

An MTH1 inhibitor is integrated with nanoparticles to enhance therapy effect of reactive oxygen species in cancer treatment.
Abstract
Although photodynamic therapy (PDT) has been clinically applied tumor hypoxia still greatly restricts the performance of this oxygen-dependent oncological treatment. The delivery of oxygen donors to tumor may produce excessive reactive oxygen species (ROS) and damage the peripheral tissues. Herein, we developed a strategy to solve the hypoxia issue by enhancing the lethality of ROS. Before PDT, the ROS-defensing system of the cancer cells was obstructed by an inhibitor to MTH1, which is a key for the remediation of ROS-caused DNA damage. As a result, both nuclei and mitochondrial DNA damages were increased, remarkably promoting cellular apoptosis. The therapeutic results demonstrated that the performance of PDT can be improved by the MTH1 inhibitor, leading to efficient cancer cell killing effect in the hypoxic tumor. This strategy makes better use of the limited oxygen, holding the promise to achieve satisfactory therapeutic effect by PDT without generating redundant cytotoxic ROS.
INTRODUCTION
Photodynamic therapy (PDT) is a clinically approved oncologic intervention approach, which requires the activation of photosensitizers by an excitation light to convert oxygen from the ground state to the cytotoxic singlet state (1). As an emerging treatment modality, PDT enjoys several merits, such as good regioselectivity, minimal invasiveness, low systematic toxicity, and high therapeutic outcome for certain types of cancer (2). Many efforts have been devoted to further improving the antitumor performance of PDT (3). For instance, to overcome the restriction of the excitation light with low penetration depth (4), photosensitizers with near-infrared excitation were developed (5, 6), rare-earth upconverting nanoparticles were used as photoconverter (7, 8), and chemiluminescence (9, 10) or bioluminescence (11) was used as excitation sources. Targeted delivery of the photosensitizers to cancer cells or subcellular organelle was achieved to improve the specificity of PDT (12, 13).
However, PDT has still yet to gain extensive acceptance due to a birth defect: As an oxygen-dependent treatment, PDT is incompatible to the hypoxic microenvironment of tumor in many cases (14, 15). To overcome this issue, oxygen suppliers such as peroxides (16), endoperoxides (17), and oxygen carriers (18, 19) were used to provide PDT with extra ammo. As light exposure at the tumor site needs to be performed repeatedly during the whole course of PDT, an adequate supply of the oxygen vector is necessary, which imposes additional requirement to the capacity of the drug carriers. In addition, due to the difficulty to accurately control the exposure dose, PDT usually suffers from toxicity by over-illumination (20). Overdosing of the oxygen vector can also result in even more singlet oxygen production in PDT and cause more harm to the peripheral tissues around the tumor. As a result, making the reactive oxygen species (ROS) generated in PDT more lethal to cancer cells can improve the therapeutic effect without producing excessive ROS, which can be achieved by sensitizing the cancer cells by hampering its ROS-defensing system.
DNA mispairing is a type of the cellular lesion caused by ROS-induced oxidative damage, which can also be induced by PDT (21). The increased ROS tension leads to the oxidation of the free deoxynucleoside triphosphate (dNTP) pool, which is incorporated into DNA and brings about mispairing to trigger programmed cell death. Although direct damage to DNA is not considered as the primary reason for PDT-induced apoptosis (22), PDT can cause the oxidation of the dNTP pool. Hirakawa et al. (23) reported the guanine-specific oxidation after PDT. Zhang and co-workers (24) also observed an increased oxidized guanine level in mitochondrial DNA (mtDNA) during PDT. The guanine oxidation product 8-oxoguanine can yield 8-oxo-2′-deoxyguanosine triphosphate (8-oxo-dG), which is responsible for mispairing of deoxynucleotide in DNA (25, 26). As a self-protecting functional protein, MTH1 can hydrolyze 8-oxo-dG to 8-oxo-2′-deoxyguanosine monophosphate and pyrophosphate, thereby sanitizing the oxidized product (26). Recently, a family of MTH1 inhibitors has been developed to exploit the potency of MTH1 as an anticancer target for chemotherapy (CHT). Once bound to the active site of MTH1, the inhibitor can successfully prevent the elimination of 8-oxo-dG and promote apoptosis on various cancer cell lines (27). However, such CHT relies on endogenous ROS to produce 8-oxo-dG. Because free dNTP is much more sensible to ROS than DNA double strand (28), it can be envisioned that PDT can work together with the inhibitor as a source of ROS to produce more 8-oxo-dG in killing cancer cells.
Here, we demonstrated that the antitumor performance of PDT can be greatly improved by the cocktail treatment with the CHT of the MTH1 inhibitor TH588. Using silica cross-linked micelles (SCLMs) as the nanocarrier, TH588 was concomitantly delivered with a photosensitizer, chlorine 6 (Ce6), to the cancer cells in vitro and to tumor in vivo. It was found that TH588 can exert synergistic effect with PDT (Fig. 1). In the combined therapy, the DNA mispairing occurred not only in cell nuclei DNA (nDNA) but also in the mtDNA, accounting for DNA damage–induced cell death (29, 30). In vitro mechanistic studies showed that the damaged DNA can trigger p53-mediated apoptosis, and the treatment also regulated the expression of corresponding pro-apoptotic and anti-apoptotic proteins. The performance of the combined therapy was also testified on nude mice with tumor xenograft from A431 cancer cell line, which evidenced that, with the sensitization of TH588, the PDT can better use the limited oxygen in the hypoxic tumor and gain superior therapeutic effect.
