

We uncovered structural and functional changes induced by mutations that dictate the DNA editing activity of ABE enzymes.
Abstract
Adenine base editors, which were developed by engineering a transfer RNA adenosine deaminase enzyme (TadA) into a DNA editing enzyme (TadA*), enable precise modification of A:T to G⋮C base pairs. Here, we use molecular dynamics simulations to uncover the structural and functional roles played by the initial mutations in the onset of the DNA editing activity by TadA*. Atomistic insights reveal that early mutations lead to intricate conformational changes in the structure of TadA*. In particular, the first mutation, Asp108Asn, induces an enhancement in the binding affinity of TadA to DNA. In silico and in vivo reversion analyses verify the importance of this single mutation in imparting functional promiscuity to TadA* and demonstrate that TadA* performs DNA base editing as a monomer rather than a dimer.
INTRODUCTION
Base editing is a new genome-editing technology that enables the conversion of one base pair into another at a genomic locus of interest through the precise chemical modification of a target nucleotide (1–4). Base editors consist of two subunits: a catalytically impaired Cas9 subunit [Cas9 nickase (Cas9n)] that acts as a DNA binding module and a single-stranded DNA (ssDNA)–specific editing enzyme subunit. The Cas9n binds to a preprogrammed genomic locus and opens the double-stranded DNA to expose a short stretch of ssDNA (5, 6). Subsequently, the ssDNA editing component carries out a chemical reaction to transform a target nucleobase into a noncanonical base (Fig. 1). Last, DNA replication or repair enzymes process the resulting mismatch into a canonical base pair to catalyze an overall base substitution reaction (1). Two types of base editors have been reported to date: cytosine base editors (CBEs), which rely on naturally occurring APOBEC enzymes (7, 8) to induce C⋮G → T:A mutations via a uracil intermediate (3), and adenosine base editors (ABEs), which use a modified version of the transfer RNA (tRNA) adenosine deaminase enzyme TadA to induce A:T → G⋮C mutations via an inosine intermediate (Fig. 1) (4). Both editors catalyze a deamination reaction at the target nucleobase and hence display considerable similarity between both the structure and mechanism of their enzymatic subunits.
Fig. 1. Mechanism of base editing by ABEs.
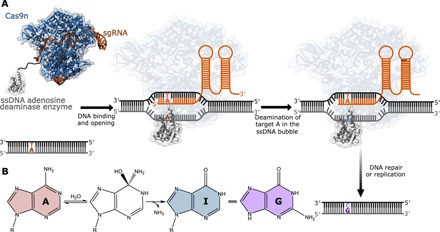
(A) A schematic representation of base editing by ABEs. The ABEs studied as a part of the current work consist of a Cas9n fused to an evolved TadA* protein. The binding of Cas9n to the target genomic locus unwinds the DNA double helix and exposes a small region of ssDNA. TadA* acts on this ssDNA and deaminates adenine (A) to form inosine (I), which is subsequently converted to guanine (G) through DNA repair and replication. (B) Overall chemical reaction catalyzed by ABEs.
Since wild-type TadA (wtTadA) was unable to perform adenosine deamination chemistry on ssDNA, despite its structural similarity to several ssDNA modifying enzymes of the APOBEC family (9), the development of ABEs required extensive protein engineering and evolution efforts. Starting with the TadA enzyme from Escherichia coli (10), which deaminates the wobble position of tRNAArg, directed evolution (11) was used to achieve efficient editing on a ssDNA substrate. Seven rounds of directed evolution identified 14 point mutations that transformed TadA into ABE7.10, which displays both high editing efficiency and broad sequence compatibility (4).
Understanding the effects of the mutations identified in TadA during the initial rounds of evolution is critical, particularly considering that expansion of the current base editing arsenal would require similar protein engineering and evolution efforts. Evolving enzymes from zero initial activity is notoriously challenging, as it requires screening an enormous sequence space for a select few mutants that impart new activity upon the enzyme of interest; evolution projects that improve upon weak initial activity see higher success rates in contrast (12). Therefore, a molecular understanding of how the initial TadA mutations gave rise to nonzero DNA editing activity would be indispensable for aiding future evolution efforts.
While the wild-type TadA enzyme does not exhibit any enzymatic activity on ssDNA when fused to Cas9n, the first two rounds of identified mutations (Asp108Asn, Ala106Val, Asp147Tyr, and Glu155Val) are responsible for imparting experimentally detectable levels of DNA editing activity to TadA*-Cas9n (* indicates incorporation of mutations) (4). Atomistic understanding of these mutations that cause the onset of detectable activity is paramount to rationally guide the development of future base editors. In this study, we use a combination of molecular dynamics (MD) simulations complemented with experimental measurements to scrutinize the structural and functional implications of these initial mutations.
RESULTS
Suppression of structural flexibility
We initiated our investigations into the effects of the TadA mutations by studying their influence on the overall structure of the protein. As the first two generations of ABE complexes are composed of a TadA monomer fused to Cas9n (the wild-type enzyme acts on tRNA as a dimer), we furthermore focused our studies on monomeric TadA mutants. In addition, while the final generation ABE7.10 construct is composed of a wtTadA-TadA* dimer fused to Cas9n, we measured the A:T to G⋮C base editing efficiency of the monomeric TadA7.10*-Cas9n construct at six different target As in human embryonic kidney (HEK) 293T cells and found no decrease in efficiency as compared to the dimer construct (Fig. 2A). These results suggest that the successive rounds of evolution performed on TadA have caused the enzyme to modify ssDNA as a monomer. Therefore, the TadA monomer is the most relevant model system with which to study the enzyme in the context of its interaction with ssDNA. Wild-type TadA consists of a five-stranded β sheet core, with five α helices wrapped around to form the active site. In addition, TadA displays a long-disordered loop (24 amino acids, residue numbers 118 to 142) that joins the β4 and β5 strands (Fig. 2, B and C) (10). We performed 500-ns all-atom MD simulations starting with the crystal structure of wild-type E. coli TadA (10) (TadA*0.1) to gain insights into the structural dynamics of the protein (see Materials and Methods). The simulations confirmed the highly fluxional nature of the β4-β5 loop in the wild-type enzyme (Fig. 2, B and C). To observe the effects associated with the mutations on the structure and dynamics of TadA, we subjected the TadA*0.1 model to sequential mutations at residues 108, 106, and 147 and 155 to yield the TadA*1.1, TadA*1.2, and TadA*2.1 mutants, respectively. MD simulations of the four TadA* mutants reveal that the most substantial structural difference between TadA*0.1 and the higher-generation TadA*s occurs in this β4-β5 loop. While TadA*0.1 displays high flexibility in this region, the first mutation (Asp108Asn) leads to restricted structural mobility of the loop, with the TadA*1.2 and TadA*2.1 following this same trend (Fig. 2B). The ubiquitous nature of this change is indicated by the reduced flexibility being observed for TadA*7.10, which harbors all the 14 mutation reported in the most evolved ABE protein (fig. S1A) (4).
