

Temperature-stable dissolving film eliminates cold-chain storage and successfully immunizes mice sublingually and buccally.
Abstract
A novel, thin-film platform that preserves live viruses, bacteria, antibodies, and enzymes without refrigeration for extended periods of time is described. Studies with recombinant adenovirus in an optimized formulation that supports recovery of live virus through 16 freeze-thaw cycles revealed that production of an amorphous solid with a glass transition above room temperature and nitrogen-hydrogen bonding between virus and film components are critical determinants of stability. Administration of live influenza virus in the optimized film by the sublingual and buccal routes induced antibody-mediated immune responses as good as or better than those achieved by intramuscular injection. This work introduces the possibility of improving global access to a variety of medicines by offering a technology capable of reducing costs of production, distribution, and supply chain maintenance.
INTRODUCTION
Vaccines have often been described as the greatest human intervention supporting global health, second only to clean drinking water (1). In 1900, 53% of deaths in the United States were due to infectious disease (2). In 2010, that number markedly fell to 3%. However, many vaccines are rarely designed to meet the needs of every global community. This is highlighted by the fact that, in 2015, more than half of the leading causes of death in low-income countries were the result of infectious disease, while less than 10% of deaths in high-income countries were attributed to similar causes (3). One of the primary reasons for this disparity is limited uptake of vaccines with nearly 20 million infants worldwide not receiving routine immunizations, such as three doses of diphtheria-tetanus-pertussis vaccine (4). To date, the vaccine requisites of developing countries have not been adequately met for a variety of reasons, including issues associated with fragile health care systems, conflict resolution, policy making, program management, financing, supply chain, and distribution across large urban areas and to the most remote locations (5). Each of these issues contributed in some part to the delay in the distribution of experimental vaccines and therapeutics against Ebola during the 2014–2016 outbreak (6, 7). The ramifications of these roadblocks were also seen in the 2016–2017 meningitis outbreak in Nigeria, where the cost and limited supply of a vaccine contributed to the mortality and spread of disease (8). A more long-term example of this effect is exemplified by the prevalence of deaths in children less than 5 years of age from rotavirus infection in India and Sub-Saharan African countries over the past decade due to their inability to access the vaccine without external subsidiaries (9).
Much of the cost of a given vaccine is product specific and dictated by the nature of the antigen, immunogen, or pathogen of interest and the complexity of large-scale production processes (10). Distribution and administration costs also substantially add to the price of a vaccine. These costs are often not product specific, as most vaccines are temperature sensitive and require cold-chain maintenance, which entails transporting, storing, and monitoring them at a recommended temperature from the point of production to the point of use (11). In total, the cost of distribution and administration often exceeds the cost of vaccine production, making them the most prohibitive barriers to global immunization campaigns. One simulation model found that of the $64 billion needed to introduce 18 vaccines to 94 countries within a 10-year time frame, $38 billion would be used to support distribution and administration alone (12). Even when developing countries can access vaccines, such as those available through the Expanded Program on Immunization (EPI) and other programs (13), maintenance and monitoring of required cold-chain conditions are not guaranteed due to varying access to equipment and resources needed to maintain optimal environmental conditions (14, 15). For example, during the inspection of vaccinating facilities in Cameroon, only 76% had a functional thermometer in use. Of those, 20% had recorded temperatures that fell outside those recommended by vaccine manufacturers. This was primarily due to vulnerabilities in the power grid and lack of alternative sources of electricity, as was also found in other regions around the globe (16, 17).
Development of a technology that could notably minimize resources needed for distribution and administration of vaccines would notably enhance access to these medicines and improve global health. Breakthroughs in formulation development and technology that have effectively stabilized live bacteria, viruses, and recombinant proteins at ambient temperatures offer a viable solution to improving global distribution of vaccines (18–20). It has been predicted that if the original 5-in-1 pentavalent vaccine was reformulated as a fully thermostable preparation, its availability in developing countries could reach nearly 100% (21). Most of the vaccines recommended for low- and middle-income countries by the World Health Organization (WHO) are packaged in glass and plastic vials, often with diluents and related materials (22). Thus, the amount of revenue and manpower required for storage and monitoring of these preparations is significant and may be prohibitive for stockpiling in preparation for widespread infectious disease outbreaks for many regions around the world. Most require reconstitution with specific solvents before administration and are predominantly given by injection (22). This requires the participation of numerous health care professionals, which adds to the cost of any vaccination campaign and can also be a source of vaccine failure and wastage due to human error in developed as well as low- and middle-income countries (23, 24). Vaccines developed for mucosal administration provide a reasonable alternative to parenteral vaccination strategies. They are needle free, eliminating the need for and associated costs of trained personnel for vaccine administration. They are also capable of inducing both localized and systemic immune responses by facilitating transport of vaccine and associated antigens throughout the mucosa-associated lymphoid tissue (25). For some pathogens, mucosal immunity may also be necessary to improve the durability or long-term protection offered by a vaccine (26). They also heighten patient acceptance and compliance in both industrialized and developing countries (25, 27). However, only four of the vaccines currently available are delivered by the mucosal route (28).
In this paper, we summarize our efforts to develop a novel platform that can reduce costs associated with storage, distribution, and administration of vaccines and other biological drug products. Initially, more than 400 formulations were screened for their ability to enhance the immune response of an adenovirus-based Ebola vaccine (29). Formulations providing favorable in vivo data were then assessed for their ability to stabilize recombinant adenoviruses in a thin, peelable film matrix (Fig. 1, A and B). One formulation was found to stabilize adenovirus for a period of 3 years at room temperature (Fig. 1C). This formulation, reconstituted and given via the respiratory route to nonhuman primates, offered full protection from a lethal dose of Ebola (30) but was never used for the primary purpose for which it was designed, immunization by the sublingual (SL) or the buccal (BU) routes. Additional studies revealed that this platform was also capable of preserving live bacteria at room temperature for 8 months with minimal loss of viability upon reconstitution (Fig. 1D). Further investigation revealed that the performance of a primary antibody (Fig. 1E) and an antibody-enzyme conjugate (Fig. 1F) embedded in our thin film at room temperature was superior to that of the same product stored in the manufacturer’s liquid formulation stored under the same conditions. Subsequent mechanistic studies with a recombinant adenoviral vector have allowed us to identify criteria vital for long-term stabilization that can be applied and adapted for other biological products. These are summarized here along with results of a pivotal proof-of-concept study illustrating the utility of this dosage form for oral immunization.
Fig. 1. Film technology stabilizes live microorganisms and biological compounds for extended periods of time at ambient temperatures.
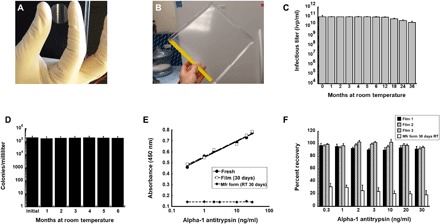
(A) Prototype unit dose film useful for the assessment of long-term stability as described in this manuscript. (B) Prototype large-scale film that can be used for stockpiling and storage and sectioned into multiple single-dose films for distribution. (C) Thirty-six-month stability profile of recombinant adenovirus in solid film matrix. Replicate films (n = 5 per time point) were stored at 20°C, reconstituted with sterile water and infectious titer assessed with a standard limiting dilution assay (19). ivp, infectious virus particles. (D) Six-month stability profile of film containing live bacteria at 20°C. Films (n = 5 per time point) were reconstituted with sterile saline, and solutions were plated on nutrient rich agar. Colonies were counted for assessment of recovery of live bacteria from the film. (E) Binding affinity of primary antibody (178260, Millipore) stabilized in thin film and stored at room temperature (RT) for 30 days is superior to that of the manufacturer’s product stored as a liquid under the same conditions. Solutions made from rehydrated films were used in an alpha-1 antitrypsin (A1AT) enzyme-linked immunosorbent assay (ELISA) assay in triplicate as described (59). Correlation coefficients (r2) for standard curves made with fresh stock stored at 4°C (fresh), antibody reconstituted from film after storage for 30 days at 20°C (film), and antibody stored in the manufacturer’s liquid formulation after storage for 30 days at 20°C (Mfr. form) were 0.99, 0.99, and 0.10 respectively. (F) Recovery of binding affinity of AP192P, a donkey anti-mouse IgG antibody, horseradish peroxidase (HRP) conjugate (Millipore) after storage in a thin film at 20°C. Percent recovery is the relative absorbance reading generated by an assay using secondary antibody from reconstituted films with that of fresh stock as supplied by the manufacturer for each given concentration of A1AT standard. Results were also compared with the manufacturer’s stock stored at RT (instead of −20°C as recommended). Each of the bars in the graph represents readings obtained from antibody recovered from three separate films in a given experiment, and data reflect the averages ± SEM of data collected from three separate experiments. Photo credit (A and B): Maria A. Croyle, The University of Texas at Austin.
