

Mechanotransduction model provides insights into integrin clustering/disassembly dynamics and associated FAK phosphorylation.
Abstract
Transduction of extracellular matrix mechanics affects cell migration, proliferation, and differentiation. While this mechanotransduction is known to depend on the regulation of focal adhesion kinase phosphorylation on Y397 (FAKpY397), the mechanism remains elusive. To address this, we developed a mathematical model to test the hypothesis that FAKpY397-based mechanosensing arises from the dynamics of nanoscale integrin clustering, stiffness-dependent disassembly of integrin clusters, and FAKY397 phosphorylation within integrin clusters. Modeling results predicted that integrin clustering dynamics governs how cells convert substrate stiffness to FAKpY397, and hence governs how different cell types transduce mechanical signals. Existing experiments on MDCK cells and HT1080 cells, as well as our new experiments on 3T3 fibroblasts, confirmed our predictions and supported our model. Our results suggest a new pathway by which integrin clusters enable cells to calibrate responses to their mechanical microenvironment.
INTRODUCTION
The ways that cells transduce and respond to their mechanical microenvironments underlie pathological remodeling in metastasis and fibrosis, and physiological remodeling in development and stem cell differentiation (1–3). A variety of potential mechanosensors are available to cells including various adhesion proteins such as integrin, focal adhesion kinase (FAK), talin, and vinculin (4). In many cells, a relatively high substrate stiffness up-regulates phosphorylation of FAK on Y397 (FAKpY397) (Fig. 1A), which affects cell behaviors including proliferation, migration, and differentiation (5). However, the molecular mechanism by which cell adhesions enable this stiffness sensing via FAKpY397 remains elusive.
Fig. 1. Integrin clustering dynamics on a plasma membrane.
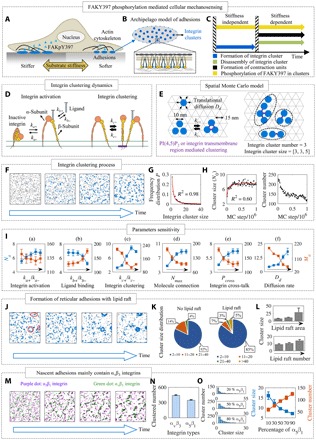
(A) Stiff substrate facilitates integrin activation and clustering. Integrin clustering then promotes the phosphorylation of FAK on the Y397 site and, thus, leads to a high level of FAKpY397 when cells are cultured on a stiff substrate. (B) Cell adhesions are loose aggregates containing tight integrin clusters with a size of ~100 nm and composed of ~20 to 50 integrin molecules. A single integrin cluster may act as a platform where intracellular biochemical signals (e.g., FAK) are activated depending on substrate stiffness. (C) Integrin clusters turn over through clustering-disassembly dynamics on the time scale of minutes, depending on the traction of actomyosin-based sarcomere-like contractile units (CUs). These dynamics are related to phosphorylation of FAKY397. (D) Integrin has two states, i.e., inactive state and active state. The talin head domain can promote the activation of inactive integrin. Active integrins from the clusters can either be connected by PI(4,5)P2 or by integrin transmembrane regions. (E) A spatial Monte Carlo algorithm was used to simulate the spatial dynamics of integrin clustering, which considered integrin activation and inactivation, diffusion, and clustering on the plasma membrane. (F) As an initial configuration, integrin molecules were randomly placed within the simulation space. During the simulation, the clustered integrin number increased and gradually reached saturation. (G) Simulated frequency distribution of integrin cluster size. (H) Evolution of cluster size and number during integrin clustering. (I) Parametric sensitivity analysis. (J) Formation of reticular adhesions (RAs) is regulated by lipid rafts. (K and L) Increases in lipid raft number and size can notably increase the integrin cluster size. (M) Time sequence of the integrin clustering in a simulation containing equal numbers of αvβ3 and α5β1 integrins. (N) αvβ3 integrins are more likely to form clusters. (O) Higher concentration of α5β1 integrins led to the formation of more and smaller clusters.
A hypothesis proposed recently is that the dynamics of nanoscale integrin clusters within focal adhesions (FAs) governs mechanosensing via FAKpY397 (6). The loose aggregates containing tight clusters of integrins with a size of ~100 nm and composed of ~20 to 50 integrin molecules (Fig. 1B), as identified by super-resolution imaging (7), are believed to be sites for FAKY397 phosphorylation. Substrate stiffness–dependent cluster dynamics affects the ability of integrin molecules to form adhesions (8) and is hypothesized to play a key role in cellular decisions about the suitability of a microenvironment for growth and differentiation. Integrin clusters turn over through clustering-disassembly dynamics on the time scale of minutes (9), but their lifetimes can be prolonged by the traction of actomyosin-based sarcomere-like contractile units (CUs) (10). These dynamics may be related to the phosphorylation of FAKY397 (Fig. 1C).
On the basis of these observations, we hypothesized that integrin clusters serve as platforms for substrate stiffness–dependent FAKY397 activation. To test our hypothesis, we developed a mathematical model integrating submodels of (i) integrin clustering, (ii) stiffness-dependent integrin cluster disassembly, and (iii) FAKY397 phosphorylation within integrin clusters. With this model, we assessed several biomechanical and biochemical processes that appear to play important roles in integrin clustering and clustered integrin–mediated FAKY397 phosphorylation.
RESULTS
Spatial Monte Carlo model of integrin clustering kinetics
To test our hypothesis, we developed a coarse-grained spatial Monte Carlo model of integrin clustering. Integrin clustering requires activation of integrin molecules to their high ligand binding affinity state by the talin head domain (11, 12), with activation and inactivation rate constants of ka+ and ka−, respectively (Fig. 1D). Activated integrins could bind to ligand with a rate of kb+ and unbind upon separating from clusters with a rate of kb− (Fig. 1D). Integrin molecules aggregate or separate from neighbors by binding or unbinding via (i) membrane-bound PI(4,5)P2 (phosphatidylinositol 4,5-bisphosphate) molecules (12) and (ii) integrin transmembrane domains (ITDs) (13) with binding and unbinding rates of kc+ and kc–, respectively (Fig. 1E). Beginning with a random distribution of integrin molecules on the membrane lattice, the number of clustered integrins increases with computational time (Fig. 1F).
To quantify these dynamics, we computed the average number of integrin molecules per cluster (“integrin cluster size,” Na) and the spatial density of integrin clusters (“integrin cluster number,” Ma) after reaching a steady state (Fig. 1E). The distribution of Na fits a power function well (red line in Fig. 1G)
| (1) |
where α and β are two fitted parameters. The growth of integrin clusters approaches a steady state according to first-order kinetics with time constant k (red line in Fig. 1H)
| (2) |
where N0 is the average cluster size during clustering process, while Na is the average cluster size at steady state. Accordingly, integrin cluster number decreases with time (Fig. 1H). Overall, these simulation results reveal that our integrin clustering submodel is adequate to replicate the process of integrin clustering.
Opposite roles of Integrin activation and integrin-ligand binding affinity in integrin clustering dynamics
Integrins can be activated by other molecules such as talin, kindlin, and their ligands (14). Because integrin activation is a prerequisite for integrin clustering, the ratio of activation rate to deactivation rate (ka+/ka−) determines the number of activated integrin molecules that contribute to integrin clustering. The simulation results show that increasing ka+/ka− will decrease Ma but increase Na (Fig. 1Ia).
Integrin-ligand binding affinity is also a key regulator of integrin clustering, with different integrins having distinct ligand binding affinity (15). Simulation results show that increase in ligand binding affinity (kb+/kb−) will decrease Na and increase Ma (Fig. 1Ib), likely because high ligand binding affinity will decrease the effective diffusion rate of integrin and, thus, inhibit integrin clustering.