Fig. 2. Structural changes in the TadA* mutants revealed through MD simulations.
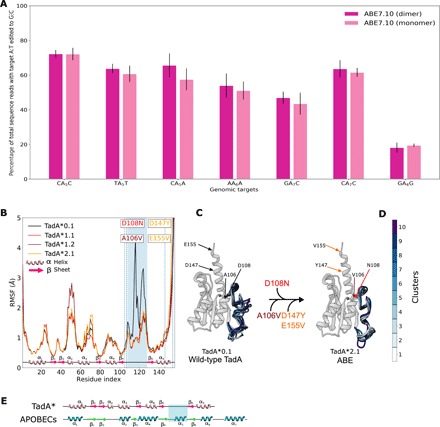
(A) The A:T to G⋮C base editing efficiency of the monomeric and dimeric ABE7.10 at six different target As in HEK293T cells. Values and error bars reflect the mean and SD of three independent biological replicates performed on different days. (B) Residue level flexibility of TadA* shown in terms of the root mean squared fluctuation (RMSF) of the Cα atoms of the peptide backbone. The β4-β5 loop region is highlighted in blue, and each mutation is indicated with its respective location in the protein. (C) Representative clusters from the trajectory of TadA*0.1and TadA*2.1 superimposed on each other, with clusters color coded as indicated in (D). (E) Comparison of the secondary structure of zinc-dependent deaminases: TadA* and APOBECs. Helices and arrows denote the α helices and β strands, respectively. The β4-β5 loop of interest in this study that interacts with the polynucleotide substrate is highlighted in both cases.
The suppression of the loop dynamics indicates that the replacement of Asp with Asn at residue number 108 of the protein is accompanied by a gain of structure. To quantify this effect in each TadA* mutant, we clustered all the conformations sampled by the β4-β5 loop throughout the simulations into 10 structural groups representative of the conformational space. Comparison of these representative clusters reveals high variability among the loop conformations sampled by TadA*0.1 [average root mean square deviation (RMSD) = 1.75 Å; table S1], while TadA*1.1 and higher display significantly smaller differences in the orientation of the β4-β5 loop across the 10 representative structural groups (average RMSD = 0.74, 0.88, and 0.624 Å for TadA*1.1, TadA*1.2, and TadA*2.1, respectively; table S1). Our simulations also indicate that this decrease in the structural flexibility of the β4-β5 loop of the TadA* mutants (Fig. 2) may be responsible for TadA* acting as a monomer to modify DNA, as it resembles the dynamics of the wtTadA dimer (fig. S2).
Interaction of TadA*s with ssDNA
Next, we sought to understand the functional significance of the ABE mutations in the context of ssDNA binding. The lack of any reported structure of the entire ABE-DNA complex in the literature precludes the use of MD simulations on the entire ABE complex. Since the system of interest is only the evolving monomeric TadA enzyme and its ssDNA target and the TadA*-Cas9n complex has a size of more than 200 kDa, we reduced our molecular model to a series of TadA* mutants in complex with a 11-mer piece of ssDNA (5′-GACTACAGACT-3′). In lieu of including Cas9n and the full R-loop portions of the ABE complex, we have imposed constraints on the 5′- and 3′-terminal nucleotides of the ssDNA, keeping them 40 Å apart [based on Protein Data Bank (PDB) ID: 5y36 (13)] to maintain its R-loop conformation throughout the entirety of the simulations (Materials and Methods). We then carried out unbiased MD simulations in which we allowed each of the four TadA* mutants to interact with the constrained ssDNA for 500 ns and looked for changes in interactions between individual TadA* residues and the nucleic acid substrate among the four mutants. Experimentally, TadA*0.1 is not competent for base editing, but the three mutants (TadA*1.1, TadA*1.2, and TadA*2.1) are. We therefore specifically focused on identifying the interactions present in only TadA*1.1 and higher, with a particular emphasis on residue 108 (Asp in TadA*0.1 and Asn in all others), as this residue is responsible for imparting the enzyme with detectable base editing activity. To gain insights into the spatial extent of the interactions at play in the binding process, we projected the interactions between the target adenosine and its 5′- and 3′-adjacent bases (TAC) and the surrounding amino acids onto asteroid diagrams (Fig. 3, A to D). In these diagrams, we use a network representation in which these three nucleotides of the DNA are depicted as the central node and the TadA* residues are the peripheral nodes. As the typical donor atom–donor hydrogen–acceptor atom distance is approximated to be 3.5 Å in globular proteins (14, 15), we defined the first interaction shell around the DNA as all amino acids within 4 Å of the three bases in the active site. The size of each node is proportional to the time individual residues spend within the 4-Å shell during the simulation. Hydrogen bonds between residues [defined as in the CPPTRAJ package (16, 17)] are depicted as arrows connecting the corresponding nodes, with the arrow size being proportional to the hydrogen-bond strength, which is defined as the number of times that the specific hydrogen bond is established (Fig. 3, A to D, and fig. S3). In the crystal structure of wild-type TadA in complex with its tRNA substrate [PDB ID: 2b3j (18)], Asp108 makes a hydrogen bond with the 2′-OH group of the 5′ flanking base. In contrast, when complexed with ssDNA, which lacks this hydrogen-bond donor, the repulsive electrostatic interactions between the negatively charged Asp108 and the phosphate backbone of the DNA favors a conformation in which Asp108 points toward the active site zinc ion (Fig. 3E). Mutating Asp108 to Asn neutralizes this repulsive interaction and causes the residue to flip into a more energetically favorable conformation in which it faces the DNA substrate and interacts with the base 5′ to the target adenosine. This conformational change allows Asn108 to form a hydrogen bond with the carbonyl at position 2 of the 5′ nucleobase when this base is a pyrimidine (Fig. 3F). This interaction between Asn108 and the 5′ pyrimidine may explain the earlier generation ABE’s strict sequence preference for a pyrimidine at this position. As subsequent mutations are introduced into TadA*, this hydrogen bond is progressively strengthened, and in the TadA*2.1 mutant, a second hydrogen bond forms between Asn108 and the phosphate backbone (Fig. 3F). We attribute this conformational switch to the hydrogen-bond donor nature of Asn as opposed to the hydrogen-bond acceptor nature of the negatively charged Asp. The Asp147Tyr and Glu155Val mutations, which are introduced as TadA*1.2 becomes TadA*2.1, do not lie within the first interaction shell, but rather cause structural rearrangements to the protein that strengthen the interactions between Lys110, Phe148, and Phe149 and the ssDNA and cause Arg153 to become a double donor (Figs. 3, D and F, and 4A).
