

Abstract
We aimed to characterize the genetics of endothelial function and how this influences risk for cardiovascular diseases such as ischemic stroke. We integrated genetic data from a study of ultrasound flow-mediated dilatation of brachial artery in adolescents from ALSPAC (Avon Longitudinal Study of Parents and Children; n=5214) with a study of ischemic stroke (MEGASTROKE: n=60 341 cases and 452 969 controls) to identify variants that confer risk of ischemic stroke through altered endothelial function. We identified a variant in PDE3A (Phosphodiesterase 3A), encoding phosphodiesterase 3A, which was associated with flow-mediated dilatation in adolescents (9–12 years of age; β[SE], 0.38 [0.070]; P=3.8×10−8) and confers risk of ischemic stroke (odds ratio, 1.04 [95% CI, 1.02–1.06]; P=5.2×10−6). Bayesian colocalization analyses showed the same underlying variation is likely to lead to both associations (posterior probability, 97%). The same variant was associated with flow-mediated dilatation in a second study in young adults (age, 24–27 years; β[SE], 0.47 [0.23]; P=0.047) but not in older adults (β[SE], −0.012 [0.13]; P=0.89). We conclude that a genetic variant in PDE3A influences endothelial function in early life and leads to increased risk of ischemic stroke. Subtle, measurable changes to the vasculature that are influenced by genetics also influence risk of ischemic stroke.
Keywords: cyclic nucleotide phosphodiesterases, type 3; genetics; genome-wide association study; stroke; vascular endothelium
Genome-wide association studies (GWAS) have identified numerous variants underlying cardiovascular diseases including stroke and coronary heart disease.1,2 However, in the main, the mechanism of these variants on disease risk has been elusive. A complementary approach to the standard GWAS design of clinical end points such as stroke is to integrate data with GWAS on intermediate phenotypes of disease risk such as carotid intima-media thickness or plaque.3 Identifying variation that is common to an intermediate phenotype and disease outcome has the potential to identify mechanism-specific associations with disease and to illuminate causal pathways.
Endothelial function is one such phenotype of importance to stroke.4 The endothelium is a group of cells lining blood vessels that functions both as a barrier, controlling the passage of materials into and out from the blood stream, and as a signal transducer, regulating vessel structure and function.5 Impaired endothelial function is involved in the initiation of atherosclerosis, as well as latter plaque instability,6 and promotion of small vessel arteriopathy.7,8 Several techniques exist for measuring endothelial function in vivo.5 One validated method, ultrasound flow-mediated dilatation (FMD), measures the response of the arterial endothelium to reactive hyperemia via inflation and subsequent deflation of a blood flow constricting cuff (sphygmomanometer).5
Here, we perform a GWAS of FMD in adolescents from ALSPAC (Avon Longitudinal Study of Parents and Children). We integrate the results with a large-scale GWAS of stroke in over 60 341 cases and 452 969 controls (MEGASTROKE)2 and use genome-wide colocalization approaches to identify genetic variation that contributes to stroke through impaired endothelial function in adolescents.
Methods
Data used in this study are available to all researchers through application to ALSPAC (http://www.bristol.ac.uk/alspac/researchers/) and UK Biobank (https://www.ukbiobank.ac.uk/register-apply/). Publicly available GWAS data pertaining to cardiovascular risk factors and traits used in this study are available from http://megastroke.org (stroke), https://www.cardiomics.net/download-data (coronary heart disease), http://www.diagram-consortium.org/downloads.html (type 2 diabetes mellitus), http://www.broadcvdi.org/ (atrial fibrillation), http://lipidgenetics.org/ (lipids), and https://conservancy.umn.edu/handle/11299/201564 (smoking status).
Study Subjects
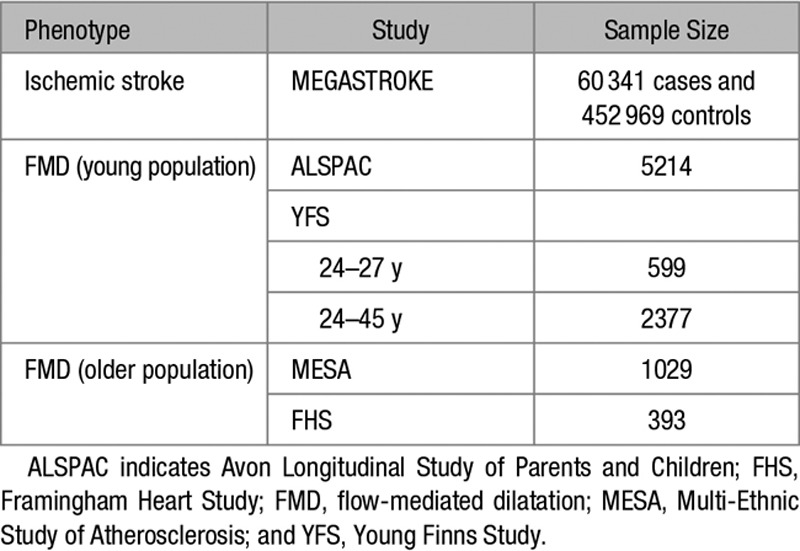
Study Populations are outlined in Table 1. Pregnant women resident in Avon, UK with expected dates of delivery April 1, 1991, to December 31, 1992, were invited to take part in ALSPAC—a prospective observational study of the genetic and environmental determinants of development and health from the prenatal period into adulthood.9–11 The initial number of pregnancies enrolled was 14 541, which was later increased to 15 247 by retrospective recruitment. A total of 15 656 fetuses were included in further studies, of which 14 889 were alive at 1 year of age. Please note that the ALSPAC study website contains details of all the data that are available through a fully searchable data dictionary and variable search tool (http://www.bristol.ac.uk/alspac/researchers/our-data/). We studied endothelial function in 7557 of these children aged between 9 and 12 years (mean [SD], 10.7 [0.25] years). Age, sex, body mass index, blood pressure, and environmental factors were collected as described previously.9 Room and skin temperatures were assessed using a commercial digital thermometer immediately before the vascular examination. Brachial artery endothelial function was successfully measured in 88% of children by FMD.11 The right brachial artery was imaged using high-resolution ultrasound (ALOKA 5500) 5 to 10 cm above the antecubital fossa with the probe held in a stereotactic clamp. Edge detection software (Brachial Tools, MIA, IA) was used to measure the brachial artery diameter at 3-second intervals throughout the 11-minute recording protocol. Brachial artery FMD was induced by a 5-minute inflation of a pneumatic cuff to 200 mm Hg, around the forearm immediately below the medial epicondyle followed by rapid deflation using an automatic air regulator (Logan Research, United Kingdom). We used FMD expressed as a percentage calculated using peak diameter in response to reactive hyperemia in relation to the baseline diameter.12
Table 1.