Lateral interaction regulated integrin clustering dynamics
Many structures and molecules, such as talin-PI(4,5)P2-lipid molecular complexes, kindlin2-PI(4,5)P2, and ITDs, are involved in integrin lateral binding (14). Although molecular mechanisms for lateral binding of integrins remain controversial, we explored the potential effects by adjusting the integrin lateral binding affinity (kc+/kc−) to simulate varying degrees of lateral binding–mediated integrin clustering. The simulation results reveal a biphasic role of lateral affinity mediated integrin clustering: Lateral affinity promotes formation of larger clusters up to a critical binding affinity but inhibits the formation of larger clusters beyond this critical binding affinity (Fig. 1Ic). The biophysical basis for this unexpected biphasic relationship is a competition between the thermodynamics of integrin clustering and the kinetics of integrin mobility. Increased lateral affinity accelerates cluster growth. However, formation of the largest clusters requires coalescence of smaller clusters via cluster diffusion. Because larger clusters diffuse more slowly on membranes, the largest clusters are inhibited by lateral affinity via a kinetic trap.
In addition to lateral binding affinity, the number of lateral links between integrins may also be limited. α-ITDs of integrin tend to form α-dimers, while β-ITDs of integrin tend to form β-trimers (13). This suggests that one integrin molecule might only be able to connect to at most three others. We therefore asked what effect such limitations on lateral links might have by considering a parameter Nmax that represents the number of integrin molecules that can connect to a single integrin molecule and ranged from 3 to 6 (the latter a limitation of the hexagonal grid). Results show that increasing this maximum molecular coordination number increases Na and decreases Ma (Fig. 1Id).
Enhanced integrin clustering via integrin cross-talk
Activated integrins can directly activate adjacent inactive integrin (16). Such integrin cross-talk produces a positive feedback loop to enhance integrin clustering. We used an activation probability parameter (0 ≤ Pcross ≤ 1) to represent different efficiencies of integrin cross-talk–induced integrin activation. Increasing integrin cross-talk increases Na and decreases Ma (Fig. 1Ie).
Lipid raft–mediated integrin clustering dynamics
Recently, Lock et al. (17) discovered a colocalization of PI(4,5)P2 in reticular adhesions (RAs), indicating that lipid rafts and PI(4,5)P2 may contribute to integrin clustering dynamics. Therefore, we used our model to investigate how both lipid raft area and number affect integrin clustering. Because integrin molecules in lipid raft regions have smaller diffusion rates, we investigated the role of diffusion rate in integrin clustering without considering confined diffusion in lipid rafts. Results show that increasing integrin diffusion rate (Dd) decreased Ma and increased Na (Fig. 1If).
To model lipid rafts containing PI(4,5)P2, we generated circular regions of the simulation domain in which integrins had lower diffusion rate. Integrin clusters in these regions were similar in shape (pore structure) to experimental observations (Fig. 1J), suggesting that lipid rafts may play roles in the formation of RAs. Results show that both the number and area of lipid rafts can notably increase the integrin cluster size (Fig. 1, K and L).
αvβ3 integrins forming clusters more readily than α5β1 integrins
Cells present a range of adhesion types. These can include nascent focal complexes (FXs), FAs, and fibrillar adhesions (FBs) (18). FXs contain more αvβ3 integrins, while more mature FAs and FBs contain more α5β1 integrins (18).
We therefore asked why these αvβ3 integrins might be favored in the earlier clusters of FXs. To investigate this, we distributed equal numbers of α5β1 and αvβ3 integrins in a simulation region and found that consistent with published experimental observations (18), most early clusters contained αvβ3 (Fig. 1, M and N). In the context of our model, this behavior can be understood as arising from the higher activation rate of αvβ3 integrins and the fact that clustering requires activation. Parametric sensitivity analysis (Fig. 1If) reveals that lower ligand binding affinity and higher diffusion rate of αvβ3 integrins were also factors.
This led us to ask what role α5β1 integrins play in regulating integrin clustering. By performing simulations with increasing percentages of α5β1 integrins, we discovered that increased concentrations of α5β1 integrins promote formation of more and smaller integrin clusters (Fig. 1O).
Key roles of α5β1 integrins in strength and lifetime of integrin clusters
Although it has been shown that cells can form more stable FAs with longer lifetime on stiffer substrate (19), the underlying molecular mechanisms remain unclear. To investigate the mechanisms, we developed a parallel molecular bond model to describe the CU stretching–dependent integrin cluster disassembly dynamics (Fig. 2A). The proposed model consists of Nint parallel integrin molecules (modeled as springs of stiffness kint) that are connected to a ligand-coated substrate (modeled as a spring of stiffness kecm) (Fig. 2B, left) (20). The integrins adhere to the ligand via catch-slip bonds or Kank-dependent slip bonds (21), and actomyosin stretching (22) is modeled as a time-varying displacement boundary condition (Fig. 2B, right).
Fig. 2. Increases of average integrin cluster size and substrate stiffness contributed to lengthened lifetimes of integrin clusters.
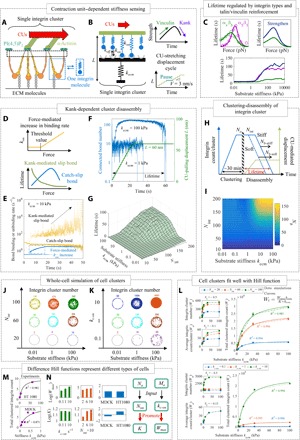
(A) An integrin cluster could bind to a substrate of ECM proteins on its extracellular end and to actomyosin-based sarcomere-like contractile units (CUs) on its intracellular end. Each integrin molecule was modeled as one spring. (B) The ECM substrate to which the integrin cluster attached was modeled as a spring, and the dynamics of the integrin cluster–ECM substrate was modeled as a catch-slip bond that was pulled by CUs with a displacement boundary condition. (C) The lifetimes of α5β1 and αvβ3 integrin clusters changed differently with substrate stiffnesses. Talin/vinculin binding can enhance cluster lifetime. (D to E) Two important assumptions: force-mediated ligand binding rate increases and Kank-mediated unbinding rate changes. (F and G) With a limited displacement of CU-mediated pulling, the lifetime of integrin clusters on a stiff substrate (~100 kPa) is longer than that on an intermediate substrate (~1 kPa). (H and I) Integrin count at the initial of computation (Nint), and substrate stiffness (kecm) affects the average integrin count (Nb) during disassembly. (J and K) Integrin clusters can rupture at the edge of the cell and then slide with inward actin flow and later rebind to the substrate. The number of clustered integrins, Wt, was obtained from the number of clustered integrins, Wn, and the average number of integrins per cluster, Wa = 50. Wt increased with substrate stiffness to a plateau value. (L) A Hill function can be used to describe the relationship between Wt and substrate stiffness. (M) The Hill function can also describe the experimental data well (5, 32), which showed that the total clustered integrin increased with increasing stiffness. (N) Average integrin count (Na) mainly influences the values of K and Wmax parameters in the Hill function, while cluster number (Ma) only influences the values of the Wmax parameter.
Adhesion strength has been observed to depend predominantly upon α5β1 integrin–ligand bonds rather than upon αvβ3 integrin–ligand bonds (23). We asked how these different integrin-ligand bonds govern integrin cluster disassembly. Results reveal a key role of this in mechanosensing. The lifetimes of α5β1-formed integrin cluster increase with increasing stiffness, but those of αvβ3-formed integrin clusters are relatively insensitive to substrate stiffness (Fig. 2C). The simulation is consistent with experimental observation (18) that FAs, with more α5β1 integrins, are more stable than FXs with more αvβ3 integrins and explains a key step in maturation of adhesions. Thus, in our cluster-disassembly submodel, only α5β1 integrin–ligand bond dynamics is used for subsequent analysis for simplicity.
