

Abstract
Summary: In 1863, Michel described a condition characterized by a total absence of differentiated inner ear structures associated with other skull base anomalies, including an abnormal course of the facial nerve and jugular veins. Michel aplasia clearly differs from Michel dysplasia, in which arrest of embryologic development occurs later. Recently, the role of otic capsule formation on mesenchymal differentiation was reported as well as the impact of the genetic deletion of the homeobox gene on the development of the ear, cranial nerves, and hindbrain. We report two patients with a total absence of inner ear structures bilaterally, illustrating the characteristic appearance of Michel aplasia and associated skull base anomalies.
Michel aplasia is a rare condition, first described by P. Michel in 1863 in the autopsy report of an 11-year-old deaf and mute boy who died in the children's hospital of Strasbourg (1). It is defined by a bilateral absence of differentiated inner ear structures (1), and persons who have this deformity have anacusis. However, a case of unilateral Michel-type aplasia has recently been reported (2). The inner ear aplasia is due to a failure of development of the otic placode (ectoderm) (3, 4), which occurs before the third week of gestation (5). The failure of development of the placode may induce an anomaly in the development of the skeletal portion of the second arch (endoderm) with nondifferentiation of the stapes and an abnormal course of the facial nerve (1, 4). Other skull base anomalies that are encountered include hypoplasia of the petrous bone, platybasia, and an aberrant course of the jugular veins (1).
We describe the CT and MR imaging features of bilateral Michel aplasia in two siblings.
Case Reports
Case 1
A psychologically normal 9-year-old girl with profound bilateral congenital sensorineural hearing loss was referred to the E.N.T. department for auditory rehabilitation by cochlear implantation. An audiogram demonstrated profound hearing loss with some residual auditory perception on low frequencies (eg, 125–1000 Hz). No facial or external ear malformations were found. An external hearing aid and early education enabled satisfactory oral communication. Speech development and scholastic achievement were satisfactory. CT scans showed platybasia and bilateral absence of the inner ear structures, associated with bilateral aplasia of the petrous apex, and nondevelopment of the internal acoustic meatus (IAM). On each side, the medial wall of the middle ear was flat and the stapes was absent. The course of the facial nerve canal was abnormal and the geniculate ganglion and the second portion of the facial nerve were absent; the facial nerve was displaced inferoposteriorly, entering through a small foramen below the level of the long process of the incus (Fig 1). T1- and T2-weighted MR images showed platybasia of the skull and a bilateral petrous apex hypoplasia. Except for an abnormally high tentorium position and anteromedial displacement of the cerebellar hemispheres around the pons and the medulla, no other brain anomalies were found. Thin, high-resolution T2-weighted MR images of the cerebellopontine angle confirmed the bilateral absence of the cochleovestibular nerve and showed only a thin linear structure, hypointense on T2-weighted images, which crossed through the mastoid bone and was attributed to the cisternal course of the facial nerve.
fig 1.
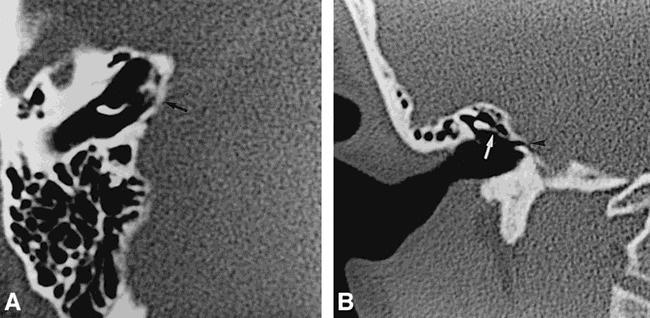
Case 1: 9-year-old girl with profound bilateral congenital sensorineural hearing loss.
A, Axial CT scan shows right petrous bone aplasia with absence of inner ear structures. The medial wall of the middle ear is flattened (arrow), being in close contact with the infratentorial nervous structures. Note normal differentiation of the malleus.
B, Coronal CT scan shows a normally developed external and middle right ear. The long process of the incus (arrow) leans against the medial middle ear wall. The oval window, the stapes, and the second portion of the facial nerve are absent. A small dehiscence of the medial wall of the middle ear (arrowhead), located at the IAM, probably corresponds to the entrance of the facial nerve.