Study Populations
Genotyping and Quality Control in ALSPAC
We used imputed genotypic data of the ALSPAC. Genotyping, quality control, and imputation has been described previously.13 GWAS data were generated by Sample Logistics and Genotyping Facilities at Wellcome Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe. Imputation was performed using the complete reference panel from the third phase of the 1000 Genomes Project.14 Five thousand two hundred ninety-seven children had both genotyping information and FMD measurements. We excluded 83 cryptically related samples with PI-HAT >0.1875, determined using PLINK2,15 meaning the final analyses were performed on 5214 samples. We analyzed all single-nucleotide polymorphisms (SNPs) that were polymorphic (minor allele frequency, >1%) in European samples and had an imputation info value >0.5. PLINK2 was used to estimate identity-by-descent and to perform ancestry-informative principal components (PC) analysis.15
MEGASTROKE Genome-Wide Summary Statistics
We used summary statistics, downloaded from http://megastroke.org, derived from MEGASTROKE—a transethnic genome-wide meta-analysis of stroke.2 Our primary analysis was for all ischemic stroke cases versus controls (60 341 cases and 452 969 controls). We also explored associations with ischemic stroke subtypes in MEGASTROKE: cardioembolic stroke (9006 cases and 426 629 controls), large artery stroke (6688 cases and 345 446 controls), and small vessel stroke (11 710 cases and 346 101 controls).
Genome-Wide Analyses in Adolescents From ALSPAC
Per-allele β-coefficients and SEs for FMD were generated in a regression model that included age, sex, room temperature, body temperature, and 8 ancestry-informative PCs. Covariates were selected based on their significant association with FMD in previous ALSPAC publications.11 For sensitivity analysis, we performed analysis of a second model that was additionally adjusted for heritable covariates: body mass index, systolic blood pressure, and diastolic blood pressure. All statistical tests conducted were 2 sided, and SNPTEST v2.5.4-beta3 was used to perform the analysis. We used a P threshold of 5.0×10−8 to determine statistical significance. We discarded significant low-frequency SNPs (minor allele frequency, <0.05) without substantial linkage disequilibrium support (1 region).
Validation Datasets
We explored validation of the FMD-associated novel SNP in independent cohorts of FMD measured in both younger and older cohorts of subjects.
We sought replication in younger subjects in YFS (Young Finns Study)—a study of young subjects (age, 24–45 years).16 The YFS cohort is a Finnish longitudinal population study sample on the evolution of cardiovascular risk factors from childhood to adulthood. In the present study, we used the variables measured in 2001 and described in detail previously.17 For these subjects, genotyping was performed in 2009 using a custom-built Illumina Human 670k BeadChip at the Welcome Trust Sanger Institute and imputed to 1000 Genomes phase 3.18 We attempted replication of a single SNP in the youngest quartile (age, 24–27 years) and overall (age, 24–45 years). All analyses included age, sex, center, and ancestry-informative PCs as covariates.
We sought replication of the novel SNP with FMD in older individuals of European ancestry in both the FHS (Framingham Heart Study) and the MESA (Multi-Ethnic Study of Atherosclerosis).
The MESA subjects analyzed were 1029: 50% men; mean age, 61 years (SD, 10 years). Flow-mediated dilation (FMD) of the brachial artery mean (SD), 4.8 (3.1). FHS subjects analyzed were 393: 38% men; mean age, 41 years (SD, 8 years). FMD of the brachial artery mean (SD), 5.9 (3.7).
