

Abstract
BACKGROUND AND PURPOSE: Cocaine and its metabolites can produce vasospasm, and cocaine-dependent patients are at increased risk for stroke. Based on previous case reports, we hypothesized that the incidence of hyperintense brain lesions observed on T2-weighted MR images would also be increased in asymptomatic cocaine-dependent individuals.
METHODS: Sixty-two male “crack” (smoked) cocaine–dependent participants ranging in age from 25 to 66 years were compared with 116 normal male control participants ranging in age from 25 to 80 years. Those with histories of neurologic symptoms or illnesses were excluded. The severity of hyperintense lesions was rated on a 0- to 3-point scale, and ratings of 3 were used in the data analysis as an indicator of a probable pathologic process. Three regions were separately rated: the cerebral white matter, insular subcortex white matter, and subcortical gray matter (basal ganglia and thalamus region).
RESULTS: Significantly increased risk of severe lesions was observed in the two white matter regions of the cocaine-dependent group (odds ratio of 16.7 and 20.3) but not in the subcortial gray matter region (odds ratio of 1.4). In the insula subcortex white matter, the risk of lesions increased with age in the cocaine-dependant sample, but remained essentially absent among normal controls through the age of 80 years. In the cerebral white matter, the relationship of age and risk of lesion among normal participants was similar in shape to that in cocaine-dependent participants, but equivalent risk was seen 20 years earlier among cocaine-dependent participants.
CONCLUSIONS: Cocaine-dependent participants had a significantly increased age-related risk of white matter damage. The possible clinical implications of this damage are discussed.
Cocaine dependence is associated with vascular events and is a common cause of clinically apparent stroke in young individuals, even in the absence of other risk factors (1–6). Vasospasm induced by cocaine and its metabolites (7, 8), with or without secondary intravascular thrombosis (9), is thought to be the principal mechanism of cocaine-induced coronary and CNS ischemic toxicity (4, 7, 9–12).
Cocaine has substantial effects on cerebral blood flow, with a 25% to 30% reduction of global cerebral blood flow being shown experimentally in human cocaine users receiving a single 40-mg IV administered cocaine infusion (13, 14). Using MR angiography, cerebral vasospasm of large arteries has been shown in humans receiving 0.2 to 0.4 mg/kg IV-administered cocaine (15). These doses are modest compared with illicit cocaine use patterns; most chronic cocaine-dependent patients use an average of 3.5 g per week, but in some individuals, the dose can range up to 10 to 30 g per day (16, 17). In rats, exposure to chronic high doses of cocaine can result in capillary disruption (18).
Studies of cerebral perfusion conducted in asymptomatic cocaine-dependent participants without clinically apparent CNS symptoms have shown widespread and frequent (70–100%) perfusion defects (19–22). Such defects can be observed even after 6 months of verified abstinence (22). These perfusion studies evaluated mostly young cohorts (mean age, 25–32 years), and some MR imaging examinations failed to reveal structural lesions corresponding to the functional deficits (19, 22).
The prevalence of ischemic brain lesions in asymptomatic cocaine-dependent participants has not been investigated in controlled studies that evaluate a wide age range. Two case report series evaluating structural abnormalities in asymptomatic cocaine-dependent participants using MR imaging detected high-intensity focal brain lesions (23, 24). Approximately 25% to 60% of cocaine-abusing patients with acute strokes have ischemic infarcts (2, 4, 10, 25). Of the patients who present with ischemic symptoms, the majority (50–80%) have infarcts involving the middle cerebral artery (MCA) (2, 4, 10). They may be older on average than those presenting with hemorrhages (1, 6) and may more commonly become symptomatic after a delay with respect to when they last used cocaine (6, 10).
Impaired endothelium-dependent vasorelaxation in long-term cocaine users (12, 26), as well as the formation of longer half-life vasoactive metabolites of cocaine (8, 27, 28), may explain the delay in time from the last cocaine administration to the occurrence of ischemic symptoms (6, 8–10). This delay between cocaine use and ischemic symptoms, combined with the possibility that ischemic lesions may be located in areas of the brain (such as the cerebral white matter) that produce less severe or no apparent symptoms, reduce the likelihood that an individual will request medical attention. This raises the possibility that the relationship between cocaine use and ischemic stroke may be under-reported compared with hemorrhagic stroke (2) and may explain why some studies have not found an association between cocaine use and clinically apparent stroke (29).
MR imaging is superior to CT for detecting small ischemic changes (25, 30–33). Variability in identifying and quantifying high-intensity lesions (34) as well as the presence of risk factors (ie, age, hypertension, diabetes) for white matter hyperintensities in asymptomatic individuals (32) require the use of matched control samples (24). In the current study, we evaluated T2-weighted MR images to assess the incidence of severe hyperintense brain lesions across a wide age range of asymptomatic cocaine-dependent and normal control participants.
Methods
Participants
Cocaine-dependent participants were recruited from patients admitted to inpatient and outpatient treatment programs and research clinics of a metropolitan Department of Veterans Affairs Medical Center. The cocaine-dependent participants were 25- to 66-year-old men with histories of cocaine dependence longer than 1 year, current cocaine dependence diagnosis by Diagnostic and Statistical Manual of Mental Disorders-IV (DSM-IV) criteria (35), and self-reported use of at least $50/week, primarily by smoking. The cocaine-dependent group reported relatively chronic illness, with a mean length of exposure to cocaine of 10.8 years (SD = 5.7 years; range, 1–30 years). Of the 62 cocaine-dependent participants, 53 (85%) were African-American, seven (11%) were Caucasian, and two (3%) were of other races.
Cocaine-dependent participants were excluded if they had other psychiatric disorders of such severity as to require the use of pharmacotherapy; met DSM-IV criteria for dependence to opiates, benzodiazepines, or other sedative-hypnotics; or had a history of clinically significant medical conditions that are also likely to result in structural brain abnormalities (stroke, transient ischemic attack, cerebral infection, hypertension, diabetes, head trauma resulting in loss of consciousness of greater than 15 minutes). The final cocaine-dependent sample included 12 (19%) participants with current alcohol dependence, 16 (26%) with histories (more than 2 years ago) of alcohol dependence, and three (5%) with past amphetamine dependence.