Fig. 1. Schematic diagram of the designed strategy for the combined therapy of CHT by MTH1 inhibitor and PDT by Ce6.
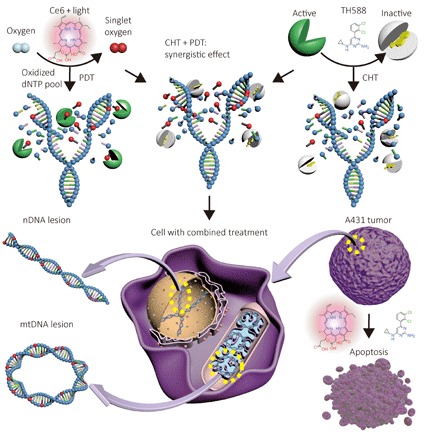
In the combined therapy, the scavenger of 8-oxo-dG is inhibited by TH588, and thus, the oxidation-induced DNA mutant can be amplified. The mutant can be found in both nDNA and mtDNA, leading to the apoptosis of the cancer cells.
RESULTS
Synthesis and characterization of the nanocarrier SCLMs
The schematic diagram for the preparation and structure of SCLMs is illustrated in Fig. 2A. SCLMs were prepared by the self-assembly of the silica precursor and the amphiphilic triblock copolymer pluronic F108 (PEO132-PPO50-PEO132, where PEO represents polyethylene oxide and PPO represents polypropylene oxide) (31). The PEO domain of the polymer can form polyethylene glycol (PEG) units on the surface, endowing the nanocarrier with stealth property. The silica shell acts as a cross-linker to hold the hydrophobic PPO core, enhancing the stability of the micellar structure to avoid the premature release of the payloads (32). It was demonstrated that SCLMs can passively target tumor xenografts through the enhanced permeability and retention effect (31). Thus, SCLMs can serve as the nanocarrier for the co-delivery of TH588 and Ce6 in the combined therapy. Transmission electron microscopy (TEM) image (Fig. 2B) shows that the fabricated SCLMs had a spherical morphology with an average size of 5 nm. The hollow structure of the silica shell can be observed from the high-resolution TEM image of the calcined sample (fig. S1A). Type IV isotherm and H2 hysteresis loop were observed from the isotherm of the N2 adsorption-desorption analysis, again revealing the hollow structure. The pore size distribution curve indicates a dual pore distribution centered at 4.4 and 1.2 nm, corresponding to the mesopores inside the silica shell and the micropores on the silica wall, respectively (fig. S1B). A hydrated size of 17 nm (Fig. 2C) was obtained for SCLMs by dynamic light scattering (DLS). The hydrated size is larger than the size observed by TEM, which is due to the PEO ligands outside the silica shell.
Fig. 2. Characterizations of the nanocarrier SCLMs and loading/release of the MTH1 inhibitor TH588 and photosensitizer Ce6.
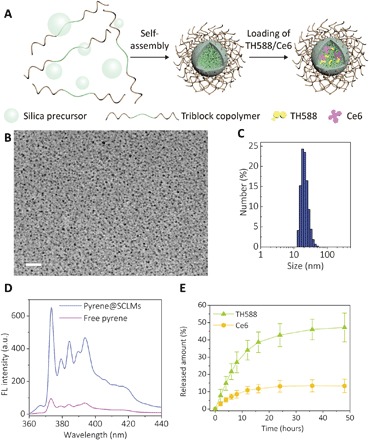
(A) Schematic illustration for the preparation and structure of SCLMs. (B) TEM image of as-prepared SCLMs. Scale bar, 50 nm. (C) Hydrated size histogram of SCLMs obtained by DLS measurement. (D) Fluorescence (FL) of 10−6 M pyrene in aqueous solution (purple line) and after being loaded in SCLMs (blue line). (E) Release profiles of TH588 and Ce6 from T&C@SCLMs under sink conditions with 1.5% Tween 80 and 5% ethanol in PBS (pH 7.4). Bars represent SD of three individual samples. a.u., arbitrary units.
The MTH1 inhibitor TH588 and the photosensitizer Ce6 were loaded into the nanocarrier with different concentrations via a solvent-assisted method (33). The nanocarrier loaded with TH588 or Ce6 as well as the nanocarrier co-loaded with TH588 and Ce6 were denoted as T@SCLMs, C@SCLMs, and T&C@SCLMs. Pyrene was used as a model molecule to validate the loading of hydrophobic guest molecules. The fluorescence intensity of pyrene increased six times, and the intensity ratio of the peaks at 374 and 384 nm changed from 1.61 to 1.46, indicating that the pyrene was located in the hydrophobic domain inside the SCLMs (Fig. 2D). As both TH588 and Ce6 cannot be solubilized in the aqueous phase, the release of TH588 and Ce6 from T&C@SCLMs was tested under a sink condition in a phosphate-buffered saline (PBS) solution with 1.5% Tween 80 and 5% ethanol. The release rate of TH588 is faster than Ce6 (Fig. 2E). The released amount of TH588 after 48 hours was nearly 50%, which was about four times more than the released amount of the loaded Ce6. The differentiated release rates of TH588 and Ce6 can be ascribed to their different hydrophobicity and molecular size. After the cellular uptake, the inhibitor TH588 would be released earlier to start its action before the consequent PDT.
