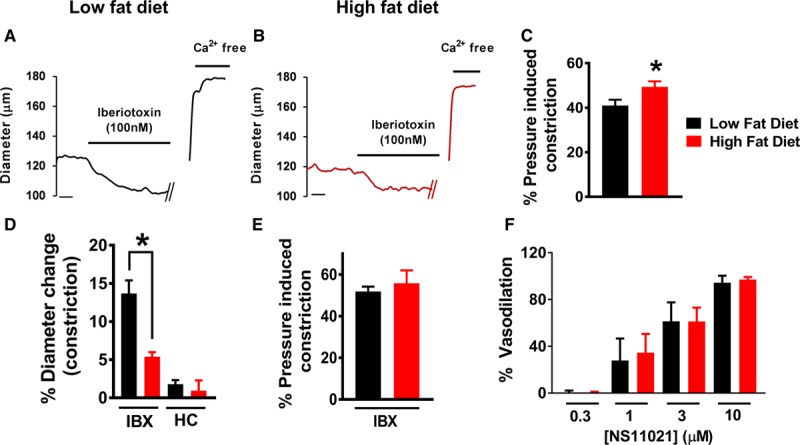
Enhanced pressure-induced constriction due to reduced large conductance Ca2+-activated potassium channel (BK) channel vasodilatory effect in high fat–fed mice. Raw pressure myography traces presented similarly to those in Figure 2. Arterial diameter demonstrates stable pressure-induced constriction before application of the drug. Data are presented showing the effects of BK channel agonist (NS11021) and blocker (IBX). A–C, Representative traces from low fat–fed (LFD) and high fat–fed (HFD) mice are shown in A (LFD) and B (HFD). C, The degree of pressure-induced constriction, calculated as a percentage of passive free diameter, was greater in HFD (n=13 arteries from 10 mice) compared with LFD arteries (LFD: n=13 arteries from 9 mice. LFD: 41±2.6% vs HFD: 49.2±2.4%; P=0.02). D, Mesenteric resistance arteries from LFD mice showed significantly greater constriction to IBX compared with those from the HFD group (LFD: 6 arteries from 4 mice. HFD: 4 arteries from 4 mice. LFD: 13.7±2.6% vs HFD: 5.2.2±1.1%; P=0.005). There was no change in lumen diameter when the arteries from either HFD or LFD mice were incubated with the TRPV4 (vanilloid 4 transient receptor potential ion channel) blocker HC-067047 (HC; LFD: 3 arteries from 3 mice. HFD: 4 arteries from 4 mice). E, When the degree of pressure-induced constriction of the arteries following incubation with IBX was compared, there was equivalence between the LFD and HFD arteries. F, Vasodilations to the BK channel agonist NS11021 were equivalent between LFD and HFD mice (n=4 arteries from 4 mice for both LFD and HFD). See Figure S5 for raw pressure myography traces showing NS11021 responses. IBX indicates Iberiotoxin.
During steady-state pressure-induced constriction, BK channels of small resistance arteries are only partially activated (to around an ED30) by Ca2+ sparks.15 In this context, BK channels can be further activated pharmacologically to induce maximal vasodilation. To assess the functional bioavailability of BK channels at steady-state pressure-induced constriction, we used NS11021, an agonist which binds the α-subunit of the BK channel. Vasodilation of pressure-constricted arteries to NS11021 was equivalent between the low fat–fed and high fat–fed arteries (Figure 3F; Figure S6).
Diet-Induced Obesity Compromises the Pressure Dependence of Ca2+ Spark Frequency in VSMC
In individual vascular myocytes, studied after enzymatic digestion of the artery, Ca2+ spark frequency is unaffected by diet-induced obesity.12,16 However, Ca2+ sparks are particularly sensitive to changes in intraluminal pressure. At low intraluminal pressure, Ca2+ spark frequency is low whereas at high intraluminal pressures, Ca2+ spark frequency rises sufficiently to trigger activation of BK channels.17 We therefore examined regulation of Ca2+ sparks between high fat–fed and low fat–fed groups both at low (20 mm Hg) and normal (80 mm Hg) physiological intraluminal pressures (Figure 4). At low intraluminal pressures, the frequency of Ca2+ sparks was the same in both groups. However, when arteries were studied at high intraluminal pressures, there was a rise in Ca2+ spark frequency only in the low fat–fed mouse mesenteric arteries (Figure 4B and 4C). There was no significant change in Ca2+ spark frequency in the high fat–fed mouse mesenteric arteries with the increase in intraluminal pressure and at this intraluminal pressure (80 mm Hg) there were significantly fewer Ca2+ sparks compared with the arteries from the low fat–fed mouse (Figure 4D through 4F; Figure S6A). There was no difference in amplitude, duration, rise-time, or decay of Ca2+ sparks between the 2 groups, either at low or elevated intraluminal pressures (Figure S6B through S6D).
Figure 4.