For the MESA cohort, analysis was restricted to subjects with European ancestry based on the self-reported race/ethnicity. MESA and FHS genotype panels are described elsewhere.19,20 Standard quality control procedures for the genotype data were performed using PLINK. We excluded SNPs with a minor allele frequency <5% or significant deviation from Hardy-Weinberg equilibrium (P<1.0×10−6). SHAPEIT and IMPUTE2 software were used to impute PDE3A (Phosphodiesterase 3A) region (chr 12; p12.2:18060181-23060180; GRCh v37), using the 1000 Genomes reference panel (GRCh v37, phase III).18 Imputed SNPs with an imputation quality estimate (R2<0.40) were removed for association tests. The first 3 PCs for population structures were derived using the smartpca script in EIGENSTRAT and included as covariates in association analyses for both MESA and FHS.21
For both cohorts, linear regression analyses assuming an additive model were computed using 2 models: model 1 that included sex, age, and PCs 1 to 3 and model 2 where body mass index, diastolic blood pressure, and systolic blood pressure at the time of the measurements were added to the covariates of model 1. Linear mixed-effects Kinship models (package lmekin in R) for family data were implemented in FHS.
Further Analysis of Novel Association
We used a Bayesian test for colocalization at the novel gene between FMD GWAS summary results and stroke to assess whether 2 association signals are consistent with a shared causal variant.22 The gwas-pw package was used to perform all analyses (https://github.com/joepickrell/gwas-pw).
Association With Cardiovascular Risk Factors, Outcomes, and Coronary Artery Expression
Using data from publicly available repositories and UK Biobank, we assessed the impact of the PDE3A rs11045239 variant on cardiovascular risk factors: systolic blood pressure (id 4080), diastolic blood pressure (id 4079), pulse pressure (derived from the former 2 variables), pulse wave arterial stiffness (id 21021), hypertension (self-reported, id 200002=1465), hypercholesterolemia (self reported: id 20002=1473), ever smoking,23 as well as cardiovascular diseases: coronary artery disease,24 type 2 diabetes mellitus,25 and atrial fibrillation.26
UK Biobank (http://www.ukbiobank.ac.uk) is a prospective study that recruited 500 000 community-dwelling participants aged 40 to 69 years from across the United Kingdom between 2006 and 2010. The study collects extensive data from questionnaires, interviews, health records, physical measures, biological samples, and imaging. For all variables we considered in this analysis, we excluded outlier readings that were >3 SDs from the median value.
The UK Biobank genotyping procedure has been described elsewhere.27 In short, 2 custom genotyping arrays were used to genotype 49 950 (UK BiLEVE Axiom Array) and 438 427 participants (UK Biobank Axiom Array).27,28 Genotype data (805 426 markers) were available for 488 377 individuals and were subsequently imputed to the Haplotype Reference Consortium reference panel (39 131 578 autosomal SNPs). Imputed genotypes were available for 487 442 individuals in this study.27 From the resulting imputed dataset, we excluded (1) individuals who did not segregate with European individuals based on PC analysis, (2) individuals with high levels of heterozygosity or missingness (>5%), and (3) individuals whose reported sex did not match with sex inferred from the genetic data.
All UK Biobank analyses included age, sex, and ancestry-informative PC as covariates. In analyses of blood pressure variables, we additionally corrected for body mass index, as has been the convention in GWAS studies.29
In addition, we investigated the association of rs11045239, and other variants in close linkage disequilibrium (r2>0.8), calculated using LDlink,30 with expression of PDE3A in coronary arteries from Genotype-Tissue Expression.31 All analyses were performed using the Genotype-Tissue Expression portal (https://gtexportal.org/home/; accessed May 14, 2019).
Standard Protocol Approvals, Registrations, and Patient Consents
Ethical approval for the study was obtained from the ALSPAC Ethics and Law Committee and the Local Research Ethics Committees. UK Biobank received ethical approval from the research ethics committee (reference 11/NW/0382). All participants provided informed consent to participate. The present analyses were conducted under UK Biobank application number 36509.
Results
We performed a genome-wide association analysis of FMD in 5214 adolescents from ALSPAC. The inflation of test statistic using genomic control was 1.014, while the linkage disequilibrium score regression intercept was 1.001 indicating no inflation of test statistics.1 We identified an SNP (rs11045239) on chromosome 12 (12:20579694) that was associated with FMD (β[SE], 0.38 [0.070]; P=3.8×10−8). The risk allele (A) had a frequency of 40% among Europeans, and the SNP was located in an intron of the PDE3A gene, which encodes phosphodiesterase 3A—a member of the cyclic GMP (cGMP)–inhibited cyclic nucleotide phosphodiesterase family.
To identify regions with evidence of shared genetic variation impacting on FMD and ischemic stroke, we performed a genome-wide Bayesian colocalization analysis.22 This analysis highlighted 1 region around the same PDE3A gene where the underlying genetic variation was highly likely to be associated with both FMD and ischemic stroke (posterior probability, 97%; Figure 1). The most likely causal SNP based on Bayesian colocalization was rs11045239, although this was not conclusive (posterior probability, 65%). The 95% credible set contained 4 other SNPs: rs12811752, rs11045244, rs7489190, and rs10841519. The same PDE3A locus was recently reported to be associated at genome-wide significance with ischemic stroke in MEGASTROKE (lead SNP, rs7304841; odds ratio, 1.05 [95% CI, 1.03–1.07]; P=4.9×10−8).2 Effects were similar across ischemic stroke subtypes: cardioembolic stroke (odds ratio, 1.04 [95% CI, 1.00–1.08]), large artery stroke (odds ratio, 1.05 [95% CI, 1.00–1.10]), and small vessel stroke (odds ratio, 1.05 [95% CI, 1.01–1.08]). No other regions were highlighted with posterior probability of shared genetic effects >50%.