Male control participants were recruited from community volunteers and ranged in age from 25 to 80 years. Older control participants, whose ages were beyond the oldest cocaine-dependent participants, were included to compare the age-related patterns of hyperintensity incidence in the two groups. Of the 116 control participants, 14 (12%) were African-American, 83 (72%) were Caucasian, and 19 (16%) were of other races. Exclusion criteria for the control participants were the same as those for the cocaine-dependent group. In addition, the control participants did not have histories of significant psychopathology or substance dependence and no first-degree relatives diagnosed with a major psychiatric disorder.
The above criteria excluded 12 cocaine-dependent participants (six with hypertension, three with diabetes, and three with histories of brain trauma with loss of consciousness of 45 minutes to 6 days) and two normal control participants (history of brain trauma with loss of consciousness of 9 and 48 hours, respectively). Written informed consent was obtained from all participants after a complete description of the study.
MR Evaluation
Imaging Protocol
MR imaging was performed using a 1.5-T instrument with an imaging protocol consisting of three sequences. Coronal pilot spin-echo images (100/30/1 [TR/TE/excitations]) with 10-mm thickness were used to evaluate symmetrical positioning of the head. If a patient's head was noticeably tilted laterally, he was repositioned and a new coronal pilot image was acquired. The image was then used to align the acquisition grid of the subsequent sagittal images. Pilot sagittal images were obtained from a second spin-echo sequence (550/26/2) with a 5-mm thickness. To obtain a true mid-sagittal image, the middle section of this series was aligned on the coronal pilot. The remaining sagittal images enabled visualization of the entire brain and were used to position the subsequent axial sequence. A transverse dual spin-echo sequence (2500/20, 90/2) with a 256 × 192-view matrix and a 25-cm field of view was used to acquire the 3-mm-thick contiguous axial images used for the ratings of hyperintensity.
MR Ratings
The images were rated visually for hyperintense lesions by an experienced radiologist with training in neuroradiology (D.B.H.), who was blinded to the subjects' clinical data. Separate ratings of the severity of hyperintensities were obtained for subcortical gray and white matter regions. Based on previous MR studies (24, 36), three regions were selected to be rated: cerebral white matter, subcortical gray matter (basal ganglia and thalamus region), and insular subcortex white matter. Representative images of hyperintense lesions exemplifying the three grades of severity are depicted in Figure 1.
fig 1.
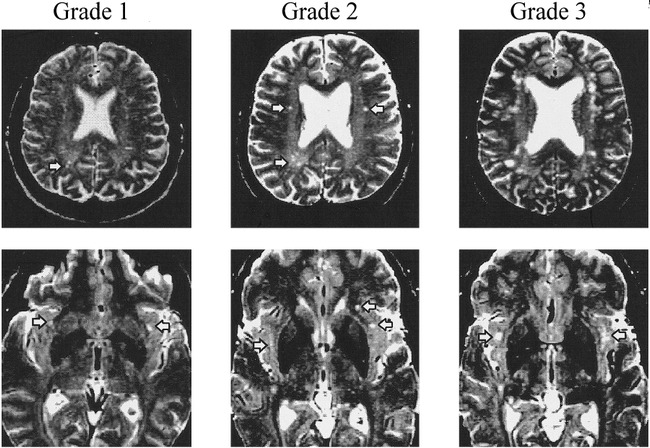
Representative images of the three grades of lesions in cerebral and insular subcortex white matter regions. Grade 0 is not included and contains no hyperintensities. Grade 1 has minimal changes, Grade 2 involves mild changes, and Grade 3 indicates moderate-to-severe changes, thought to be significant indicators of a possible pathologic process. Arrowheads point to rated T2-weighted hyperintensities.
Top panel, Cerebral white matter hyperintensities (Grade 3 image has too many large hyperintensities to be defined by arrowheads).
Lower panel, hyperintensities in the insular subcortex white matter region.
The rater selected abnormal foci and avoided including expected hyperintensities, such as those produced by partial volume effects of sulcal CSF or uniform round cortical vessels appearing in typically anticipated positions within sulci. Only hyperintensities identified on both early and late echo images were considered in the rating, which was based on intensity, size, and confluence of lesions.
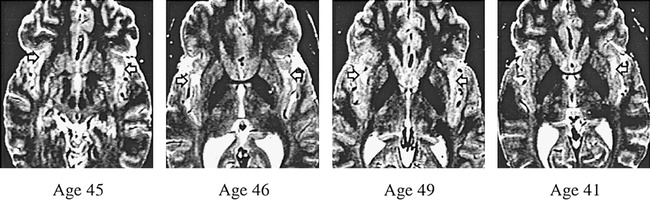
The absence of hyperintensities in a particular brain region was rated as zero. A rating of 1 involved minimal changes, which are rarely mentioned in radiologic reports because they are generally deemed to be of no significance. A rating of 2 was defined as definite brain tissue changes of mild severity and of questionable significance. Moderate to severe changes, thought to be significant indicators of a possible pathologic process, received a rating of 3. In asymptomatic older normal individuals, low-grade hyperintensities (ratings of 1 and 2) have not been correlated with cognitive or blood flow changes whereas lacunae (rating of 3) were associated with these changes (37, 38). Therefore, the data were analyzed using the severity ratings of 3 only. Representative images of cerebral and insular subcortex white matter lesions from images receiving a rating of 3 are shown in Figures 2 and 3.
fig 2.