Case 2
A 16-year-old boy (a brother of the patient described in case 1) was referred on the same day with the same signs and symptoms as his sister. CT scans similarly showed bilateral inner ear aplasia associated with absence of the stapes. Both petrous apices were hypoplastic and both IAMs were absent. Moreover, on the left side, the course of the internal carotid artery was aberrant, bulging into the middle ear and associated with a persistent stapedial artery. Concomitantly, a well-circumscribed left-sided petrous bone defect communicated via a lateral dehiscence with the horizontal portion of the intrapetrous internal carotid artery. This bony defect was of low signal intensity on T1- and T2-weighted MR images and had high signal intensity on gradient-echo T2-weighted images. These anomalies were related to a carotid fenestration resulting in two vascular pathways: a persistent stapedial artery and an aberrant course of the internal carotid artery (Fig 2), uniting at the horizontal segment. Furthermore, MR images showed anomalies of the cervicooccipital junction. The foramen magnum was unusually large. The posterior fossa was small, with low-lying transverse sinuses and a small jugular foramen. The cerebellar hemispheres extended anteromedially around the pons and the medulla and there was bilateral cerebellar tonsillar herniation and platybasia. During a palatine tonsillectomy performed under general anesthesia, a concomitant right middle ear exploration confirmed the absence of the round and oval windows and of the stapes. The incus and malleus were normal.
fig 2.
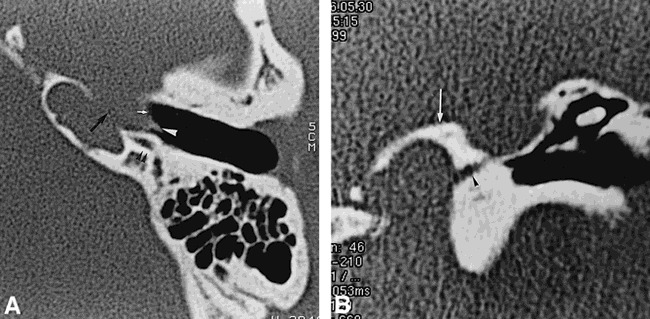
Case 2: 16-year-old boy, brother of girl in case 1, also with profound bilateral congenital sensorineural hearing loss.
A, Axial CT scan shows an aberrant course of the left internal carotid artery, which bulges into the middle ear (white arrow). Dehiscence of the lateral wall of the petrous bone adjacent to the horizontal carotid artery is present (black arrow). A small lateral tract corresponds to a persistent stapediohyoid artery (white arrowhead). The facial nerve passes behind the internal carotid artery (black arrowheads).
B, Coronal CT scan shows enlargement of inferior tympanic canaliculus (arrowhead). Arrow indicates jugular foramen.
Discussion
Inner ear anomalies responsible for congenital sensorineural hearing loss were classified by Schuknecht, according to their frequencies, into Mondini, Scheibe, Michel, and Alexander forms (6). These anomalies are rare and result from a complete or partial failure of development of the inner ear structures during the first weeks of embryonic life (7, 8). Two different congenital anomalies, such as Mondini and Michel, may affect each ear of the same individual. Other congenital anomalies, such as congenital preauricular and cervical fistulas and renal anomalies (branchio-oto-renal syndrome) may be associated with inner ear anomalies (9, 10). Factors predisposing to these anomalies include exposure to thalidomide (11, 12), congenital cytomegalovirus infection (13), and genetic disorders (14). Some cases of Michel aplasia have been reported in association with tracheoesophageal (10), cardiac (11, 12), or extremity (7, 10, 15) malformations and with facial palsy (12).
Development of the inner ear structures and of the stapes occurs during the first 18 to 28 days of embryonic life (2.5–5 mm). Derived from ectoderm, the otic placode appears dorsal to the second branchial arch during the third week. Each placode invaginates and becomes an otic pit, the mouth of which closes to form a rounded auditory vesicle. At the periphery, the mesenchyma is converted into the cartilaginous otic capsule, which ossifies, forming the bony labyrinth. Failure of development of the otic placode prior to this crucial stage results in complete membranous and osseous labyrinthine aplasia, namely Michel aplasia (3). Thus, Michel aplasia differs from Michel dysplasia in that developmental arrest occurs in the latter, which explains some development of the semicircular canal and vestibule (6, 7). Developmental failure of the inner ear occurring between the 21st and 47th days of embryologic life (so-called pre-Mondini labyrinthine anomalies) results in less severe inner ear anomalies, such as amorphotic labyrinthine sac and “common cavity” deformity. These anomalies are known for their potential risk of fistulous communication with the subarachnoid space (6, 7, 16, 17). Recently, Jackler et al (5) described a classification of congenital inner ear malformations based on their radiologic appearance in concordance with the failure of anatomic development and the presence or absence of inner ear structures.
In keeping with Michel's description (1), our two patients with bilateral inner ear aplasia had associated anomalies of the promontory, the stapes, and the cochleovestibular and facial nerves.
Absence of the round and oval windows is associated with inner ear aplasia, indicating failure of otic differentiation (6, 7). Experimental studies with hybrid mice have shown the role of the otocyst as an inductor of chondrogenesis in the mesenchyma and in the formation of the otic capsule (18). The concomitant absence of the stapes may be attributed to the lack of formation of the oval window (7), as the theory of dual origin of the stapes suggests.