Figure 1.
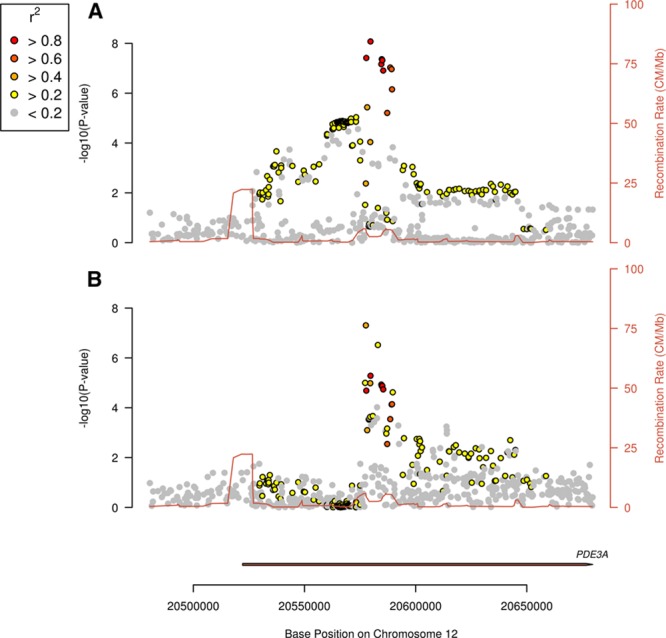
Colocalization of Genetic Associations with Flow Mediated Dilation and Ischaemic Stroke at PDE3A Locus. Plots of −log10(P) for association of single-nucleotide polymorphisms (SNPs) in the PDE3A gene region. Each point indicates an SNP association with the trait, with color indicating correlation (r2) with the lead SNP, rs11045239. Red line indicates recombination rate (CM/Mb) at this region of the genome. A, SNP associations with flow-mediated dilatation by chromosome position. B, SNP associations with ischemic stroke by chromosome position.
Having established that genetic variation in a single locus in PDE3A is associated with both ischemic stroke and FMD, we sought validation of the association in other cohorts with FMD data. As no other cohorts with genetic data and FMD measurement in adolescents exist to our knowledge, we sought replication in adult populations. In the YFS, we first analyzed the youngest individuals (the first quartile, 24–27 years; n=599) who most closely resemble the discovery cohort. In these individuals, there was evidence at nominal significance that the variant was associated with disease (β[SE], 0.47 [0.23]; P=0.047). Conversely, when considering the whole cohort (age, 24–45 years; n=2337), we did not observe a significant effect (β[SE], 0.14 [0.13]; P=0.25). We also explored whether the same variant was associated with FMD in older individuals from the FHS and MESA (n=1186). We found no evidence of an association in this older group (β[SE], −0.012 [0.13]; P=0.89). These results might indicate that the genetic variant identified in the study has different influence on endothelial function in adolescents compared with adults.
To establish whether variation in PDE3A was associated solely with endothelial function, or influenced other cardiovascular pathways, we looked at association of the PDE3A rs11045239 SNP with other cardiovascular risk factors and outcomes in large publicly available datasets and UK Biobank. The PDE3A rs11045239 variant was not associated with any other related cardiovascular traits or outcomes (all P>0.05; Figure 2), suggesting the association with ischemic stroke is not mediated through alternative cardiovascular pathways.
Figure 2.
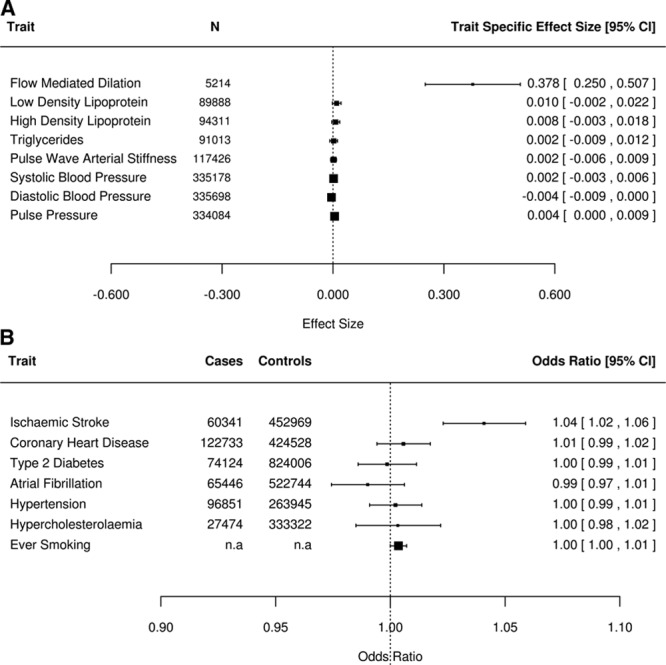
Associations with PDE3A rs11045239 single-nucleotide polymorphism. Plot of the association of each risk allele (effect size and 95% CI) of the PDE3A rs11405239 single- nucleotide polymorphism on (A) cardiovascular risk factors and (B) cardiovascular disease outcomes and binary risk factor traits. Exact numbers of cases and controls were not available in publicly available data for ever smoking.