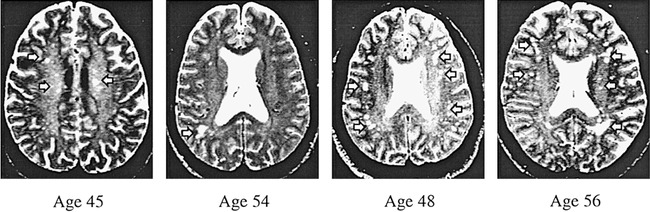
Axial T2-weighted spin-echo images (2500/90/2) of four cocaine-dependent participants. These are examples of severe cerebral white matter lesions (arrows), graded as 3 on an increasing severity scale of 0 to 3. These participants were randomly selected from those with cerebral white matter ratings of 3
To assess inter-rater reliability, the severity of the hyperintensities in a subset of 59 images was independently rerated by the MR researcher (G.B.). Inter-rater reliability coefficients (intraclass r) were highly statistically significant (P < .0001) for the hyperintensity severity ratings of the three regions and the summary composite variable: (rxx = .94, F = 30.6) for cerebral white matter, (rxx =.91, F = 20.2) for insular subcortex white matter, (rxx = .89, F = 17.8) for subcortical gray matter, and (rxx = .94, F = 34.8) for the composite score of the two white matter regions.
Data Analyses
The rates of occurrence of severe lesions (radiologist's rating of 3) among cocaine-dependent and normal participants were compared in multiple logistic regression analyses (SAS Proc CATMOD). The models included age, diagnosis, and their interaction. These were calculated in three separate regions (cortical white matter, insular subcortex white matter, and subcortical gray matter). A summary composite variable was also defined as an indicator of severe lesions in either of the two white matter regions. Odds ratios were computed to describe the strength of association between diagnosis and pathologic lesions (these estimates were computed at age 50 years, approximately the mean age of the sample). Follow-up multiple logistic regression analyses were conducted to evaluate the effects of race and years of drug-using experience (within the cocaine-dependent sample). Race was coded as a dichotomy (African-American versus non-African-American). The impact of race on the results was investigated because, despite the exclusion of participants with diagnosed hypertension, African-Americans have been reported to have higher blood pressure than non-African-Americans and hypertension may contribute to cerebrovascular disease (29). To analyze years of drug-using experience, normal participants were coded zero. Because the diagnostic group was also included in the statistical model, this statistical device yields regression estimates for that variable equivalent to what would be obtained if the cocaine-dependent group were analyzed separately.
Results
In the white matter region of the insula subcortex, the interaction of age by diagnosis was statistically significant (χ2 = 4.26, df = 1, P = .039), indicating that the nature of the relationship between increasing age and risk of lesion was not the same in the two samples. As shown in Figure 4, the risk of lesions in this region increased with age in the cocaine-dependent sample but remained essentially absent among normal individuals through the entire age range studied. Estimated at age 50 years, the probability of a lesion being present in this region was 20.3 times greater among cocaine-dependent participants (χ2 = 12.11, df = 1, P = .0005).
fig 4.
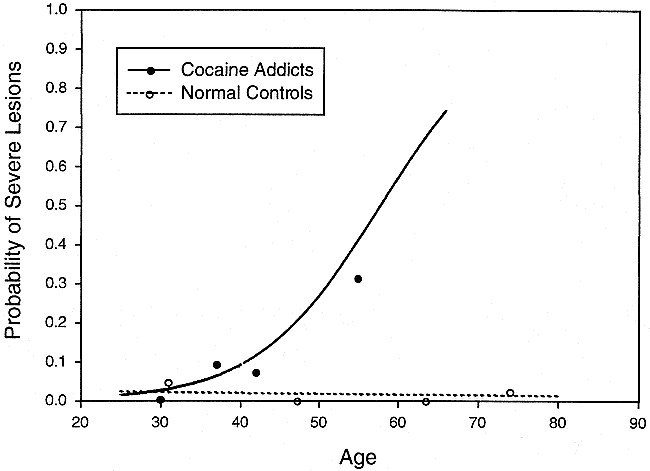
Percent of participants with severe lesions in the insular subcortex white matter versus age. Cocaine-dependent participants had a significantly increased incidence of severe insular subcortex white matter lesions compared with normal control participants. The age dependence of the lesion incidence was significantly different in the two groups (eg, the shapes of the two lines differ). The lines are derived from logistic regression analysis of 62 cocaine addicts and 116 normal control participants. The raw data from each group were divided into age quartiles and are displayed at the median age of the quartile. The sample sizes of the age quartiles (in order of ascending age) were 10, 22, 14, and 16 for the cocaine group and 28, 29, 28, and 31 for the normal group. Severe lesions graded as grade 3 based on a 0 to 3 severity scale
In the cerebral white matter, only the diagnosis had a significant effect (χ2 = 5.1, df = 1, P = .024). Figure 5 illustrates that the relationship between age and risk of lesion among control participants is similar in shape to that of cocaine-dependent participants, but equivalent risk is seen among cocaine-dependent participants approximately 20 years earlier (odds ratio = 16.7).
fig 5.