Considering that talin/vinculin binding reinforces clutch link upon mechanical stretch. Thus, we used a different catch-bond dynamics curve of α5β1 integrin–ligand bond with a higher rupture force to represent the talin/vinculin reinforcement function (24). The simulations show that reinforcement of talin/vinculin can increase the lifetime of clusters on the stiffer substrate (Fig. 2C).
Kank-dependent integrin cluster disassembly dynamics
Connections between the extracellular matrix (ECM) and the intracellular cytoskeleton are composed of several force-sensitive molecules acting as important force-transmission links for cells. Integrin-fibronectin and vinculin-actin links show catch-bond dynamics, while actin-talin links shows slip bond dynamics (8, 25, 26). These slip bonds are strengthened by force-induced recruitment of vinculin due to the exposure of vinculin binding sites on talin molecules. Central to these cell-adhesion dynamics is a role of Kank (27), which likely binds with the R7-R8 domain of the talin rod (i.e., C-terminal rod-domain) to form an “FA belt” at the edge of an FA that transforms integrin-fibronectin catch-bond dynamics into slip bond dynamics (21). This provides a reasonable molecular mechanism of reduced cell adhesion strength after Kank recruitment (Fig. 2B, right).
In response to actomyosin stretching, integrin clusters could be stretched to a maximum length of 50 to 60 nm (22). We therefore modeled CU stretching–dependent integrin cluster disassembly dynamics using the above spring model by incorporating the following two factors: (i) force-mediated increase in integrin binding rate (kon) via force-induced local ligand recruitment (15) and (ii) Kank-mediated slip bond dynamics (Fig. 2D). With these two assumptions, simulations reveal that (i) kon increases with time, (ii) unbinding rate caused by Kank-mediated slip bond also increases with time, (iii) unbinding rate caused by catch-slip bond gradually decreases exponentially with time during disassembly of clusters (Fig. 2E).
The simulations indicate that integrin cluster lifetimes are greater on stiff (~100 kPa) than on intermediate stiffness substrate (~1 kPa) (Fig. 2F), and are smallest on compliant substrate (~0.01 kPa), which is consistent with experimental observations that integrin cluster lifetime increases with increasing substrate stiffness (7) (Fig. 2G). Thus, a balance between substrate stiffness and Nint controls the dynamic lifetimes of integrin cluster. The mechanisms described in this section serve as a tool for integrin cluster–mediated mechanosensing. The underlying factors are that force-mediated binding increases over time to a steady state, while Kank-mediated unbinding gradually increases, resulting in the disassembly of integrin clusters. Lifetimes of large integrin clusters on stiffer substrate are lengthened by the greater probability of integrin rebinding.
Integrin clustering occurs immediately when the cell contacts with a substrate, and the average integrin count per cluster lastly reaches Na in the steady state. We assumed that about 30 min later, the CUs form and pull the clusters (10), which leads to the disassembly of clusters and the average connected bond number per cluster is Nb during disassembly (Fig. 2H). Simulation results reveal that Nb increases with both substrate stiffness and Nint (Fig. 2I).
A Hill-type function between cluster-mediated cell adhesion and substrate stiffness
The cluster-mediated cell adhesions are determined by the stress-free cluster formation and the subsequent cluster disassembly upon CU stretching (6). Then, we used whole-cell simulation (28) to assess how integrin cluster dynamics varies with (i) Nint, (ii) the rate of integrin cluster formation kc-on (kc-on = cMa, where c is a correction factor), which is proportional to the steady-state integrin cluster number Ma during the integrin clustering, and (iii) substrate stiffness. In this simulation, integrin clusters appear at the cell edge in a stiffness-independent fashion and then migrate toward the cell center until their disassembly in a stiffness-dependent fashion.
Simulations show higher integrin cluster number on the cell at an elevated kecm, and kc-on (Fig. 2, J and K). Increasing substrate stiffness increases the total number of clustered integrins (Wt) and integrin clusters on a cell (Wn) and the average number of integrin molecules per cluster on a cell (Wa) (Fig. 2L). Binding statistics in both simulations and in the reported data follows a relationship vaguely analogous to a first-order Hill function, so that the relative fraction of clustered integrin molecules is
| (3) |
where the constant Wmax is the expected value of Wt on an infinite stiff substrate, and the ratio K/kecm determines the range of stiffness sensitivity for cells. For example, the range over which integrin clustering on a cell shifts from 10 to 90% of the maximum potential adhesion size corresponds to K/9 ≤ kecm ≤ 9K, which is a measure of cell sensing to a specific substrate stiffness. K/9 ≤ kecm ≤ 9K. Both Wa and Wn increase with Nint, while only Wn is sensitive to kc-on.
To test the capability of Eq. 3 in explaining and predicting the range of stiffness sensitivity for cells through the integrin cluster–mediated adhesion dynamics, we calculated Wmax and K by fitting existing experimental data (Fig. 2M) (5). We found that Madin-Darby canine kidney (MDCK) cells and HT1080 cells have similar values of Wmax but very different stiffness-sensing ranges, where HT1080 cells have a much larger K than MDCK cells (Fig. 2N).
In general, two parameters, Na and Ma, obtained from the spatial Monte Carlo model of integrin clustering can be used as a basis (Nint and kc-on, respectively) for modeling the cluster disassembly. The simulation reveals that Nint can notably broaden the stiffness sensing ranges (i.e., a larger K) (Fig. 2N). However, the kc-on can improve the ability of forming integrin cluster of cells (i.e., a larger Wmax) (Fig. 2N).
Linearly relationship between FAKpY397 and clustered integrin number
To study the role of FAK binding in integrin-mediated mechanotransduction (29), we simulated the effect of integrin clustering on FAKY397 phosphorylation. We performed a spatially particle-based simulation (30) with excluded volume interactions on a regular three-dimensional (3D) lattice (Fig. 3A). Integrin and FAK were modeled as spheres with the same diameter l, which also defined the spacing of the close-packed square lattice. Periodic boundary conditions were applied for the membrane edges and reflective boundary conditions for the top and bottom surfaces. In our model, FAKY397 can be activated by the integrin cytoplasmic tail according to experimental observations (Fig. 3B) (31). We then studied the effects of different factors on FAKY397 phosphorylation, including FAK/integrin association and disassociation rate constants kin and kout, the activation rate constant of FAKY397 in clusters ke+, the deactivation rate constant of activated FAKY397 in cytoplasm ke−, and the number of FAKY397 in cytoplasm NFAK, Wa, and Wn (Fig. 3C).
Fig. 3. The level of FAKpY397 is linearly related to clustered integrin.
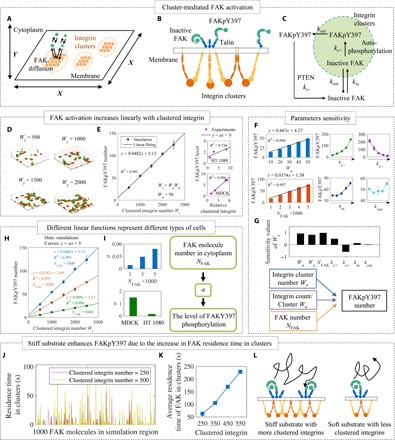
(A) Schematics of spatial arrangement of integrin and FAK molecules. FAK can diffuse freely in the cytoplasm, and integrin clusters are fixed on the membrane. (B) FAK can be activated by integrin cytoplasmic domains in integrin clusters. (C) The association and disassociation of FAK with integrin clusters are described by kin and kout, respectively. The activation and deactivation of FAK in clusters or cytoplasm through dephosphorylation by phosphatase and tensin homolog (PTEN) are described by ke+ and ke−, respectively. (D and E) The number of FAKpY397 is linearly related to clustered integrin number. The linear relationship can also be used to describe the experimental data well (5, 32). (F and G) Parameter sensitivity analysis indicates that the activation and deactivation rates, integrin cluster size/number, and FAK number can notably influence the level of FAKY397 phosphorylation. (H and I) The FAK number in the cytoplasm can increase the FAK activation efficiency with fixed clustered integrin number. (J) The residence time for individual FAK molecules within a simulation region, which (K) increased as the number of clustered integrins increased. (L) This enabled mechanotransduction by increasing the likelihood of FAK rebinding to integrin via talin molecules, as shown in this schematic.