RESULTS
To identify factors key for recovery and preservation of virus infectivity in a novel thin film dosage form, over 30 combinations of buffers and excipients were evaluated according to a fractional factorial design. Excipients used in our multicomponent films fall into three specific categories: base, binder, and surfactant. For simplicity, multicomponent formulations will be referred to by number throughout the text as they are listed in table S1. Preliminary studies identified the importance of solvent, base concentration, the presence of binders and surfactants, and final formulation pH on recovery of infectious virus upon reconstitution of films. Additional studies illustrate the role film components play in the preservation of live virus during exposure to stressors such as repeated freeze-thaw cycles and elevated temperatures. The final series of studies summarized here aim to identify mechanistic explanations for virus stabilization during the film-forming process and illustrate the utility of this dosage form for oral immunization.
Identifying key formulation components for stability: Solvent, excipient selection, and pH
Initial screening studies were designed to evaluate the role of several different aqueous solvent systems in the maintenance of infectivity of live, recombinant adenovirus during the drying process. Films prepared in unbuffered formulations were least effective at preserving virus titer (Fig. 2). Infectious virus was not recovered from films produced with the lowest amount of base excipient in the absence of buffering agents (Fig. 2A). Use of phosphate-buffered saline (PBS) to prepare films containing the lowest concentration of base significantly improved recovery of infectious virus particles (ivp) (73%; Fig. 2A). PBS also improved recovery of virus from films made with medium (92%; Fig. 2B) and high (75%; Fig. 2C) base concentrations with respect to those prepared in an unbuffered solvent. Preparations made with tris buffer with low and medium base concentrations demonstrated recoveries of live virus of greater than 95% (Fig. 2, A and B). Preparations made with this buffer experienced a drop in pH of ~1.1 units regardless of base concentration during the drying process, while those made with PBS and unbuffered solution demonstrated drops in pH of 2.5 and 3 units, respectively.
Fig. 2. Solvent and excipient combinations that preserve virus recovery from film matrix maintain optimal pH during drying.
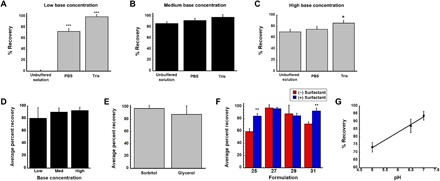
Recovery of virus from films containing 0.5% (low, A), 1.5% (medium, B), and 3.0% (high, C) concentrations of base excipient prepared in three different solvent systems was evaluated using an infectious titer assay. Once an optimal solvent system was selected, stepwise addition of excipients to the film identified an optimal base excipient concentration (D) and binder (E). Additional screening revealed that surfactant further improved recovery of infectious virus from the matrix (F). Analysis of data collected during each phase of screening revealed that formulations that maintained a final dried pH of 7.0 exhibited the highest recovery of infectious virus from the film (G). In each panel (A to G), data represent the average ± SD of a minimum of three films per condition. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed Student’s t test. Formulations are summarized in the figure according to the numbers assigned in table S1.
Since films prepared with tris buffer were the most efficient in maintaining virus infectivity during the drying process, a second series of screening studies was initiated to identify the impact base concentration had on virus recovery during drying (Fig. 2D). Films prepared with the lowest base concentration were able to retain 80 ± 17% of the original titer after drying, while those prepared with moderate and high base concentrations recovered 90 ± 6.5 and 93 ± 5.4% of ivp, respectively. With the realization that films containing base alone could not support full recovery of infectious virus upon reconstitution, two different binders were added to the medium base formulation and evaluated for their ability to improve infectious titer after drying. The average recovery of films prepared with sorbitol was 97 ± 4.1% (Fig. 2E). Films prepared with glycerol maintained 88 ± 14% of the original virus titer.
In a final effort to further improve recovery of infectious virus from films after drying, surfactant was added to tris-buffered preparations containing either base formulation alone or each of the binding agents described above (Fig. 2F). Addition of surfactant significantly improved recovery of infectious titer in films containing only the medium concentration of base from 59 ± 4.7% (formulation 25, table S1) to 84 ± 1%. A similar effect was seen with the highest base concentration with recovery increasing from 72 ± 3.6% (formulation 31) to 93 ± 4.6%. When sorbitol was added to the medium base preparation (formulation 27), recovery of infectious virus rose from 96 ± 3.4% to 97 ± 2.1%. The surfactant affected recovery in a slightly opposite manner when glycerol was also present in the formulation as the original virus concentration fell from 88 ± 14% to 85 ± 1.0% (formulation 30). Aggregate analysis of data collected during the screening of solvent, base, and binder formulations revealed that there was a direct correlation between virus recovery and the pH of the film after drying (r2 = 0.996; Fig. 2G). Film preparations capable of recovering more than 90% of the original virus titer after drying had final pH values within the range of 6.5 to 7.4. Because of the high variability, flexibility, and difficulty in handling films prepared with the low and high concentrations of base excipient, their use was excluded from the remainder of the studies outlined here.
Role of surfactant on the rate of release of virus from films
Films prepared with the base excipient alone released virus at a rate of 6.3 × 106 ivp/ml per minute after placement in PBS at 37°C with gentle agitation (Fig. 3A). Addition of sorbitol increased this rate twofold. When surfactant was also included in the formulation, the rate increased 10-fold to 6.0 × 107 ivp/ml per minute. The entire amount of infectious virus added to films containing both surfactant and sorbitol was released within 5 min (Fig. 3B). Films prepared with base excipient alone (formulation 25) released 90% of the dose in 5 min, and only 97% of the total dose after 2 hours. The addition of sorbitol (formulation 27) resulted in 88% of dose released after 5 min but only marginally improved release to 98% of the full dose after 2 hours.
Fig. 3. Release of virus from the film matrix is tunable.

(A) Virus release rate from films prepared by stepwise addition of excipients. Films containing 1.25 × 1012 vp were placed in warmed (37°C) PBS with gentle agitation, and samples were collected over a period of 2 hours. The concentration of infectious virus was determined by a standard infectious titer assay. (B) Cumulative release profiles of films containing base, binder, and surfactant combinations. Data collected during the dissolution of each film was normalized with that generated from virus placed in the correlating liquid formulation and collected over 20-min intervals to account for any loss attributable to agitation and extended exposure to heat. In each panel, data represent the average ± SD of a minimum of three films per condition. *P < 0.05, two-tailed Student’s t test. Formulations are summarized in the figure according to the numbers assigned in table S1.
Mechanical properties of films
During the screening process, the mechanical properties of each film formulation were also assessed as the ductility of a film dictates not only the feasibility of production but also the ease at which it can be handled and manipulated (31). Varying the amount of base, binder, and surfactant did not significantly affect the measured elongation of any of the films when a uniform force was applied to them (fig. S1). Evaluation of tensile strength of films revealed that glycerol was the only excipient that significantly altered the mechanical properties of the films, making them prone to tearing (fig. S2). Thus, because of the difficulty in processing and handling films containing glycerol, sorbitol was selected as the most optimal binder to be used for the remainder of the studies summarized in this manuscript. Since the surfactant further improved recovery of virus from the film, a short-term stability study was developed with formulations 27 and 28.
Impact of rehydration medium on recovery of virus infectivity from films
Solutions prepared from solubilized films have the potential to be used for administration of vaccines and other biologicals by various routes, which may dictate the nature of the solvent used for rehydration. After the initial screening studies were complete, a small pilot study was initiated to determine whether certain solvents, commonly used for reconstitution of drug products, would be compatible with the film platform. Solvents evaluated were as follows: sterile water for injection, sterile saline, 100 mM PBS (pH 7.4), and 10 mM tris buffer (pH 8.1). Cell culture medium [Dulbecco’s modified Eagle’s medium (DMEM) containing 2% fetal bovine serum (FBS)] was used as a control solvent for comparison. Statistical analysis revealed that there was no significant difference between the recovery of infectious virus with respect to solvent used for reconstitution in films prepared with the same formulation and that contained the same amount of virus before drying (fig. S3). Assessment of film formulations before drying revealed that reducing the amount of base excipient with subsequent addition of binder and surfactant significantly reduced the viscosity of each preparation (fig. S4). When virus was added, the viscosity of each liquid film formulation fell to half of its initial value. Viscosities of solutions made from reconstituted films were also significantly less than those made before drying. Because the film platform can be used for a variety of applications, the volume of diluent can be altered to support different routes of administration or to concentrate the virus. Reducing the reconstitution volume by half increased the solution viscosity fourfold.