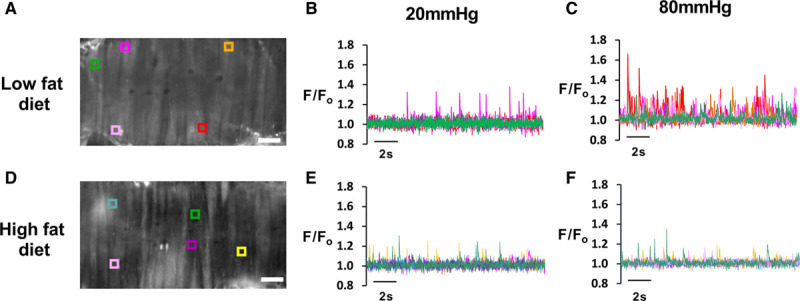
Differential pressure regulation of Ca2+ spark frequency in high fat–fed and low fat–fed arteries. A, Confocal image of an intact pressurized mesenteric artery from a low-fat diet mouse with representative F/Fo traces (B and C) derived from regions of interest (ROI) as shown on the image at 20 mmHg (B) and 80 mmHg (C) of intraluminal pressure. D, Confocal image of artery from high-fat diet mice with changes in F/Fo at ROI at both 20 mmHg (E) and 80 mmHg (F) intraluminal pressure (for quantification of Ca2+ spark characteristics, see Figure S6). F/Fo indicates fluorescence intensities relative to baseline.
Role of Oxidant Activation of Protein Kinase G in Ca2+ Spark Vasoregulation in Obesity
Our data indicate that obesity interrupts the pathway through which intraluminal pressure within resistance arteries maintains physiological Ca2+ spark frequency. We have previously investigated this pathway in health and found it dependent on oxidant activation of PKG.18 We were able to delineate this mechanism through use of a genetically modified knock-in mouse model whereby the capacity of PKG to be oxidized is disabled through an amino acid substitution (Cysteine to Serine at residue 42, the PKG [C42S]KI mouse), although the capacity to be activated by cGMP is maintained. Similar to mesenteric arteries from the mice on a high-fat diet in this study, PKG[C42S]KI arteries are unable to increase Ca2+ spark frequency when intraluminal pressure is raised.18 Subsequently, there is increased pressure-induced constriction of the PKG[C42S]KI arteries due to lack of BK channel activity. To clarify the involvement of oxidant-activated PKG in the development of obesity-related small artery dysfunction, we placed PKG[C42S]KI and matched wild type (WT) mice on a 6-month high-fat diet and assessed mesenteric resistance artery contractility. Notably, there was no difference in the degree of pressure-induced constriction between the WT and the PKG[C42S]KI arteries (Figure 5A through 5C). Furthermore, and also consistent with an effect of the high-fat diet primarily on BK channel vasodilation, the constriction to the BK channel blocker Paxilline was also equivalent between the WT and the PKG[C42S]KI arteries (Figure 5D). We also determined whether high-fat diet altered PKG oxidative dimerization using a western blot approach. Arteries from only WT mice on both low- and high-fat diets were studied and Figure 5E indicates equivalence in mesenteric artery PKG dimerization following incubation with exogenous H2O2 between the high fat–fed and low fat–fed diet mice.
Figure 5.
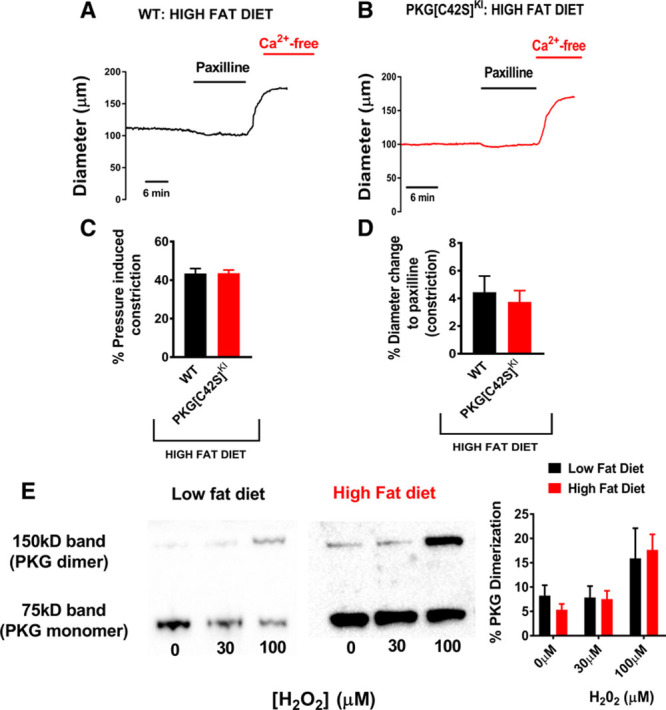
Contribution of oxidant-activated PKG to large conductance Ca2+-activated potassium channel (BK) channel dysfunction in high-fat feeding. A, Representative traces from third-order mesenteric arteries from high fat–fed WT and high fat–fed PKG[C42S]KI mice showing basal pressure–induced constriction and incubation with Paxilline. B, There is equivalence in the degree of pressure-induced constriction of the mesenteric arteries between the WT and PKG[C42S]KI mice (WT: n=8 arteries from 4 mice. PKG[C42S]KI: n=5 arteries from 4 mice). C, There is equivalence in the responses to Paxilline between WT (n=5 arteries from 3 mice) and PKG[C42S]KI (n=4 arteries from 3 mice) mouse third order mesenteric arteries. D, Representative Western blot protocols showing the monomer (lower band) and dimer (upper band) formation of PKG following exposure to H2O2 from mesenteric arteries from low fat–fed and high fat–fed arteries from WT mice. There was no difference in the percentage of total PKG dimerization between the low fat–fed (n=5 from 5 mice) and the high fat–fed (n=13 from 13 mice for baseline and 100 µM H2O2, n=7 from 7 mice for 30 µM H2O2) groups. PKG indicates Protein Kinase G; and WT, wild type.