Consistent with experimental observations (5, 32), simulation results predicted a linear relationship between the number of FAKpY397 and the number of clustered integrin molecules Wt (Fig. 3, D and E)
| (4) |
where a represents the activation efficiency of FAKY397 for clustered integrins, and b represents the baseline activation of FAKY397 that is independent on clustered integrins. FAKpY397 level increases with increasing activation rate ke+, association rate kin, and total level of cellular FAK (NFAK), and decreases with increasing deactivation rate ke−. FAKpY397 levels are insensitive to the dissociation rate kout (Fig. 3F).
Of these, Wa, Wn, and NFAK show significant influence on FAKY397 activation (Fig. 3G). With fixed kecm, increase in NFAK notably enhances the level of FAKY397 phosphorylation (Fig. 3H). The fitting parameter a of experiments indicates that MDCK cells have a larger FAKY397 phosphorylation rate (a larger a) than HT1080 cells and, therefore, is more sensitive to softer substrate (Fig. 3I).
An interesting paradox arises from the recent discovery that FAK association and dissociation with integrin clusters are not influenced by the substrate stiffness (33). How then can stiffness-dependent FAKY397 activation arise in the simulations? Our simulations predicted an unusual behavior showing increasing average residence time for FAK molecules as the number of integrin clusters increases (Fig. 3, J and K), indicating an increased probability of FAK molecules rebinding with integrins via talin on stiffer substrate (Fig. 3L). This mechanism enables a form of mechanosensing in that it led to higher FAKpY397 concentrations on integrin clusters forming on stiff substrata than on those forming on compliant substrata.
Negative feedback regulation mediated integrin clustering
Phosphorylation of paxillin can promote the formation of FXs and FAs, while the pPaxillin/FAK complex can inhibit the formation of FXs and FAs, thus resulting in a negative feedback loop for integrin clustering (34). To evaluate the effects of negative feedback regulation on integrin clustering, we developed an integrated signaling network model containing a serials of ordinary differential equation (ODE) equations. First, we developed a Kank-mediated cluster disassembly ODE model (model I) to integrate our previous three submodels (fig. S1, A and B). The simulation results show that high substrate stiffness promotes integrin clustering and FAKY397 phosphorylation (fig. S1, C and D). The relationship between integrin cluster lifetime and substrate stiffness can also be described by a Hill function (fig. S1E). FAKpY397 levels also increased linearly with clustered integrins (fig. S1F). These two results are consistent with our previous three submodels.
Then, we expanded model I by adding pPaxillin/FAK-mediated negative feedback regulation (model II; fig. S1G). The simulation results show that clustered integrin increases with time and then decreases due to pPxillin-FAK complex–mediated cluster disassembly (fig. S1H). Clustered integrin also increases with substrate stiffness (fig. S1I). FAKpY397 levels also increased linearly with increasing clustered integrin (fig. S1J).
To model the promotion of cluster disassembly by FAKpY397 (35), we modulated the assembly/disassembly rate (k3f/k3r and k4f/k4r, from 10 to 0.1). Results reveal that FAKpY397 activation reduces the clustered integrin number via increasing pPaxillin/FAK complexes (fig. S1H). The reduced number of clustered integrins may be attributed to the decrease in integrin cluster lifetime as induced by FAKpY397 activation.
We next expanded model II to simulate negative feedback of tensioned integrin clusters that undergo FAK signaling, which would negatively affect affinity parameters within the integrin cluster itself (model III, fig. S1K). The simulation results show that FAKpY397 is independent of the clustered integrins and maintains a high FAKpY397 level (~0.4) with different numbers of clustered integrins (fig. S1K).
Validation of mathematical models using 3T3 fibroblasts
According to Eqs. 2 to 4, we can predict the integrin cluster–mediated FAKpY397 activation by the following equation (fig. S1L, see the Supplementary Materials for more details)
| (5) |
The mathematical model for FAKpY397 levels has four parameters (a, b, Wmax, and K) that must be fitted to the experimental data. This model was fitted to match data from the literatures on stiffness-dependent FAK activation in HT1080 cells and MDCK cells, and we also validated the model by testing against the area of FAs and stiffness-dependent FAK phosphorylation levels in 3T3 fibroblasts. Our simulations predicted FAKpY397 levels for cells in a stable, spread state. We therefore performed cell-spreading experiments to determine the time required for cell area to reach this stable state. These experiments show that the cell area reaches a stable state after ~10 hours for the entire range of substrata considered (fig. S2). Our calculations of levels of cell adhesions and FAKpY397 were, thus, appropriate for times longer than 10 hours. Immunofluorescence images and quantitative results show increasing level of the area of FAs and Y397-phosphorylated FAK with increasing substrate stiffness (Fig. 4, A, B, and D). Also, quantitative data from Western blot images show a Hill-type relationship between substrate stiffness and FAKpY397 level, in a way that is well predicted by the model (Fig. 4, C and E). Consistent with other types of cells, our experiments show that 3T3 fibroblasts also exhibit a linear relationship between FAKpY397 and area of FAs (Fig. 4F). These data support the model and the assumptions underlying it and suggest that comparison of model parameters can lend insight into the mechanisms by which cells can tune their responses to substrate of different stiffness.
Fig. 4. An integrated computational model to analyze mechanochemical conversion in different types of cells.
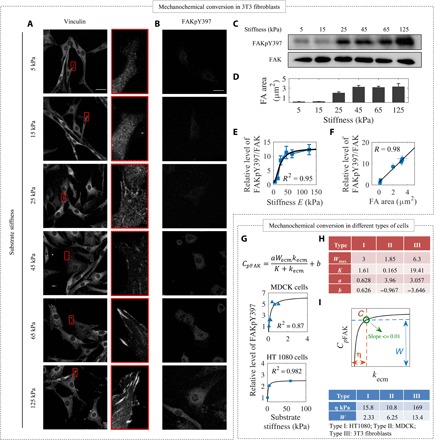
(A) FAs of 3T3 fibroblasts on substrates with stiffness ranging between 5 and 125 kPa. Scale bars, 30 μm (data shown represent one of three independent experiments). Vinculin was detected after 3 days of cell culture. (B) 3T3 fibroblasts cultured on substrata of different stiffness were immunostained for Y397-phosphorylated FAK. Scale bar, 30 μm (data shown represent one of three independent experiments). FAKpY397 was detected after 3 days of cell culture; note that cell spreading reaches a stable level after 10 hours (cf. fig. S2). Increased FAKpY397 levels were found on stiff substrata as reflected by increased fluorescence intensity. (C) Cell lysate was analyzed by Western blot analysis with antibodies for detecting specific FAK phosphorylation site on Y397 (data shown represent one of three independent experiments). (D) The average FA area increases with substrate stiffness (n = 4, 3, 23, 31, 50, and 41 adhesions for increasing stiffness and measured in 10 to 15 cells). (E) Quantification of the Western blot data in 3T3 fibroblasts supports the relationship between stiffness and FAKpY397 predicted by our model (pooled from n = 3 independent experiments). (F) Relative level of FAKpY397 increases linearly with the area of FAs. (G and H) Different types of cells have different values of K and Wmax. The value of K in 3T3 fibroblasts is larger than that in HT1080 cells and MDCK cells, meanwhile the value of Wmax in 3T3 fibroblasts is also larger than that in HT1080 cells and MDCK cells (5, 32). (I) The mechanosensing range varies with cell type. C point is used to analyze the mechanosensing ranges of different types of cells. At the C point, the slope of the curve is equal to 0.01 depending on the special algebraic expression. Within the range denoted by η, the phosphorylation of FAKY397 is sensitive to changes in substrate stiffness. Another parameter, W, represents the magnitude of the activation of the FAKY397.