Stability of adenovirus in an optimized film formulation
The durability of virus stabilized in the optimized film formulation was evaluated by placing films containing 1.25 × 1012 virus particles (vp) in a temperature-controlled chamber set at 37°C. Infectious titer was then evaluated in films reconstituted with sterile media at set time points for 14 days (Fig. 4A). Virus at the same concentration was also stored under the same conditions in a traditional standard liquid formulation [100 mM PBS (pH 7.0), 10% v/v glycerol, control A) commonly used to compare novel formulations (32, 33) and titer assessed over time for comparison. The amount of infectious virus recovered from films stored at 37°C was ~ 97 ± 2.6% of the original titer throughout the 14-day study period. Infectious virus could not be detected in the control A preparation within 24 hours and for the remainder of the study. A longer evaluation of virus stability revealed that films could maintain 100 ± 2.2% of their original titer for 84 days at 4°C (Fig. 4B). This was in stark contrast to preparations containing the same amount of virus in PBS alone (control B formulation) in which recovery fell to 67 ± 0.85% of its original titer after 25 days at 4°C. Infectious virus could not be detected in either of the control formulations after 48 days at 4°C.
Fig. 4. Optimized thin film matrix enhances adenovirus stability at elevated temperatures and under environmental stressor conditions.
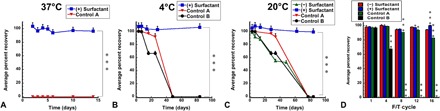
Films containing 1.25 × 1012 vp were prepared in batch and either stored in controlled environmental chambers held at 37°C (A), 4°C (B), and 20°C (C) for 84 days or subjected to a series of 16 freeze-thaw (F/T) cycles (D). Replicates (at least three per time point) were reconstituted, and live virus concentration was assessed by a standard infectious titer assay (19). Virus was placed at the same concentration in two standard liquid formulations, and infectious titers under each storage condition were also assessed for comparison. In each panel, control A formulation consisted of 100 mM PBS (pH 7.4) and 10% glycerol, and control B formulation consisted of 100 mM PBS alone. In each panel, data represent the average ± SD. *P < 0.05, ***P < 0.001, two-tailed Student’s t test. Formulations are summarized in the figure according to the numbers listed in table S1.
To determine the role the surfactant played in virus stability, films containing 1.25 × 1012 vp were prepared and stored at 20°C. Infectious titer was compared over time to preparations stored in the same manner containing the same amount of virus in liquid control A and control B formulations (Fig. 4C). All preparations retained full recovery of infectious virus during the first 7 days of storage except for films that did not contain surfactant (92 ± 4.1%). Films containing surfactant did not experience a significant drop in virus infectivity throughout the study period. These films contained 100 ± 0.14% of their original titer after storage for 84 days at 20°C. Infectious virus could not be detected in either of the control formulations or in films without surfactant at the 84-day time point.
The robust stability profile of films containing surfactant stimulated further evaluation of the ability of this preparation to protect virus under different environmental stressors. Films prepared with and without surfactant were exposed to a series of 16 freeze-thaw cycles by freezing samples at −80°C and then thawing them to 20°C. Replicate films were collected at different intervals, rehydrated, and infectious titer compared with liquid formulations containing PBS and 10% glycerol (control A) or PBS alone (control B) that underwent the same freeze-thaw schedule. By the fourth freeze-thaw cycle, infectious titer of virus formulated in PBS alone fell to 83 ± 0.4%, while all other preparations retained more than 95% of their original titer (Fig. 4D). Infectious virus could not be detected in the control B formulation by the eighth freeze-thaw cycle, while 91 ± 0.08% of the original titer was detected in the control A preparation. By the 16th freeze-thaw cycle, the infectious titer of films prepared without surfactant fell to 94 ± 0.5% of their original titer, while those containing surfactant did not demonstrate a notable drop in titer (100 ± 0.7% recovery) after completing the entire freeze-thaw series.
Stability of adenovirus in rehydrated films
Solutions prepared from films could be used for a variety of applications, such as nasal, oral, or ocular delivery of biologicals. Thus, the physical stability of virus in rehydrated films prepared with and without surfactant was assessed at 4° and 20°C for a period of 84 days. Infectious titer was assessed over time and compared to virus prepared at the same concentration in control A and control B formulations. Infectious titer of all preparations remained at 97% of the original concentration or higher after 14 days at 4°C, except the control B preparation, which fell to 67 ± 1.3% of the original titer and remained at this level through the 25-day time point (Fig. 5A). Infectious virus could not be detected in either control formulation after the 48-day time point. In sharp contrast, rehydrated films containing surfactant retained 100 ± 0.6% of their original titer, while those without surfactant retained 93 ± 2.0% infectivity at the 84-day time point.
Fig. 5. Adenovirus stability profiles of liquid film formulations during long-term storage.
Virus (1.25 × 1012 vp) was placed in formulations 27 and 28 and stored in the liquid form in controlled environmental chambers held at 4°C (A) and 20°C (B) for 84 days. Replicate samples (n = 3) were collected at each time point, and live virus concentration was assessed by a standard infectious titer assay (19). Virus at the same concentration was also placed in two standard liquid formulations, and infectious titers under each storage condition were also assessed for comparison. Control A formulation consisted of 100 mM PBS (pH 7.4) and 10% glycerol. Control B formulation consisted of 100 mM PBS alone. In each panel, data represent the average ± SD. *P < 0.05, ***P < 0.001, two-tailed Student’s t test.
The infectious titer of virus stored in control buffer A fell to 89 ± 0.06% of the original titer when stored at 20°C for 14 days and to 67 ± 4.0% by day 42 (Fig. 5B). Infectious virus could no longer be detected in the control B preparation after 25 days at room temperature. Rehydrated films that did not contain surfactant retained 95 ± 0.53% of their original infectious virus concentration for 84 days at 20°C, while those containing surfactant did not demonstrate a notable loss of viral titer (100 ± 0.53% recovery) throughout the entire study period. A final series of studies designed to identify the limits at which this formulation stabilized live virus was conducted at 40° and 50°C, temperatures that can potentially be reached in cargo compartments during transportation and in the heat of many developing countries (34). At 40°C, liquid formulations containing surfactant maintained 91+ 0.95% of their original titer for 5 days, while formulations without surfactant fell to 82 + 1.6% of their original concentration within 3 days (fig. S5A). Infectious virus could no longer be detected in control formulations after 24 hours at 40°C. Recovery of infectious virus from the film matrix was significantly affected at temperatures above 37°C as films containing surfactant displayed 74± 0.98% of their original titer after 5 days at 40°C and 73± 1.3% of their original titer after 2 days at 50°C (fig. S5, C and D). During these studies, environmental monitors indicated that relative humidity (RH) significantly fell below 25% when temperature was increased to 50°C. This prompted a pilot study to determine how humidity affects stability of virus in the film matrix during accelerated stability studies. When samples were stored at 40°C and RH reduced to levels at and below what was observed for the 50°C study, virus titer was reduced by 88% after 3 days under these conditions (fig. S6). Additional studies involving assessment of humidity and various packaging systems on viral stability in the film matrix are currently underway.
Physical characterization of thin films
Visualization of films by scanning electron microscopy (SEM) revealed that the surfaces of those prepared with the base formulation were porous and contained small pockets of varying size throughout the three-dimensional matrix (Fig. 6, A to C). When films containing virus were dried in the optimized formulation containing sorbitol and surfactant, the surface of the film assumed a glassy state in which vp were evenly suspended (Fig. 6D, arrows). Differential scanning calorimetry (DSC) was used to analyze samples in a stepwise fashion to evaluate the amorphous quality of films (fig. S7). The absence of an observable endotherm in each scan indicates that none of the film components melt at temperatures below 150°C. The glass transition temperature (Tg’) of films prepared with base formulation alone was 148.5°C. Addition of sorbitol to the base did not significantly affect Tg’ (148.4°C), while addition of surfactant to the base and binder fostered a slight shift in Tg’ (146.5°C). The optimized film formulation containing virus had a Tg’ of 149.7°C, which correlated with the SEM image of a glassy surface when drying was complete. The results obtained from SEM and DSC were further supported by wide-angle x-ray diffraction patterns (fig. S8). Individual components of the film, most notably sorbitol, were crystalline substances in the dry state and contained diffraction peaks of varying intensity (fig. S8A). However, films made with the optimized combination of base, sorbitol, and surfactant exhibited a single broad peak of relatively low intensity, characteristic of amorphous compounds (fig. S8D). Inclusion of virus in the optimized film platform did not disrupt the amorphous state of the final product (fig. S8E).
Fig. 6. Excipient combinations transform porous film base into an amorphous solid.