Fig. 3. Analyses of the TadA*-DNA contacts.
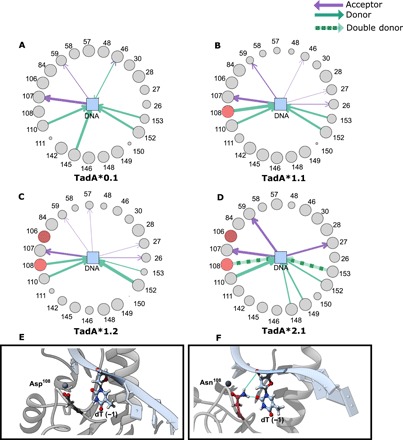
Asteroid plots for (A) TadA*0.1-ssDNA, (B) TadA*1.1-ssDNA, (C) TadA*1.2-ssDNA, and (D) TadA*2.1-ssDNA complexes. Details of the conformational change of residue 108 when it is mutated from Asp (TadA*0.1) (E) to Asn (TadA*1.1and later) (F).
Fig. 4. Asteroid plot analysis of second-generation mutations.
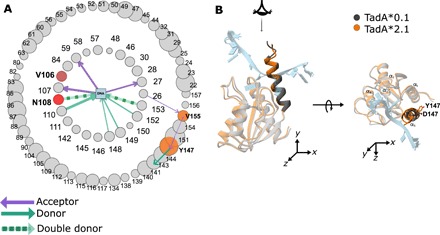
(A) The first and second interaction shell around the three nucleotides in the active site of the TadA*2.1-ssDNA complex. The size of the node corresponds to the time in which the amino acid resides in the first/second shell. First round mutations are red, and second round mutations are orange. (B) Structural overlay of average structure of TadA*0.1-ssDNA and TadA*2.1-ssDNA complexes. This α5 helix has been highlighted to depict its overall movement toward the active site upon Asp147Tyr mutation.
Analyses of mutations in the α5 helix
To better understand the effects of the second-generation mutations (Asp147Tyr and Glu155Val), which are located outside of the 4-Å primary interaction shell, we expanded our analysis of the TadA*-ssDNA simulations to include the secondary interaction shell, which encompasses all residues within 4 Å of the primary interaction shell residues. Analogous to Fig. 3, individual residues are represented by nodes whose sizes are proportional to the number of frames in the MD trajectory in which the residue lies within the specific shell, with hydrogen bonds between residues depicted as arrows between the interacting nodes, and the arrow size being proportional to the hydrogen-bond strength (Fig. 4A). We found that while Asp147Tyr and Glu155Val do not belong to the primary interaction shell, they do influence the manner in which the primary shell residues interact with the ssDNA. Mutation of Asp147 to Tyr abrogates a salt bridge between itself and Arg150 (primary interaction shell) that exists in TadA*0.1 (Fig. 2A). This lost interaction results in the movement of the entire α5 helix toward the active site (Fig. 4B), causing residues 150 to 153 to considerably spend more time within the primary interaction shell and increasing the strength of the hydrogen bonds between residues 148, 149, and 153 and the ssDNA (Figs. 3, A and D and 4A, and fig. S4A). Moreover, the Asp147Tyr and Glu155Val mutations, which convert negatively charged residues into neutral amino acids in the α5 helix, increase the positive charge density on the surface of the TadA*2.1 (fig. S4, B and C), potentially enhancing the electrostatic interactions of the TadA* with the negatively charged ssDNA.
Differential binding of TadA*s to ssDNA
After qualitatively observing the interactions between the TadA* residues and the ssDNA, we sought to quantify the thermodynamics of ssDNA binding by the four TadA* mutants. To this end, we performed umbrella sampling simulations to determine the potential of mean force (PMF) associated with the binding process. In this analysis, the PMF is calculated as a function of the relative distance between the centers of mass of the ssDNA and the TadA* mutants (ξ, collective variable), which we vary from 10 to 30 Å (Fig. 5, A and B). The PMF profile describing the binding of TadA*0.1 to ssDNA has a minimum at ξ = 20 Å and shows a relatively small (17 kcal/mol) dissociation energy as the ssDNA is moved away from the protein to ξ = 30 Å. Once the Asp108Asn mutation in TadA*1.1 has been introduced, the PMF minimum slightly shifts toward the active site (to ξ = 18 Å), and we observe the free energy of binding increase to 42 kcal/mol as ξ is increased to 30 Å (Fig. 5C). The PMF profiles calculated for the binding of TadA*1.2 and TadA*2.1 to ssDNA maintain this increased slope for ξ larger than 20 Å, implying that the single Asn108 mutation is effectively responsible for increasing the binding free energy by ≈20 kcal/mol. For ξ values less than 20 Å, the PMF profiles become sequentially more repulsive with subsequent generations, demonstrating a tighter binding of the ssDNA to the TadA*. We repeated the binding free energy calculations with a different sequence of ssDNA that lacks 5′-pyrimidine (5′-GTCAAGAAAC-3′) and again observed mutation-dependent TadA*-ssDNA binding but to a lesser extent of only 10 kcal/mol for this substrate (fig. S5). These results are in agreement with experimental observations that these early generation ABE mutants had a strong preference for YAC (Y = pyrimidine) sequence motifs. These findings highlight the importance of the Asp108Asn mutation in imparting functional promiscuity to the TadA* enzyme toward ssDNA editing (4) through an increase in the free energy of binding. While the binding affinity is not a direct measure of the editing efficiency, our analyses of the TadA*-ssDNA complexes demonstrate that the initial Asp108Asn mutation, which plays a critical role in the onset of the DNA editing capability of the ABEs, leads to increased binding between the TadA* and the ssDNA substrate. We speculate that higher-generation mutations take advantage of this increased binding to improve the kinetics of base editing and broaden the substrate sequence scope.