Hill parameters determined cellular mechanosensing
Comparing parameters for the three different cell types reveals that 3T3 fibroblasts have higher levels of FAKY397 phosphorylation on stiffer substrate than the other two cell types, indicating that 3T3 fibroblasts are tuned to respond to stiffer microenvironments (Fig. 4E). We got the fitting parameters (a, b, Wmax, and K) from Fig. 4, E and G and found that 3T3 fibroblasts have the highest Wmax and K than other two types of cells (Fig. 4H). MDCK cells show the highest FAK397 activation efficiency of FAKY397 (a) among the three cell types. 3T3 fibroblasts and MDCK cells show negative values of b, indicating that their FAKY397 activation may not happen on soft substrates. As discussed below, the results, interpreted through the lens of the model, suggest that by tailoring the concentration of FAK and integrin molecules and by controlling molecular transport and diffusion rates, cells can tailor cell-specific responses to substrate stiffness.
DISCUSSION
A new pathway for mechanochemical conversion
Our model of mechanochemical conversion by integrin clustering requires the integration of three processes to predict cellular mechanosensing, i.e., integrin clustering on the plasma membrane, stiffness-dependent disassembly of integrin clusters, and FAKY397 phosphorylation within these integrin clusters. Spatial Monte Carlo simulation shows that the size distribution of integrin clusters obeys the power function (Eq. 2). Our model predicted, correctly, that the lifetime of these clusters increases monotonically with substrate stiffness over the physiological stiffness range following the Hill-type function (Eq. 3).
These results suggest that, for a cell, the maximum possible density of clustered integrins on an infinitely stiff substratum, Wmax, is mainly influenced by the integrin number on the membrane, while the cell-specific optimum stiffness, K, is mainly influenced by integrin cluster size.
Recently, experimental observations show that substrate viscosity also plays a key role in the formation of FAs (36). We also used our model to investigate the role of substrate viscosity in integrin cluster disassembly (fig. S3). We found that the increase in viscosity can notably decrease the lifetime of the integrin cluster mainly because of increasing concentration of stress on the interface of cell ECM.
Our model supports the hypothesis that a single integrin cluster independently acts as a platform for intracellular FAKY397 activation. FAKY397 phosphorylation requires stable integrin cluster and is affected by the size distribution and lifetime of integrin clusters. Our spatially resolved simulations provide a structural explanation of how the number of FAKpY397 molecules increases with the number of clustered integrin molecules. Results predict a linear relationship between the concentration of FAKpY397 and the number of clustered integrin molecules (Eq. 4). Last, result suggests a Hill-type function for the relationship between substrate stiffness and the concentration of FAKpY397 (Eq. 5). The model, thus, provides a potential mechanistic explanation of how substrate stiffness can be sensed by cells and converted into intracellular biochemical signals through integrin clustering and FAK phosphorylation in three steps: (i) activation of single integrins by talin or fibronectin is a precursor for integrin clustering; (ii) myosin-mediated intracellular tension regulates strength and lifetime of mechanical bonds at the adhesion-ECM interface and, hence, the disassembly of integrin clusters; (iii) and cytoplasmic integrin tails in integrin clusters can bind to FAK and induce a partially open FAK conformation that exposes the Y397 autophosphorylation site. Thus, integrin clusters can sense ECM stiffness by dynamically changing their numbers and lifetime, as they serve as a platform for activation of mechanosensing molecules.
These results also can be validated by other experimental data from the literature. For example, the model suggests how the integrin clustering enhances FAKpY397 activity in the experiments of Paszek et al. (29), who studied a β1 integrin mutant (β1 V737N) with increased transmembrane affinity. Cells with β1 V737N have elevated FAKpY397 even on compliant substrate, as would be expected in our model from the increase in Wt. Wei et al. (5) showed that integrin clustering rather than activation leads to FAKY397 phosphorylation, which is consistent with our model’s prediction.
Two mechanisms exist for FAKY397 phosphorylation: tensioned integrin–mediated FAKY397 activation and recruitment of FAK into FAs without the requirement of tension (37). Here, our model is suitable for the background of clustered integrin–mediated requirement and activation of FAKY397 without tension acting directly on FAK molecules. Our model suggests that increasing substrate stiffness increases the lifetime and size of integrin clusters, increasing FAK397 residence time and, hence, the probability of FAKY397 phosphorylation.
Integrin clusters mediated mechanosensitive information processing
Our results suggest that integrin clusters serve as machinery for mechanotransduction by efficiently converting information about substrate stiffness into biochemical signaling (FAKpY397). This is similar to roles of other membrane protein clusters such as Ras clusters, which convert extracellular biochemical signals into the mitogen-activated protein kinase (MAPK) signaling cascade (38). For integrin cluster–mediated FAKY397 phosphorylation, the molecular mechanism suggested by our model relates to the close packing of integrins enabled by clustering. The increase in spatial density of clustered integrins prolongs the reaction time for FAKY397 phosphorylation in integrin clusters by increasing the probability of FAK rebinding to integrins via talin. Because stiffer substrate promotes more and longer-lived integrin clusters, FAKpY397 concentrations increase with substrate stiffness. Integrin clusters, thus, appear to provide a mechanism for cell mechanosensing via FAKY397 phosphorylation.
Distinct mechanosensitive ranges in different cell types
The way that FAKY397 phosphorylation increases with substrate stiffness follows distinct Hill-type function in different types of cells (Fig. 4, E and G). To compare the mechanosensing ranges, we defined the point C for each cell type at which the slope of each curve is 0.01 (the rate of relative change in concentration of FAKY397 phosphorylation in response to different substrate stiffnesses) (Fig. 4, E and G). Point C corresponds to a substrate stiffness (kecm = η) and a concentration of FAKpY397 (W), where the level of FAKY397 phosphorylation in cells shows significant changes with substrate stiffness within the stiffness range of 0 to η kPa. It is noted that FAKY397 phosphorylation is most sensitive to substrate stiffness for kecm < η. Meanwhile, W represents the magnitude of FAKY397 phosphorylation, which can be used for signal transmission. Both η and W for 3T3 fibroblasts are larger than those for HT1080 and MDCK cells, which is consistent with the observation that 3T3 fibroblasts can effectively sense stiffer substrate stiffness than MDCK and HT1080 cells.
Mechanisms by which cell-specific mechanosensing can occur include variations in the relationship between actin flow and substrate stiffness, dynamics of actin-talin slip bonds, and dynamics of fibronectin-integrin catch-slip bonds. Here, we show that the catch-slip bond can enable different cell types to present unique stiffness ranges for mechanosensing through integrin clustering dynamics and the special way that fibronectin-integrin bonds are stabilized by force. Variations of talin and vinculin dynamics and integrin expression among cell types enable this cell-specific stiffness sensing ranges via integrin clustering dynamics.
Potential applications of Hill-type stiffness FAKpY397 algebraic expression
The central role of integrin clusters in regulating FAKp397 would be expected to propagate downstream, including Src activation by FAK phosphorylation, RhoA activation of mDia and ROCK, mDia and ROCK promotion of actomyosin-based stress fibers and contraction, and contraction-induced promotion of YAP/TAZ migration into the nuclear envelope and gene regulation (39). The FAK-RhoA-mDia/ROCK-YAP/TAZ cascade can further interact with other signaling pathways, such as MRTF and transforming growth factor–β cascades (40,41).