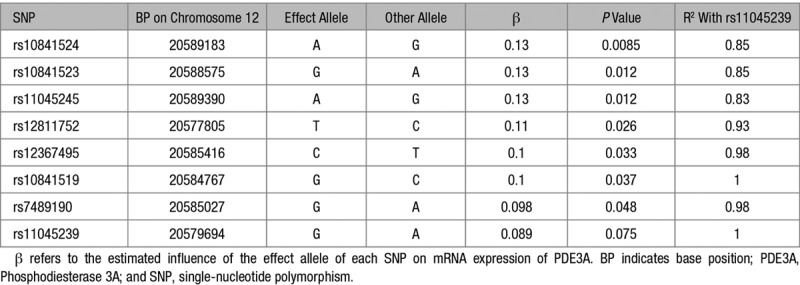
In addition, we looked at the association of PDE3A SNP rs11045239 and its proxies with mRNA expression of PDE3A in coronary arteries from Genotype-Tissue Expression. Although rs11045239 was not significantly associated with PDE3A expression (P=0.075), all proxy SNPs showed association at P<0.05 (Table 2). The A allele association with increased FMD and risk of stroke was associated with lower levels of PDE3A. Therefore, although not conclusive, this finding is consistent with variants in the PDE3A locus influencing expression of PDE3A in relevant tissues.
Table 2.
Association of SNPs in Linkage Disequilibrium With rs11045239 With mRNA Expression of PDE3A in Coronary Arteries From Genotype-Tissue Expression (n=152)
Discussion
We identified a genetic variant in the phosphodiesterase 3A gene that was associated at genome-wide significance with both FMD in early life and ischemic stroke. Colocalization analyses indicated that the same genetic variation in PDE3A associated with ischemic stroke is also associated with FMD, thereby showing the shared association is not merely coincidental. The association with variants in PDE3A was consistent across all stroke subtypes, which are presumed to have distinct etiologies. It, therefore, seems probable that the variant acts via a risk factor common to all subtypes. Exploring association of the same genetic variant with multiple other cardiovascular risk factors and outcomes, we could find no association that the variant also acts via other independent processes. There was some suggestion that variants at the PDE3A locus influence expression of PDE3A in coronary arteries, although this was not conclusive. An interpretation consistent with these data is that the associated genetic locus influences expression of PDE3A in arterial tissues, which leads to altered endothelial function and subsequent risk of ischemic stroke. Mediation analysis might help to shed light on whether this is a potential causal pathway.
Although it is not possible to determine the exact mechanism by which the PDE3A variant confers risk of ischemic stroke and alters endothelial function without further experimental data, some speculation on potential pathways is warranted. In vascular smooth muscle, phosphodiesterases play a key role in the NO/cGMP pathway—one of the most important regulators of vascular smooth muscle contraction and platelet activation. NO activates sGC (soluble guanylyl cyclase), which activates cGMP, activating many signaling molecules, in particular, PRKG (protein kinase G), which in turn promotes vascular smooth muscle contraction. Phosphodiesterases decompose cGMP, as well as cAMP, into GMP and AMP, respectively. This has the effect of monitoring the influence of cGMP on downstream processes such as smooth muscle contraction and platelet activation. Inhibition of phosphodiesterases has, therefore, been a target of multiple pharmaceuticals, with the intended effect of prolonging the influence of cGMP and promoting vasodilation. One such pharmaceutical cilostazol is a selective inhibitor of phosphodiesterase type 3 and is used primarily to treat peripheral vascular disease. Studies in East Asian populations suggest it ameliorates endothelial dysfunction and reduces recurrent stroke rates in patients with previous stroke,32 although not all studies have been positive,33 and it is currently being trialed for use to prevent recurrence after lacunar stroke in European populations.34 Off-label studies have also suggested it has the potential to prevent progression of intracranial arterial stenosis.35 However, PDE3A is also expressed in cardiac muscle, and whether the variant might influence stroke risk (particularly of cardioembolic source) via altered expression in the heart cannot be ruled out.
Why we detected an association of the PDE3A locus with endothelial function in adolescents, but not in adults, deserves further consideration. Aging has a considerable impact on endothelial biology36,37; normal endothelial function in children has a different molecular basis to the (dysfunctional) endothelium of older adults. In particular, decreases in NO production, expression of endothelial NO synthase,38 expression of cell adhesions molecules such as ICAM1 (intercellular adhesion molecule 1) and growth factors such as VEGF (vascular endothelial growth factor), as well as increases in levels of endothelin with age, contribute significant changes to endothelial biology. Whether these factors lead to a fundamental change in the relationship between the PDE3A locus and endothelial function, or simply influence the signal-to-noise ratio and, therefore, our ability to detect an effect, remains unclear. Nevertheless, the fact that the same PDE3A locus has a subsequent influence on stroke risk suggests that the changes that occur in adolescence have a lasting impact on the vasculature and subsequent pathological processes.
Our study has limitations. We demonstrate that genetic variation that influences levels of PDE3A influences endothelial function and subsequently, ischemic stroke. As such, we demonstrate a pathway by which risk of stroke conferred. However, we cannot rule out other horizontal pathways by which the PDE3A locus also leads to risk of ischemic stroke. However, we note that our analysis of other cardiovascular risk factors showed no association. As we were not able to assess endothelium-independent vasodilation in response to a nitrate, we cannot be absolutely conclusive that our FMD measure results reflect endothelium-dependent vasodilation. Rather, it is possible that they reflect nonspecific alterations in vascular reactivity.