Fig. 5. ABE mutations lead to an increase in TadA* binding to the ssDNA.
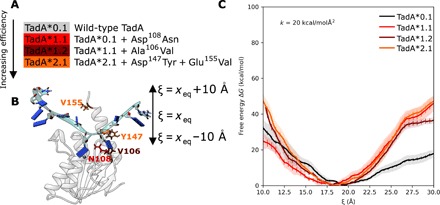
(A) List of early generation mutations in TadA that were analyzed in this study. (B) The model of the TadA*-ssDNA complex simulated to determine the binding energy profile of the TadA* mutants. The binding-unbinding event was monitored using the collective variable (ξ) defined as the distance between the center of mass of the protein and DNA. (C) The free-energy profile of binding of the ssDNA to various TadA*s. For each TadA*-ssDNA complex, the average PMF is shown as a function of the continuously changing ξ values. The shaded regions around individual curves depict the standard deviation for four independent replicates of the umbrella sampling simulations. The error bars associated with the mean PMFs indicate the error calculated using the block-averaging method.
Reversion analysis of Asn108 mutation
To confirm the crucial role played by Asn108 in ssDNA editing by ABE, we subjected the higher generation of TadA* mutants (TadA*1.2 and TadA*2.1) to reversion analysis of this mutation. Specifically, by mutating Asn108 back to Asp108 in both TadA*1.2 and TadA*2.1, we generated two new TadA mutants, TadA*1.2(N108D) and TadA*2.1(N108D), respectively (Fig. 6A).
Fig. 6. Significance of Asn108 for base editing.
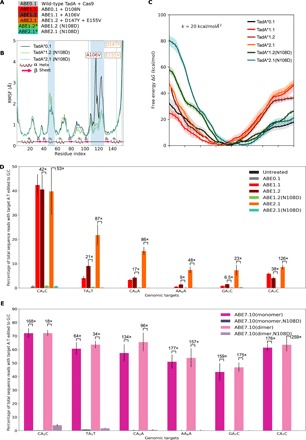
(A) ABE constructs created by reverting the Asp108Asn mutation in the higher generation ABEs. (B) RMSF of the Cα atoms of the TadA*1.2(N108D) and TadA*2.1(N108D) enzymes. (C) The free-energy profile of binding of the hybrid TadA*s to ssDNA. The shaded regions around individual curves depict the SD for four independent replicates of the umbrella sampling calculations. The error bars associated with the mean PMFs indicate the error calculated using block-averaging method. (D and E) A:T to G⋮C base editing efficiencies in HEK293T cells by the various ABEs at six different target As. Fold-decrease values upon reversion analysis of the Asp108Asn mutation are indicated above the bars. Values and error bars reflect the mean and SD of three independent biological replicates performed on different days.
To disentangle the structural contribution of Ala106Val, Asp147Tyr, and Glu155Val from that of Asp108Asn, we monitored the structural flexibility of TadA*1.2(N108D) and TadA*2.1(N108D) (Fig. 6B). We observed the maintenance of the β4-β5 loop stabilization, suggesting that the Ala106Val mutation is also sufficient to induce this change in structural flexibility (fig. S6). We also observed a slight increase in the flexibility of the α2 helix due to this mutation, but upon introduction of the round two mutations, this is lost. To complement these structural studies, we also characterized the binding free energy in the TadA*1.2(N108D)-ssDNA and TadA*2.1(N108D)-ssDNA complexes. Unlike the structural results and despite having respectively one and three mutations that were experimentally found to be favorable for ssDNA editing, TadA*1.2(N108D) and TadA*2.1(N108D) produced PMF profiles that are significantly different from those of their parent mutants (Fig. 6C). In particular, both PMFs closely follow the corresponding profile obtained for TadA*0.1 for ξ values larger than 20 Å yet are considerably more repulsive for ξ values smaller than 20 Å. We performed analogous reversion analysis for the TadA*7.10 (which contains all 14 identified mutations) and observed qualitatively similar trends for the TadA*7.10(N108D) (fig. S7A).
These differences demonstrate weaker binding between the ssDNA and ABE mutants lacking the Asn108 mutation. To confirm our computational results, we generated the ABE1.2(N108D) and ABE2.1(N108D) constructs and experimentally measured their respective A:T to G⋮C base editing efficiencies using high-throughput sequencing (HTS) alongside ABE0.1, ABE1.2, and ABE2.1 in HEK293T cells at six different targets. Reversion of Asn108 mutation to Asp led to an average decrease in the A:T to G⋮C base editing efficiency of 22-fold (ranging from 6.5- to 42-fold) and 70-fold (ranging from 22.6- to 126-fold) for ABE1.2 and ABE2.1, respectively (Fig. 6D). It is notable that even the presence of all three Ala106Val, Asp147Tyr, and Glu155Val mutations was not sufficient to restore editing activity with Asp at position 108; both ABE1.2(N108D) and ABE2.1(N108D) induced average A:T to G⋮C base editing efficiencies of 0.36 and 0.29% across all six editable As, as compared to 3.6 and 16.8% for their respective parental mutants. Reversion of the Asn108 mutation in the ABE7.10 background displayed a similar trend. Replacement of Asn108 with Asp in both monomeric and dimeric ABE7.10 decreased the A:T to G⋮C base editing efficiency by an average factor of 146-fold (ranging from 67- to 176-fold) and 123-fold (ranging from 35-fold to 259-fold), respectively (Fig. 6E). This indicates that the presence of 13 higher generation mutations, independently of being installed in the monomeric or dimeric construct, cannot compensate for the loss of the Asn108 mutation. The importance of residue Asn108 in ABE7.10 was also recognized in the experimental study by Rees et al. (19), where radical substitutions of Asn108 with Phe, Trp, and Met were found to result in complete abolishment of any DNA editing activity at all target adenosines except when the target nucleobase was at position 5 within the protospacer. However, conservative substitutions of Asn108 with Gln, and Lys, resulted in decreased DNA editing efficiencies for these mutants, albeit in a sequence-dependent manner and to a much smaller extent than the substitution with Asp (19). The results of this study thus provide further support of the hydrogen-bonding analysis presented here, which emphasizes the requirement of a positive charge density, either in the form of a hydrogen-bond donor as Asn (Fig. 4) or Gln (19) or a positively charged residue as Lys (19) for enabling the ssDNA activity of TadA*. Collectively, these data demonstrate the drastic effects a single atom substitution (from N to O) can have on protein function and highlight the complexity of protein sequence-structure-function relationships.