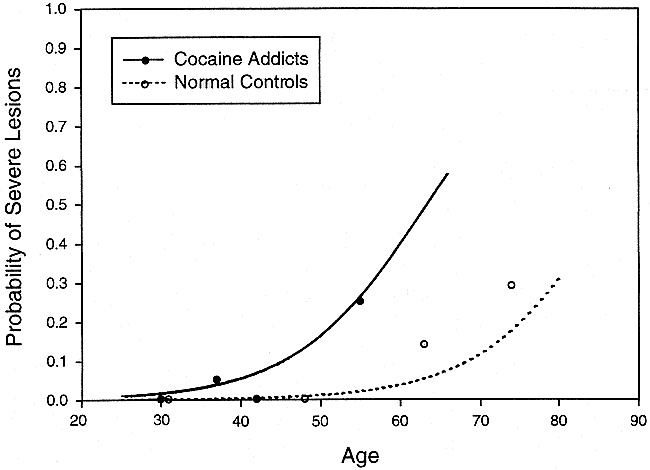
Percent of participants with severe lesions in the cerebral white matter versus age. Cocaine-dependent participants have a significantly increased incidence of severe cerebral white matter lesions compared with normal control participants. The lines are derived from logistic regression analysis of 62 cocaine addicts and 116 normal control participants. The raw data from each group were divided into age quartiles and are displayed at the median age of the quartile. The sample sizes of the age quartiles (in order of ascending age) were 10, 22, 14, and 16 for the cocaine group and 28, 29, 28, and 31 for the normal group. Severe lesions graded as grade 3 based on a 0 to 3 severity scale
The analysis of the composite variable based on lesion in either region yielded the most significant diagnostic group differences (χ2 = 14.5, df = 1, P = .0001, odds ratio = 20.7). The apparent difference in shape of these functions displayed in Figure 6 did not reach statistical significance (interaction χ2 = 2.15, df = 1, P = .14). In the subcortical gray matter, no effects were statistically significant (all, P > .7). The addition of the indicators, race and years of cocaine use, did not alter these results. Neither of those variables was significantly related to risk of lesion in any area.
fig 6.
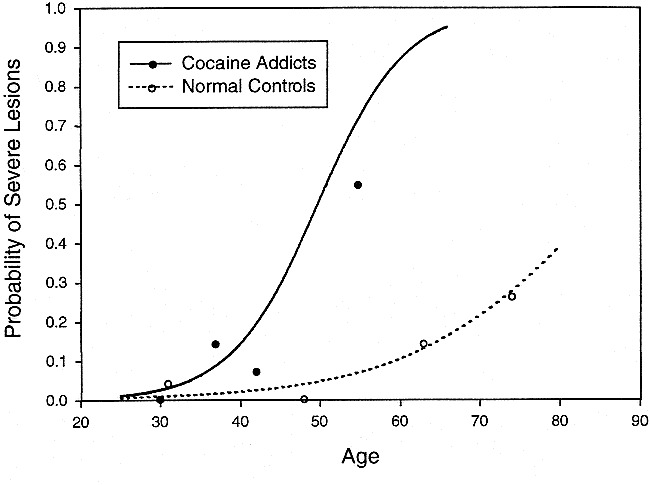
Percent of participants with severe lesions in the insular subcortex or cerebral white matter versus age. When the combined incidence of white matter lesions (cerebral or insular subcortex white matter) is used in the analysis, cocaine-dependent participants have a markedly significantly increased incidence of severe white matter lesions compared with normal control participants. The lines are derived from logistic regression analysis of 62 cocaine addicts and 116 normal control participants. The raw data from each group were divided into age quartiles and are displayed at the median age of the quartile. The sample sizes of the age quartiles (in order of ascending age) were 10, 22, 14, and 16 for the cocaine group and 28, 29, 28, and 31 for the normal group. Severe lesions graded as grade 3 based on a 0 to 3 severity scale
Finally, two of the older control participants (age 68 and 79 years, respectively) had small cortical infarcts but did not show evidence of lesions with severity ratings of 3 in either white matter regions. None of the cocaine-dependent participants had evidence of cortical infarcts.
Discussion
The MR data suggest that cocaine-dependent participants without clinically apparent cerebrovascular symptoms have an increased risk of white matter damage when compared with age-matched normal control participants. The increased frequency of lesions was strongly age-related and limited to white matter regions. The cerebral hemispheres and insula white matter lesion distribution is consistent with reports that the majority (50–80%) of cocaine-dependent patients presenting with infarctions had MCA involvement (2, 4, 10). Almost all of the lesions appeared hyperintense on the T2-weighted images, consistent with the premise that transient occlusion of arteries in the territory of the MCA can cause incomplete infarction that usually does not progress to frank cavitation and appear as hyperintensities (39, 40). In the absence of pathologic or angiographic proof, however, other mechanisms causing such lesions cannot be ruled out.
In this asymptomatic cocaine-dependent population, no cortical lesions were noted. Almost all lesions occurred in the territories supplied by either the medullary arteries (vessels that perforate the cortex and extend toward the lateral ventricles) or the perforating (lenticulostriate) arteries of the MCA (39, 41). These arteries usually have singular territories and do not anastomose even when (as in the case of the insula and its subcortical white matter) multiple branches of the MCA supply the region (39, 41–43). Although the multiple source arrangement of the insula region may offer some protection from hypotension (42), it clearly offers no protection from the effects of cocaine, as shown by the fact that insula lesions were observed almost exclusively in cocaine-dependent participants. This observation lends support to vasoconstriction of individual arteries as a possible mechanism underlying the lesions.
Cocaine's very short (40–50 minutes) plasma half-life can, in large part, be attributed to its immediate redistribution into tissues, particularly in the human brain (44), which poses no barrier to this lipophilic molecule (45). With its short half-life, the vasoconstrictive properties of cocaine cannot readily explain the cerebrovascular symptoms that occur hours to days after use (6, 8–10). In vivo, benzoylecgonine (BE), the primary pharmacologically active metabolite of cocaine, has a 6-hour half-life (44, 46, 47) and is a potent neurotoxin, causing agitation, seizures, and vasoconstriction (8, 48–51). Unlike cocaine, the uptake of BE into the brain is very low (52), but butyrylcholine esterase, the enzyme that catalyzes the metabolism of cocaine to BE, is abundantly present in cerebral white matter (30, 46, 53–55) as well as in plasma and in the liver. Because of its long half-life, BE may accumulate in brain tissue in higher concentrations than cocaine (45), resulting in a similar if not greater level of neurotoxicity (27), and it could trigger vasoconstriction, resulting in hyperintensities (8). In addition, elevated BE levels deep in the white matter (beyond the muscular arteriole level) may be neurotoxic through other (non-vasoconstrictive) mechanisms such as direct toxicity to brain cells or the vascular endothelium (18, 42).