Perspectives
In conclusion, we show that genetic variation in phosphodiesterase 3A leads to altered endothelial function in early life, mostly likely via expression of PDE3A, and also leads to increased risk of ischemic stroke, thus elucidating a pathway by which risk of disease is likely conferred.
Acknowledgments
We are extremely grateful to all the families who took part in ALSPAC (Avon Longitudinal Study of Parents and Children), the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses. Data used in this work were obtained from UK Biobank (data application 36509). We are grateful to UK Biobank for making the data available and to all UK Biobank study participants, who generously donated their time to make this resource possible. H.S. Markus and M. Traylor conceived the study and obtained funding. M. Traylor and A. Amin Al Olama designed the analysis plan; M. Traylor, A. Amin Al Olama, S. Marini, J. Chung, and L.-P. Lyytikäinen performed the statistical analyses. M. Traylor and A. Amin Al Olama wrote the first draft of the manuscript. L.-P. Lyytikäinen, T. Lehtimäki, and O.T. Raitakari contributed data. All authors read and approved the final manuscript.
Sources of Funding
This study was supported by the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 667375. The UK Medical Research Council and Wellcome (grant reference No. 102215/2/13/2) and the University of Bristol provide core support for ALSPAC (Avon Longitudinal Study of Parents and Children). This publication is the work of the authors, and M. Traylor and H.S. Markus will serve as guarantors for the contents of this article. This work was supported by a British Heart Foundation Programme Grant (RG/16/4/32218). H.S. Markus is supported by a National Institute for Health Research (NIHR) Senior Investigator award, and his work is supported by the Cambridge Universities NIHR Comprehensive Biomedical Research Centre. C.D. Anderson is supported by National Institutes of Health R01NS103924 and K23NS086873. YFS (Young Finns Study) has been financially supported by the Academy of Finland: grants 286284, 134309 (Eye), 126925, 121584, 124282, 129378 (Salve), 117787 (Gendi), and 41071 (Skidi); the Social Insurance Institution of Finland; Competitive State Research Financing of the Expert Responsibility area of Kuopio, Tampere and Turku University Hospitals (grant X51001); Juho Vainio Foundation; Paavo Nurmi Foundation; Finnish Foundation for Cardiovascular Research; Finnish Cultural Foundation; Sigrid Juselius Foundation; Tampere Tuberculosis Foundation; Emil Aaltonen Foundation; Yrjö Jahnsson Foundation; Signe and Ane Gyllenberg Foundation; Diabetes Research Foundation of Finnish Diabetes Association; EU Horizon 2020 (grant 755320 for TAXINOMISIS); European Research Council (grant 742927 for MULTIEPIGEN project); and Tampere University Hospital Supporting Foundation. Availability of data and materials: ALSPAC: http://www.bristol.ac.uk/alspac/researchers/; MEGASTROKE GWAS: http://megastroke.org; Coronary Artery Disease GWAS: https://www.cardiomics.net/download-data; Type 2 Diabetes GWAS: http://www.diagram-consortium.org/downloads.html; Atrial Fibrillation GWAS: http://www.broadcvdi.org/; UK Biobank: https://www.ukbiobank.ac.uk/; Smoking Status: https://conservancy.umn.edu/handle/11299/201564.
Disclosures
C.D. Anderson has consulted for ApoPharma, Inc. The other authors report no conflicts.
Footnotes
These authors contributed equally to this work.
Novelty and Significance
What Is New?
Using a large-scale genetic study of stroke (MEGASTROKE) and a study of endothelial function in adolescents (ALSPAC [Avon Longitudinal Study of Parents and Children]), we identified a genetic locus in the PDE3A gene, encoding phosphodiesterase 3A, that was associated with both endothelial function and ischemic stroke. The genetic variant was not associated with any other cardiovascular risk factor suggesting its influence on stroke acts solely through altered endothelial function.
What Is Relevant?
Our results highlight a pathway that in adolescence is influenced by genetic variation, which influences risk of stroke in later life.
Summary
Genetic variation in PDE3A influences risk of stroke through altered endothelial function, as measured by flow-mediated dilatation.
