

Abstract
Summary: We report an unusual case of extraventricular (“cerebral”) neurocytoma with ganglion cells located in the right temporal lobe in a 9-year-old girl with complex partial seizures and precocious puberty. CT showed a calcified mass with central cystic zones. MR imaging showed a markedly hyperintense predominately solid tumor on both T1- and T2-weighted images, without appreciable contrast enhancement. Cerebral neurocytomas are histologically benign and radical surgery is curative; they should be included in the differential diagnosis of temporal lobe tumors in children.
Central neurocytoma, first described by Hassoun et al in 1982 (1), is a rare neuronal tumor of the CNS accounting for 0.25% to 0.5% of all CNS tumors (2). The term central underscores the fact that most neurocytomas are located within the ventricular system, at or near the midline (1, 3). However, several reports on extraventricular neurocytomas have appeared in the neuropathologic literature in the past few years, and the term cerebral neurocytoma was introduced to describe such locations (4). Histologically, increasing evidence suggests that cerebral neurocytomas are probably different from central neurocytomas in their pronounced tendency to ganglionic or glial differentiation (5); the term ganglioneurocytoma has been introduced to describe the presence of ganglion cells within an extraventricular neurocytic tumor (5). We recently encountered one neurocytic tumor of the right temporal lobe in a 9-year-old girl with complex partial seizures and precocious puberty. The tumor displayed unprecedented features on MR images.
Case Report
A 9-year-old left-handed girl was admitted to our institution after a 2.5-year history of complex partial seizures. Her birth and postnatal development were uneventful until age 7 years, when four episodes of absence occurred and treatment with valproate sodium (600 mg per day) was started. Precocious puberty was noted at age 8.5 years, and the girl was placed on treptoreline (3.75 mg per month). She remained well until 2 months before this admission, when a new episode of absence occurred. This was followed, 1 month later, by an episode of verbal automatism.
On admission, physical examination revealed hypertrophic pubic hairs and breast development corresponding to age 11 to 12 years, according to Tanner's scale. Neurologic examination was normal. An electroencephalogram showed hypostructured fundamental activity in the right cerebral hemisphere. Spike-wave–like elements at 1 to 2 cycles per second were registered in the right temporal derivations, with occasional contralateral diffusion.
A CT scan (Fig 1A) showed a mass in the right temporal lobe with marked punctate calcifications and central areas of low density, consistent with foci of cystic change. The solid part of the mass was isodense with gray matter. The tumor was clearly separated from the adjacent temporal horn. Contrast material was not administered. On MR images (Fig 1B–D), the mass was centered in the white matter of the right temporal lobe and was clearly demarcated from adjacent gray matter; it displayed a peripheral solid part and a central cystic core. The solid part was markedly hyperintense relative to gray matter, both on spin-echo (SE) and turbo inversion-recovery T1-weighted images, as well as on SE proton density–weighted images. It was hyperintense also on SE and fast-SE T2-weighted images. On gradient-echo T2*-weighted images, scattered hypointense spots were noted in the solid part of the tumor. Gadoteric acid was administered intravenously at a dose of 0.1 mmol/kg body weight; no enhancement was appreciable. Perilesional edema was absent, and the mass effect was negligible. Midline structures were not shifted; the hypothalamus and the pituitary gland and stalk were normal.
fig 1.
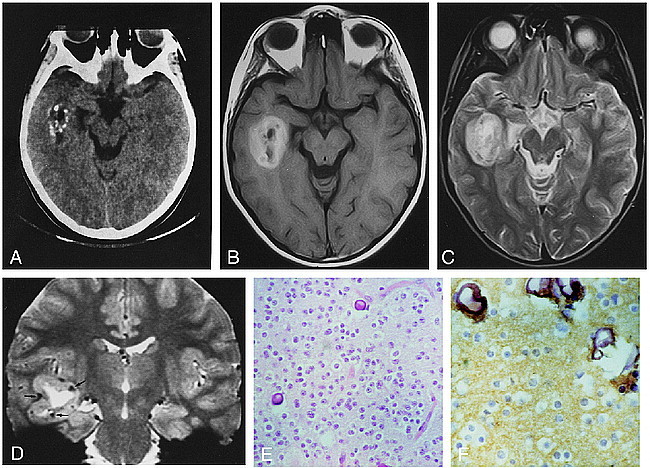
9-year-old girl with history of complex partial seizures.