High Fat Feeding and Structural Changes to the Resistance Artery Wall
Assessment of the wall thickness and lumen diameter in the absence of pressure-induced constriction (in Ca2+ free PSS) at varying intraluminal pressure allowed quantification of standard structural assessment of the mesenteric arteries. Although remodeling of the wall has been previously documented in resistance arteries in obesity,19,20 we did not see any change in the wall-to-lumen ratio, wall thickness, the cross sectional area (Figure S7) or the stress to strain ratio (Figure S8) between arteries from low fat–fed mice and high fat–fed mice.
Discussion
The principal finding from this study was that in this mouse model of obesity-driven hypertension, increased pressure-induced constriction of mesenteric resistance arteries was associated with failure of VSMC Ca2+ spark–induced BK channel vasodilation. In contrast, both the endothelial vasodilatory function and the structure and distensibility of the resistance artery wall were unaffected. In health, VSMC Ca2+ spark frequency is dramatically increased by intraluminal pressure within resistance arteries and specifically, it was this pressure dependence of Ca2+ sparks which was disabled by the high-fat diet. The uncoupling of this intraluminal pressure–driven mechanotransduction pathway by obesity appeared to be at the level of the sarcoplasmic reticulum, downstream of pressure-induced oxidative activation of PKG. Our observations reinforce the critical role of the Ca2+ spark-BK channel vasodilatory pathway in the development of obesity-related hypertension and suggest novel targets for therapeutic intervention.
Increased contractility of small resistance arteries in the Metabolic Syndromes represents a composite phenotype of diverse ion channel dysfunction. A reduction in the activity of vasodilatory K+ channels such as the voltage-gated K+ channel Kv2.121 or the BK channel22 is accompanied by overactivity of procontractile Ca2+ channels, specifically the L-type Ca2+ channel.23 Of these divergent targets, the BK channel is regarded as particularly promising due to its important contribution toward the resting tone of arteries and broad pharmacological profile. Furthermore, BK channel dysfunction has been observed in almost all metabolic syndromes: diabetes mellitus,16 obesity,12,24 dyslipidemias,25 and hypertension.26,27 From a mechanistic perspective, the most convincing evidence to date suggests that BK dysfunction is due to reduction in Ca2+ sensitivity from failure of the β subunit integration into the channel.12,16 In this article, for the first time, we show that BK channel dysfunction in resistance arteries is also associated with failure of intraluminal pressure–induced Ca2+ spark vasoregulation.
Ca2+ sparks have been studied in humans16,28 and in animal models of hypertension,26,27 obesity12 and diabetes mellitus,29–31 and overall, spark frequency within VSMC is unchanged12,26,27,29,31 or even increased30 compared with control groups. However, these cardiometabolic studies have been almost exclusively undertaken by imaging single VSMCs, isolated from native resistance arteries. To date, there has only been one study which studied Ca2+ sparks in intact pressurized resistance arteries, comparing high fat–fed diabetic dyslipidemic swine against controls.25 In contrast to the single cell studies, there was a trend toward a reduction in Ca2+ spark frequency in the arteries from overweight diabetic swine compared with control swine.25 Our study confirms that in murine diet–induced obesity with hypertension, the capacity to increase VSMC Ca2+ spark frequency in response to intraluminal pressure is lost. However, to uncover this deficit, it was necessary to study Ca2+ spark characteristics in intact arteries at both low and high intraluminal pressures. This observation may account for apparently contradictory observations whereby Ca2+ sparks are normal in isolated VSMC from obese diabetic models12,29–31 but reduced in pressurized arteries.25 Thus, when Ca2+ spark frequency is studied in isolated VSMC, lack of mechanical stretch or tension across the individual cell would not reveal damage to the coupling, which we have described and therefore there is equivalence between the metabolic and control groups.12,29–31 However, if our intact artery preparation were to be replicated in the diabetic studies hitherto described, we would predict that the pressure dependence of Ca2+ spark vasodilation would be compromised in tandem with BK channel dysfunction.12,16
We previously identified that the mechanism by which intraluminal pressure triggers Ca2+ spark vasodilation hinges on oxidant activation of Protein Kinase G (PKG).18 Oxidant generation within myocytes in response to mechanical stretch occurs in both the heart32 and resistance arteries across multiple territories.18,33–35 In resistance arteries, oxidants induce a conformational change to PKG through disulphide bond formation at appositioned cysteine residues.36,37 Thus activated, PKG regulates Ca2+ spark release from the sarcoplasmic reticulum.18 We were able to delineate this pathway through the use of a genetically modified mouse: the PKG[C42S]KI mouse. In this mouse model, a cysteine at position 42 is switched to a serine residue and as a result PKG cannot undergo oxidative activation.37 The PKG[C42S]KI mouse is hypertensive and in addition there is enhanced pressure induced constriction of the mesenteric arteries which is attributable to a failure of pressure-induced Ca2+ spark vasodilation.18 The mesenteric small artery phenotype of the PKG[C42S]KI mouse is therefore strikingly similar to that of the high fat–fed mouse in this study; an increase in pressure-induced constriction due to BK channel dysfunction. Therefore, to clarify whether oxidant-activated PKG plays a role in the development of small artery dysfunction in high-fat feeding, we placed PKG[C42S]KI mice and matched WT on a high-fat diet and assessed pressure-induced constriction in mesenteric resistance arteries. Consistent with our hypothesis that high-fat feeding interrupts the intraluminal pressure-Ca2+ spark vasodilatory axis, pressure-induced constriction was increased in WT arteries but there was no increase in the myogenic constriction of the PKG[C42S]KI arteries in response to the high-fat diet, judged against our previous work.18 Furthermore, both the high fat–fed WT arteries and high fat–fed PKG[C42S]KI arteries constricted equivalently to Paxilline. Our findings align closely to those by Rudyk et al38 who also studied high-fat feeding of PKG[C42S]KI mice and WT counterparts. Thus, while the obesogenic diet increased blood pressure in WT mice to the level seen in the PKG[C42S]KI mice at baseline, there was no rise in the blood pressure of the PKG[C42S]KI mice in response to the diet.38 The lack of effect of the high-fat diet on either pressure-induced constriction (Figure 5) or blood pressure in the PKG[C42S]KI mice38 further emphasizes the central role of BK channel dysfunction in the development of obesity-induced hypertension.