References
- 1.Howson JMM, Zhao W, Barnes DR, Ho WK, Young R, Paul DS, Waite LL, Freitag DF, Fauman EB, Salfati EL, et al. CARDIoGRAMplusC4D; EPIC-CVD. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat Genet. 2017;49:1113–1119. doi: 10.1038/ng.3874. doi: 10.1038/ng.3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, Rutten-Jacobs L, Giese AK, van der Laan SW, Gretarsdottir S, et al. AFGen Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium; International Genomics of Blood Pressure (iGEN-BP) Consortium; INVENT Consortium; STARNET; BioBank Japan Cooperative Hospital Group; COMPASS Consortium; EPIC-CVD Consortium; EPIC-InterAct Consortium; International Stroke Genetics Consortium (ISGC); METASTROKE Consortium; Neurology Working Group of the CHARGE Consortium; NINDS Stroke Genetics Network (SiGN); UK Young Lacunar DNA Study; MEGASTROKE Consortium. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–537. doi: 10.1038/s41588-018-0058-3. [Google Scholar]
- 3.Franceschini N, Giambartolomei C, de Vries PS, Finan C, Bis JC, Huntley RP, Lovering RC, Tajuddin SM, Winkler TW, Graff M, et al. MEGASTROKE Consortium. GWAS and colocalization analyses implicate carotid intima-media thickness and carotid plaque loci in cardiovascular outcomes. Nat Commun. 2018;9:5141. doi: 10.1038/s41467-018-07340-5. doi: 10.1038/s41467-018-07340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flammer AJ, Anderson T, Celermajer DS, Creager MA, Deanfield J, Ganz P, Hamburg NM, Lüscher TF, Shechter M, Taddei S, et al. The assessment of endothelial function: from research into clinical practice. Circulation. 2012;126:753–767. doi: 10.1161/CIRCULATIONAHA.112.093245. doi: 10.1161/CIRCULATIONAHA.112.093245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–1295. doi: 10.1161/CIRCULATIONAHA.106.652859. doi: 10.1161/CIRCULATIONAHA.106.652859. [DOI] [PubMed] [Google Scholar]
- 6.Halcox JP, Donald AE, Ellins E, Witte DR, Shipley MJ, Brunner EJ, Marmot MG, Deanfield JE. Endothelial function predicts progression of carotid intima-media thickness. Circulation. 2009;119:1005–1012. doi: 10.1161/CIRCULATIONAHA.108.765701. doi: 10.1161/CIRCULATIONAHA.108.765701. [DOI] [PubMed] [Google Scholar]
- 7.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markus HS. Genes, endothelial function and cerebral small vessel disease in man. Exp Physiol. 2008;93:121–127. doi: 10.1113/expphysiol.2007.038752. doi: 10.1113/expphysiol.2007.038752. [DOI] [PubMed] [Google Scholar]
- 9.Boyd A, Golding J, Macleod J, Lawlor DA, Fraser A, Henderson J, Molloy L, Ness A, Ring S, Davey Smith G. Cohort profile: the ‘children of the 90s’–the index offspring of the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 2013;42:111–127. doi: 10.1093/ije/dys064. doi: 10.1093/ije/dys064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraser A, Macdonald-Wallis C, Tilling K, Boyd A, Golding J, Davey Smith G, Henderson J, Macleod J, Molloy L, Ness A, et al. Cohort profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int J Epidemiol. 2013;42:97–110. doi: 10.1093/ije/dys066. doi: 10.1093/ije/dys066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donald AE, Charakida M, Falaschetti E, Lawlor DA, Halcox JP, Golding J, Hingorani AD, Smith GD, Deanfield JE. Determinants of vascular phenotype in a large childhood population: the Avon Longitudinal Study of Parents and Children (ALSPAC). Eur Heart J. 2010;31:1502–1510. doi: 10.1093/eurheartj/ehq062. doi: 10.1093/eurheartj/ehq062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris RA, Nishiyama SK, Wray DW, Richardson RS. Ultrasound assessment of flow-mediated dilation. Hypertension. 2010;55:1075–1085. doi: 10.1161/HYPERTENSIONAHA.110.150821. doi: 10.1161/HYPERTENSIONAHA.110.150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kemp JP, Medina-Gomez C, Estrada K, St Pourcain B, Heppe DH, Warrington NM, Oei L, Ring SM, Kruithof CJ, Timpson NJ, et al. Phenotypic dissection of bone mineral density reveals skeletal site specificity and facilitates the identification of novel loci in the genetic regulation of bone mass attainment. PLoS Genet. 2014;10:e1004423. doi: 10.1371/journal.pgen.1004423. doi: 10.1371/journal.pgen.1004423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raitakari OT, Juonala M, Rönnemaa T, Keltikangas-Järvinen L, Räsänen L, Pietikäinen M, Hutri-Kähönen N, Taittonen L, Jokinen E, Marniemi J, et al. Cohort profile: the cardiovascular risk in Young Finns Study. Int J Epidemiol. 2008;37:1220–1226. doi: 10.1093/ije/dym225. doi: 10.1093/ije/dym225. [DOI] [PubMed] [Google Scholar]
- 17.Juonala M, Viikari JS, Laitinen T, Marniemi J, Helenius H, Rönnemaa T, Raitakari OT. Interrelations between brachial endothelial function and carotid intima-media thickness in young adults: the cardiovascular risk in Young Finns Study. Circulation. 2004;110:2918–2923. doi: 10.1161/01.CIR.0000147540.88559.00. doi: 10.1161/01.CIR.0000147540.88559.00. [DOI] [PubMed] [Google Scholar]