According to Michel's autopsy findings (1), the cochleovestibular nerve is absent, probably as a result of absence of the otocyst, which arrests formation of the acoustic ganglion and nerves (16). The facial nerve, which is normally developed, follows an abnormal course; therefore, the IAM is either absent or very small if it carries the facial nerve. The facial nerve may penetrate directly into the temporal bone, passing through the mastoid bone into the stylomastoid foramen (case 2) or into the middle ear through a small IAM or a single foramen (case 1). In all cases, neither the labyrinthine or tympanic portions of the facial nerve nor the greater petrosal nerve recess or pyramidal eminence are developed. Thus, the absence of the otic capsule is associated with developmental anomalies of the second branchial arch. However, normal function of the seventh nerve indicates that development of the facial nerve is independent of development of the inner ear.
Michel aplasia may also be associated with skull base and vascular anomalies: platybasia, an abnormal course of the transverse sinus and jugular veins, and craniocervical junction anomalies (1). Furthermore, we hypothesize that arterial developmental anomalies (eg, aberrant carotid artery, persistent stapedial artery, or carotid artery fenestration) (19) may also be encountered. In our patients, the persistent stapedial artery ended at the mastoid portion of the facial nerve, as the geniculate ganglion was missing.
Because mesenchymal differentiation of petrous bone formation occurs at the same time as inner ear formation, hypoplasia or aplasia of the petrous apex may occur. The role of the otocyst in mesenchyma differentiation and in otic formation is known (18). Recently, an experimental study on targeted disruption of the mouse homeobox gene Hox-1.6 showed the genetic interaction on CNS development with associated defects in the formation of the inner ear and the cochlear and vestibular nerve, and with distortion of the pons (20). These anomalies partly demonstrated the relationship between the gene Hox-1.6 and rhombomeres 4 through 7, which are associated with neural migration into the second branchial arch. Surprisingly, the cochleovestibular nuclei were present in the genetically experimentally induced inner ear aplasia of mice (18).
In the hypoplastic petrous apex, areas of sclerotic bone can be observed in the temporal bone, and Michel aplasia may mimic labyrinthitis ossificans (17, 21). Flattening of the medial middle ear wall due to absence of development of the oval and round windows is the most characteristic diagnostic feature of Michel aplasia (6, 21).
Michel aplasia and associated findings differ from other severe inner ear dysplasia, sometimes described as Michel dysplasia (6, 22) or the so-called pre-Mondini syndromes. Michel aplasia contrasts with inner ear abnormalities in that it occurs later in embryologic development, when communication between the subarachnoid spaces and the inner ear leads to secondary CSF or perilymphatic fistula (17). Despite the close association between the middle ear and the subarachnoid spaces, Michel aplasia is not associated with a higher risk for meningitis, as no communication between the middle ear and the subarachnoid spaces exists.
The identification of Michel aplasia is of paramount importance in patients being assessed for possible cochlear implant surgery. Contrary to other congenital malformations, such as the Mondini and so-called pre-Mondini anomalies, which may profit from cochlear implants, in Michel aplasia the absence of the inner ear rules out the possibility of any cochlear implantation. The findings in our two patients clearly establish the need for imaging studies before electrical stimulation of the round window so that the risk of unnecessary general anesthesia can be avoided.
Surprisingly, our two patients had some auditory rehabilitation with an external ear device. The benefit of vibrotactile devices in Michel aplasia was previously reported by Brookhouser (23). We hypothesize that external sound may be directly transmitted to the brain stem by the facial nerve or by bone conduction. Moreover, our report suggests that despite the inner ear and cochlear nerve aplasia, the cochlear nuclei may be functional, as previous experimental work has reported (18).
Conclusion
Michel aplasia is a rare congenital inner ear anomaly defined by the absence of inner ear structures. Associated skull base anomalies should be identified, as they can lead to potential complications, especially if a surgical procedure of the middle ear is considered. Finally, the occurrence of bilateral inner ear aplasia in two siblings suggests a genetic origin due to new mutations, inherited as an autosomal dominant trait (23).
Acknowledgments
We thank W. Grauer and K. Shehata for their valuable contribution in clarifying the manuscript and P. Lasjaunias and J. Vignaud for their advice, especially as regards the embryologic development of the inner ear and cerebral vasculature.
Footnotes
Presented at the annual meeting of the American Society of Head and Neck Radiology, Toronto, Ontario, May 1997.
Address reprint requests to Kathlyn Marsot-Dupuch, MD, Service de Radiologie, Hôpital Saint-Antoine, 75012 Paris, France.