Why increased rates of hyperintensities are observed only in the white matter regions of the cerebrum and insula, but not in the gray matter region of the basal ganglia and thalamus, is unknown. One possibility is that the enzymatic metabolism of cocaine to BE by butyrylcholine esterase may place white matter regions, where high enzyme activity is found in oligodendroglial cells, at especially high risk for vasoconstriction. Even though brain white matter has significantly higher levels of butyrylcholine esterase than cortical gray matter (56–59), subcortical gray matter structures have also been reported to have substantial levels of butyrylcholine esterase; however, enzyme activity in the subcortical gray matter is observed in neurons (56). Another possible explanation for the apparent differential susceptibility of white matter to develop hyperintensities involves our participant selection. For example, hyperintensities in the subcortical gray matter/internal capsule regions may be more likely to result in noticeable symptoms (60) and more likely to develop into intraparenchymal hemorrhages, which are most often symptomatic (4, 5, 25). Thus, many cocaine-dependent individuals with subcortical gray matter region hyperintensities would not be present in the current sample of asymptomatic participants.
Another important observation was the striking age-related pattern of the increase in white matter lesions, which was not explained by the number of years of cocaine use. In animal models, experimental transient MCA occlusion causes damage limited to the lateral aspect of the striatum (61, 62) and insula (63). Such transient occlusion has been shown to result in age-related deficits in locomotion and passive avoidance as well as altered pharmacologic responses to dopaminergic agents (61, 62). These studies support the possibility that cocaine-dependent patients who suffer from severe lesions in the MCA territory may have subtle neurocognitive deficits (22, 64–66). Individuals with these subtle deficits may not respond to treatment or be less likely to quit cocaine use or both and could therefore be over-represented in the older cocaine-dependent population.
The current cross-sectional study cannot readily answer questions relating to differential age-related sample attrition. To address such issues, prospective studies are needed. Several additional limitations of the study must be acknowledged. First, meaningful data regarding female addicts were not available, and the results cannot be generalized to female cocaine-dependent patients. Second, there is no certainty that the increased hyperintensities are related to cocaine use alone. For example, cocaine is sometimes “cut” with amphetamines and is often used in conjunction with other drugs of abuse, such as alcohol, producing other toxic metabolites, such as cocaethylene. Even diet, lifestyle, race, higher rates of tobacco dependence, and past IV drug use in the cocaine-dependent participants, or subtle differences in blood pressure, may have synergistically contributed to the observed differences in hyperintensities. Third, because the extent of drug abuse in the control group was documented by history alone, it is possible that their drug use was more extensive than reported. To the extent that this level of drug abuse would have resulted in an increase of hyperintensities in the control group, it would also have reduced the significance of the differences between the two groups.
Finally, it is possible that in this cross-sectional sample, the age-related increase in hyperintensities in the cocaine-dependent sample may be an underestimate because of differential mortality. Sudden, unexpected death resulting from cocaine use in young, otherwise healthy individuals occurs in an idiosyncratic manner and is commonly thought to be arrhythmogenic in nature, although the exact mechanism of death is rarely documented (67). Cocaine-related sudden death is a significant cause of mortality among young adults (68, 69), and in those who use cocaine and alcohol concomitantly, risk of sudden death is especially increased (16). Cardiac mortality is the most common cause of death in stroke patients and is associated with an annual cardiac mortality of 5% to 10% (70, 71). The insula region is involved in human heart rate regulation, and its damage may encourage a proarrhythmic state (72). Thus, if the kind of insular subcortex white matter lesions we observed in the cocaine-dependent population increases risk of sudden death, the frequency of the subinsular lesions we detected may be an underestimation because of selective mortality.
These findings have several important possible implications. If the possibility that premature appearance of severe white matter lesions may be associated with drug use was mentioned in the differential-diagnosis section of radiologic reports, it could help referring physicians to address the issue of covert cocaine use and its life-threatening risks with their patients better. As the cocaine-dependent population ages, an accelerated rate of white matter disease may have important epidemiologic implications. For example, the rate of dementia may increase as a result of the epidemic of crack cocaine use, which began in the mid-1980s (73, 74). Finally, the presence of white matter lesions in cocaine-dependent patients may affect their prognosis. Even though the relationship of hyperintensities to treatment outcome is unknown, it is possible that medications that prevent or ameliorate the vascular/perfusion effects of cocaine may be useful in the treatment of certain subgroups of cocaine-dependent patients.
Conclusion
The MR data suggest that cocaine-dependent participants without clinically apparent cerebrovascular symptoms have an increased frequency of severe T2 hyperintense lesions. The incidence of these lesions is strongly age-related and limited to the cerebral and insular subcortex white matter regions.
fig 3.
Axial T2-weighted spin-echo images (2500/90/2) of four cocaine-dependent participants. These are examples of severe insular subcortex white matter lesions (arrows), graded as 3 on an increasing severity scale of 0 to 3. These participants were randomly selected from those with cerebral white matter ratings of 3
Acknowledgments
This work was supported by the Medication Development Division of the National Institute on Drug Abuse (1YO1 DA 50038), Research Grant AG-11595 from the National Institute of Aging, the Research and Psychiatry Services of the Department of Veterans Affairs, and the Marie Wilson Howells Endowment. The authors thank Sun Sook Hwang for data management and statistical support, Marguerite Callinan and Ewa Witt for coordinating the study, Yolanda Yamat for technical assistance, and Jeffrey Cummings, MD, for reviewing the manuscript.
Footnotes
The data herein were presented in part at the 149th Annual Meeting of the American Psychiatric Association, New York, 1996.
Address reprint requests to George Bartzokis, MD, North Little Rock VA Medical Center (116A/NLR), 2200 Fort Roots Drive, Building 170, North Little Rock, AR 72114.