- 18.Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berardi C, Larson NB, Decker PA, Wassel CL, Kirsch PS, Pankow JS, Sale MM, de Andrade M, Sicotte H, Tang W, et al. Multi-ethnic analysis reveals soluble L-selectin may be post-transcriptionally regulated by 3’UTR polymorphism: the Multi-Ethnic Study of Atherosclerosis (MESA). Hum Genet. 2015;134:393–403. doi: 10.1007/s00439-014-1527-0. doi: 10.1007/s00439-014-1527-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jun G, Asai H, Zeldich E, Drapeau E, Chen C, Chung J, Park JH, Kim S, Haroutunian V, Foroud T, et al. PLXNA4 is associated with Alzheimer disease and modulates tau phosphorylation. Ann Neurol. 2014;76:379–392. doi: 10.1002/ana.24219. doi: 10.1002/ana.24219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 22.Pickrell JK, Berisa T, Liu JZ, Ségurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet. 2016;48:709–717. doi: 10.1038/ng.3570. doi: 10.1038/ng.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, Datta G, Davila-Velderrain J, McGuire D, Tian C, et al. 23andMe Research Team; HUNT All-In Psychiatry. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–244. doi: 10.1038/s41588-018-0307-5. doi: 10.1038/s41588-018-0307-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122:433–443. doi: 10.1161/CIRCRESAHA.117.312086. doi: 10.1161/CIRCRESAHA.117.312086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50:1505–1513. doi: 10.1038/s41588-018-0241-6. doi: 10.1038/s41588-018-0241-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roselli C, Chaffin MD, Weng LC, Aeschbacher S, Ahlberg G, Albert CM, Almgren P, Alonso A, Anderson CD, Aragam KG, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–1233. doi: 10.1038/s41588-018-0133-9. doi: 10.1038/s41588-018-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bycroft C, Freeman C, Petkova D, et al. Genome-wide genetic data on ~500,000 UK Biobank participants. bioRxiv. 2017 doi: 10.1101/166298. [DOI] [Google Scholar]
- 28.Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, Bossini-Castillo L, Obeidat M, Henry AP, Portelli MA, Hall RJ, et al. Understanding Society Scientific Group; Geisinger-Regeneron DiscovEHR Collaboration. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet. 2017;49:416–425. doi: 10.1038/ng.3787. doi: 10.1038/ng.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warren HR, Evangelou E, Cabrera CP, Gao H, Ren M, Mifsud B, Ntalla I, Surendran P, Liu C, Cook JP, et al. International Consortium of Blood Pressure (ICBP) 1000G Analyses; BIOS Consortium; Lifelines Cohort Study; Understanding Society Scientific group; CHD Exome+ Consortium; ExomeBP Consortium; T2D-GENES Consortium; GoT2DGenes Consortium; Cohorts for Heart and Ageing Research in Genome Epidemiology (CHARGE) BP Exome Consortium; International Genomics of Blood Pressure (iGEN-BP) Consortium; UK Biobank CardioMetabolic Consortium BP Working Group. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat Genet. 2017;49:403–415. doi: 10.1038/ng.3768. [Google Scholar]
- 30.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. doi: 10.1093/bioinformatics/btv402. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shinohara Y, Katayama Y, Uchiyama S, Yamaguchi T, Handa S, Matsuoka K, Ohashi Y, Tanahashi N, Yamamoto H, Genka C, et al. CSPS 2 Group. Cilostazol for prevention of secondary stroke (CSPS 2): an aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol. 2010;9:959–968. doi: 10.1016/S1474-4422(10)70198-8. doi: 10.1016/S1474-4422(10)70198-8. [DOI] [PubMed] [Google Scholar]
- 33.Kim BJ, Lee EJ, Kwon SU, Park JH, Kim YJ, Hong KS, Wong LKS, Yu S, Hwang YH, Lee JS, et al. PICASSO Investigators. Prevention of cardiovascular events in Asian patients with ischaemic stroke at high risk of cerebral haemorrhage (PICASSO): a multicentre, randomised controlled trial. Lancet Neurol. 2018;17:509–518. doi: 10.1016/S1474-4422(18)30128-5. doi: 10.1016/S1474-4422(18)30128-5. [DOI] [PubMed] [Google Scholar]
- 34.Blair GW, Appleton JP, Law ZK, Doubal F, Flaherty K, Dooley R, Shuler K, Richardson C, Hamilton I, Shi Y, et al. Preventing cognitive decline and dementia from cerebral small vessel disease: the LACI-1 Trial. Protocol and statistical analysis plan of a phase IIa dose escalation trial testing tolerability, safety and effect on intermediary endpoints of isosorbide mononitrate and cilostazol, separately and in combination. Int J Stroke. 2018;13:530–538. doi: 10.1177/1747493017731947. doi: 10.1177/1747493017731947. [DOI] [PubMed] [Google Scholar]
- 35.Kwon SU, Cho YJ, Koo JS, Bae HJ, Lee YS, Hong KS, Lee JH, Kim JS. Cilostazol prevents the progression of the symptomatic intracranial arterial stenosis: the multicenter double-blind placebo-controlled trial of cilostazol in symptomatic intracranial arterial stenosis. Stroke. 2005;36:782–786. doi: 10.1161/01.STR.0000157667.06542.b7. doi: 10.1161/01.STR.0000157667.06542.b7. [DOI] [PubMed] [Google Scholar]
- 36.Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res. 2005;66:286–294. doi: 10.1016/j.cardiores.2004.12.027. doi: 10.1016/j.cardiores.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 37.Lundberg MS, Crow MT. Age-related changes in the signaling and function of vascular smooth muscle cells. Exp Gerontol. 1999;34:549–557. doi: 10.1016/s0531-5565(99)00036-4. doi: 10.1016/s0531-5565(99)00036-4. [DOI] [PubMed] [Google Scholar]
- 38.Sverdlov AL, Ngo DT, Chan WP, Chirkov YY, Horowitz JD. Aging of the nitric oxide system: are we as old as our NO? J Am Heart Assoc. 2014;3:e000973. doi: 10.1161/JAHA.114.000973. doi: 10.1161/JAHA.114.000973. [DOI] [PMC free article] [PubMed] [Google Scholar]