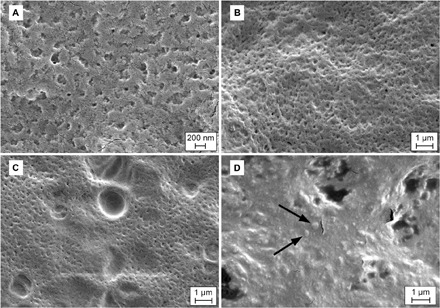
(A) Scanning electron micrograph of porous surface of thin film consisting only of base excipient (formulation 25, table S1). (B) Surface of two component film (formulation 14) in the absence of virus. (C) Large noncrystalline pockets in film (formulation 27), which foster stabilization of vp. (D) Adenovirus particles (arrows) suspended in amorphous film matrix of optimized formulation 28. Perforations in the film surface are artifacts from extended electron bombardment on the surface of the thin film.
Fourier transform infrared (FTIR) spectroscopy was used to identify bond interactions between each of the components of the optimized film and the adenovirus. The most pronounced change in bond formation in films prepared with and without virus was found for nitrogen-hydrogen bonds at the wave number 3300 (Fig. 7). Absorbance at this wavelength increased twofold when virus was added to base formulation alone. A similar trend was noted when virus was added to the formulation containing base and sorbitol. No significant difference in absorbance was noted at this wavelength when virus was added to the optimized film formulation, indicating that direct binding of vp to the surfactant may be a key factor for stabilization in our optimized thin film matrix.
Fig. 7. FTIR spectroscopy scans suggest that nitrogen-hydrogen bonds between virus and surfactant may be key for stabilization in thin film matrix.
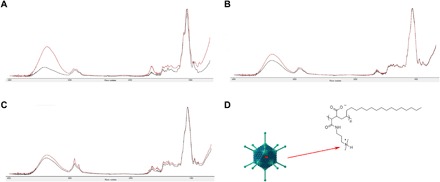
Infrared absorption spectra in the 4000 to 800 cm−1 region for films containing (A) base alone (formulation 25), (B) base and sorbitol (formulation 27), and (C) optimized formulation 28. In each panel, black traces represent film in the absence of adenovirus. Red traces represent film containing 1.25 × 1012 particles of adenovirus. The most prominent change in spectra in the nitrogen-hydrogen region (wave number 3300) can be seen in scans where surfactant was present (C). (D) The proposed interaction between the cationic region of the surfactant and the negatively charged glutamic acid residues of the adenovirus hexon proteins. Virus image adapted from: Splettstoesser, T. A simplified 3D-generated structure of the adenovirus. Wikimedia Commons accessed 5 March 2019; available at: https://commons.wikimedia.org/wiki/File:Adenovirus_3D_schematic.png.
In vivo assessment of film performance
H1N1 influenza virus stabilized in the optimized thin film matrix (formulation 28) was administered to the BU and SL mucosa of BALB/c mice. Films dissolved within 30 to 60 s after placement in the mouth. Mice given the same dose of unformulated virus by the intranasal or intramuscular routes were included for comparison. Analysis of serum collected 28 days after immunization revealed that there was no significant difference in the level of anti-influenza immunoglobulin G (IgG) antibodies between all treatment groups (Fig. 8A) and a control antibody that had a defined protective neutralizing antibody titer. Animals immunized by the BU route had significantly higher levels of neutralizing antibodies with respect to those immunized by intramuscular injection (P < 0.001; Fig. 8B). The formulation was well tolerated, and there was no visible irritation to the BU or SL mucosa after administration and dissolution of films. These observations are in line with a cytotoxicity study conducted on TR146 cells, an in vitro model of the human SL and BU mucosa (35), where each formulation component with and without virus failed induce measurable toxicity over a 2-hour exposure time (fig. S9).
Fig. 8. Immunogenicity profiles of thin film vaccine validate use in oral immunization strategies.
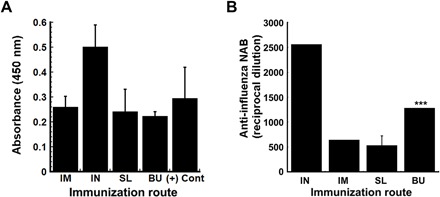
Individual samples of heat-inactivated serum collected 28 days after immunization from BALB/c mice were evaluated for influenza-specific IgG by ELISA. The average optical density read from samples obtained from each treatment group are presented to serve as a measure of relative antibody concentration. Each assay was validated by readings obtained from a sample collected from mice immunized with the same strain of influenza with an established anti-influenza antibody titer of 1:40 (+ control, Emory). Readings obtained from samples collected from mice given saline (negative controls) were subtracted from all absorbances (A). Neutralization capacity of antibodies was assessed by serial dilution of heat-inactivated serum with a fixed amount of H1N1 influenza virus before infection of MDCK (Madin-Darby canine kidney) cells. The reciprocal dilution plotted for each treatment group reflects the dilution at which the ability of the virus to infect target cells was reduced by 50% (B). In each panel, results are expressed as average values ± SEM and are representative of groups containing four mice per immunization route. ***P < 0.001, two-tailed Student’s t test. IM, intramuscular; IN, intranasal; SL, sublingual; BU, buccal.
DISCUSSION
In a recent publication, Kristensen et al., in collaboration with the Expanded Program on Immunization, surveyed 158 managers of immunization campaigns in low- and middle-income countries to identify characteristics of vaccines they believed would suit logistical and infrastructure challenges faced within their countries (36). Respondents valued characteristics that prevent heat damage, vaccine wastage, and simplified vaccine delivery. The novel platform described here meets most of those expressed needs. With respect to heat stability, we have shown that a recombinant adenovirus-based vaccine can be stored at ambient temperature for a significant period of time, which is quite noteworthy (Fig. 1C). One configuration of the film can accommodate a single dose of vaccine (Fig. 1A) and is space saving as we have estimated that 350,000 doses of the current BCG (bacille Calmette-Guérin) (10/20) vaccine, when incorporated in our film, would hold the space of 600 8.5 × 11–inch sheets and weigh ~3 kg, while the same number of vaccine doses in the current platform and packaging (vial and ampule of diluent) would require space a little larger than an American football field (5749 m2) for storage and weigh 2730 kg (37). This platform may also offer additional space-saving capability by accommodating more than one antigen, as we have also demonstrated that we can also effectively stabilize live bacteria, antibodies, and enzymes at ambient temperatures in the film matrix (Fig. 1, D to F). While pivotal studies investigating the amount of antigen that can be effectively loaded in a single-use film are ongoing, we have shown that our process is capable of providing an even distribution of virus throughout films of varying size (fig. S10), that the amount of virus embedded in a film does not affect recovery upon reconstitution, and that there is a direct correlation (r2 = 0.994; fig. S11) between the amount of virus added to and recovered from our optimized film matrix, indicating that vaccines and other biologicals can be stabilized in large sheets that can be divided into various sizes/doses or single unit dose films that contain a defined amount of antigen.
The third most valued characteristic of a vaccine in low- and middle-income countries was that it should be easy to use and understand (36). Vaccines stabilized within our film matrix could be self-administered orally in a needle-free manner. Because most influenza vaccines currently on the market are given by injection, we believe that the intramuscular route is the true yardstick of success for our formulated film-based vaccine as it represents the most clinically relevant route of immunization for influenza. In this report, we have shown that administration of an antigen stabilized in the film matrix by the SL and BU routes is possible in a rodent model and can induce antibody levels comparable to administration of the same antigen by intramuscular injection (Fig. 8). While these levels may seem low with respect to those observed after immunization by the nasal route, it is important to note that detectable serum antibody titers equal to or above 1:40, as determined by hemagglutination inhibition or neutralizing antibody assays, which we have surpassed here (SL, 1:533; BU, 1:1280; Fig. 8), are considered predictive of a favorable clinical outcome following exposure to influenza (38–40). It is also important to realize that the virus used in our studies, the mouse adapted PR8 strain, preferentially replicates in the airways, supporting a significantly stronger response after intranasal administration in contrast to that obtained from the other routes of administration (41). For this reason, animals given the virus by the intranasal route were also included in our study to validate assays for assessment of the antibody-mediated immune response by the novel SL and BU routes. A comprehensive analysis of the immunogenicity profiles of film-stabilized vaccines administered by each route, including mucosal and systemic T cell responses, is currently underway as are studies to further strengthen the immune response by including various permeability enhancers and adjuvants in the film matrix. This platform is also amenable to a variety of methods of administration as we have previously shown with regard to the respiratory route in primates (30). Stability data also suggest that vaccine waste would be minimal if the film was used as a reconstituted solution as virus in rehydrated films was stable for more than 2 months at ambient temperature and for 5 days at 40°C (Fig. 5 and fig. S5).