DISCUSSION
Enhancing our understanding of how an enzyme’s sequence influences its function will help increase the success of future directed evolution projects. Although the mutations discovered using directed evolution are exceptional at enhancing the particular enzymatic property being pursued, these mutations are difficult to predict and require considerable experimental resources. As the development of future base editors will likely involve additional directed evolution efforts (20, 21), maximizing our understanding of the outcomes of previous studies on this front will aid in these future studies. This work is an a posteriori study using a combination of computational simulations and experimental measurements to understand the mutations generated during the directed evolution of ABEs (4). We have additionally carried out MD simulations of TadA* and TadA*-ssDNA models to explore how the initial mutations accumulated during directed evolution give rise to ssDNA editing by the ABE enzyme. Installation of the Asp108Asn mutation in the TadA*0.1 to generate TadA*1.1 leads to a significant decrease in the flexibility of the β4-β5 loop of the TadA (Fig. 2). This loop is known to both impart sequence specificity to the wild-type TadA enzyme through interactions with the nucleobases immediately upstream of the target A base and also serve as the dimerization interface between the individual TadA proteins (18). Our simulations indicate that the structural dynamics of TadA* mutants (Fig. 2) resembles that of the wtTadA dimer (fig. S2), which may explain how the TadA* enzymes are performing DNA base editing as monomers. The changes observed in the dynamics of the β4-β5 loop therefore may help broaden the substrate scope of the TadA* enzymes to include both tRNA and ssDNA. In addition, as the TadA* mutants were evolved to function as monomers, this change in the dynamics may be increasing the enzyme’s affinity for ssDNA at the expense of protein dimerization. This is proven to be the case, as we experimentally observe that the TadA enzyme works as a monomer when acting on ssDNA, a finding that represents a key step in characterizing the mechanism of base editing by ABE (Fig. 2). This is an unexpected result that fundamentally changes our understanding of how ABEs function and will likely affect future ABE engineering and optimization studies.
Intriguingly, loss of conformational flexibility in the β4-β5 loop of TadA* appears to make the overall structure of the protein more analogous to the APOBEC family of proteins (Fig. 2E). APOBEC enzymes are a class of proteins that have cytidine deamination activity on both ssDNA and ssRNA (7, 8) and were repurposed into the original CBEs. The inherent nature of the APOBECs to edit a broad range of nucleotide targets is preserved in the CBEs, which have been shown to exhibit considerable off-target DNA and RNA activities due to the APOBEC1 portion of the base editor (22, 23). This dual-substrate specificity of APOBECs has been attributed to specific conformations of the active site loop (α1-β1 loop, β2-α2 loop, and β4-α5 also referred to as the loop 1, loop 3, and loop 7, respectively) that interacts with the 5′ flanking base of the substrate nucleotide using both experiments and simulations (24–27). Both TadA and the APOBEC enzymes share a core five-stranded β sheet structural element surrounded by α helices. The β4-β5 loop serves the same functional purpose in both enzymes, but the length of this loop is substantially longer in TadA, and in the APOBECs, it assumes a definite α-helical secondary structure (Fig. 2E and fig. S8).
The gain in structure of this loop in TadA may contribute to the gain of ssDNA editing capability by TadA* (7, 28), but it is not solely responsible for this activity. The TadA*1.2(N108D) enzyme retains reduced mobility in the β4-β5 loop yet displays wild-type like ssDNA binding affinity according to our simulations and nearly undetectable base editing efficiencies in our experimental work. Note that the Ala106Val mutation causes a substantial gain in mobility of the α2 helix (Figs. 2A and 6B), which is canceled out when the Asp147Tyr and Glu155Val mutations are incorporated. The α2 helix of TadA aligns with the β2-α2 active site loop of the APOBECs (Fig. 2E and fig. S8), which lacks secondary structure and has been shown to be responsible for sequence specificity of the enzymes.
Our simulations show that when wild-type TadA interacts with ssDNA, the absence of a hydrogen-bond donor (in the form of the 2′-OH group of the ribose sugar in RNA) for Asp108 causes this residue to flip into an energetically unfavorable conformation away from the negatively charged DNA backbone. This unfavorable conformation is responsible for the lack of ssDNA editing by the wild-type enzyme, as the presence of all other 13 favorable mutations, and the favorable interactions they bring with them, is not enough to compensate for the strained configuration that Asp108 is forced to adopt when in the presence of DNA rather than RNA. However, upon neutralization of this negative charge when Asp108 is mutated to Asn (a single atom substitution from O to N), the residue can now rotate back into a more energetically favorable position, allowing for the enzyme to interact with ssDNA. This rotation toward the ssDNA substrate also allows for the formation of a hydrogen bond between residue 108 in TadA* and the ssDNA (the −1 nucleotide in Fig. 3, D and E). This hydrogen bond further strengthened in TadA*2.1, where Asn108 becomes a double hydrogen-bond donor, interacting with the phosphate backbone. The phosphate backbone is a structural element common to both DNA and RNA, suggesting that in the process of acquiring ssDNA editing capabilities, TadA* may not surrender its native RNA editing functionality. This has been confirmed by previous reports of off-target RNA editing by ABE enzymes (19, 23). Furthermore, it was recently found that removing wtTadA from ABE7.10 does not suppress its RNA deamination activity, which demonstrates that the Asp108Asn mutation supports RNA binding by TadA* (29).