We also established that PKG remained amenable to oxidative dimerization, albeit by incubation with H2O2 rather than by intraluminal pressure as we have previously shown.18 We propose, therefore, that diet-induced obesity uncouples the pathway by which intraluminal pressure triggers Ca2+ sparks through interference at a point downstream of the oxidation of PKG (Figure 6). Whether this point of uncoupling is the engagement of oxidized PKG with the sarcoplasmic reticulum, intraluminal pressure–induced Ca2+ loading of the sarcoplasmic reticulum or release of Ca2+ sparks via the RyR (ryanodine receptor) remains to be elucidated and will form part of subsequent studies. From a translational perspective, however, specific compounds have been developed which can vasodilate arteries via direct oxidative activation of PKG,39 and it will be instructive to establish whether these may improve Ca2+ spark-BK vasoregulation in obesity-related hypertension. Conversely, however, activation of VSMC PKG by adipose-derived oxidants has also been implicated in microvascular dysfunction,40 suggesting that tight regulation of oxidant activation of PKG is necessary for vascular homeostasis.
Figure 6.
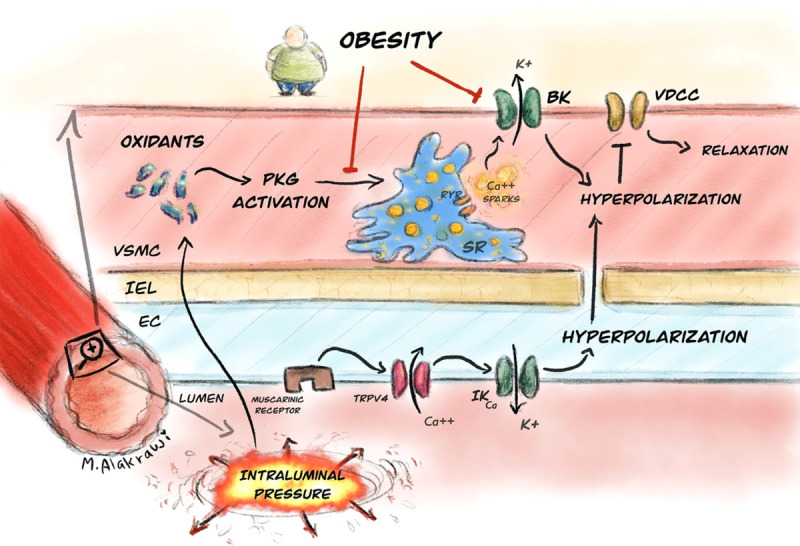
Cartoon illustrating proposed pathway by which obesity influences vascular tone of resistance arteries. Intraluminal pressure generates oxidants which activate PKG. Thus, activated PKG targets the sarcoplasmic reticulum to increase Ca2+ spark frequency and these activate BK channels to trigger vasodilation through hyperpolarization. While PKG can still undergo oxidative activation in obesity, the capacity of the SR to generate Ca2+ sparks is abrogated. In tandem, work from Navedo et al has shown that obesity also has a direct effect on the BK channel directly, limiting the Ca2+ sensitivity. In our model, obesity had no effect on the endothelial muscarinic/TRPV4/IKCa vasodilatory pathway. BK indicates large conductance Ca2+-activated potassium channel. EC, endothelial cell; IEL, internal elastic lamina; IKCa, intermediate conductance calcium-activated potassium channel; PKG, Protein Kinase G; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; TRPV4, vanilloid transient receptor potential 4 channel; VDCC, voltage-dependent Ca2+ channel; and VSMC, vascular smooth muscle cell.