In vitro 1O2 production was then evaluated by 1,3-diphenylisobenzofuran (DPBF) probe. As shown in fig. S2, upon the irradiation of 660-nm light, TH588 with the only absorption band at 300 nm (fig. S2A) produced no 1O2, and the bleaching dynamics of DPBF by Ce6 was almost unchanged with the addition of TH588 (fig. S2, B to D). Therefore, in the combined therapy, only Ce6 is responsible for the generation of 1O2.
In vitro therapeutic effect of the combined therapy
The enhanced therapeutic effect of the combined therapy was first demonstrated in vitro with human epidermoid carcinoma cell line A431 using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. T@SCLMs, C@SCLMs, and T&C@SCLMs were incubated with the cells at designed concentrations. First, the effect of CHT by TH588 was tested, and only a moderate anticancer activity was observed. The half-maximal inhibitory concentration (IC50) values for the cells after 24- and 48-hour incubation were found to be 39 and 25 μM, respectively (Fig. 3A). It should be noted that the IC50 values are higher than the reported data using free TH588 with 0.2% dimethyl sulfoxide (DMSO) (27), perhaps due to shorter exposure time and sustained release of TH588 from the nanocarrier. Under the dark conditions, both MTT and calcein-AM (acetoxymethyl)/PI (propidium iodide) costaining assay showed no obvious cytotoxicity for the cells incubated with C@SCLMs, indicating good biocompatibility of the nanocarrier and the photosensitizer (fig. S3). The single PDT treatment by C@SCLMs showed an IC50 value of 1.1 μM Ce6 after 5-min exposure by a 660-nm light-emitting diode (LED) light with a power density of 20 mW cm−2. In the combined therapy, the cells were incubated in a culture medium with T&C@SCLMs containing 10 μM TH588 and different concentrations of Ce6 for 24 hours and then irradiated under the same condition. Compared with PDT, the anticancer effect of the combined therapy was obviously improved, with an IC50 value as low as 0.6 μM Ce6 (Fig. 3B). The improvement was more notable at high concentrations of Ce6. In the combined therapy at 4.0 μM Ce6, the viability of the A431 cells was only 9%, which was about one-third value of the PDT case. These results indicate that the MTH1 inhibitor can sensitize the cancer cells to PDT, leading to a synergistic therapeutic effect.
Fig. 3. MTH1 inhibitor TH588 improves the in vitro anticancer activity of PDT.
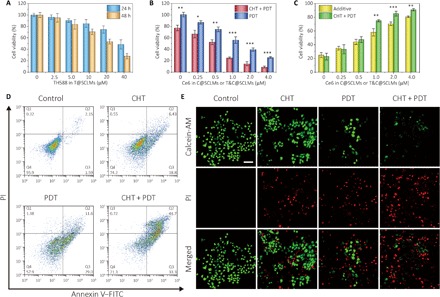
(A) Cell viability of A431 cells incubated with T@SCLMs containing 2.5 to 40 μM TH588 for 24 and 48 hours. (B) Cell viability of A431 cells after the treatment of PDT and the combined therapy. The cells were incubated with C@SCLMs containing 0.25 to 4.0 μM Ce6 (PDT group) or C&T@SCLMs with 0.25 to 4.0 μM Ce6 and 10 μM TH588 (combined therapy group) for 24 hours and irradiated using a 660-nm LED light with a power density of 20 mW cm−2 for 30 s. Data show the mean value and SD of eight individual samples (*P < 0.05, **P < 0.01, ***P < 0.005). (C) Therapeutic efficacy for PDT and combined therapy, and calculated additive therapeutic efficacy. (D) Results of flow cytometric apoptosis analysis. A431 cells were treated by CHT, PDT, and combined therapy and costained with annexin V–FITC (fluorescein isothiocyanate)/PI. (E) Live/dead cell imaging of A431 cells costained with calcein-AM (live cells, green) and PI (dead cells, red). Scale bar, 100 μm.
To quantitatively analyze the synergistic effect between the CHT and PDT, the additive therapeutic effect (Tadditive) and the therapeutic effect of the combined treatment (Tcombined) were calculated according to the following equations (34)
| (1) |
| (2) |
where fCHT, fPDT, and fcombined stand for the percentage of the viable cells after the treatment of CHT, PDT, and combined therapy, respectively. As shown in Fig. 3C, Tadditive was comparable with Tcombined when Ce6 was lower than 0.5 μM. However, for combined treatment with Ce6 concentration above 1.0 μM, it is evident that Tcombined is higher than Tadditive, indicating the underlying synergistic effect between the MTH1 inhibitor–based CHT and PDT. The flow cytometry results of cell apoptosis assay using annexin V/PI costaining showed the same tendency to the MTT analysis (Fig. 3D). The terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay also revealed more apoptotic cells after the combined therapy (fig. S4), which was in good accordance with the calcein-AM/PI costaining results of live/dead cells (Fig. 3E).