Thirteen of the current WHO-recommended vaccines are distributed as solid products, primarily as lyophilized powders (22, 28). While this method of stabilization has significantly extended the shelf life of vaccines, it remains limited by high production costs necessary to acquire specialized equipment, long processing times of more than 48 hours in some cases, and susceptibility of vaccine candidates to the process stressors of freezing and drying (42). Thus, there is a growing demand in the pharmaceutical and other industries for novel technologies that stabilize proteins and other biologicals through a nonthermal approach (43). While films for oral use are a relatively new dosage form, there are currently 13 approved products available in the United States and Europe, which contain small molecules for over-the-counter use (44). Standard methods for manufacturing films also require the use of specialized equipment for hot melt extrusion or thermal printing and involve use of volatile solvents to accelerate drying (45), which can substantially compromise the integrity of live organisms and other antigens and the subsequent potency of a vaccine. With the intent to develop technology that would be easily transferrable to facilities in resource-poor countries, our approach is different in that we identified combinations of excipients that could effectively stabilize a live virus during the film-forming process at ambient temperatures, under aseptic conditions in the absence of heat and other external stressors without using highly technical equipment and a drying process, which was complete within a single working day. This also holds promise for outbreak and pandemic scenarios as it offers a means for minimizing bottlenecks for production and distribution of large volumes of vaccines and biologicals and, as we have shown here, is flexible for adaptation to different types of products.
In the Kristensen study, resistance to freezing was one of the top 10 features of an ideal vaccine (36). Loss of vaccine potency to freezing is a serious threat to global health as one study noted that, in low-income countries, vaccines were more likely to be exposed to temperatures below recommended ranges during storage, while vaccines were more likely to be exposed to freezing temperatures during transit in higher-income countries (46). Our optimized thin-film matrix successfully protected the adenovirus from disruption during thermocycling for a series of 16 freeze-thaw cycles. This is a significant finding as the most promising formulations described in the literature to date are liquid preparations capable of protecting the virus from 5 and 12 cycles, respectively (47, 48). It also suggests that this optimal combination of excipients suspended the virus in a matrix capable of protecting it from a series of environmental stressors.
While conducting accelerated stability studies, we made several very interesting observations that illustrate the novelty of our dosage form and how it does not follow the traditional concepts commonly taught with respect to stabilization of biologicals in the dried state. For a lyophilized product, it is standard practice to create dried powder with limited residual moisture content and to keep it in a moisture-free environment to prevent water sorption and degradation via hydrolysis. Our optimized formulation demonstrated adequate stability for 3 days at 40°C under high humidity (97%), but when humidity dropped to 20% and to 0% at the same temperature, we could not recover any infectious virus from the sample at the same time point (fig. S6). This leads us to believe that the film creates a very different microenvironment for the adenovirus, compared with that of a lyophilized powder. RH has been reported to affect the stability of a variety of viruses by controlling the amount of water retained, the concentration of solutes, and its pH within the microenvironment of aerosol droplets with some viruses (like the adenovirus) stable in high-humidity environments (80% RH) and others (for example, influenza) stable at lower humidities (20% RH) (49). While the studies outlined in this manuscript identified a formulation that provides a favorable microenvironment for the adenovirus within the film at certain temperatures and RH environments above 60%, it failed to do so when the RH fell below 25%. Films used for each of the stability studies summarized here were packaged in particle-free resealable plastic bags under aseptic conditions. Alternative packaging systems that can maintain a favorable internal humidity level when stored in low-humidity environments are currently under evaluation in our laboratory to address this important issue.
Our efforts in assessing the stability of virus under stressed conditions also led to the realization that current guidelines set by both the WHO and the International Conference on Harmonization (ICH) specify that long-term and accelerated stability studies be conducted at temperatures ranging from 25° to 40°C and at 60 to 75% RH are relevant for both hot and dry and hot and humid climates (50). Our results suggest that stability data collected from biological preparations stored in low-humidity environments might provide results different than those performed according to WHO/ICH guidelines and, if implemented routinely, could improve product stability as they may encounter RH environments as low as 10% in homes and distribution centers around the globe on a seasonal basis (51) and during transit in arid climates when RH of 10 to 30% are experienced during daylight hours (52). It also poses the question about the stability profiles of lyophilized products, which were not previously evaluated under low-humidity conditions and how they would perform under these conditions. Additional accelerated stability studies above and below 25% RH are also underway with a panel of different viruses in our film matrix and will be the subject of an upcoming manuscript.
One of the pivotal findings of this study was the correlation of pH within the film matrix with virus recovery upon drying and with long-term stability (Fig. 2). This principle was previously established within the pH range of 6 to 8 for recombinant adenovirus-based products prepared in solid and liquid dosage forms (19, 53) and suggests virus degradation within the film matrix may also be pH mediated. None of the aqueous solvents evaluated in our study were capable of fully maintaining the original pH of the formulation during the film-forming process. This was probably due to loss of original pH of the preparation due to the concentration of buffering agents and their interaction with other components of the formulation in a manner that prevented them from maintaining pH during the vitrification process (54). One of the prime constituents of successful formulations identified in these studies was the base excipient. A polymer capable of forming strong intramolecular hydrogen bonds (55) played a key role in maintaining structural confirmation of the virus capsid. This was evident during the rate of release studies, as films containing this excipient alone failed to completely release the entire dose of virus originally loaded in the film (Fig. 3). It is also important to note that this excipient significantly enhanced the viscosity of each preparation in the liquid state (fig. S4), suggesting that its ability to minimize molecular motility both in the solid and liquid formulations was also key to maintaining long-term stability at ambient temperatures.
Sorbitol and glycerol, predominately used in film formulations as plasticizers (56), affected virus stability in the film matrix very differently. Preparations containing glycerol took longer to dry, indicating that this reagent had a higher capacity for retaining water within the film matrix. We suggest that sorbitol more efficiently contributed to the development of the three-dimensional matrix of the film through strong interactions with the other excipients, replacing their interactions with water, thus accelerating the drying process and leaving the virus suspended in the highly vitreous, yet flexible matrix of the film. The amorphous nature of the film was first visualized by electron microscopy (Fig. 6) and further confirmed by x-ray diffraction (fig. S8). Calorimetric assessment of the optimized film matrix indicates that it has a Tg’ above 140°C, suggesting that at each of the temperatures used in the assessment of adenovirus stability, none of the excipients were susceptible to crystallization, which could compromise the adenovirus capsid and facilitate precipitation of buffer components and other excipients, allowing the virus to remain stably suspended in an amorphous polymer matrix during long-term storage. We have shown previously that addition of a zwitterionic surfactant to our preparations at the concentration used here significantly improves the potency of an adenovirus-based Ebola vaccine (29, 30) and attributed it to the fact that it is capable of significantly reducing the energy barrier for the virus to cross the lipid bilayer of the cell membrane (57). Here, we show that it is paramount for conferring stability of virus in the film matrix and rehydrated films when stored at ambient and elevated temperatures (Figs. 4 and 5). FTIR analysis revealed that nitrogen-hydrogen bond formation was most prevalent between this excipient and the virus, suggesting that integrity of the virus capsid was maintained through interaction of the positively charged amine group on the surfactant with the negatively charged glutamic acid residues located on the adenovirus hexon protein (58). Despite this, rate of release studies indicate that the presence of the surfactant was key in achieving rapid and complete release of an infectious dose of virus from the film (Fig. 3). We envision a model in which the size of the surfactant incorporated within the amorphous network formed by it and the other excipients of the optimized formulation facilitated rapid hydration of the film and subsequent release of virus through a series of hydrophobic and hydrophilic interactions. Additional mechanistic studies are currently underway to evaluate the strength of the virus-surfactant interaction in both the liquid and solid states and to determine whether it also is necessary for stabilization of other viruses within the film matrix.
This paper is the first to identify essential parameters for effective stabilization of a live virus in a thin film matrix prepared in aqueous formulations under ambient conditions. We believe these findings will serve as the basis for accelerated design of strategies for thermostabilization of other biologicals like therapeutic antibodies and enzymes in a similar dosage form. The platform described here that effectively stabilizes adenovirus at ambient and elevated temperatures, releases virus efficiently, and facilitates significant immunogenic responses in mice is notably distinct from all other vaccine platforms described to date. This approach, designed with developing countries in mind, holds great promise to enhance global access to vaccines and other biological medicines by reducing costs associated with their production, distribution, and supply chain maintenance. The impact of this is significant given that the WHO estimates that nearly 50% of lyophilized (freeze-dried) and 25% of liquid vaccines are discarded each year due to disruption of the cold chain (21).
MATERIALS AND METHODS
Materials
Dulbecco’s PBS, Trizma base [2-amino-2-(hydroxymethyl)-1,3-propanediol], potassium ferricyanide, potassium ferrocyanide, glutaraldehyde (grade I, 25% in water), FBS (qualified, U.S. origin), glycerol, and d-sorbitol [USP (United States Pharmacopeia) grade] were purchased from Sigma-Aldrich (St. Louis, MO). Poly(maleic anhydride-alt-1-octadecene) substituted with 3-(dimethylamino)propylamine was purchased from Anatrace (Maumee, OH). DMEM was purchased from Mediatech (Manassas, VA). Hydroxypropyl methylcellulose 4*KM was provided by the Dow Chemical Company (Midland, MI). Penicillin (10,000 IU) and streptomycin (10,000 μg/ml) were purchased from Gibco Life Technologies (Grand Island, NY). 5-Bromo-4-chloro-3-indolyl-β-d-galactoside (X-gal) was purchased from Gold Biotechnology (St. Louis, MO). All other chemicals were of analytical reagent grade and purchased from Thermo Fisher Scientific (Pittsburgh, PA) unless specified otherwise.