References
- 1.Michel P. Mémoire sur les anomalies congénitales de l'oreille interne. Gazette Méd de Strasbourg 1863;23:55-58 [Google Scholar]
- 2.Cruz M, Biscoito L, Branco G. Estudo por CT de 17 casos de anomalia congenital coclear. Acta Med Port 1993;6:371-375 [PubMed] [Google Scholar]
- 3.Sadler TW. Langman's Medical Embryology 5th ed. Baltimore: Williams & Wilkins; 1985;311-319
- 4.Larsen WJ. Embryologie Humaine Bruxelles: De Boeck & Lancier; 1996;352-359
- 5.Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope 1987;97(Suppl 40):2-14 [DOI] [PubMed] [Google Scholar]
- 6.Schuknecht HF. Mondini dysplasia: a clinical and pathological study. Ann Otol Rhinol Laryngol 1980;89(Suppl 65):1-23 [PubMed] [Google Scholar]
- 7.Kavanagh KT, Magill HL. Michel dysplasia: common cavity inner ear deformity. Pediatr Radiol 1989;19:343-345 [DOI] [PubMed] [Google Scholar]
- 8.Cho H, Nakai Y, Ezaki Y, Maruoka K, Miki Y, Konishi K. Light and electron microscopic studies of a case with simultaneous Mondini and Michel deformities of the inner ears. Arch Oto-Rhino-Laryngol 1987;243:361-365 [DOI] [PubMed] [Google Scholar]
- 9.Desnos J, Larget-Piet L, Riberi P, Cleirens P, Beucher A. Le syndrome malformatif branchio-oto-rénal. Ann Otolaryngol Chir Cervicofac 1979;96:849-861 [PubMed] [Google Scholar]
- 10.Isenberg SF, Tubergen LB. Unilateral complete aplasia of the inner ear with associated tracheo-oesophageal fistula, report of a case. Otolaryngol Head Neck Surg 1979;87:435-439 [DOI] [PubMed] [Google Scholar]
- 11.Rosendal T. Thalidomide and aplasia: hypoplasia of the otic labyrinth (letter). Lancet 1963;7283:724-725 [DOI] [PubMed] [Google Scholar]
- 12.Jorgensen MB, Kristensen HK, Buch NH. Thalidomide induced aplasia of the inner ear. J Laryngol 1964;78:1095-1101 [DOI] [PubMed] [Google Scholar]
- 13.Bouman NM, Kyrby-Keyser L, Dolan K. Mondini dysplasia and congenital cytomegalovirus infection. J Pediatr 1994;124:71-78 [DOI] [PubMed] [Google Scholar]
- 14.Steel KP, Bock GR. Hereditary inner ear abnormalities in animals: relationships with human abnormalities. Arch Otol-Rhinol-Laryngol 1983;109:22-29 [DOI] [PubMed] [Google Scholar]
- 15.Keeney G, Gebarski S, Brinderg JA. CT of severe inner ear anomalies, including aplasia in a case of Wildervanck syndrome. AJNR Am J Neuroradiol 1992;13:201-202 [PMC free article] [PubMed] [Google Scholar]
- 16.Lyndsay JR. Profound childhood deafness. Ann Otol Rhinol Laryngol 1973;82:1-121 [PubMed] [Google Scholar]
- 17.Phelps PD, King A, Michaels L. Cochlear dysplasia and meningitis. Am J Otol 1994;15:551-557 [PubMed] [Google Scholar]
- 18.Van de Water TR, Mc Phee JR. Determinants of otic capsule formation. Laryngoscope 1987;97:315-322 [PubMed] [Google Scholar]
- 19.Koenigsberg RA, Zito JL, Patel M, Swartz J, et al. Fenestration of the internal carotid artery: a rare mass of the hypotympanum associated with persistence of the stapedial artery. AJNR Am J Neuroradiol 1995;16:908-910 [PMC free article] [PubMed] [Google Scholar]
- 20.Chisaka O, Musci TS, Capecchi M. Developmental defects of the ear, cranial nerves and hindbrain resulting from targeted disruption of the mouse homeobox gene Hox-1.6. Nature 1992;355:516-520 [DOI] [PubMed] [Google Scholar]
- 21.Weissman JL. Hearing loss. Radiology 1996;199:593-611 [DOI] [PubMed] [Google Scholar]
- 22.Ramsey R. Neuroradiology 3rd ed. Philadelphia: Saunders; 1994;655
- 23.Brookhouser PE. Sensorineural hearing loss in children. In: Cummings CW, Fredrikson JM, Harker LA, Krause CJ, Schuller DE, eds. Otolaryngology-Head and Neck Surgery 2nd ed. St Louis: Mosby-Year Book; 1993;3080-3102