A, On unenhanced CT scan, the lesion is centered in the right temporal lobe. Several calcified spots are visible in the peripheral part of the tumor. Central hypodense zones are consistent with cystic change. The noncalcified tumor parts are isodense with, and barely discernible from, adjacent brain. The apex of the right temporal horn is widened.
B, Axial SE T1-weighted image (600/16/2) shows the tumor mass centered in the white matter of the right temporal lobe. The lesion has a central hypointense core and a markedly hyperintense peripheral portion.
C, Axial SE T2-weighted image (2300/90/1) shows a markedly hyperintense peripheral portion. No perilesional edema is visible.
D, Coronal gradient-echo T2*-weighted image (1033/42/3; 20° flip angle) perfectly depicts the location of the mass in the right temporal lobe. The lesion has a large central cystic core and a peripheral solid part that is hyperintense to gray matter. Scattered, markedly hypointense foci in the peripheral part of the tumor (arrows) are consistent with calcified spots seen on the CT scan (see A).
E, Proliferation of round cells with clear cytoplasm associated with calcification (hematoxylin-eosin, ×25 magnification).
F, Intense staining with synaptophysin in the fibrillary background (×40 magnification).
At surgery, the tumor was clearly demarcated from the surrounding brain parenchyma, had a soft consistency, and bled moderately; a radical excision was performed. Histologic examination (Fig 1E and F) revealed a proliferation of cells with large, clear cytoplasm and a round nucleus. The cellularity was low to moderate and the tumor cells were embedded in a neuropillike background; some areas showed greater cellularity with limited intervening matrix. The vascularization was rich, with focal hyalinization of the blood vessels. There was no evidence of necrosis, hemorrhage, or endothelial proliferation. Extensive calcification forming psammomatous bodies was present both in a perivascular disposition and in the tumoral stroma. Mitotic figures were rare. MIB-1 (Ki67) was used as a proliferation marker to calculate the growth fraction; about 1% of the tumor cells were MIB-1 immunoreactive. In addition, ganglion cells were focally observed throughout the tumor. These larger cells showed large cytoplasm and one or two large nuclei with nucleolus. Nonneoplastic, reactive astrocytes were also present. Immunohistochemical studies displayed intense synaptophysin and neuron-specific enolase staining in the fibrillary background. Glial fibrillary acidic protein stained the reactive astrocytes.
The postoperative course was uneventful; seizures were controlled with medication. No evidence of disease was detected on follow-up MR imaging performed 6 months postoperatively.
Discussion
The classical site for neurocytomas is within the ventricular system, at or near the midline (3). Extraventricular (cerebral) neurocytomas are definitely rarer, and the few available reports focus mainly on their histopathologic, rather than neuroradiologic, appearance. Cerebral neurocytomas are composed primarily of monotonous small round cells in a fibrillary matrix (2, 4). Tumor cells are immunoreactive toward synaptophysin and neuron-specific enolase, which identifies their neuronal nature (2). Although tumors showing clinically malignant behavior have been reported (6), the proliferative activity of cerebral neurocytoma is usually low, ranging from 1% to 1.5% as measured by the murine monoclonal antibody MIB-1, which accounts for their relatively good prognosis (5). Recently, Giangaspero et al (5) described a group of 11 patients, aged 5 to 63 years, who had extraventricular neurocytic tumors with ganglionic and astrocytic differentiation; a total resection was performed in seven cases, and a subtotal removal or stereotactic biopsy was accomplished in the remaining four. Except for one patient who died postoperatively, all patients were alive at follow-up ranging from 6 to 80 months; however, only those patients who underwent radical surgery had no evidence of disease at follow-up, whereas two tumors recurred with the same histologic picture after subtotal surgery and despite postoperative radiotherapy. Therefore, radical surgery appears to be the most critical factor in determining favorable outcome.