Adenovirus production and purification
First-generation adenovirus serotype 5 expressing Escherichia coli β-galactosidase under the control of a cytomegalovirus promoter was amplified in human embryonic kidney 293 cells (ATCC CRL-1573) and purified from secondary lysates according to established methods (19). Preparations with a ratio of infectious to physical viral particles of 1:100 were used in the studies summarized here.
Formulation screening
Base formulations were prepared in bulk with various solvents. Additional excipients were added to base and homogenized before addition of virus. A virus concentration of 1.25 × 1012 vp per film was selected so that subtle changes in infectious titer could be detected with a standard limiting dilution/infectious titer assay and histochemical staining (19). Films were dispensed into 1-ml unit dose molds (or 100-μl unit dose for mouse studies) using an E3 Repeater pipette (Eppendorf, Hauppauge, NY) and dried in 8 hours (1-ml film) and 4 hours (100-μl film) under ambient temperature and pressure (20°C, 1 atm) and aseptic conditions. Once dry, films were reconstituted in and infectious titer assays performed on HeLa cells (ATCC# CCL-2). Percent recovery was calculated as
where t = 1 is the infectious titer of a film that was reconstituted after drying, and t = 0 is the infectious titer of virus in the same formulation before drying.
Rate of release analysis
Films were placed in sterile chambers containing 1 ml of saline (pH 7) warmed to 37°C. Buffers in each chamber were stirred (60 rpm), and 10 μl of samples was collected every 5 min for a period of 2 hours. Equal amounts of blank buffer were added to each chamber after sampling to maintain a constant volume. Samples were also taken of virus containing liquid film formulations that did not undergo drying and of virus in PBS alone, which were also stirred at 37°C. Results from these controls were used to normalize data for changes in titer due to temperature, physical agitation, and formulation effects.
Short-term stability: Solid state
Virus-containing films were prepared in bulk and in parallel with the following liquid formulations: control buffer A (10% glycerol/PBS) and control buffer B (PBS). A fraction of each production lot was stored at 4°, 20°, and 37°C. Samples stored at 4° and 20°C were reconstituted and infectious titers were assessed on days 1, 3, 7, 14, 28, 48, and 84, while the titers of those stored at 37°C were assessed on days 1 to 7, 10, and 14.
Liquid state
Virus was added to replicate sterile containers containing liquid formulations and stored with replicate control A and control B preparations at 4° and 20°C. Infectious titer for each preparation was assessed on the same schedule as described for films at each respective storage temperature.
Freeze-thaw studies
Films were prepared in bulk and placed at −80°C once drying was complete. After 24 hours, all samples were thawed to room temperature (20°C). Replicate (n = 3) samples were reconstituted and infectious titer of virus was assessed, while remaining films were returned to the freezer. This cycle was repeated daily for 16 days, with infectious titer assays performed on replicate films on days 2 to 8, 12, and 16.
Characterization of film properties
Scanning electron microscopy
Films were prepared and, when drying was complete, they were coated with 12-nm platinum (Pt)/palladium (Pd) alloy in a Cressington 208HR sputter coater and subsequently photographed on a Zeiss Supra 40V scanning electron microscope (Zeiss, Jena, Germany).
FTIR spectroscopy
FTIR spectroscopy was conducted on films prepared with and without virus using a Nicolet iS 50 FT-IR Spectrometer (Thermo Fisher Scientific, Waltham, MA). Films were placed under the probe, and scans were conducted in the 4000 to 800 cm−1 region using an ATR (attenuated total reflectance) method with a diamond/ZnSe prism.
In vivo assessment of film performance
All procedures were approved by the Institutional Animal Care and Use Committees at The University of Texas at Austin and are in accordance with the guidelines established by the National Institutes of Health for the humane treatment of animals.
Immunization
Six-week-old male BALB/c mice were obtained from the Jackson laboratory (Bar Harbor, ME). Animals were housed in a temperature-controlled, 12-hour light-cycled facility at the Animal Research Center of The University of Texas at Austin. Mice were given free access to standard rodent chow (Harlan Teklad, Indianapolis, IN) and tap water. Animals were anesthetized by a single intraperitoneal injection of a 3.9:1 mixture of ketamine (100 mg/ml, Putney, Portland, ME) and xylazine (100 mg/ml, Sigma-Aldrich, St. Louis, MO). Once deep plane anesthesia was achieved, animals were immunized with 2000 CEID50 (Chicken Embryo Infectious Dose 50% endpoint) of influenza A virus, A/Puerto Rico/8-9VMC3/1934 (H1N1) (BEI Resources, Manassas, VA) by various routes. Intramuscular injection involved direct injection of virus diluted in saline into each gastrocnemius muscle located on the hindlimb (50 μl per muscle). Nasal immunization was performed by slowly dripping virus diluted in saline into each nostril (10 μl per nostril) using a standard micropipette (Gilson, Middleton, WI). For SL immunization, the SL epithelium was swabbed dry and 20 μl of saline was added to act as an adhesive for the film. Each film was subsequently placed under the tongue using sterile forceps. For BU immunization, the upper part of the check pouch was swabbed dry and 10 μl of saline was added to each cheek to act as an adhesive for the film. Half of each film was placed on the upper part of each cheek pouch using sterile forceps. For both SL and BU doses, dissolution time was complete within 5 min without the animals swallowing or chewing the film. Animals given the vaccine by the nasal, SL, or BU routes were maintained in an upright position for 30 min after immunization to minimize choking and accidental swallowing of the vaccine.
Neutralizing antibody assay
Anti-influenza antibody (neutralizing antibody) titers were assessed in serum samples collected 28 days after vaccination. Heat-inactivated serum was diluted in MEM in twofold increments starting from a 1:20 dilution. Each dilution was mixed with A/Puerto Rico/8-9VMC3/1934 (H1N1) for 1 hour at 37°C and added to MDCK (Madin-Darby canine kidney) cells in Zero-Serum Medium PSGA (Penicillin, Streptomycin, Gentamicin, and Ampicillin) (Quidel, San Diego, CA) on 96-well plates. Two hours later, 100 μl of MEM supplemented with 20% FBS was added to each well. Cells were incubated an additional 24 hours and visualized by histochemical staining. For each sample, the serum dilution that corresponded to a 50% reduction in viral expression was obtained by the method of Reed and Muench as described previously (29). The absence of neutralization in samples containing medium only (negative control) and FBS (serum control) were criteria for qualification of each assay.
Characterization of influenza-specific antibodies
To assess anti-influenza immunoglobulin levels in serum, Immulon 2 HB plates (Thermo Fisher Scientific, Pittsburgh, PA) were coated with influenza A virus, A/Puerto Rico/8-9VMC3/1934 (H1N1) (1 μg per well) in bicarbonate buffer (pH 9.5) overnight at 4°C. Plates were washed three times with PBS containing 0.05% Tween 20 and blocked in PBS containing 1% bovine serum albumin (Sigma-Aldrich) for 1 hour at room temperature. Heat-inactivated serum samples were diluted 1:5 in sterile PBS. Fifty microliters of each dilution was added to the antigen-coated plates for 4 hours at room temperature. Plates were washed three times with PBS containing 0.05% Tween 20 and incubated with horseradish peroxidase–conjugated goat anti-mouse IgG (1:2000; Southern Biotechnology Associates, Birmingham, AL) in separate wells for 2 hours at room temperature. Plates were washed, and 100 μl of substrate solution [o-phenylenediamine (0.4 mg/ml) (Sigma) in 50 mM phosphate-citrate buffer (pH 5.0) with 0.03% (v/v) hydrogen peroxide] was added to each well. The plate was incubated at room temperature for 10 min, and optical densities were read at 450 nm on a microplate reader (GloMax-Multi+ Detection System, Promega, Madison, WI).
Statistical analysis
Statistical analysis of data was performed using JMP (JMP Statistical Software from SAS, Cary, NC). Differences with respect to treatment were calculated using unpaired two-tailed Student’s t tests. Differences were determined to be significant when the probability of chance explaining the results was reduced to less than 5% (P < 0.05).