The effect of the combined therapy to other kinds of cancerous cell lines was also examined (fig. S5). It is clear that the effectiveness of PDT can be enhanced by TH588 in the case of human cervical cancer cell line (HeLa), human breast cancer cell line (MCF-7), human colorectal cancer cell line (HCT116), and murine melanoma cell line (B16F10). The synergistic effect of the combined therapy was also obtained using these different cell lines. These in vitro experimental results evidence that the 8-oxo-dG inhibitor MTH1 can successfully improve the performance of PDT to cancerous cells. In normal cells, MTH1 is nonessential (35). The 8-oxo-dG lesions in nDNA can be amended through the base excision repair by 8-oxoguanine DNA glycosylase and MutY homolog DNA glycosylase (36). Thus, their ROS-defensing system cannot be completely affected by the MTH1 inhibitor. The MTT results of human dermal fibroblasts revealed that the pretreatment of TH588 failed to sensitize noncancerous cells (fig. S6). Therefore, the specificity of PDT to cancer cells can be enhanced by the combined therapy.
Mechanism study of the combined therapy
To directly verify that more DNA mutants caused by 8-oxo-dG were accumulated in the cells during the combined therapy, avidin–Alexa Fluor 488 was used to stain A431 cells after the treatment. As avidin has special affinity to 8-oxo-dG, the DNA mutant can thus be visualized by green fluorescence. To localize the lesion, the nuclei and mitochondria of the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) and MitoTracker Red, respectively. The resulting confocal images are shown in Fig. 4A. It is obvious that, among the four groups, the cells treated by combined therapy presented the most 8-oxo-dG accumulation, which was 2.1 and 3.5 times higher than that solely treated by CHT and PDT, respectively (Fig. 4B). This result suggests that, when the cells were sensitized by TH588 and followed by PDT, much more 8-oxo-dG can be retained in the cells to induce the DNA mutant. Under the synergistic effect, the inhibition of MTH1 leads to the disruption to the ROS-defensing system of the cancer cells, and PDT actively produces ROS to promote the apoptotic response. Thereby, the two treatment approaches can benefit by each other.
Fig. 4. Enhanced accumulation of 8-oxo-dG in the TH588-assisted PDT is mainly located at mtDNA.
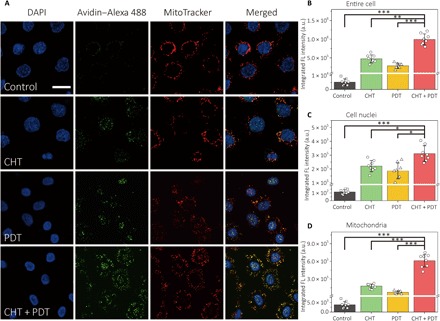
(A) Colocalization of 8-oxo-dG and mitochondria. A431 cells were subjected to different treatments and costained with DAPI, avidin–Alexa Fluor 488, and MitoTracker Red. Scale bar, 20 μm. Integrated fluorescence intensity from (B) the entire cell, (C) the nuclear region, and (D) the mitochondrial region. The integrated fluorescence intensity was obtained by analyzing the colocalized green/blue and green/red areas by ImageJ. Data show the mean value and SD of eight individual cells (*P < 0.05, **P < 0.01, ***P < 0.005).
From the overlapped areas between the green fluorescence of Alexa Fluor 488 and DAPI/MitoTracker Red, it was concluded that 8-oxo-dG located not only inside the nuclei but also in the mitochondria of the cells. Because 8-oxo-dG can be incorporated into mtDNA (37) and MTH1 is also involved in the repair of mtDNA mutant (38), both nDNA and mtDNA mutants were found in cells from all the three treatment groups (Fig. 4A). We further quantified the mutant by calculating the integrated intensity of green fluorescence in nuclear regions (Fig. 4C) and mitochondrial regions (Fig. 4D). Although the combined treatment caused the DNA mutant to be 41% higher than CHT with regard to the nuclei, the difference between the two sets of data is less obvious than the case of mitochondria. For the cells treated by the combined therapy, 8-oxo-dG in the mitochondrial region is 2.4 times higher than the CHT treatment, revealing that the additional DNA mutant caused by PDT is mainly located at mtDNA, also capable of resulting in programmed cell death (39). When the cells were pretreated by a mitochondria-targeted antioxidant mitoquinone (MitoQ), the integrated intensity of green fluorescence decreased by 60% under the combined treatment (fig. S7). In contrast, the fluorescence decrement in the nuclear region is less notable. Meanwhile, the annexin V–FITC (fluorescein isothiocyanate)/PI costaining assay by flow cytometry showed that the ratio of the apoptotic A431 cells between PDT and combined therapy reduced notably (fig. S8), suggesting that the mutant in mtDNA contributed to the combined therapy through the mitochondrial apoptotic pathway.
Next, the effect of the combined therapy on the modulation of pro-apoptotic and anti-apoptotic member proteins was assessed. The expression of some marker proteins in the apoptosis signaling pathway was investigated by Western blot analysis. The expression results are shown in Fig. 5A, and the quantification results are shown in Fig. 5 (B to G). p53 is known as an inducing factor of apoptosis upon the DNA damage. The mutation induced by the MTH1 inhibitor can trigger the p53-mediated apoptosis on cancer cells. An up-regulation of p53 for all the three treatment groups was observed, where the combined treatment was most efficient (Fig. 5B). Similar overexpression tendency also took place for the cleaved caspase-3 (Fig. 5, C and D), which is involved in the execution for the apoptosis process. Therefore, it is evident that the improved therapeutic effect of the combined treatment is relevant to the p53-mediated apoptosis.