Several terms have been coined to describe these entities, such as ganglioneurocytoma and ganglioglioneurocytoma (5). However, according to Funato et al (2), the term ganglioneurocytoma should only be used when extensive ganglionic differentiation is present throughout the tumor, excluding those tumors in which such differentiation is restricted to focal areas, such as in our case. Regardless of the adopted terminology, it appears that neoplasms with neurocytic components can be found throughout the CNS (5), and that their identification broadens the spectrum of neuronal and mixed neuronal-glial tumors.
While intraventricular neurocytomas usually present with headache and raised intracranial pressure (3), the presentation of cerebral neurocytoma varies according to tumor location. Almost 20% of patients with complex partial seizures have an underlying primary brain tumor (7). In a study by Wolf and Wiestler (8), ganglioglioma, pilocytic astrocytoma, oligodendroglioma, anaplastic astrocytoma, and dysembryoplastic neuroepithelial tumor were the neoplasms most commonly associated with complex partial seizures. However, a case of cerebral neurocytoma of the temporal lobe presenting with long-standing complex partial seizures has also been reported (7). The cause of precocious puberty in our case remained unexplained. Isosexual precocious puberty may be the presenting sign of hamartoma of the tuber cinereum, hypothalamic glioma, and pineal gland tumors; however, the sellar/suprasellar and pineal regions were normal in our patient. Only rarely may hemispheric tumors be responsible for precocious puberty, probably because of raised intracranial pressure and hydrocephalus producing compression of the hypothalamus with premature activation of its pathways (9). However, there were no signs of increased intracranial pressure or hydrocephalus in our patient, and the mass effect produced by the tumor was very limited without distortion of midline structures. On the other hand, the possibility that neuronal tumors may induce puberty by producing gonadotropin-releasing factors is speculative and, to our knowledge, has not been reported.
While the neuroradiologic appearance of intraventricular neurocytomas has been described in detail (3), only few CT and MR descriptions of cerebral neurocytoma can be found in articles dealing primarily with their neuropathologic features; we are aware of only five reported cases of cerebral neurocytoma located in the temporal lobe (4, 5, 7, 10). Cerebral neurocytomas have been described as hypodense or isodense relative to gray matter on CT scans (4); as for intraventricular neurocytomas, coarse or punctate calcification is common. On MR images, cerebral neurocytomas appear hypointense on T1-weighted sequences and hyperintense on T2-weighted studies (4); contrast enhancement is mild, and occasionally rimlike (6, 10). Most tumors are solid, but largely cystic masses with mural nodules may be found (2). In our case, the signal behavior was different in that the peripheral solid portion appeared hyperintense on T1-, proton density–, and T2-weighted images. Hyperintensity was marked also on inversion-recovery T1-weighted images, paralleling that of white matter. The causes for the high signal intensity of brain tumors on T1-weighted images are various, and include subacute-chronic blood (methemoglobin), melanin, naturally occurring ions (manganese, iron, copper) associated with necrosis and calcification, very high protein concentration, fat, and flow-related enhancement in tumor vessels (11). While calcification was exquisitely depicted on CT scans and T2*-weighted MR images, the absence of blood degradation by-products, melanin, and fat was expected on the basis of the pattern of CT attenuation and signal behavior in the other MR sequences obtained, and was eventually confirmed by histology. Therefore, the short T1 probably resulted from marked paramagnetic effects generated by ions associated with calcification. Although calcified regions in brain tumors may show contrast enhancement on MR images (12), this was not found in our case, probably because of the marked intensity of calcium-related paramagnetic effects, impairing any further significant shortening of T1 by contrast material.