References
- 1.Levine SR, Welch KM. Cocaine and stroke. Stroke 1988;19:779-783 [DOI] [PubMed] [Google Scholar]
- 2.Jacobs IG, Roszler MH, Kelly JK, Klein MA, Kling GA. Cocaine abuse. Neurovascular complications. Radiology 1989;70:223-227 [DOI] [PubMed] [Google Scholar]
- 3.Kokkinos J, Levine SR. Stroke. Neurol Clin 1993;11:577-590 [PubMed] [Google Scholar]
- 4.Daras M, Tuchman AJ, Koppel BS, Samkoff LM, Weitzner I, Marc J. Neurovascular complications of cocaine. Acta Neurol Scand 1994;90:124-129 [DOI] [PubMed] [Google Scholar]
- 5.Nolte KB, Brass LM, Fletterick CF. Intracranial hemorrhage associated with cocaine abuse. A prospective autopsy study. Neurology 1996;46:1291-1296 [DOI] [PubMed] [Google Scholar]
- 6.Fessler RD, Esshaki CM, Stankewitz RC, Johnson RR, Diaz FG. The neurovascular complications of cocaine. Surg Neurol 1997;47:339-345 [DOI] [PubMed] [Google Scholar]
- 7.He GQ, Zhang A, Altura BT, Altura BM. Cocaine-induced cerebrovasospasm and its possible mechanism of action. J Pharmacol Exp Ther 1994;268:1532-1539 [PubMed] [Google Scholar]
- 8.Madden JA, Konkol RJ, Keller PA, Alvarez TA. Cocaine and benzoylecgonine constrict cerebral arteries by different mechanisms. Life Sci 1995;56:679-686 [DOI] [PubMed] [Google Scholar]
- 9.Konzen JP, Levine SR, Garcia JH. Vasospasm and thrombus formation as possible mechanisms of stroke related to alkaloidal cocaine [comments]. Stroke 1995;26:1114-1118 [DOI] [PubMed] [Google Scholar]
- 10.Levine SR, Brust JC, Futrell N, et al. Cerebrovascular complications of the use of the “;t1crack” form of alkaloidal cocaine [comment]. N Engl J Med 1990;323:699-704 [DOI] [PubMed] [Google Scholar]
- 11.Isner JM, Chokshi SK. Cocaine and vasospasm [editorial comment]. N Engl J Med 1989;321:1604-1606 [DOI] [PubMed] [Google Scholar]
- 12.Havranek EP, Nademanee K, Grayburn PA, Eichhorn EJ. Endothelium-dependent vasorelaxation is impaired in cocaine arteriopathy. J Am Coll Cardiol 1996;28:1168-1174 [DOI] [PubMed] [Google Scholar]
- 13.Wallace EA, Wisniewski G, Zubal G, et al. Acute cocaine effects on absolute cerebral blood flow. Psychopharmacology 1996;28:17-20 [DOI] [PubMed] [Google Scholar]
- 14.Kukes TJ, Kaufman MJ, Levin JM, et al. Cocaine-induced cerebral blood volume reduction in females. Proceedings of the 59th Annual Scientific Meeting, College on Problems of Drug Dependence. Nashville, Tennessee. Book of Abstracts 1997;86: [Google Scholar]
- 15.Kaufman MJ, Levin JM, Ross M, et al. Cocaine-induced cerebral vasospasm in humans. Detection with magnetic resonance angiography. Proceedings of the 59th Annual Scientific Meeting, College on Problems of Drug Dependence. Nashville, Tennessee. Book of Abstracts 1997;8: [Google Scholar]
- 16.Gawin FH, Kleber HD. Medical Management of Cocaine Withdrawal.. New York: Werber/Mystad Associates; 1986;
- 17.Weiss RD, Gawin FH. Protracted elimination of cocaine metabolites in long-term high-dose cocaine abusers. Am J Med 1988;85:879-880 [DOI] [PubMed] [Google Scholar]
- 18.Barroso-Moguel R, Villeda-Hernandez J, Mendez-Armenta M, Rios C. Brain capillary lesions produced by cocaine in rats. Toxicol Lett 1997;92:9-14 [DOI] [PubMed] [Google Scholar]
- 19.Volkow ND, Mullani N, Gould KL, Adler S, Krajewski K. Cerebral blood flow in chronic cocaine users. A study with positron emission tomography. Br J Psychiatry 1988;152:641-648 [DOI] [PubMed] [Google Scholar]
- 20.Tumeh SS, Nagel JS, English RJ, Moore M, Holman BL. Cerebral abnormalities in cocaine abusers. Demonstration by SPECT perfusion brain scintigraphy. Work in progress. Radiology 1990;176:821-824 [DOI] [PubMed] [Google Scholar]
- 21.Weber DA, Franceschi D, Ivanovic M, et al. SPECT and planar brain imaging in crack abuse. Iodine-123-iodoamphetamine uptake and localization. J Nucl Med 1993;34:899-907 [PubMed] [Google Scholar]
- 22.Strickland TL, Mena I, Villanueva-Meyer J, et al. Cerebral perfusion and neuropsychological consequences of chronic cocaine use. J Neuropsychiatry Clin Neurosci 1993;5:419-427 [DOI] [PubMed] [Google Scholar]
- 23.Volkow ND, Valentine A, Kulkarni M. Radiological and neurological changes in the drug abuse patient. A study with MRI. J Neuroradiol 1988;15:288-293 [PubMed] [Google Scholar]
- 24.Bartzokis G, Beckson M, Ling W. Clinical and MRI evaluation of psychostimulant neurotoxicity. NIDA Res Monogr 1996;163:300-317 [PubMed] [Google Scholar]
- 25.Brown E, Prager J, Lee HY, Ramsey RG. CNS complications of cocaine abuse. Prevalence, pathophysiology, and neuroradiology. AJR Am J Roentgenol 1992;159:137-147 [DOI] [PubMed] [Google Scholar]
- 26.Copeland JR, Willoughby KA, Police RJ, Ellis EF. Repeated cocaine administration reduces bradykinin-induced dilation of pial arterioles. Am J Physiol 1996;271:H1576-H1583 [DOI] [PubMed] [Google Scholar]