Fig. 5. Improved therapeutic effect of TH588-assisted PDT is related to p53-mediated cell apoptosis and the inhibition of anti-apoptotic p21.
(A) Effect of different treatments on the expression of apoptosis signaling protein p53, cleaved caspase-3, caspase-3, BAX, Bcl-2, and p21. A431 cells were prepared after the treatment of CHT, PDT, or combined therapy. The lysates of cells were then analyzed by Western blot. (B to G) Quantification for the expression of corresponding proteins from (A) by the intensity of the chemiluminescence from each protein band. The intensities of the bands were analyzed by ImageJ. Data show the mean value and SD of three individual experiments (**P < 0.01, ***P < 0.005). (H) Proposed molecular mechanism for the combined therapy. TH588 can sensitize the cancer cells by inhibiting MTH1 and stifling the defensing system to 8-oxo-dG of the cancer cells. The ROS generated during PDT can raise the level of intracellular 8-oxo-dG, causing the DNA mutant in both nDNA and mtDNA for p53-mediated cell apoptosis.
The cyclin-dependent kinase (CDK) inhibitor p21 is a downstream protein of p53. For A431 cells subjected to different treatments, the expression of p21 showed very different tendency from p53 (Fig. 5E). The cells treated by TH588 showed 1.2 times higher p53 than the control cells, although it was the most amount of p21 expression among the four groups of cells. A down-regulation of p21 was observed for cells treated by PDT. For the combined treatment with the greatest p53 up-regulation, the cells exhibited even less p21 than sole CHT. It has been reported that p21 has multiple roles and functions in cellular apoptosis, and a prevailing understanding for p21 is that it acts as an anti-apoptotic agent after the DNA damage (40). By arresting the cell cycle of the cells with DNA damage in the G0-G1 phase, p21 allows the cells to repair the damaged DNA before its proliferation (41). With CHT of TH588, the cancer cells overexpress p21 and the cell cycle is thereby stagnated (fig. S9). On the other hand, PDT was found to reduce the expression of p21 (Fig. 5E). Therefore, the combined therapy of CHT and PDT can also promote the apoptosis progress by inhibiting the p21 expression.
The expression of apoptosis regulator protein BAX and Bcl-2 was also investigated. Both BAX and Bcl-2 are involved in mitochondria-mediated apoptosis, where Bcl-2 plays an anti-apoptotic role to inhibit the release of cytochrome c from mitochondria. The pro-apoptotic protein BAX can permeabilize the mitochondrial membrane and release cytochrome c to activate the caspases. It can be seen from Fig. 5 (F and G) that the expression of BAX and Bcl-2 was similar between CHT and PDT. In contrast, by using combined therapy, the cells obviously expressed more BAX and less Bcl-2, indicating that the damage to mitochondria was amplified in the combined therapy.
In vivo performance of the combined therapy on hypoxic tumor
The tumor hypoxia is one of the main translational barriers for PDT. Hence, it is necessary to test the performance of the TH588-sensitized PDT on solid tumor to demonstrate whether such strategy is applicable to the hypoxic tumor microenvironment. To this end, the combined therapy was carried out on nude mice with A431 solid tumor xenograft using SCLMs as the delivery vehicle. The biodistribution of SCLMs was demonstrated by ex vivo imaging of the tumor mass and the main organs harvested from the tumor-bearing mice (fig. S10A). Distinct fluorescence signal can be detected from the tumor even after 48 hours after injection, indicating good passive tumor-targeting capability of the nanocarrier. The injected SCLMs were mainly excreted through the hepatobiliary route (31), so the liver also exhibited strong fluorescence, which decreased along with time. For the tumor therapy, mice injected with saline were used as the control group, and the other two groups of mice treated by sole CHT or PDT were set as the negative control. An additional group of mice was injected with SCLMs to test the effect of the blank nanocarrier to the tumor. As the nanocarrier bioavailability for in vivo animal experiments is usually lower than that for the in vitro cell experiments, TH588 (50 μM) and Ce6 (20 μM) with higher concentrations were used for this in vivo experiment. For each group, intravenous administration of therapeutic agents was repeated every 5 days. The tumor mass for mice in each group was exposed to a 660-nm LED light under a power density of 20 mW cm−2 for 10 min every day. The control group was also irradiated to exclude the interference by light exposure. The obtained results are generally consistent with the in vitro experiments.