The differential diagnosis with other brain tumors arising in the temporal lobes in children includes ganglioglioma, dysembryoplastic neuroepithelial tumor, pilocytic astrocytoma, oligodendroglioma, and pleomorphic xanthoastrocytoma. Both pilocytic astrocytoma and pleomorphic xanthoastrocytoma usually present as cystic lesions with a mural nodule, and usually are not largely calcified; moreover, pleomorphic xanthoastrocytoma is exceedingly rare in children (11). The dysembryoplastic neuroepithelial tumor is classically a cortical mass that may involve the adjacent white matter; however, in the current case, the cortical gray matter was clearly spared. Both ganglioglioma and oligodendroglioma frequently show cystic change and calcification, and surrounding edema is usually low or even absent. Our neuroradiologic differential diagnosis included ganglioglioma, oligodendroglioma, and dysembryoplastic neuroepithelial tumor, and the eventual histologic diagnosis came as a surprise. Therefore, we suggest that cerebral neurocytoma should be considered among the uncommon causes of an intraaxial temporal lobe mass in children with complex partial seizures, and that such a diagnosis should at least be suspected in the presence of a coarsely calcified, partially cystic tumor in that location.
Footnotes
Address reprint requests to Paolo Tortori-Donati, MD, Department of Pediatric Neuroradiology, Children's Hospital and Scientific Institute “Giannina Gaslini,” Largo G. Gaslini 5, I-16148 Genoa, Italy.
References
- 1.Hassoun J, Gambarelli D, Grisoli F, et al. Central neurocytoma: an electron-microscopic study of two cases. Acta Neuropathol (Berl) 1982;56:151-156 [DOI] [PubMed] [Google Scholar]
- 2.Funato H, Inoshita N, Okeda R, Yamamoto S, Aoyagi M. Cystic ganglioneurocytoma outside the ventricular region. Acta Neuropathol (Berl) 1997;94:95-98 [DOI] [PubMed] [Google Scholar]
- 3.Goergen SK, Gonzales MF, McLean CA. Intraventricular neurocytoma: radiologic features and review of the literature. Radiology 1992;182:787-792 [DOI] [PubMed] [Google Scholar]
- 4.Nishio S, Takeshita I, Kaneko Y, Fukui M. Cerebral neurocytoma: a new subset of benign neuronal tumors of the cerebrum. Cancer 1992;70:529-537 [DOI] [PubMed] [Google Scholar]
- 5.Giangaspero F, Cenacchi G, Losi L, Cerasoli S, Bisceglia M, Burger PC. Extraventricular neoplasms with neurocytoma features: a clinicopathological study of 11 cases. Am J Surg Pathol 1997;21:206-212 [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto T, Komori T, Shibata N, Toyoda C, Kobayashi M. Multifocal neurocytoma/gangliocytoma with extensive leptomeningeal dissemination in the brain and spinal cord. Am J Surg Pathol 1996;20:363-370 [DOI] [PubMed] [Google Scholar]
- 7.Rabinowicz AL, Abrey LE, Hinton DR, Couldwell WT. Cerebral neurocytoma: an unusual cause of refractory epilepsy: case report and review of the literature. Epilepsia 1995;36:1237-1240 [DOI] [PubMed] [Google Scholar]
- 8.Wolf HK, Wiestler OD. Surgical pathology of chronic epileptic seizure disorders. Brain Pathol 1993;3:371-380 [DOI] [PubMed] [Google Scholar]
- 9.Barkovich AJ. Pediatric Neuroimaging.. 2nd ed. New York: Raven Press; 1995:379–380
- 10.Kim DH, Suh YL. Pseudopapillary neurocytoma of temporal lobe with glial differentiation. Acta Neuropathol (Berl) 1997;94:187-191 [DOI] [PubMed] [Google Scholar]
- 11.Atlas SW, Lavi E. Intra-axial brain tumors. In: Atlas SW, ed. Magnetic Resonance Imaging of the Brain and Spine. 2nd ed. Philadelphia: Lippincott-Raven; 1996:317–422
- 12.Tsuchiya K, Makita K, Furui S, Nitta K. MRI appearances of calcified regions within intracranial tumors. Neuroradiology 1993;35:341-344 [DOI] [PubMed] [Google Scholar]