In our study, the Ca2+ spark-BK dysfunction in the mesenteric arteries from the high fat–fed mice was not accompanied by discernible damage to the arterial wall structure or the endothelial vasodilatory pathways. In this regard, our findings contrast with much of the published work around small artery structure and function in the Metabolic Syndromes. Indeed, both human and animal studies have demonstrated endothelial dysfunction in obesity, as characterized by a reduction in the dilation to cumulative concentrations of muscarinic agonists.10,11,19,20,41,42 The majority of these published studies use agonists to preconstrict the artery before assessment of the endothelial dependent dilation. In our study, we used intraluminal pressure alone to trigger constriction and this approach results in a smaller degree of constriction compared with that elicited pharmacologically. It may be that the different cellular pathways involved in these 2 approaches (pressure versus agonist induced constriction) are responsible for the divergence in vasodilation seen. Consistent with this, equivalent vasodilatory responses to acetylcholine between high fat–fed and low fat–fed groups have been described in rat skeletal muscle arteries constricted with intraluminal pressure.8 Nevertheless, in alternate models of obesity43 or hypertension,14 the relaxation of mesenteric arteries to muscarinic agonists is reduced even when the constriction is to pressure alone. Endothelial dysfunction of arteries in obesity is also commonly associated with abnormal wall structure, predominantly wall hypertrophy19,20 and it is established that there is a strong link between these 2 pathologies.44 The preservation of endothelial function and normal wall structure in our obese hypertensive model suggests that fundamental pathogenic processes are absent and future comparative work could yield important insights in this area of vascular endothelium to wall crosstalk.
Although the preservation to endothelial function and structure in the arteries from the high fat–fed mice from this study diverges from existing work,19,20 the increase in pressure-induced constriction due to BK channel dysfunction replicates work from other groups.12,16 While previously, BK dysfunction has been attributed to a reduction in Ca2+ sensitivity of the channel, we now add an additional appreciation for the loss of intraluminal pressure-induced Ca2+ spark vasoregulation (Figure 6). Our work is in mouse mesenteric arteries and the observations need to be replicated in human arteries and also divergent circulations. But if our findings are consistent, pressure dependence of VSMC Ca2+ sparks becomes a potentially modifiable factor to restore BK channel function in obesity and diabetes mellitus and thus lower blood pressure. In this setting, the involvement of oxidant activation of PKG as a key regulator of Ca2+ spark vasoregulation provides layers of tissue specific targets and excitingly, novel compounds are emerging which show promise.
Perspectives
Microvascular dysfunction in the setting of obesity remains a clinical area stubbornly devoid of targeted therapy. This therapeutic vacuum is starkly at odds with the rising prevalence of obesity-related cardiovascular disease. Obesity-related hypertension exemplifies this point well. Thus, there is a linear relationship between weight and blood pressure2,3 but compared with essential lean hypertension, obesity-related hypertension is harder to treat effectively and results in more microvascular target organ damage.45 The impact of this cardiometabolic combination is devastating as a quarter of adults are diagnosed with hypertension and a third of adults are overweight or obese.46 Hypertension is also a leading cause of stroke, heart disease, dementia, and chronic kidney disease, and all of these conditions have microvascular etiologies. However, the only currently available small artery targeted therapies are the calcium channel blockers such as Amlodipine, which have diverse side effects limiting compliance and effectiveness. More focussed therapies are therefore needed.
In this regard, the increased contractile tone of resistance arteries has long been coveted as a potential druggable target in the search for more specific cardiovascular therapies for patients with the metabolic syndromes. One avenue of great interest has been the observed reduction in activity of a vascular smooth muscle cell vasodilatory ion channel, the large conductance Ca2+ activated K+ channel (BK). However, this channel is widespread throughout the body and as such direct pharmacological manipulation would have wide ranging side effects. Under steady-state conditions, BK channel function is maintained by brief tightly controlled Ca2+ release events from the sarcoplasmic reticulum within vascular smooth muscle. We show here that diet-induced obesity uncouples the intricate multilayered intracellular pathway by which intraluminal pressure within resistance arteries regulates Ca2+ spark vasoregulation. Corollary evidence that this uncoupling contributes to the development of obesity-related hypertension is presented and we propose that this pathway now represents a novel tissue-specific target for future work aimed at restoring BK channel function in obesity.
Acknowledgments
We gratefully acknowledge the Biological Services Facility at the University of Manchester and Mariam Alakrawi, who drew the graphical abstract.
Sources of Funding
This article was funded by British Heart Foundation, UK: PG/18/7/33535, PG/15/109/31931, FS/14/26/30767, FS/12/81/29882, CH/2000004/12801.
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/HYPERTENSIONAHA.119.13540.
Novelty and Significance
What Is new?
This study demonstrates that obesity compromises one of the principal vasodilatory pathways in small resistance arteries: the ability of intraluminal pressure to trigger Ca2+ spark vasodilation. Intraluminal pressure activation of Ca2+ sparks is dependent on oxidant activation of Protein Kinase G, and we show specifically that obesity interrupts the vasodilatory pathway at a point distal to the engagement of Protein Kinase G with the sarcoplasmic reticulum.
What Is Relevant?
There are no molecularly targeted therapies for obesity-related hypertension despite it being the principal cause of elevated blood pressure. This study identifies a potential new avenue for future investigation and treatment of obesity-related hypertension: the pathway by which intraluminal pressure within small resistance arteries activates a vasodilatory coupling between vascular smooth muscle cell Ca2+ signals (Ca2+ sparks) and plasma membrane–bound K+ channels (BK channels). The pathway: intraluminal pressure-generation of oxidants—activation of Protein Kinase G—release of Ca2+ sparks from the sarcoplasmic reticulum—activation of BK channels, is complex but by this token also potentially druggable. In particular, novel orally bioavailable activators of PKG recently have been developed in tandem with strategies to modulate arterial redox states and it may be that in the future these may be useful adjuncts to the traditional less-specific antihypertensive drugs.