Figure 6A shows the development of the tumor size during the 20-day treatment. The effect of the blank nanocarrier SCLMs was also investigated. Although the average tumor size and weight were slightly lower than that of the PBS control group, no statistical difference can be obtained between these two groups. Compared with the control group, all the treatment groups exhibited active therapeutic results. No distinct variation of body weights was found for the mice (fig. S10B). However, the average tumor sizes for CHT and PDT groups were 2.3 and 1.4 times larger than the original tumors. For the group of combined therapy, the average tumor volume shrunk substantially by 54% after the treatment. The resected tumor after the combined therapy also showed the lowest weight (Fig. 6B) and size (Fig. 6C). Last, we examined the apoptotic tumor cells in the tumor tissue by TUNEL staining (Fig. 6D). For the control group and the blank nanocarrier group, almost no apoptotic signal was detected. For the treatment groups, it can be seen that the tumor cells from the T&C@SCLMs group had the greatest amount of DNA fragmentation (red fluorescence), which is representative of cell apoptosis. The ratio of the integrated intensity between red fluorescence and blue fluorescence for the combined treatment is 1.87, which is much higher than sole PDT (1.02) and CHT (0.37), confirming its superior antitumor activity (Fig. 6E). It is noteworthy that, on the basis of the tumor size or weight, the therapeutic effect of the combined treatment is not statistically better than the additive therapeutic effect of CHT and PDT. However, the synergistic effect can be evidenced by the results of the TUNEL assay. According to the confocal images of TUNEL staining, the ratios of the apoptotic cells were 31.2% for CHT, 14.8% for PDT, and 60.7% for the combined treatment (Fig. 6F). The additive therapeutic effect for the two single treatments was calculated to be 41.4%, which is almost one-third lower than that of the combined treatment. Together, these results demonstrated that the MTH1 inhibitor was able to improve the in vivo performance of PDT. In vivo studies were carried out to assess the biocompatibility of SCLMs and the nanocarrier loaded with the two therapeutic agents. With an exposure dose of 0.1 g kg−1 and an administration frequency of 5 days, no obvious lesion was found from the tissue sections from the main organs of mice stained with hematoxylin and eosin (H&E; fig. S10C). The histological results preliminarily revealed the good biocompatibility of the nanocarrier and validated the biosafety of the combined treatment.
Fig. 6. In vivo antitumor effect of PDT can be greatly promoted by the combination therapy.
(A) Growth rate of A431 tumor xenograft after the treatment of PBS control, blank nanocarrier (SCLMs), CHT, PDT, and combined therapy (CHT + PDT). Data show the mean value and SD of five individual tumors from mice (**P < 0.01, ***P < 0.005). (B) Tumor weights and (C) photos of the tumor mass for each group. Photo credit (C): Junyao Li, China Pharmaceutical University. (D) TUNEL assay of tissue slices collected from the tumor mass on mice after different treatment modalities. The blue fluorescence from DAPI indicates cell nuclei, and the red fluorescence from Cy3 indicates apoptotic cells. Scale bar, 100 μm. (E) Ratios of the integrated intensity between the red fluorescence and the blue fluorescence in the images of TUNEL staining. (F) Ratios of the apoptotic cells determined by fluorescent TUNEL assay. The data were obtained by analyzing three independent confocal images of TUNEL assay with ImageJ.
DISCUSSION
One of the main obstacles that encumber the performance of PDT is the contradiction between the hypoxia microenvironment of tumor and its oxygen reliance. The tumor hypoxia restricts the quantity of the ROS generated during PDT and thus constrains the therapeutic effect. Fortunately, with the development of effective approaches in the tool kit of cancer treatment, cocktail therapy with superior therapeutic effect can be designed by compensating for the disadvantage of one treatment by the other. Here, we reported an innovative strategy to address the hypoxia problem of PDT by enhancing the cytotoxicity of ROS to cancer cells. MTH1 is a vital protein for cancer cells to withstand ROS-induced DNA damage caused by 8-oxo-dG, which is a promising target for CHT. In our strategy, PDT was combined with CHT of the MTH1 inhibitor TH588 to make the ROS generated in PDT more effective in killing cancer cells. Synergistic effect was achieved by the combined therapy to different cancerous cell lines. Compared with sole PDT and CHT, the combined therapy markedly amplifies both nDNA and mtDNA damage. Oncologic outcome on nude mice further validated the effectiveness of the combined therapy. In conclusion, our strategy could more effectively use the limited oxygen in hypoxic solid tumor and greatly improve the therapeutic performance of PDT combined with CHT. The present combined therapy is not a simple stack of different treatment modalities, but makes the sum greater than the parts. These findings are expected to promote the clinical translation of PDT and offer a reference to design complementary combination therapies for efficient cancer treatment.
MATERIALS AND METHODS
Synthesis of SCLMs
The preparation protocol of SCLMs was previously described in a published work (31). In a typical synthesis, pluronic copolymer F108 (0.25 g) and dichloromethane (100 μl) were codissolved in HCl solution (7.5 ml, 2.0 M) at room temperature. The mixture was sonicated for 2 min and stirred for 30 min. Afterward, tetraethoxysilane (268 μl) was added to the homogenous solution and stirred for another 15 min before the addition of dimethyldimethoxysilane (40 μl). After another 3 hours of reaction, the solution was collected and dialyzed using a membrane with a molecular weight cutoff (MWCO) of 25,000 in deionized water (1 liter) for five times to remove the impurities. The added dichloromethane was removed at 50°C under 150-mbar vacuum. As the centrifugation method cannot separate SCLMs from aqueous solution, the concentration of SCLMs was determined by lyophilization. For the characterization of the hollow structured silica shell, the freeze-dried SCLMs were calcined in a furnace at 550°C in air for 3 hours.
Determination of the 1O2 production
DPBF was used to estimate the ROS generation from the photosensitizer Ce6 under the irradiation of LED light. TH588 was also introduced to validate that no additional ROS can be generated by the MTH1 inhibitor. DPBF (100 μM), TH588 (0.2 μM), and/or Ce6 (0.02 μM) were dissolved in ethanol. To monitor ROS generation, the mixture was irradiated by a 660-nm LED light with a power density of 20 mW cm−2 at room temperature, and the absorption of DPBF at 411 nm was recorded every 10 s from 0 to 150 s with an ultraviolet-visible spectrophotometer.