Supplementary Material
Acknowledgments
We thank A. M. Wagner of the Institute for Biomaterials, Drug Delivery and Regenerative Medicine at The University of Texas at Austin and E. Maier, Ph.D., of the Dell Pediatric Research Institute for assistance with characterization of films, and T. N. Doan from the Croyle laboratory for assistance with RH studies. We also acknowledge the gift of positive control serum for use in the anti-influenza IgG enzyme-linked immunosorbent assay provided by I. Skountzou at Emory University School of Medicine. Funding: This project was supported by a multicenter U01 grant from the National Institute of Allergy and Infectious Disease (U01 AI 078045/MAC), a Texas Health Catalyst award from The University of Texas at Austin Dell Medical School (to M.A.C.), a UT Mentoring Diversity Fellowship (to M.A.C./I.B.), a Williams and McGinity Graduate Fellowship (to I.B.), and a Kuhn Family Scholars Award (to I.B.). Author contributions: I.B., S.C.S., and M.A.C. designed the project. I.B. conceptualized and designed the experiments and performed the stability, rate of release, FTIR, and DSC studies and the analysis. D.K.R. prepared samples for SEM, collected images, and participated in their analysis in conjunction with S.C.S., I.B., and M.A.C. I.B. performed cell culture and adenovirus amplification. S.C.S. assisted with preparation of the figures and the Supplementary Materials. I.B. and M.A.C. wrote and edited the manuscript. Competing interests: M.A.C. and S.C.S. are inventors on several patent applications related to this work filed by The University of Texas at Austin (no. 9,675,550; filed 25 January 2013, no. 9,974,850; filed: 25 March 2016). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the manuscript and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The authors agree that all reasonable requests for materials used in the studies summarized in this manuscript will be fulfilled and will require establishment of a material transfer agreements between interested parties and The University of Texas at Austin.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaau4819/DC1
Supplementary Materials and Methods
Fig. S1. Complexity of a formulation does not significantly impact elongation of thin films.
Fig. S2. Tensile strength of thin films is influenced by certain excipient combinations.
Fig. S3. Viable adenovirus can be recovered from films reconstituted with a variety of diluents.
Fig. S4. Virus, excipients, and solvent volume significantly affect viscosity of solutions made from reconstituted films.
Fig. S5. Adenovirus stability profiles of rehydrated and solid films at elevated temperatures.
Fig. S6. Recovery of live adenovirus from the film matrix at elevated temperature is significantly affected by environmental humidity.
Fig. S7. DSC analysis reveals that thin films are amorphous solids across a wide temperature range.
Fig. S8. X-ray scanning diffraction reveals films as amorphous solids.
Fig. S9. Cytotoxicity profile of thin film vaccine validates safety of dosage form.
Fig. S10. Virus is evenly distributed throughout the film matrix.
Fig. S11. The amount of virus embedded in film matrix does not affect recovery of virus from the film matrix.
Table S1. Summary of formulations.
REFERENCES AND NOTES
- 1.Greenwood B., The contribution of vaccination to global health: Past, present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones D. S., Podolsky S. H., Greene J. A., The burden of disease and the changing task of medicine. N. Engl. J. Med. 366, 2333–2338 (2012). [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization, The top 10 causes of death: Leading causes of death by economy income group (January 2018); http://www.who.int/mediacentre/factsheets/fs310/en/index1.html [accessed 30 August 2019].
- 4.Hardt K., Bonanni P., King S., Santos J. I., El-Hodhod M., Zimet G. D., Preiss S., Vaccine strategies: Optimising outcomes. Vaccine 34, 6691–6699 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Hill A. B., Kilgore C., McGlynn M., Jones C. H., Improving global vaccine accessibility. Curr. Opin. Biotechnol. 42, 67–73 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Carter R. J., Idriss A., Widdowson M. A., Samai M., Schrag S. J., Legardy-Williams J. K., Estivariz C. F., Callis A., Carr W., Webber W., Fischer M. E., Hadler S., Sahr F., Thompson M., Greby S. M., Edem-Hotah J., Momoh R. M., McDonald W., Gee J. M., Kallon A. F., Spencer-Walters D., Bresee J. S., Cohn A., Hersey S., Gibson L., Schuchat A., Seward J. F., Implementing a multisite clinical trial in the midst of an Ebola outbreak: Lessons learned from the Sierra Leone trial to introduce a vaccine against Ebola. J. Infect. Dis. 217( suppl. 1), S16-S23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schopper D., Ravinetto R., Schwartz L., Kamaara E., Sheel S., Segelid M. J., Ahmad A., Dawson A., Singh J., Jesani A., Upshur R., Research ethics governance in times of Ebola. Public Health Ethics 10, 49–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaker R., Fayad D., Dbaibo G., Challenges and opportunities for meningococcal vaccination in the developing world. Hum. Vaccin. Immunother. 14, 1084–1097 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tate J. E., Burton A. H., Boschi-Pinto C., Parashar U. D.; World Health Organization–Coordinated Global Rotavirus Surveillance Network , Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000-2013. Clin. Infect. Dis. 62( suppl. 2), S96–S105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plotkin S., Robinson J. M., Cunningham G., Iqbal R., Larsen S., The complexity and cost of vaccine manufacturing-An overview. Vaccine 35, 4064–4071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lloyd J., Cheyne J., The origins of the vaccine cold chain and a glimpse of the future. Vaccine 35, 2115–2120 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Portnoy A., Ozawa S., Grewal S., Norman B. A., Rajgopal J., Gorham K. M., Haidari L. A., Brown S. T., Lee B. Y., Costs of vaccine programs across 94 low- and middle-income countries. Vaccine 33( suppl. 1), A99–A108 (2015). [DOI] [PubMed] [Google Scholar]
- 13.D. R. Feikin, B. Flannery, M. J. Hamel, M. Stack, P. M. Hansen, Vaccines for Children in Low- and Middle-Income Countries, in Reproductive, Maternal, Newborn, and Child Health: Disease Control Priorities, Third Edition (Volume 2), R. E. Black, R. Laxminarayan, M. Temmerman, N. Walker, Eds. (The International Bank for Reconstruction and Development/The World Bank, Washington, DC, 2016), pp. 187–205. [Google Scholar]
- 14.Ashok A., Brison M., LeTallec Y., Improving cold chain systems: Challenges and solutions. Vaccine 35, 2217–2223 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Adhikari B., Mishra S. R., Babu Marahatta S., Kaehler N., Paudel K., Adhikari J., Raut S., Earthquakes, fuel crisis, power outages, and health care in Nepal: Implications for the future. Disaster Med. Public Health Prep. 11, 625–632 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Yakum M. N., Ateudjieu J., Pélagie F. R., Walter E. A., Watcho P., Factors associated with the exposure of vaccines to adverse temperature conditions: The case of North West region, Cameroon. BMC. Res. Notes 8, 277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitamura T., Bouakhasith V., Phounphenghack K., Pathammavong C., Xeuatvongsa A., Norizuki M., Okabayashi H., Mori Y., Machida M., Hachiya M., Assessment of temperatures in the vaccine cold chain in two provinces in Lao People's Democratic Republic: A cross-sectional pilot study. BMC. Res. Notes 11, 261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.M. Y. Chan, T. S. Dutil, R. M. Kramer, Lyophilization of adjuvanted vaccines: Methods for formulation of a thermostable freeze-dried product, in Vaccine Adjuvants: Methods and Protocols, C. B. Fox, Ed. (Springer New York, 2017), pp. 215–226. [DOI] [PubMed] [Google Scholar]
- 19.Croyle M. A., Cheng X., Wilson J. M., Development of formulations that enhance physical stability of viral vectors for gene therapy. Gene Ther. 8, 1281–1290 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Cardoso F. M. C., Petrovajová D., Horňáková T., Viral vaccine stabilizers: Status and trends. Acta Virol. 61, 231–239 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Lee B. Y., Cakouros B. E., Assi T.-M., Connor D. L., Welling J., Kone S., Djibo A., Wateska A. R., Pierre L., Brown S. T., The impact of making vaccines thermostable in Niger's vaccine supply chain. Vaccine 30, 5637–5643 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.World Health Organization (WHO), Summary of WHO position papers—Recommendations for routine immunization (WHO, Geneva) April 2018 update; http://www.who.int/immunization/policy/Immunization_routine_table1.pdf?ua=1 [accessed 30 August 2019].
- 23.Shastay A., Administering just the diluent or one of two vaccine components leaves patients unprotected. Home Healthc. Now. 34, 218–220 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Duttagupta C., Bhattacharyya D., Narayanan P., Pattanshetty S. M., Vaccine wastage at the level of service delivery: A cross-sectional study. Public Health 148, 63–65 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Shakya A. K., Chowdhury M. Y. E., Tao W., Gill H. S., Mucosal vaccine delivery: Current state and a pediatric perspective. J. Control. Release 240, 394–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cross R. W., Mire C. E., Feldmann H., Geisbert T. W., Post-exposure treatments for Ebola and Marburg virus infections. Nat. Rev. Drug Discov. 17, 413–434 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Schulze K., Ebensen T., Riese P., Prochnow B., Lehr C.-M., Guzmán C. A., New horizons in the development of novel needle-free immunization strategies to increase vaccination efficacy. Curr. Top. Microbiol. Immunol. 398, 207–234 (2016). [DOI] [PubMed] [Google Scholar]
- 28.World Health Organization, International Travel and Health. Chapter 6. Vaccine-Preventable Diseases and Vaccines. 2017 Update (WHO Press, Geneva, Switzerland), pp. 80–140.