- 27.Lin Y, Leskawa KC. Cytotoxicity of the cocaine metabolite benzoylecgonine. Brain Res 1994;643:108-114 [DOI] [PubMed] [Google Scholar]
- 28.Andrews P. Cocaethylene toxicity. J Addict Dis 1997;16:75-84 [DOI] [PubMed] [Google Scholar]
- 29.Qureshi AI, Akbar MS, Czander E, Safdar K, Janssen RS, Frankel MR. Crack cocaine use and stroke in young patients. Neurology 1997;48:341-345 [DOI] [PubMed] [Google Scholar]
- 30.Awad IA, Johnson PC, Spetzler RF, Hodak JA. Incidental subcortical lesions identified on magnetic resonance imaging in the elderly. II. Postmortem pathological correlations. Stroke 1986;17:1090-1097 [DOI] [PubMed] [Google Scholar]
- 31.Brown JJ, Hesselink JR, Rothrock JF. MR and CT of lacunar infarcts. AJR Am J Roentgenol 1988;151:367-372 [DOI] [PubMed] [Google Scholar]
- 32.Mineura K, Sasajima H, Kikuchi K, et al. White matter hyperintensity in neurologically asymptomatic subjects. Acta Neurol Scand 1995;92:151-156 [DOI] [PubMed] [Google Scholar]
- 33.Fazekas F, Fazekas G, Schmidt R, Kapeller P, Offenbacher H. Magnetic resonance imaging correlates of transient cerebral ischemic attacks. Stroke 1996;27:607-611 [DOI] [PubMed] [Google Scholar]
- 34.Yetkin FZ, Haughton VM, Fischer ME, et al. High-signal foci on MR images of the brain: observer variability in their quantification. AJR Am J Roentgenol 1992;159:185-188 [DOI] [PubMed] [Google Scholar]
- 35. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders.. 4th ed. Washington, DC: American Psychiatric Press; 1994;
- 36.Goldstein IB, Bartzokis G, Hance DB, Shapiro D. Relationship between blood pressure and subcortical lesions in healthy elderly people. Stroke 1998;29:765-772 [DOI] [PubMed] [Google Scholar]
- 37.Kobari M, Meyer JS, Ichijo M, Oravez WT. Leukoaraiosis. Correlation of MR and CT findings with blood flow, atrophy, and cognition. AJNR Am J Neuroradiol 1990;11:273-281 [PMC free article] [PubMed] [Google Scholar]
- 38.Fushimi H, Inoue T, Yamada Y, Udaka F, Kameyama M. Asymptomatic cerebral small infarcts (lacunae), their risk factors and intellectual disturbances. Diabetes 1996;45[suppl 3]:S98-S100 [DOI] [PubMed] [Google Scholar]
- 39.Bogousslavsky J, Regli F. Centrum ovale infarcts. Subcortical infarction in the superficial territory of the middle cerebral artery [comments]. Neurology 1992;42:1992-998 [DOI] [PubMed] [Google Scholar]
- 40.Garcia JH, Lassen NA, Weiller C, Sperling B, Nakagawara J. Ischemic stroke and incomplete infarction. Stroke 1996;27:761-765 [DOI] [PubMed] [Google Scholar]
- 41.Shellshear JL. The subcortical gray matter arteries of the forebrain and their functional significance. J Anat 1921;55:27-35 [PMC free article] [PubMed] [Google Scholar]
- 42.Moody DM, Bell MA, Challa VR. Features of the cerebral vascular pattern that predict vulnerability to perfusion or oxygenation deficiency. An anatomic study. AJNR Am J Neuroradiol 1990;11:431-439 [PMC free article] [PubMed] [Google Scholar]
- 43.Damasio H. A computed tomographic guide to the identification of cerebral vascular territories. Arch Neurol 1983;40:138-142 [DOI] [PubMed] [Google Scholar]
- 44.Saady JJ, Bowman ER, Aceto MD. Cocaine, ecgonine methyl ester, and benzoylecgonine plasma profiles in rhesus monkeys. J Anal Toxicol 1995;19:571-575 [DOI] [PubMed] [Google Scholar]
- 45.Spiehler VR, Reed D. Brain concentrations of cocaine and benzoylecgonine in fatal cases. J Forensic Sci 1985;30:1003-1011 [PubMed] [Google Scholar]
- 46.Isenschmid DS, Fischman MW, Foltin RW, Caplan YH. Concentration of cocaine and metabolites in plasma of humans following intravenous administration and smoking of cocaine [comments]. J Anal Toxicol 1992;16:311-314 [DOI] [PubMed] [Google Scholar]
- 47.Cone EJ, Hillsgrove M, Darwin WD. Simultaneous measurement of cocaine, cocaethylene, their metabolites, and “crack” pyrolysis products by gas chromatography-mass spectrometry. Clin Chem 1994;40:1299-1305 [PubMed] [Google Scholar]
- 48.Benowitz NL. Clinical pharmacology and toxicology of cocaine. Pharmacol Toxicol 1993;72:3-12 [published erratum appears in Pharmacol Toxicol 1993;72: 343 ] [DOI] [PubMed] [Google Scholar]
- 49.Erzouki HK, Baum I, Goldberg SR, Schindler CW. Comparison of the effects of cocaine and its metabolites on cardiovascular function in anesthetized rats. J Cardiovasc Pharmacol 1993;22:557-563 [DOI] [PubMed] [Google Scholar]
- 50.Schuelke GS, Konkol RJ, Terry LC, Madden JA. Effect of cocaine metabolites on behavior. Possible neuroendocrine mechanisms. Brain Res Bull 1996;39:43-48 [DOI] [PubMed] [Google Scholar]
- 51.Hedaya MA, Pan WJ. Cocaine and alcohol interactions in naive and alcohol-pretreated rats. Drug Metab Dispos 1996;24:807-812 [PubMed] [Google Scholar]