While one may expect only residues in the first shell (that interact directly with the ssDNA) to be primarily responsible for enhancing the thermodynamics and kinetics of ssDNA editing by TadA*, 6 of the 14 overall mutations accumulated during directed evolution actually reside in the second shell of the enzyme (fig. S1). In addition to electrostatic contributions, through our simulations, we observed that the Asp147Tyr and Glu155Val mutations, both of which reside in the α5 helix (fig. S4B), cause structural rearrangements in the protein, effectively initiating a chain reaction that strengthens the interactions between a variety of primary shell residues and the ssDNA substrate. Note that nearly half (6 of 14) of the ABE7.10 mutations are located in the α5 helix, highlighting the significance of understanding its role in ssDNA editing. These enhanced hydrogen-bonding interaction between the TadA* residues and the ssDNA, caused in aggregate by all four mutations, and the now-favorable conformation of the residue 108 when it is Asn, also translate into an increased free energy binding of the TadA*s to ssDNA (Fig. 5 and fig. S5). Upon reversion of Asn108 to Asp, however, even in the presence of the three other advantageous mutations (Ala106Val, Asp147Tyr, and Glu155Val), we observe a marked decrease in the binding affinity of TadA*1.2(N108D) and TadA*2.1(N108D) to ssDNA (Fig. 6C). On the basis of these observations, we speculate that the Asp108Asn mutation may play a bipartite role: It affords structural rigidity to the region of the enzyme responsible for sequence specificity and increases the binding affinity of the TadA enzyme to ssDNA through hydrogen-bonding interactions. However, the hydrogen bonds that Asn108 forms with the 5′ nucleobase and the phosphate backbone are not its only contribution to the onset of DNA editing activity by ABEs. Simulations and experiments verify that reversion of Asn108 back to Asp from higher-generation ABEs leads to nearly complete loss in the base editing activities of higher ABE mutants (Fig. 6), despite the presence of up to 13 other beneficial mutations in TadA* that have created additional hydrogen-bonding interactions between TadA* and the ssDNA (Figs. 3D and 4). It is likely that the increased conformational strain imposed on the Asp108 residue when it must flip around to point away from the DNA backbone is energetically unfavorable enough to preclude ssDNA binding even with these additional favorable hydrogen-bonding interactions.
This study provides the first insights into the mechanism of base editing by ABEs, beginning with the observation that the TadA* enzyme acts a monomer to modify ssDNA. The results presented in this study additionally provide an explanation of the structural and functional roles of the initial TadA mutations identified in the evolution of ABE. We anticipate that this atomistic understanding of previous successful directed evolution experiments will enable the prediction of new mutations and lead to the rational engineering of future base editors.
MATERIALS AND METHODS
Computer simulations
The crystal structure of E. coli TadA enzyme (PDB ID: 1z3a) was used to define the initial coordinates for TadA*0.1 (10). The TadA*1.1, TadA*1.2, TadA*2.1, TadA*1.2(N108D), and TadA*2.1(N108D) mutants were prepared by inducing virtual mutations to the TadA0.1 structure using the mutagenesis plugin available in PyMOL (30). We then combined the crystal structure of E. coli TadA enzyme with the tRNA substrate from its structural homolog from Staphylococcus aureus [PDB ID: 2b3j (18)] to prepare the TadA*-ssDNA complexes. The remodeling of the tRNA structure by the removal of the 2′ hydroxyl groups and all changes in the sugar pucker of the nucleotide backbone were carried out using the swapna command in the Chimera software (31). Moreover, since the tRNA structure was crystallized bearing nebularine, a nonhydrolyzable adenosine analog (18), we used the swapna command to substitute nebularine with adenine. To unpair the 3′ and 5′ ends of the hairpin loop, we used steered MD simulations using the exposed ssDNA nucleotides of the ternary complex of the cryo–electron microscopy structure of CRISPR-Cas9 [PDB ID: 5y36 (13)] as a reference structure (fig. S9). This yielded the TadA*0.1-ssDNA complex as illustrated in fig. S9. Similarly, the complexes of TadA*1.1, TadA*1.2, TadA*2.1, TadA*1.2(N108D), and TadA*2.1(N108D) mutants with ssDNA were developed using the mutagenesis plugin of PyMOL (30). All crystallographic water molecules within 3-Å distance of the protein/protein-ssDNA surface were preserved during the modeling process, and each of the systems was solvated using a truncated octahedral box of TIP3P water molecules (32). All titratable residues were assigned protonation states at pH 7 as predicted by the H++ server (33, 34). Varying number of Na+ ions were added to each system to maintain charge neutrality. The protein and the DNA atoms were represented using the Amber ff14SB force field and the bsc1 parameters, respectively (35–37). All MD simulations were performed under periodic boundary conditions using the CUDA accelerated version of PMEMD implemented in Amber18 suite of programs (38–40). The structures were first relaxed using a combination of steepest descent and conjugate gradient minimization. This was followed by a 1-ns heating to 298.15 K and 10-ns equilibration under harmonic restraints. Subsequently, we removed all restraints (except on the 5′ and 3′ termini of the substrate DNA sequence) and carried out 500-ns unbiased MD simulations for the six TadA* mutants and corresponding TadA*-ssDNA complexes. Additional details of this protocol can be found in Supplementary Materials and Methods. Table S2 summarizes all the simulations that were carried out during this study.
We calculated the free-energy binding profiles of the TadA*-ssDNA complexes along the collective variable corresponding to the distance between the centers of mass of the protein and the ssDNA substrate. For each TadA*-ssDNA complex, the PMF along this collective variable was calculated using umbrella sampling simulations. Starting from the equilibrated TadA*-ssDNA structures, we conducted four independent sets of umbrella sampling simulations for all of the six TadA*-ssDNA complexes, and the final PMFs were reconstructed using the weighted histogram analysis method (WHAM) algorithm (41). Additional error analysis was carried out using a custom block averaging script based on the method described by Zhu and Hummer (42).
The CPPTRAJ module implemented within Amber18 was used to analyze all the MD trajectories (16, 17). The root mean squared fluctuation of the ABE mutants and clustering of configurations from each MD trajectory were calculated, with respect to the Cα atoms of the protein backbone. We identified the primary and secondary interaction shells and the associated H-bonding network using the mask and hbond keywords of CPPTRAJ, respectively (see the Supplementary Materials for details). The PDB2PQR webserver, in conjunction with the APBS server, was used to calculate the electrostatic maps for the ABE0.1 and ABE2.1 models (43). The visualization of the MD trajectories was rendered using Chimera, and data were plotted using Matplotlib (44).