- 29.Choi J. H., Schafer S. C., Freiberg A. N., Croyle M. A., Bolstering components of the immune response compromised by prior exposure to adenovirus: Guided formulation development for a nasal Ebola vaccine. Mol. Pharm. 12, 2697–2711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi J. H., Jonsson-Schmunk K., Qiu X., Shedlock D. J., Strong J., Xu J. X., Michie K. L., Audet J., Fernando L., Myers M. J., Weiner D., Bajrovic I., Tran L. Q., Wong G., Bello A., Kobinger G. P., Schafer S. C., Croyle M. A., A single dose respiratory recombinant adenovirus-based vaccine provides long-term protection for non-human primates from lethal Ebola infection. Mol. Pharm. 12, 2712–2731 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Preis M., Knop K., Breitkreutz J., Mechanical strength test for orodispersible and buccal films. Int. J. Pharm. 461, 22–29 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Pelliccia M., Andreozzi P., Paulose J., D'Alicarnasso M., Cagno V., Donalisio M., Civra A., Broeckel R. M., Haese N., Jacob Silva P., Carney R. P., Marjomäki V., Streblow D. N., Lembo D., Stellacci F., Vitelli V., Krol S., Additives for vaccine storage to improve thermal stability of adenoviruses from hours to months. Nat. Commun. 7, 13520 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Croyle M. A., Yu Q.-C., Wilson J. M., Development of a rapid method for the PEGylation of adenoviruses with enhanced transduction and improved stability under harsh storage conditions. Hum. Gene Ther. 11, 1713–1722 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Mercier S., Villeneuve S., Mondor M., Uysal I., Time–temperature management along the food cold chain: A review of recent developments. Compr. Rev. Food Sci. Food Safety 16, 647–667 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Rupniak H. T., Rowlatt C., Lane E. B., Steele J. G., Trejdosiewicz L. K., Laskiewicz B., Povey S., Hill B. T., Characteristics of four new human cell lines derived from squamous cell carcinomas of the head and neck. J. Natl. Cancer Inst. 75, 621–635 (1985). [PubMed] [Google Scholar]
- 36.Kristensen D. D., Bartholomew K., Villadiego S., Lorenson K., What vaccine product attributes do immunization program stakeholders value? Results from interviews in six low- and middle-income countries. Vaccine 34, 6236–6242 (2016). [DOI] [PubMed] [Google Scholar]
- 37.World Health Organization, How to Calculate Vaccine Volumes and Cold Chain Capacity Requirements (WHO Press, Geneva, Switzerland, 2017). [Google Scholar]
- 38.Liebowitz D., Lindbloom J. D., Brandl J. R., Garg S. J., Tucker S. N., High titre neutralising antibodies to influenza after oral tablet immunisation: A phase 1, randomised, placebo-controlled trial. Lancet Infect. Dis. 15, 1041–1048 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Koopman G., Mooij P., Dekking L., Mortier D., Nieuwenhuis I. G., van Heteren M., Kuipers H., Remarque E. J., Radošević K., Bogers W. M. J. M., Correlation between virus replication and antibody responses in macaques following infection with pandemic influenza A virus. J. Virol. 90, 1023–1033 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pillet S., Kobasa D., Meunier I., Gray M., Laddy D., Weiner D. B., von Messling V., Kobinger G. P., Cellular immune response in the presence of protective antibody levels correlates with protection against 1918 influenza in ferrets. Vaccine 29, 6793–6801 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Samet S. J., Tompkins S. M., Influenza pathogenesis in genetically defined resistant and susceptible murine strain. Yale J. Biol. Med. 90, 471–479 (2017). [PMC free article] [PubMed] [Google Scholar]
- 42.Werk T., Ludwig I. S., Luemkemann J., Huwyler J., Mahler H.-C., Haeuser C. R., Hafner M., New processes for freeze-drying in dual-chamber systems. PDA J. Pharm. Sci. Technol. 70, 191–207 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Mirmoghtadaie L., Shojaee Aliabadi S., Hosseini S. M., Recent approaches in physical modification of protein functionality. Food Chem. 199, 619–627 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Morales J. O., Brayden D. J., Buccal delivery of small molecules and biologics: Of mucoadhesive polymers, films, and nanoparticles. Curr. Opin. Pharmacol. 36, 22–28 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Fonseca-Santos B., Chorilli M., An overview of polymeric dosage forms in buccal drug delivery: State of art, design of formulations and their in vivo performance evaluation. Korean J. Couns. Psychother. 86, 129–143 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Hanson C. M., George A. M., Sawadogo A., Schreiber B., Is freezing in the vaccine cold chain an ongoing issue? A literature review. Vaccine 35, 2127–2133 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Evans R. K., Nawrocki D. K., Isopi L. A., Williams D. M., Casimiro D. R., Chin S., Chen M., Zhu D. M., Shiver J. W., Volkin D. B., Development of stable liquid formulations for adenovirus-based vaccines. J. Pharm. Sci. 93, 2458–2475 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Renteria S. S., Clemens C. C., Croyle M. A., Development of a nasal adenovirus-based vaccine: Effect of concentration and formulation on adenovirus stability and infectious titer during actuation from two delivery devices. Vaccine 28, 2137–2148 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Price R. H. M., Graham C., Ramalingam S., Association between viral seasonality and meteorological factors. Sci. Rep. 9, e929 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.World Health Organization Expert Committee on Specifications for Pharmaceutical Preparations, Fifty Second Report on Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products, WHO Technical Report Series Report No.: 1010, Geneva, Switzerland (2018), pp. 309–351.
- 51.Nguyen J. L., Dockery D. W., Daily indoor-to-outdoor temperature and humidity relationships: A sample across seasons and diverse climatic regions. Int. J. Biometeorol. 60, 221–229 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walsberg G. E., Small mammals in hot deserts: Some generalizations revisited. Bioscience 50, 109–120 (2000). [Google Scholar]
- 53.Rexroad J., Martin T. T., McNeilly D., Godwin S., Middaugh C. R., Thermal stability of adenovirus type 2 as a function of pH. J. Pharm. Sci. 95, 1469–1479 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Gómez G., Pikal M. J., Rodríguez-Hornedo N., Effect of initial buffer composition on pH changes during far-from-equilibrium freezing of sodium phosphate buffer solutions. Pharm. Res. 18, 90–97 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Kadajji V. G., Betageri G. V., Water soluble polymers for pharmaceutical applications. Polymers 3, 1972–2009 (2011). [Google Scholar]
- 56.Gheribi R., Puchot L., Verge P., Jaoued-Grayaa N., Mezni M., Habibi Y., Khwaldia K., Development of plasticized edible films from Opuntia ficus-indica mucilage: A comparative study of various polyol plasticizers. Carbohydr. Polym. 190, 204–211 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Lu Y., Li J., Lu D., The mechanism for the complexation and dissociation between siRNA and PMAL: A molecular dynamics simulation study based on a coarse-grained model. Mol. Simul. 43, 1385–1393 (2017). [Google Scholar]
- 58.Karlin S., Brendel V., Charge configurations in viral proteins. Proc. Natl. Acad. Sci. U.S.A. 85, 9396–9400 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le H. T., Yu Q.-C., Wilson J. M., Croyle M. A., Utility of PEGylated recombinant adeno-associated viruses for gene transfer. J. Control. Release 108, 161–177 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaau4819/DC1
Supplementary Materials and Methods
Fig. S1. Complexity of a formulation does not significantly impact elongation of thin films.
Fig. S2. Tensile strength of thin films is influenced by certain excipient combinations.
Fig. S3. Viable adenovirus can be recovered from films reconstituted with a variety of diluents.
Fig. S4. Virus, excipients, and solvent volume significantly affect viscosity of solutions made from reconstituted films.
Fig. S5. Adenovirus stability profiles of rehydrated and solid films at elevated temperatures.
Fig. S6. Recovery of live adenovirus from the film matrix at elevated temperature is significantly affected by environmental humidity.
Fig. S7. DSC analysis reveals that thin films are amorphous solids across a wide temperature range.
Fig. S8. X-ray scanning diffraction reveals films as amorphous solids.
Fig. S9. Cytotoxicity profile of thin film vaccine validates safety of dosage form.
Fig. S10. Virus is evenly distributed throughout the film matrix.
Fig. S11. The amount of virus embedded in film matrix does not affect recovery of virus from the film matrix.
Table S1. Summary of formulations.