Although details about individual pathways are well known, little is known about their interactions. The Hill-type form of our model suggests that certain features of integrin cluster–based mechanosensing propagate far down the signaling cascade. Two kinds of phenomenological models from the literature describe the relationship between substrate stiffness (mechanical force) and intracellular signaling molecules (FAK or ROCK): A Hill-type relationship and a log-linear relationship. The Hill-type form of our model provides a first principles explanation of how these qualitative phenomenological models can arise from integrin clusters and FAKpY397.
In this study, we developed a mathematical model to test the hypothesis that FAKpY397-based mechanosensing arises from the dynamics of nanoscale integrin clustering, stiffness-dependent disassembly of integrin clusters, and FAKY397 phosphorylation within integrin clusters. Our model successfully predicts the relationships between substrate stiffness and FAKpY397 levels and, thus, provides a physical foundation for understanding the role of integrin clusters in mechanochemical conversion. The model provides mechanistic insight into how substrate stiffness can be sensed by the dynamics of integrin clusters in a cell-specific fashion and how these dynamics can translate into intracellular signals through FAKY397 phosphorylation.
MATERIALS AND METHODS
Spatial Monte Carlo model of integrin clustering
We developed a multistep process of integrin clustering. In our model, single integrin has two states, i.e., inactive state where integrin has a low affinity with ligands, which are assumed to be uniformly distributed in our simulation domain, and active state where integrin has a high affinity with ligands. The configuration transition of integrin from the inactive state to the active state is the prerequisite for integrin clustering. The molecular mechanisms underlying the lateral clustering of integrins remain controversial. The integrin clustering submodel applied in this work was based on three clustering mechanisms: ITD-mediated clustering, PI(4,5)P2-mediated clustering, and integrin cross-talk–mediated clustering. We note that many other mechanisms have been proposed, and the rationale for focusing on these three is described in the Supplementary Materials. We then built a coarse-grained model to simulate the integrin clustering process based on the multistep processes as described above.
We modeled the integrin clustering via a spatial Monte Carlo algorithm. All reactions including integrin activation and inactivation, integrin clustering and disassociation, and translational diffusion of integrin on the membrane were modeled on a 2D lattice plane with periodic boundary conditions. The integrin molecules were randomly placed in the lattice domains for the initial configuration. The simulation was implemented by randomly choosing an integrin molecule (an occupied lattice site), and then an event was determined (chemical reactions or translational diffusion) based on the probabilities. An event was chosen by calculating the probability distribution for all possible events
| (6) |
where i is the integrin site and M is the event; is the transition rate for M event; and σtot is defined as
| (7) |
where σD is the transition probability of translational diffusion, and σR is the transition probability of chemical reactions. All transition probabilities are shown in table S1. After integrin clustering, the clustered integrins (three or more integrin molecules) will stop diffusion. The plasma membrane was modeled as a 2D triangle lattice with each pixel being 15 nm in size. We set the integrin density as 500/μm2. Each integrin is simplified as a circular with a radius of 10 nm.
The dynamical parameters of the α5β1 and αvβ3 integrins were taken from the literature. Four dynamical parameters differ for α5β1 and αvβ3 integrins: (i) the diffusion coefficients of integrins (~0.1 μm2/s for α5β1 integrins and ~0.3 μm2/s for αvβ3 integrins); (ii) the activation rate of αvβ3 integrin is 10-fold higher than that of α5β1 integrin, as deduced from free-energy analysis; (iii) the intrinsic ligand binding affinity of the α5β1 integrin is higher than that of the αvβ3 integrin; and (iv) α5β1 integrins have longer lifetime than αvβ3 integrins, in response to the same bond tension. The two types of integrins with different dynamical parameters were dispersed within simulation regions. Note that, because lateral clustering of integrin remains controversial, we assumed that different integrins had the same lateral clustering affinity.
Stretchable parallel spring model of integrin cluster disassembly
The integrin clusters are connected to the actin cytoskeleton and ECM molecules (i.e., fibronectin). The integrin clusters and ECM are simplified as a stretchable parallel spring model in which Nint springs (integrin molecules, kint) are arranged in parallel along one spring (ECM, kecm). The fibronectin-integrin links are described as the catch-slip bond with dissociation rate koff as
| (8) |
The first term is identical to the slip portion of the catch-slip bond, and the second term is the catch portion of the catch-slip bond. ka, kb, Fa, and Fb are fitted parameters from existing experiments and are listed in table S2. At low force range (e.g., <~15 to 20 pN), the catch portion dominates and the dissociation rate decreases with increasing force. At high force range, the slip portion dominates and the dissociation rate increases with increasing force. The Kank-mediated slip bond is
| (9) |
The force from F acting on each of the closed bonds changed dynamically and follows mechanical equilibrium for the spring system
| (10) |
where kint is the stiffness of the integrin, kecm is the stiffness of the ECM, L is the distance between the ECM and the integrin molecules, and n is the number of closed bonds upon stretching. The whole spring system is pulled by actomyosin-based sarcomere-like CUs. According to the experimental observation, the displacement is applied to an integrin cluster as a function of time [L(t)]. We specified this displacement as a piecewise constant function, starting at L(0) = 5 nm, and with an increasing rate of 3 nm/s (2 to 3 step/s; 1.2 nm per step in experiments) to 60 nm as estimated from experimental values.
The substrate viscosity represents the resistance to diffusion of ligands on the substrate (36). The relationship between diffusion rate and substrate viscosity follows
| (11) |
where D is the diffusion coefficient, kB is Boltzmann’s constant, T is the absolute temperature, ηm is the substrate viscosity, R is the radius of a single lipid, and λ is the characteristic length.
We simulated the disassembly dynamics of integrin clusters using a Gillespie algorithm as follows:
1) If all springs are disassociated from actin and ECM, then the integrin cluster size is set to zero and the simulation terminates.
2) All springs are associated with actin and ECM as an initial configuration.
3) The force on each integrin is calculated by mechanical equilibrium.
4) Calculating the times of all types of chemical reactions.
5) The reaction with shortest time is implemented.
6) Update the displacement if the maximum value is not reached.
7) Return to step 1.
Whole-cell simulation of cell clusters
We assumed that integrin cluster formation rate is proportional to the integrin cluster number during the integrin formation phase in the spatial Monte Carlo model. The basic formation rate (kon = cMa ∝ 1/texp) was calculated according to experiments. Again, using the Gillespie algorithm, an integrin cluster is formed in the cell edge (a random angle) and then disappears based on its size and substrate stiffness. Note that all the formed clusters obey the power distribution according to the spatial Monte Carlo model. On the basis of the previous model, the integrin cluster will move toward the cell center as
| (12) |
where V0 = 3, c0 = 2, and c1 = 0.1. These steps are repeated until a steady state is reached. It has been shown that small clusters containing only one or two molecules are not experimentally detectable. So, we defined a minimum integrin cluster size of 10 molecules. We only included these detectable integrin clusters in our simulation. In the whole-cell simulation, the model did not actually simulate the dynamics of the entire FA directly; instead, it only simulated individual clusters that formed in a whole cell. The model did not simulate how these clusters aggregated to form FA.