Summary
Obesity increases small artery contractility by disabling the pressure dependence of vascular smooth muscle cell Ca2+ sparks, thus raising blood pressure.
References
- 1.Dam R, Robinson HA, Vince-Cain S, Heaton G, Greenstein A, Sperrin M, Hassan L. Engaging parents using web-based feedback on child growth to reduce childhood obesity: a mixed methods study. BMC Public Health. 2019;19:300. doi: 10.1186/s12889-019-6618-3. doi: 10.1186/s12889-019-6618-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Narkiewicz K. Obesity and hypertension–the issue is more complex than we thought. Nephrol Dial Transplant. 2006;21:264–267. doi: 10.1093/ndt/gfi290. doi: 10.1093/ndt/gfi290. [DOI] [PubMed] [Google Scholar]
- 3.Crump C, Sundquist J, Winkleby MA, Sundquist K. Interactive effects of physical fitness and body mass index on the risk of hypertension. JAMA Intern Med. 2016;176:210–216. doi: 10.1001/jamainternmed.2015.7444. doi: 10.1001/jamainternmed.2015.7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juonala M, Magnussen CG, Berenson GS, Venn A, Burns TL, Sabin MA, Srinivasan SR, Daniels SR, Davis PH, Chen W, et al. Childhood adiposity, adult adiposity, and cardiovascular risk factors. N Engl J Med. 2011;365:1876–1885. doi: 10.1056/NEJMoa1010112. doi: 10.1056/NEJMoa1010112. [DOI] [PubMed] [Google Scholar]
- 5.Pérez GJ, Bonev AD, Nelson MT. Micromolar Ca(2+) from sparks activates Ca(2+)-sensitive K(+) channels in rat cerebral artery smooth muscle. Am J Physiol Cell Physiol. 2001;281:C1769–C1775. doi: 10.1152/ajpcell.2001.281.6.C1769. doi: 10.1152/ajpcell.2001.281.6.C1769. [DOI] [PubMed] [Google Scholar]
- 6.Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627–9632. doi: 10.1073/pnas.0801963105. doi: 10.1073/pnas.0801963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi: 10.1126/science.1216283. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howitt L, Sandow SL, Grayson TH, Ellis ZE, Morris MJ, Murphy TV. Differential effects of diet-induced obesity on BKCa {beta}1-subunit expression and function in rat skeletal muscle arterioles and small cerebral arteries. Am J Physiol Heart Circ Physiol. 2011;301:H29–H40. doi: 10.1152/ajpheart.00134.2011. doi: 10.1152/ajpheart.00134.2011. [DOI] [PubMed] [Google Scholar]
- 9.Frisbee JC. Enhanced arteriolar alpha-adrenergic constriction impairs dilator responses and skeletal muscle perfusion in obese Zucker rats. J Appl Physiol (1985) 2004;97:764–772. doi: 10.1152/japplphysiol.01216.2003. doi: 10.1152/japplphysiol.01216.2003. [DOI] [PubMed] [Google Scholar]
- 10.Subramanian R, MacLeod KM. Age-dependent changes in blood pressure and arterial reactivity in obese Zucker rats. Eur J Pharmacol. 2003;477:143–152. doi: 10.1016/j.ejphar.2003.08.003. doi: 10.1016/j.ejphar.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Rizzoni D, De Ciuceis C, Porteri E, Semeraro F, Rosei EA. Structural alterations in small resistance arteries in obesity. Basic Clin Pharmacol Toxicol. 2012;110:56–62. doi: 10.1111/j.1742-7843.2011.00786.x. doi: 10.1111/j.1742-7843.2011.00786.x. [DOI] [PubMed] [Google Scholar]
- 12.Nystoriak MA, Nieves-Cintrón M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell’acqua ML, et al. AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res. 2014;114:607–615. doi: 10.1161/CIRCRESAHA.114.302168. doi: 10.1161/CIRCRESAHA.114.302168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chadha PS, Haddock RE, Howitt L, Morris MJ, Murphy TV, Grayson TH, Sandow SL. Obesity up-regulates intermediate conductance calcium-activated potassium channels and myoendothelial gap junctions to maintain endothelial vasodilator function. J Pharmacol Exp Ther. 2010;335:284–293. doi: 10.1124/jpet.110.167593. doi: 10.1124/jpet.110.167593. [DOI] [PubMed] [Google Scholar]
- 14.Sonkusare SK, Dalsgaard T, Bonev AD, Hill-Eubanks DC, Kotlikoff MI, Scott JD, Santana LF, Nelson MT. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci Signal. 2014;7:ra66. doi: 10.1126/scisignal.2005052. doi: 10.1126/scisignal.2005052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wellman GC, Nelson MT. Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium. 2003;34:211–229. doi: 10.1016/s0143-4160(03)00124-6. doi: 10.1016/s0143-4160(03)00124-6. [DOI] [PubMed] [Google Scholar]