- 52.Gatley SJ, Yu DW, Fowler JS, et al. Studies with differentially labeled [11C]cocaine, [11C]norcocaine, [11C]benzoylecgonine, and [11C]- and 4’-[18F]fluorococaine to probe the extent to which [11C]cocaine metabolites contribute to PET images of the baboon brain. J Neurochem 1994;62:1154-1162 [DOI] [PubMed] [Google Scholar]
- 53.Mendal B, Rudney H. Butyrylcholine estrase in the white matter. J Biochem 1953;43:59-63 [Google Scholar]
- 54.Lumsden CE. The problem of correlation of quantitative methods and tissue morphology in the central nervous system. The distribution of cholinesterases. In: Richter D, ed Metabolism of the Nervous System New York: Plenum Press; 1957;91-100
- 55.Carmona GN, Baum I, Schindler CW, et al. Plasma butyrylcholine esterase activity and cocaine half-life differ significantly in rhesus and squirrel monkeys. Life Sci 1996;59:939-943 [DOI] [PubMed] [Google Scholar]
- 56.Friede RL. A comparative histochemical mapping of the distribution of butyrylcholine esterase in the brains of four species of mammals, including man. Acta Anat (Basel) 1967;66:161-177 [DOI] [PubMed] [Google Scholar]
- 57.Atack JR, Perry EK, Bonham JR, Candy JM, Perry RH. Molecular forms of acetylcholinesterase and butyrylcholine esterase in the aged human central nervous system. J Neurochem 1986;47:263-277 [DOI] [PubMed] [Google Scholar]
- 58.Jbilo O, Bartels CF, Chatonnet A, Toutant JP, Lockridge O. Tissue distribution of human acetylcholinesterase and butyrylcholine esterase messenger RNA. Toxicon 1994;32:1445-1457 [DOI] [PubMed] [Google Scholar]
- 59.Wright CI, Geula C, Mesulam MM. Neurological cholinesterases in the normal brain and in Alzheimer's disease. Relationship to plaques, tangles, and patterns of selective vulnerability. Ann Neurol 1993;34:373-384 [DOI] [PubMed] [Google Scholar]
- 60.Damasio AR, Damasio H, Rizzo M, Varney N, Gersh F. Aphasia with nonhemorrhagic lesions in the basal ganglia and internal capsule. Arch Neurol 1982;39:15-24 [DOI] [PubMed] [Google Scholar]
- 61.Borlongan CV, Cahill DW, Sanberg PR. Locomotor and passive avoidance deficits following occlusion of the middle cerebral artery. Physiol Behav 1995;58:909-917 [DOI] [PubMed] [Google Scholar]
- 62.Borlongan CV, Martinez R, Shytle RD, Freeman TB, Cahill DW, Sanberg PR. Striatal dopamine-mediated motor behavior is altered following occlusion of the middle cerebral artery. Pharmacol Biochem Behav 1995;52:225-229 [DOI] [PubMed] [Google Scholar]
- 63.Sette G, Baron JC, Young AR, et al. In vivo mapping of brain benzodiazepine receptor changes by positron emission tomography after focal ischemia in the anesthetized baboon. Stroke 1993;24:2046-2057 [DOI] [PubMed] [Google Scholar]
- 64.O'Malley S, Adamse M, Heaton RK, Gawin FH. Neuropsychological impairment in chronic cocaine abusers. Am J Drug Alcohol Abuse 1992;18:131-144 [DOI] [PubMed] [Google Scholar]
- 65.Berry J, van Gorp WG, Herzberg DS, et al. Neuropsychological deficits in abstinent cocaine abusers. Preliminary findings after two weeks of abstinence. Drug Alcohol Depend 1993;32:231-237 [DOI] [PubMed] [Google Scholar]
- 66.Hoff AL, Riordan H, Morris L, et al. Effects of crack cocaine on neurocognitive function. Psychiatry Res 1996;60:167-176 [DOI] [PubMed] [Google Scholar]
- 67.Bauman JL, Grawe JJ, Winecoff AP, Hariman RJ. Cocaine-related sudden cardiac death. A hypothesis correlating basic science and clinical observations. J Clin Pharmacol 1994;34:902-911 [DOI] [PubMed] [Google Scholar]
- 68.Marzuk PM, Tardiff K, Leon AC, et al. Fatal injuries after cocaine use as a leading cause of death among young adults in New York City. N Engl J Med 1995;332:1753-1757 [DOI] [PubMed] [Google Scholar]
- 69.Shen WK, Edwards WD, Hammill SC, Bailey KR, Ballard DJ, Gersh BJ. Sudden unexpected nontraumatic death in 54 young adults. A 30-year population-based study. Am J Cardiol 1995;76:148-152 [DOI] [PubMed] [Google Scholar]
- 70.Hass WK, Easton JD, Adams HPJ, et al. A randomized trial comparing ticlopidine hydrochloride with aspirin for the prevention of stroke in high-risk patients. Ticlopidine Aspirin Stroke Study Group [see comments]. N Engl J Med 1989;321:501-507 [DOI] [PubMed] [Google Scholar]
- 71. North American Symptomatic Carotid Endarterectomy Trial Collaborators. Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis [see comments]. N Engl J Med 1991;325:445-453 [DOI] [PubMed] [Google Scholar]
- 72.Oppenheimer SM, Kedem G, Martin WM. Left-insular cortex lesions perturb cardiac autonomic tone in humans. Clin Auton Res 1996;6:131-140 [DOI] [PubMed] [Google Scholar]
- 73.van Swieten JC, Staal S, Kappelle LJ, Derix MM, van Gijn J. Are white matter lesions directly associated with cognitive impairment in patients with lacunar infarcts? J Neurol 1996;243:196-200 [DOI] [PubMed] [Google Scholar]
- 74.Pasquier F, Leys D. Why are stroke patients prone to develop dementia? J Neurol 1997;244:135-142 [DOI] [PubMed] [Google Scholar]