Spatially particle-based simulation of FAKY397 activation in integrin clusters
We performed spatially resolved simulations with excluded volume interactions on a regular 3D lattice. We defined that all molecules have the same diameter l, and let this diameter define the lattice spacing, so that neighboring molecules are contacted with each other on the lattice. Integrin molecules in a cluster are placed in contact in a square arrangement on the membrane (note that the shapes of cluster will not influence the results). The membrane is modeled as x-y plane and extends for a length X in each direction with periodic boundaries. The cytoplasm has the depth Y with reflective boundaries at the top and bottom surfaces. N integrin clusters are randomly fixed on the membrane, and inactive FAK is initialized randomly in the simulation space with a diffusion coefficient D. At each time step, particles are looped over, with each particle diffusing to a neighboring site or participating in a reaction with a certain probability. We assumed that FAK can be activated by integrin cytoplasmic domain. We described the activation of FAK within integrin cluster in a simple reversible reaction. The association and disassociation of FAK with the integrin cluster are described by kin and kout, respectively. The activation (deactivation) of FAK in the cluster (cytoplasm) is described by ke+ (ke−).
Negative feedback regulation of integrin clustering
Phosphorylation of paxillin can promote the formation of FXs and FAs, while pPaxillin/FAK complexes can inhibit the formation of FXs and FAs. This results in a negative feedback loop for integrin clustering. To evaluate the effects of such negative feedback regulation on integrin clustering, we developed an integrated signaling network model containing a series of ordinary differential equations (see model equations and parameters in the Supplementary Materials).
Hydrogel preparation and cell culture
To prepare the substrates with different stiffnesses, 3 g of gelatin (type A, Sigma-Aldrich, USA) was dissolved in 10 ml of ultrapure water at 50°C under stirring to obtain a 30 weight % (wt %) gelatin aqueous solution. Microbial transglutaminase (0.4 g; mTG, Pangbo, China) was dissolved in 10 ml of ultrapure water at room temperature to obtain a 4 wt % aqueous solution. Gelatin and mTG aqueous solutions were filtered and then mixed with different concentration, respectively (gelatin aqueous solution: 10, 15, 10, 10, 15, and 20%; mTG aqueous solution: 0.5, 0.5, 1.5, 2, 2, and 2%) in petri dishes at 37°C for 3 hours to obtain hydrogels with gradient stiffness of 5, 15, 25, 45, 65, and 125 kPa, respectively. The stiffness was measured by an atomic force microscope (Agilent Technologies, USA). After soaking in phosphate-buffered saline for 24 hours, 3T3 cells were seeded onto gelatin hydrogels at a concentration of 1 × 106 cells/ml.
Western blotting analysis
FAKY397 phosphorylation was detected after 2 hours of cell seeding. Total proteins were extracted with a mammalian protein extraction kit (CWBIO, China) according to its protocol. The protein samples were separated by SDS–polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes. The membranes were then blocked with 5% bovine serum albumin at room temperature for 1 hour, followed by incubating with anti–phospho-FAKY397 and anti-FAK antibody (1:1000; Abcam, UK) at 4°C overnight. After washing with triethanolamine buffered saline and polysorbate 20 (TBST), the membranes were incubated with secondary antibodies (1:5000; Cell Signaling, USA) at room temperature for 1 hours. The relative integrated density of each protein band was determined using an Odyssey infrared imaging system (LI-COR, USA).
Immunofluorescence staining and quantification
3T3 cells were fixed in 4% paraformaldehyde for 20 min and permeabilized in 0.1% Triton X-100 for 10 min. The cells were then blocked with 5% goat serum and incubated with anti–phospho-FAKY397 antibody (Abcam, UK) and vinculin antibody (Cell Signaling Technology, USA) at 4°C overnight. Then, a DyLight 488 goat anti-rabbit polyclonal antibody (Zuangzhibio, China) was used as secondary antibody. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. FAKY397 phosphorylation and vinculin were detected after 3 days of cell culture. ImageJ software was used to quantify cell adhesions from imaging data. Briefly, the pixel intensity was extracted for each adhesion, which was then converted to a length in ImageJ by selecting the cell adhesion profile for each adhesion and setting minimal adhesion size to 0.15 μm2. Last, cell adhesion areas were calculated using ImageJ.
Sensitivity analysis of the model parameters
The sensitivity value S of Na and Ma to a parameter p is defined in Eq. 13, where p is the base parameter value, Na is the average integrin cluster size, and Ma is the cluster number at the base parameter value
| (13) |
The S value is equivalent to the slope of Na and Ma versus p on a log-log plot and represents the fold change in Na and Ma resulting from a fold change in a parameter value. The S value was calculated by plotting Na and Ma at 0.1-, 0.3-, 1-, 3-, and 10-fold changes of the base parameter on a log-log scale. A line was fitted to the data points with the slope of the line taken to be the S value. For some parameters (such as integrin density, diffusion constant), the allowable range did not allow for the extreme fold changes, so in these cases, the maximum or minimum fold change of the parameter was used to calculate the sensitivity instead.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (11772253, 11972280, and 11532009), Natural Science Basic Research Plan in Shaanxi Province of China (2018JM1007), the Shaanxi Province Youth Talent Support Program, the Young Talent Support Plan of Xi’an Jiaotong University, the National Institutes of Health (U01EB016422 and R01HL109505), and the NSF Science and Technology Center for Engineering Mechanobiology (CMMI 1548571). Author contributions: M.L. and F.X. designed the study, in consultation with G.H., Y.L., G.M.G., M.R.K.M., and T.J.L. B.C. and W.W. performed the modeling and experiments, respectively. B.C. and W.W. collected and analyzed the data. M.L., F.X., B.C., and G.M.G. prepared the manuscript. All authors read and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All reagents, data, and computer code for this study are available upon request from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaax1909/DC1
Note S1. The ordinary differential equations for integrin clustering, cluster disassembly, and FAKY397 activation.
Note S2. Assumptions.
Note S3. Limitations and potential problems.
Note S4. Quantified relationship between substrate stiffness and FAKY397 phosphorylation.
Table S1. The plasma membrane events and transition rates.
Table S2. Baseline model parameters.
Table S3. Conservation equations.
Table S4. Equations describing integrin activation and clustering and FAK-Src interactions.
Table S5. Equations describing negative feedback regulation.
Fig. S1. Negative feedback ODE model.
Fig. S2. Cell-spreading experiments.
Fig. S3. The effect of substrate viscosity on the integrin cluster lifetime.
Fig. S4. The comparison of Odde’s and our model.
Fig. S5. Sliding assumption of intact FA.