- 16.Nieves-Cintrón M, Syed AU, Buonarati OR, Rigor RR, Nystoriak MA, Ghosh D, Sasse KC, Ward SM, Santana LF, Hell JW, et al. Impaired BKCa channel function in native vascular smooth muscle from humans with type 2 diabetes. Sci Rep. 2017;7:14058. doi: 10.1038/s41598-017-14565-9. doi: 10.1038/s41598-017-14565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaggar JH. Intravascular pressure regulates local and global Ca(2+) signaling in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. 2001;281:C439–C448. doi: 10.1152/ajpcell.2001.281.2.C439. doi: 10.1152/ajpcell.2001.281.2.C439. [DOI] [PubMed] [Google Scholar]
- 18.Khavandi K, Baylie RA, Sugden SA, Ahmed M, Csato V, Eaton P, Hill-Eubanks DC, Bonev AD, Nelson MT, Greenstein AS. Pressure-induced oxidative activation of PKG enables vasoregulation by Ca2+ sparks and BK channels. Sci Signal. 2016;9:ra100. doi: 10.1126/scisignal.aaf6625. doi: 10.1126/scisignal.aaf6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grassi G, Seravalle G, Brambilla G, Facchetti R, Bolla G, Mozzi E, Mancia G. Impact of the metabolic syndrome on subcutaneous microcirculation in obese patients. J Hypertens. 2010;28:1708–1714. doi: 10.1097/HJH.0b013e32833af3c9. doi: 10.1097/HJH.0b013e32833af3c9. [DOI] [PubMed] [Google Scholar]
- 20.Grassi G, Seravalle G, Scopelliti F, Dell’Oro R, Fattori L, Quarti-Trevano F, Brambilla G, Schiffrin EL, Mancia G. Structural and functional alterations of subcutaneous small resistance arteries in severe human obesity. Obesity (Silver Spring) 2010;18:92–98. doi: 10.1038/oby.2009.195. doi: 10.1038/oby.2009.195. [DOI] [PubMed] [Google Scholar]
- 21.Nieves-Cintrón M, Nystoriak MA, Prada MP, Johnson K, Fayer W, Dell’Acqua ML, Scott JD, Navedo MF. Selective down-regulation of KV2.1 function contributes to enhanced arterial tone during diabetes. J Biol Chem. 2015;290:7918–7929. doi: 10.1074/jbc.M114.622811. doi: 10.1074/jbc.M114.622811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rusch NJ. BK channels in cardiovascular disease: a complex story of channel dysregulation. Am J Physiol Heart Circ Physiol. 2009;297:H1580–H1582. doi: 10.1152/ajpheart.00852.2009. doi: 10.1152/ajpheart.00852.2009. [DOI] [PubMed] [Google Scholar]
- 23.Nystoriak MA, Nieves-Cintron M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, Grandi E, Fernandes JD, Forbush K, Hofmann F, et al. Ser1928 phosphorylation by pka stimulates the l-type ca2+ channel cav1.2 and vasoconstriction during acute hyperglycemia and diabetes. Science signaling. 2017;10:eaaf9647. doi: 10.1126/scisignal.aaf9647. doi: 10.1126/scisignal.aaf9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borbouse L, Dick GM, Asano S, Bender SB, Dincer UD, Payne GA, Neeb ZP, Bratz IN, Sturek M, Tune JD. Impaired function of coronary BK(Ca) channels in metabolic syndrome. Am J Physiol Heart Circ Physiol. 2009;297:H1629–H1637. doi: 10.1152/ajpheart.00466.2009. doi: 10.1152/ajpheart.00466.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mokelke EA, Dietz NJ, Eckman DM, Nelson MT, Sturek M. Diabetic dyslipidemia and exercise affect coronary tone and differential regulation of conduit and microvessel K+ current. Am J Physiol Heart Circ Physiol. 2005;288:H1233–H1241. doi: 10.1152/ajpheart.00732.2004. doi: 10.1152/ajpheart.00732.2004. [DOI] [PubMed] [Google Scholar]
- 26.Amberg GC, Bonev AD, Rossow CF, Nelson MT, Santana LF. Modulation of the molecular composition of large conductance, Ca(2+) activated K(+) channels in vascular smooth muscle during hypertension. J Clin Invest. 2003;112:717–724. doi: 10.1172/JCI18684. doi: 10.1172/JCI18684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amberg GC, Santana LF. Downregulation of the BK channel beta1 subunit in genetic hypertension. Circ Res. 2003;93:965–971. doi: 10.1161/01.RES.0000100068.43006.36. doi: 10.1161/01.RES.0000100068.43006.36. [DOI] [PubMed] [Google Scholar]
- 28.Wellman GC, Nathan DJ, Saundry CM, Perez G, Bonev AD, Penar PL, Tranmer BI, Nelson MT. Ca2+ sparks and their function in human cerebral arteries. Stroke. 2002;33:802–808. doi: 10.1161/hs0302.104089. doi: 10.1161/hs0302.104089. [DOI] [PubMed] [Google Scholar]
- 29.McGahon MK, Dash DP, Arora A, Wall N, Dawicki J, Simpson DA, Scholfield CN, McGeown JG, Curtis TM. Diabetes downregulates large-conductance Ca2+-activated potassium beta 1 channel subunit in retinal arteriolar smooth muscle. Circ Res. 2007;100:703–711. doi: 10.1161/01.RES.0000260182.36481.c9. doi: 10.1161/01.RES.0000260182.36481.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong L, Zheng YM, Van Riper D, Rathore R, Liu QH, Singer HA, Wang YX. Functional and molecular evidence for impairment of calcium-activated potassium channels in type-1 diabetic cerebral artery smooth muscle cells. J Cereb Blood Flow Metab. 2008;28:377–386. doi: 10.1038/sj.jcbfm.9600536. doi: 10.1038/sj.jcbfm.9600536. [DOI] [PubMed] [Google Scholar]