Cloning
All ABE plasmids were constructed using USER cloning (45) with pCMV_ABEmax (Addgene plasmid no. 112095) as a template using Phusion U Hot Start Polymerase (Thermo Fisher Scientific). Complete sequences of ABE’s are listed in the Supplementary Materials. All single guide RNA (sgRNA) expression plasmids were generated using blunt-end cloning (3) with pFYF1230 (Addgene plasmid no. 47511) as a template using Phusion High-Fidelity DNA Polymerase (New England BioLabs). Complete protospacer/protospace adjacent motif (PAM) sequences are listed in table S3. All DNA vector amplification was carried out using NEB 10-β competent cells (New England BioLabs). All plasmids were purified using the ZymoPURE II Plasmid Midiprep Kit (Zymo Research).
Cell culture
HEK293T cells (American Type Culture Collection, CRL-3216) were maintained in high glucose Dulbecco’s modified Eagle’s medium supplemented with GlutaMAX (Thermo Fisher Scientific), 10% (v/v) fetal bovine serum (Thermo Fisher Scientific), and penicillin-streptomycin (100 μg/ml; Thermo Fisher Scientific) at 37°C with 5% CO2.
Transfections
HEK293T cells were seeded in 48-well VWR Multiwell Cell Culture Plates at a density of 150,000 cells per well in 250 μl of media without penicillin-streptomycin. Four hours after plating, 1000 ng of ABE plasmid and 250 ng of sgRNA plasmid were transfected using 1.5 μl of Lipofectamine 2000 (Thermo Fisher Scientific) per well according to the manufacturer’s protocol.
High-throughput DNA sequencing of genomic DNA
Transfected cells were rinsed with phosphate-buffered saline (150 μl/well; Thermo Fisher Scientific) 5 days after transfection. Cells were lysed on the plate by addition of 100 μl of lysis buffer [10 mM tris (pH 7.5), 0.1% SDS, and proteinase K (25 μg/ml)]. Lysed cells were then heated at 37°C for 1 hour, followed by 80°C for 20 min. Genomic loci of interest were polymerase chain reaction (PCR) amplified with Phusion High-Fidelity DNA Polymerase (New England BioLabs) according to the manufacturer’s protocol using the primers indicated in table S4, 1 μl of genomic DNA mixture as a template, and 26 or fewer rounds of amplification. Unique forward and reverse combinations of Illumina adapter sequences were then appended with an additional round of PCR amplification with Phusion High-Fidelity DNA Polymerase (New England BioLabs) according to the manufacturer’s protocol using 1 μl of round 1 PCR mixture as a template and 15 rounds of amplification. The products were gel purified and quantified using the NEBNext Ultra II DNA Library Prep Kit for Illumina. Samples were then sequenced on an Illumina MiniSeq according to the manufacturer’s protocol.
HTS data analysis
Sequencing reads were demultiplexed in MiniSeq Reporter (Illumina), and individual FASTQ files were analyzed using a previously reported MATLAB script (4).
Supplementary Material
Acknowledgments
We thank M. Norman for a Director's Discretionary Allocation on the Comet GPU cluster at the San Diego Supercomputer Center. K.L.R. thanks C. Egan and E. Lambros for helpful discussions. Funding: This research was supported by the University of California San Diego and the NIH through grant no. 1R21GM135736-01. All computer simulations used resources of the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF through grant no. ACI-1548562. Author contributions: The manuscript was written through contributions of all authors. K.L.R. performed all computer simulations. K.L.R., A.C.K., and F.P. conceptualized and designed the research. Competing interests: A.C.K. is a consultant of Pairwise Plants and Beam Therapeutics, companies that are developing and using base editing technologies. All other authors declare that they have no competing interests. Data and materials availability: HTS data have been deposited in the National Center for Biotechnology Information Sequence Read Archive database under accession code PRJNA590028. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaaz2309/DC1
Supplementary Materials and Methods
Fig. S1. Asteroid plot for TadA*7.10-ssDNA.
Fig. S2. Comparison of the structural flexibility of the TadA monomer with TadA dimer.
Fig. S3. Percentage contact and the fractional H-bonding between the three nucleotides and the first interaction shell amino acids.
Fig. S4. Asteroid plot for TadA*0.1-ssDNA complex.
Fig. S5. Mutations lead to an increase in TadA* binding to the ssDNA (AAG).
Fig. S6. Structural importance of Ala106Val mutation.
Fig. S7. PMF of the TadA*-ssDNA complexes calculated using steered MD simulations.
Fig. S8. Comparison of the structural flexibility of the ecTadA with hAPOBEC3A.
Fig. S9. Modeling of TadA*-ssDNA.
Fig. S10. Umbrella sampling data and biased statistics.
Table S1. Comparison of RMSD of the representative clusters.
Table S2. Summary of the systems modeled and the types of simulations conducted in this study.
Table S3. DNA sequences used for simulations and in mammalian tissue culture experiments.
Table S4. First round genomic DNA PCR sequences.