REFERENCES AND NOTES
- 1.Huang G., Li F., Zhao X., Ma Y., Li Y., Lin M., Jin G., Lu T. J., Genin G. M., Xu F., Functional and biomimetic materials for engineering of the three-dimensional cell microenvironment. Chem. Rev. 117, 12764–12850 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng X.-Q., Lee P. V. S., Lim C. T., Preface: Molecular, cellular, and tissue mechanobiology. Acta Mech. Sinica 33, 219–221 (2017). [Google Scholar]
- 3.Cheng B., Lin M., Huang G., Li Y., Ji B., Genin G. M., Deshpande V. S., Lu T. J., Xu F., Cellular mechanosensing of the biophysical microenvironment: A review of mathematical models of biophysical regulation of cell responses. Phys. Life Rev. 22-23, 88–119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moreno-Layseca P., Icha J., Hamidi H., Ivaska J., Integrin trafficking in cells and tissues. Nat. Cell Biol. 21, 122–132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei W.-C., Lin H.-H., Shen M.-R., Tang M.-J., Mechanosensing machinery for cells under low substratum rigidity. Am. J. Physiol. Cell Physiol. 295, C1579–C1589 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Changede R., Sheetz M., Integrin and cadherin clusters: A robust way to organize adhesions for cell mechanics. Bioessays 39, 1–12 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Changede R., Xu X., Margadant F., Sheetz M. P., Nascent integrin adhesions form on all matrix rigidities after integrin activation. Dev. Cell 35, 614–621 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Choquet D., Felsenfeld D. P., Sheetz M. P., Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell 88, 39–48 (1997). [DOI] [PubMed] [Google Scholar]
- 9.Choi C. K., Vicente-Manzanares M., Zareno J., Whitmore L. A., Mogilner A., Horwitz A. R., Actin and α-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 10, 1039–1050 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saxena M., Liu S., Yang B., Hajal C., Changede R., Hu J., Wolfenson H., Hone J., Sheetz M. P., EGFR and HER2 activate rigidity sensing only on rigid matrices. Nat. Mater. 16, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo B.-H., Springer T. A., Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 18, 579–586 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cluzel C., Saltel F., Lussi J., Paulhe F., Imhof B. A., Wehrle-Haller B., The mechanisms and dynamics of αvβ3 integrin clustering in living cells. J. Cell Biol. 171, 383–392 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li R., Mitra N., Gratkowski H., Vilaire G., Litvinov R., Nagasami C., Weisel J. W., Lear J. D., Degrado W. F., Bennett J. S., Activation of integrin αIIbβ3 by modulation of transmembrane helix associations. Science 300, 795–798 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Calderwood D. A., Campbell I. D., Critchley D. R., Talins and kindlins: Partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503–517 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elosegui-Artola A., Bazellières E., Allen M., Andreu I., Oria R., Sunyer R., Gomm J., Marshall J. F., Jones J. L., Trepat X., Roca-Cusachs P., Rigidity sensing and adaptation through regulation of integrin types. Nat. Mater. 13, 631–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye F., Kim S.-J., Kim C., Intermolecular transmembrane domain interactions activate integrin αIIbβ3. J. Biol. Chem. 289, 18507–18513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lock J. G., Jones M. C., Askari J. A., Gong X., Oddone A., Olofsson H., Göransson S., Lakadamyali M., Humphries M. J., Strömblad S., Reticular adhesions are a distinct class of cell-matrix adhesions that mediate attachment during mitosis. Nat. Cell Biol. 20, 1290–1302 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Zamir E., Katz M., Posen Y., Erez N., Yamada K. M., Katz B.-Z., Lin S., Lin D. C., Bershadsky A., Kam Z., Geiger B., Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts. Nat. Cell Biol. 2, 191–196 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Trichet L., Le Digabel J., Hawkins R. J., Vedula S. R. K., Gupta M., Ribrault C., Hersen P., Voituriez R., Ladoux B., Evidence of a large-scale mechanosensing mechanism for cellular adaptation to substrate stiffness. Proc. Natl. Acad. Sci. U.S.A. 109, 6933–6938 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seifert U., Rupture of multiple parallel molecular bonds under dynamic loading. Phys. Rev. Lett. 84, 2750–2753 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Sun Z., Tseng H.-Y., Tan S., Senger F., Kurzawa L., Dedden D., Mizuno N., Wasik A. A., Thery M., Dunn A. R., Fässler R., Kank2 activates talin, reduces force transduction across integrins and induces central adhesion formation. Nat. Cell Biol. 18, 941–953 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolfenson H., Meacci G., Liu S., Stachowiak M. R., Iskratsch T., Ghassemi S., Roca-Cusachs P., O’Shaughnessy B., Hone J., Sheetz M. P., Tropomyosin controls sarcomere-like contractions for rigidity sensing and suppressing growth on soft matrices. Nat. Cell Biol. 18, 33–42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roca-Cusachs P., Gauthier N. C., del Rio A., Sheetz M. P., Clustering of α5β1 integrins determines adhesion strength whereas αvβ3 and talin enable mechanotransduction. Proc. Natl. Acad. Sci. U.S.A. 106, 16245–16250 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng B., Lin M., Li Y., Huang G., Yang H., Genin G. M., Deshpande V. S., Lu T. J., Xu F., An integrated stochastic model of matrix-stiffness-dependent filopodial dynamics. Biophys. J. 111, 2051–2061 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang D. L., Bax N. A., Buckley C. D., Weis W. I., Dunn A. R., Vinculin forms a directionally asymmetric catch bond with F-actin. Science 357, 703–706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong F., Garcia A. J., Mould A. P., Humphries M. J., Zhu C., Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 185, 1275–1284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rafiq N. B. M., Nishimura Y., Plotnikov S. V., Thiagarajan V., Zhang Z., Shi S., Natarajan M., Viasnoff V., Kanchanawong P., Jones G. E., Bershadsky A. D., A mechano-signalling network linking microtubules, myosin IIA filaments and integrin-based adhesions. Nat. Mater. 18, 638–649 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Walcott S., Kim D.-H., Wirtz D., Sun S. X., Nucleation and decay initiation are the stiffness-sensitive phases of focal adhesion maturation. Biophys. J. 101, 2919–2928 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paszek M. J., Zahir N., Johnson K. R., Lakins J. N., Rozenberg G. I., Gefen A., Reinhart-King C. A., Margulies S. S., Dembo M., Boettiger D., Hammer D. A., Weaver V. M., Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Mugler A., Bailey A. G., Takahashi K., ten Wolde P. R., Membrane clustering and the role of rebinding in biochemical signaling. Biophys. J. 102, 1069–1078 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamadi A., Bouali M., Dontenwill M., Stoeckel H., Takeda K., Rondé P., Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J. Cell Sci. 118, 4415–4425 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Friedland J. C., Lee M. H., Boettiger D., Mechanically activated integrin switch controls α5β1 function. Science 323, 642–644 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Stutchbury B., Atherton P., Tsang R., Wang D.-Y., Ballestrem C., Distinct focal adhesion protein modules control different aspects of mechanotransduction. J. Cell Sci. 130, 1612–1624 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zaidel-Bar R., Milo R., Kam Z., Geiger B., A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 120, 137–148 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Ballestrem C., Hinz B., Imhof B. A., Wehrle-Haller B., Marching at the front and dragging behind: Differential αvβ3 integrin turnover regulates focal adhesion behavior. J. Cell Biol. 155, 1319–1332 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett M., Cantini M., Reboud J., Cooper J. M., Roca-Cusachs P., Salmeron-Sanchez M., Molecular clutch drives cell response to surface viscosity. Proc. Natl. Acad. Sci. U.S.A. 115, 1192–1197 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seong J., Tajik A., Sun J., Guan J., Humphries M. J., Craig S. E., Shekaran A., García A. J., Lu S., Lin M. Z., Wang N., Wang Y., Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc. Natl. Acad. Sci. U.S.A. 110, 19372–19377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian T., Harding A., Inder K., Plowman S., Parton R. G., Hancock J. F., Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 9, 905–914 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Liu J., Zhao X., Wang K., Zhang X., Yu Y., Lv Y., Zhang S., Zhang L., Guo Y., Li Y., A novel YAP1/SLC35B4 regulatory axis contributes to proliferation and progression of gastric carcinoma. Cell Death Dis. 10, 452 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Speight P., Kofler M., Szászi K., Kapus A., Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFβ-regulated Smad3. Nat. Commun. 7, 11642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piersma B., Bank R. A., Boersema M., Signaling in fibrosis: TGD-β, wnt, and yap/taz converge. Front. Med. 2, 59–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaax1909/DC1
Note S1. The ordinary differential equations for integrin clustering, cluster disassembly, and FAKY397 activation.
Note S2. Assumptions.
Note S3. Limitations and potential problems.
Note S4. Quantified relationship between substrate stiffness and FAKY397 phosphorylation.
Table S1. The plasma membrane events and transition rates.
Table S2. Baseline model parameters.
Table S3. Conservation equations.
Table S4. Equations describing integrin activation and clustering and FAK-Src interactions.
Table S5. Equations describing negative feedback regulation.
Fig. S1. Negative feedback ODE model.
Fig. S2. Cell-spreading experiments.
Fig. S3. The effect of substrate viscosity on the integrin cluster lifetime.
Fig. S4. The comparison of Odde’s and our model.
Fig. S5. Sliding assumption of intact FA.