- 31.Rueda A, Fernández-Velasco M, Benitah JP, Gómez AM. Abnormal Ca2+ spark/STOC coupling in cerebral artery smooth muscle cells of obese type 2 diabetic mice. PLoS One. 2013;8:e53321. doi: 10.1371/journal.pone.0053321. doi: 10.1371/journal.pone.0053321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 33.Nowicki PT, Flavahan S, Hassanain H, Mitra S, Holland S, Goldschmidt-Clermont PJ, Flavahan NA. Redox signaling of the arteriolar myogenic response. Circ Res. 2001;89:114–116. doi: 10.1161/hh1401.094367. doi: 10.1161/hh1401.094367. [DOI] [PubMed] [Google Scholar]
- 34.Ren Y, D’Ambrosio MA, Liu R, Pagano PJ, Garvin JL, Carretero OA. Enhanced myogenic response in the afferent arteriole of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;298:H1769–H1775. doi: 10.1152/ajpheart.00537.2009. doi: 10.1152/ajpheart.00537.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, Harrison DG, Bhatnagar A American Heart Association Council on Basic Cardiovascular Sciences. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the American Heart Association. Circ Res. 2016;119:e39–e75. doi: 10.1161/RES.0000000000000110. doi: 10.1161/RES.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schröder E, Browning DD, Eaton P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 37.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med. 2012;18:286–290. doi: 10.1038/nm.2603. doi: 10.1038/nm.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudyk O, Eaton P. Examining a role for PKG Iα oxidation in the pathogenesis of cardiovascular dysfunction during diet-induced obesity. Free Radic Biol Med. 2017;110:390–398. doi: 10.1016/j.freeradbiomed.2017.07.007. doi: 10.1016/j.freeradbiomed.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burgoyne JR, Prysyazhna O, Richards DA, Eaton P. Proof of principle for a novel class of antihypertensives that target the oxidative activation of PKG Iα (Protein Kinase G Iα). Hypertension. 2017;70:577–586. doi: 10.1161/HYPERTENSIONAHA.117.09670. doi: 10.1161/HYPERTENSIONAHA.117.09670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen Dinh Cat A, Antunes TT, Callera GE, Sanchez A, Tsiropoulou S, Dulak-Lis MG, Anagnostopoulou A, He Y, Montezano AC, Jaisser F, et al. Adipocyte-specific mineralocorticoid receptor overexpression in mice is associated with metabolic syndrome and vascular dysfunction: role of redox-sensitive PKG-1 and Rho kinase. Diabetes. 2016;65:2392–2403. doi: 10.2337/db15-1627. doi: 10.2337/db15-1627. [DOI] [PubMed] [Google Scholar]
- 41.Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW, Malik RA, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119:1661–1670. doi: 10.1161/CIRCULATIONAHA.108.821181. doi: 10.1161/CIRCULATIONAHA.108.821181. [DOI] [PubMed] [Google Scholar]
- 42.Schofield I, Malik R, Izzard A, Austin C, Heagerty A. Vascular structural and functional changes in type 2 diabetes mellitus: evidence for the roles of abnormal myogenic responsiveness and dyslipidemia. Circulation. 2002;106:3037–3043. doi: 10.1161/01.cir.0000041432.80615.a5. doi: 10.1161/01.cir.0000041432.80615.a5. [DOI] [PubMed] [Google Scholar]
- 43.Haddock RE, Grayson TH, Morris MJ, Howitt L, Chadha PS, Sandow SL. Diet-induced obesity impairs endothelium-derived hyperpolarization via altered potassium channel signaling mechanisms. PLoS One. 2011;6:e16423. doi: 10.1371/journal.pone.0016423. doi: 10.1371/journal.pone.0016423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dzau VJ, Gibbons GH. Endothelium and growth factors in vascular remodeling of hypertension. Hypertension. 1991;18(5)(suppl):III115–III121. doi: 10.1161/01.hyp.18.5_suppl.iii115. doi: 10.1161/01.hyp.18.5_suppl.iii115. [DOI] [PubMed] [Google Scholar]
- 45.Schillaci G, Pirro M, Vaudo G, Gemelli F, Marchesi S, Porcellati C, Mannarino E. Prognostic value of the metabolic syndrome in essential hypertension. J Am Coll Cardiol. 2004;43:1817–1822. doi: 10.1016/j.jacc.2003.12.049. doi: 10.1016/j.jacc.2003.12.049. [DOI] [PubMed] [Google Scholar]
- 46.Narkiewicz K. Diagnosis and management of hypertension in obesity. Obes Rev. 2006;7:155–162. doi: 10.1111/j.1467-789X.2006.00226.x. doi: 10.1111/j.1467-789X.2006.00226.x. [DOI] [PubMed] [Google Scholar]