Loading of TH588 and Ce6 to SCLMs
TH588 and Ce6 were dissolved in DMSO to form a solution with TH588 and Ce6 concentrations of 25 and 20 mM, respectively. Then, the DMSO solutions containing TH588 or Ce6 in designed amounts were separately added to a solution with SCLMs (10 mg ml−1) under vigorous sonication to load the hydrophobic molecules into the PPO core of SCLMs. The SCLMs loaded with TH588 and Ce6 were separately denoted as T@SCLMs and C@SCLMs. The SCLMs co-loaded with TH588 and Ce6 were denoted as T&C@SCLMs. Because the DMSO content in the final solutions was very low, the solutions were used for biological studies without the removal of DMSO.
In vitro release of TH588 and Ce6
The release rate of TH588 and Ce6 was tested under sink conditions in a PBS solution (pH 7.4) with 1.5% Tween 80 and 5% ethanol. Aqueous solution (5 ml) containing T&C@SCLMs (10 mg ml−1) with TH588 (50 μM) and Ce6 (20 μM) was sealed in a dialysis membrane with MWCO of 25,000. The dialysis bag was then soaked in the PBS solution (45 ml) under stirring in the dark. Solution (0.5 ml) was taken at designed time. The concentrations of TH588 and Ce6 were determined by high-performance liquid chromatography.
Cell culture
The cells were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) in a humidified atmosphere under 5% CO2 at 37°C.
Cell viability assay
Cell viability was measured using human epidermoid cancer cell line A431, human cervical cancer cell line HeLa, human breast cancer cell line MCF-7, human colorectal cancer cell line HCT116, and murine melanoma cell line B16F10 by MTT assay. Each group of cancer cells was seeded at a density of 104 cells per well in a 96-well cell culture plate with DMEM (100 μl). After 12 hours, the culture medium was replaced by fresh DMEM (95 μl), and aqueous solution (5 μl) containing T@SCLMs, C@SCLMs, or T&C@SCLMs (10 mg ml−1) with designed concentrations of TH588 and Ce6 was then added to each well. The final concentration of TH588 in the CHT group and combined therapy group was 10 μM, and Ce6 was 0 to 4.0 μM. After 24-hour incubation, the medium was replaced by fresh culture medium. The cells were irradiated using a 660-nm LED light with a power density of 20 mW cm−2 for 30 s. The cells were placed in the incubator for another 1 hour, and then MTT solution (100 μl, 0.5 mg ml−1) in DMEM was added to each well, followed by incubation at 37°C for 4 hours. The culture medium was removed, and the blue product was dissolved in DMSO (100 μl). The absorption value at 570 nm was measured using a Molecular Devices SpectraMax M4 plate reader, and the absorbance at 690 nm was subtracted as the background. Cell viability was calculated using the following formula
Apoptosis assay
Cellular apoptosis was estimated using an annexin V–FITC/PI apoptosis detection kit. Following the manufacturer’s protocol, the apoptotic cell distributions were determined. A431 cells were seeded in six-well plates at 2.0 × 105 cells per well for 48 hours, and then the medium was replaced with fresh medium (1.9 ml) together with T@SCLMs (100 μl, 10 mg ml−1) containing TH588 (200 μM), C@SCLMs (100 μl, 10 mg ml−1) containing Ce6 (40 μM), or T&C@SCLMs (100 μl, 10 mg ml−1) containing TH588 (200 μM) and Ce6 (40 μM). After incubation for 24 hours, the cells were washed twice with fresh medium. The plate was irradiated using a 660-nm LED light for 30 s. After an additional 1 hour of incubation in the dark, the cells were trypsinized, harvested, washed with PBS, and suspended in the binding buffer (195 μl) in the analysis kit. The cells were then co-incubated under dark conditions at room temperature for 15 min by adding annexin V–FITC (5 μl) and PI (10 μl) to the buffer. The cells were analyzed with a MACSQuant flow cytometer. The data were analyzed and processed with FlowJo 7.6. In another two groups, the cells were pretreated with or without the mitochondria-targeted antioxidant MitoQ (0.2 μM) for 24 hours before the addition of drug-containing SCLMs. The following procedures were the same as described above.
Calcein-AM/PI staining assay
A431 cells were seeded in a 24-well plate at a density of 1 × 104 cells per well and incubated for 24 hours. The medium was replaced with fresh medium (0.95 ml), together with T@SCLMs (50 μl, 10 mg ml−1) containing TH588 (200 μM), C@SCLMs (50 μl, 10 mg ml−1) containing Ce6 (40 μM), or T&C@SCLMs (50 μl, 10 mg ml−1) containing TH588 (200 μM) and Ce6 (40 μM). Following 24-hour incubation, the cells were washed twice. The plate was irradiated using a 660-nm LED light for 30 s. After another 1 hour of incubation, the cells were washed twice with PBS and costained with calcein-AM and PI.