Supplementary sequences
REFERENCES AND NOTES
- 1.Rees H. A., Liu D. R., Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19, 770–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komor A. C., Badran A. H., Liu D. R., Editing the genome without double-stranded DNA breaks. ACS Chem. Biol. 13, 383–388 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komor A. C., Kim Y. B., Packer M. S., Zuris J. A., Liu D. R., Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaudelli N. M., Komor A. C., Rees H. A., Packer M. S., Badran A. H., Bryson D. I., Liu D. R., Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishimasu H., Ran F. A., Hsu P. D., Konermann S., Shehata S. I., Dohmae N., Ishitani R., Zhang F., Nureki O., Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang F., Taylor D. W., Chen J. S., Kornfeld J. E., Zhou K., Thompson A. J., Nogales E., Doudna J. A., Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conticello S. G., The AID/APOBEC family of nucleic acid mutators. Genome Biol. 9, 229 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris R. S., Petersen-Mahrt S. K., Neuberger M. S., RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 10, 1247–1253 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Rubio M. A. T., Pastar I., Gaston K. W., Ragone F. L., Janzen C. J., Cross G. A. M., Papavasiliou F. N., Alfonzo J. D., An adenosine-to-inosine tRNA-editing enzyme that can perform C-to-U deamination of DNA. Proc. Natl. Acad. Sci. U.S.A. 104, 7821–7826 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J., Malashkevich V., Roday S., Lisbin M., Schramm V. L., Almo S. C., Structural and kinetic characterization of Escherichia coli TadA, the wobble-specific tRNA deaminase. Biochemistry 45, 6407–6416 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Arnold F. H., Design by directed evolution. Acc. Chem. Res. 31, 125–131 (1998). [Google Scholar]
- 12.Brustad E. M., Arnold F. H., Optimizing non-natural protein function with directed evolution. Curr. Opin. Chem. Biol. 15, 201–210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huai C., Li G., Yao R., Zhang Y., Cao M., Kong L., Jia C., Yuan H., Chen H., Lu D., Huang Q., Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat. Commun. 8, 1375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mills J. E. J., Dean P. M., Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J. Comput. Aided Mol. Des. 10, 607–622 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Baker E. N., Hubbard R. E., Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 44, 97–179 (1984). [DOI] [PubMed] [Google Scholar]
- 16.Roe D. R., Cheatham T. E. III, PTRAJ and CPPTRAJ: Software for processing and analysis of molecular synamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Roe D. R., Cheatham T. E. III, Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data. J. Comput. Chem. 39, 2110–2117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Losey H. C., Ruthenburg A. J., Verdine G. L., Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol. 13, 153–159 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Rees H. A., Wilson C., Doman J. L., Liu D. R., Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv. 5, eaax5717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang T., Badran A. H., Huang T. P., Liu D. R., Continuous directed evolution of proteins with improved soluble expression. Nat. Chem. Biol. 14, 972–980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thuronyi B. W., Koblan L. W., Levy J. M., Yeh W.-H., Zheng C., Newby G. A., Wilson C., Bhaumik M., Shubina-Oleinik O., Holt J. R., Liu D. R., Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 37, 1070–1079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gehrke J. M., Cervantes O., Clement M. K., Wu Y., Zeng J., Bauer D. E., Pinello L., Joung J. K., An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol. 36, 977–982 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grünewald J., Zhou R., Garcia S. P., Iyer S., Lareau C. A., Aryee M. J., Joung J. K., Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi K., Carpenter M. A., Banerjee S., Shaban N. M., Kurahashi K., Salamango D. J., McCann J. L., Starrett G. J., Duffy J. V., Demir Ö., Amaro R. E., Harki D. A., Harris R. S., Aihara H., Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 24, 131–139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi K., Demir Ö., Carpenter M. A., Wagner J., Kurahashi K., Harris R. S., Amaro R. E., Aihara H., Conformational switch regulates the DNA cytosine deaminase activity of human APOBEC3B. Sci. Rep. 7, 17415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holden L. G., Prochnow C., Chang Y. P., Bransteitter R., Chelico L., Sen U., Stevens R. C., Goodman M. F., Chen X. S., Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature 456, 121–124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salter J. D., Bennett R. P., Smith H. C., The APOBEC protein family: United by structure, divergent in function. Trends Biochem. Sci. 41, 578–594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conticello S. G., Langlois M.-A., Neuberger M. S., Insights into DNA deaminases. Nat. Struct. Mol. Biol. 14, 7–9 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Grünewald J., Zhou R., Iyer S., Lareau C. A., Garcia S. P., Aryee M. J., Joung J. K., CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 37, 1041–1048 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.W. L. DeLano, The PyMOL Molecular Graphics System, Version 1.8 (Schrödinger LLC, 2015).
- 31.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 33.Gordon J. C., Myers J. B., Folta T., Shoja V., Heath L. S., Onufriev A., H++: A server for estimating p Ka s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33, W368–W371 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anandakrishnan R., Aguilar B., Onufriev A. V., H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–W541 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., Simmerling C., ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ivani I., Dans P. D., Noy A., Pérez A., Faustino I., Hospital A., Walther J., Andrio P., Goñi R., Balaceanu A., Portella G., Battistini F., Gelpí J. L., González C., Vendruscolo M., Laughton C. A., Harris S. A., Case D. A., Orozco M., Parmbsc1: A refined force field for DNA simulations. Nat. Methods 13, 55–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salomon-Ferrer R., Götz A. W., Poole D., Le Grand S., Walker R. C., Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Salomon-Ferrer R., Case D. A., Walker R. C., An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3, 198–210 (2013). [Google Scholar]
- 39.Case D. A., Cheatham T. E. III, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., Woods R. J., The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York, P. A. Kollman, AMBER 2018 (University of California, 2018).
- 41.Kumar S., Rosenberg J. M., Bouzida D., Swendsen R. H., Kollman P. A., The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 13, 1011–1021 (1992). [Google Scholar]
- 42.Zhu F., Hummer G., Convergence and error estimation in free energy calculations using the weighted histogram analysis method. J. Comput. Chem. 33, 453–465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L. E., Brookes D. H., Wilson L., Chen J., Liles K., Chun M., Li P., Gohara D. W., Dolinsky T., Konecny R., Koes D. R., Nielsen J. E., Head-Gordon T., Geng W., Krasny R., Wei G.-W., Holst M. J., McCammon J. A., Baker N. A., Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunter J. D., Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007). [Google Scholar]
- 45.Badran A. H., Guzov V. M., Huai Q., Kemp M. M., Vishwanath P., Kain W., Nance A. M., Evdokimov A., Moshiri F., Turner K. H., Wang P., Malvar T., Liu D. R., Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance. Nature 533, 58–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaaz2309/DC1
Supplementary Materials and Methods
Fig. S1. Asteroid plot for TadA*7.10-ssDNA.
Fig. S2. Comparison of the structural flexibility of the TadA monomer with TadA dimer.
Fig. S3. Percentage contact and the fractional H-bonding between the three nucleotides and the first interaction shell amino acids.
Fig. S4. Asteroid plot for TadA*0.1-ssDNA complex.
Fig. S5. Mutations lead to an increase in TadA* binding to the ssDNA (AAG).
Fig. S6. Structural importance of Ala106Val mutation.
Fig. S7. PMF of the TadA*-ssDNA complexes calculated using steered MD simulations.
Fig. S8. Comparison of the structural flexibility of the ecTadA with hAPOBEC3A.
Fig. S9. Modeling of TadA*-ssDNA.
Fig. S10. Umbrella sampling data and biased statistics.
Table S1. Comparison of RMSD of the representative clusters.
Table S2. Summary of the systems modeled and the types of simulations conducted in this study.
Table S3. DNA sequences used for simulations and in mammalian tissue culture experiments.
Table S4. First round genomic DNA PCR sequences.
Supplementary sequences