Fluorescence imaging of the DNA mutant
The incorporation of 8-oxo-dG in DNA was imaged by Alexa Fluor 488–conjugated avidin based on the high binding affinity of avidin to 8-oxo-dG. After incubation with MitoTracker Red CMXRos (200 nM) at 37°C for 30 min, the cells were treated by the same protocol in the calcein-AM/PI staining assay and then fixed in 4% paraformaldehyde at room temperature for 30 min, followed by washing twice with TBST (tris-buffered saline with 0.1% Triton X-100). Blocking was performed at room temperature for 2 hours using a 15% FBS solution with TBST. The cells were then incubated in the blocking solution containing Alexa Fluor 488–conjugated avidin (10 μg ml−1) at 37°C for 1 hour. Subsequently, the cells were washed twice by TBST for 5 min and then imaged with a confocal microscope. The mutant in nuclei and mitochondria was estimated by calculating the intensity of green fluorescence from overlapped green/blue and green/red regions, respectively. The data were analyzed with ImageJ 1.41 software. Each dataset was collected from eight individual cells.
TUNEL staining
TUNEL staining of A431 cells and the tumor tissue slices was performed using the ApopTag In Situ Apoptosis Detection Kit. After staining, the samples were imaged using a confocal microscope.
Western blot
A431 cells were treated with CHT, PDT, or the combined therapy. Cell lysates (approximately 30 μg of protein) were solubilized in SDS–polyacrylamide gel electrophoresis (PAGE) containing 12% gel at 120 V for 50 min. Then, SDS-PAGE was transferred onto a nitrocellulose membrane at 300 mA for 35 min. The membranes were blocked with TBS containing 0.1% Tween 20 and 5% fat-free milk at room temperature for 2 hours. The membranes were incubated with rabbit antibodies against p53, p21, BAX, Bcl-2, cleaved caspase-3, caspase-3, and α-tubulin at 4°C for another 12 hours, and then incubated in a peroxidase-conjugated affinipure goat anti-rabbit immunoglobulin G at room temperature for 2 hours. The intensity of the band was analyzed and quantified with ImageJ software using densitometry. The quantification of other proteins was determined by calculating the relative intensity to α-tubulin.
Cell cycle analysis
The cell cycle was analyzed according to the manufacturer’s protocol of the Cell Cycle and Analysis Kit. A431 cells were seeded in a six-well plate at 2.0 × 105 cells per well for 48 hours, and then the medium was replaced with fresh medium (1.9 ml) together with T@SCLMs (100 μl, 10 mg ml−1) containing TH588 (200 μM). After 24-hour treatment, the cells were harvested and washed with cold PBS and then fixed in 70% ice-cold ethanol at 4°C for 24 hours. After that, the fixed cells were washed gently with cold PBS twice and incubated with staining buffer (0.5 ml), ribonuclease (RNase) A (10 μl), and PI (25 μl) in the dark at 37°C for 30 min. The stained cells were analyzed using MACSQuant. Data analysis was performed with ModFit LT 5.0 software.
In vivo antitumor test
Female nude mice (4 weeks, 18 to 20 g) were purchased from the Animal Center of Nanjing University. Animal procedures were in agreement with the approved protocols by the Institutional Animal Care and Use Committee of China Pharmaceutical University. A431 cells (2 × 106) in DMEM (100 μl) were subcutaneously injected to the back of each nude mouse on the right side. When the tumor volume reached ∼70 mm3, T@SCLMs containing TH588 (50 μM), C@SCLMs containing Ce6 (20 μM), or T&C@SCLMs containing TH588 (50 μM) and Ce6 (20 μM) with the SCLM concentration of 10 mg ml−1 were injected via tail vein in a total volume of 0.2 ml. The mice injected with PBS (0.2 ml) were used as the control group. The nanocarrier SCLMs (0.2 ml) with a concentration of 10 mg ml−1 were also injected as an individual negative control group. At 24 hours after the injection, mice were irradiated with a 660-nm LED light at a power density of 20 mW cm−2 for 10 min per day during the treatment. The injection was repeated at the 5th, 10th, and 15th day of the treatment. Tumor size and body weight of mice were monitored every day. The treatment was continued for 20 days. The mice were executed at the 21st day, and the tumors from each group of mice were harvested to take the images. Tumor slices with a thickness of 10 μm were taken from the tumors and subjected to the TUNEL staining assay.
Biodistribution of SCLMs
To track the biodistribution of SCLMs, near-infrared fluorescent dye Cy5.5 was covalently labeled to SCLMs using a method described in our previous work (31). Cy5.5-labeled SCLMs (0.2 ml) with a concentration of 10 mg ml−1 were intravenously injected to mice with A431 tumor xenograft. The tumor mass and main organs (heart, liver, spleen, lung, and kidney) were harvested at designed time. The ex vivo imaging of the organs was performed with a Vilber FUSION FX7 imaging system (λex = 680 nm, λem = 750 ± 10 nm).
Histological study
T@SCLMs containing TH588 (50 μM), C@SCLMs containing Ce6 (20 μM), or T&C@SCLMs containing TH588 (50 μM) and Ce6 (20 μM) with an SCLM concentration of 10 mg ml−1 was intravenously injected to nude mice. The control group was injected with PBS (0.2 ml) only. The injection was repeated every 5 days for three times. At the 21th day, the mice were executed. The main organs of the mice (heart, liver, spleen, lung, and kidney) were harvested for H&E staining.
Supplementary Material
Acknowledgments
Funding: This research is supported by the Singapore National Research Foundation Investigatorship (NRF-NRFI2018-03) and the Singapore Agency for Science, Technology and Research (A*STAR) AME IRG grant (A1883c0005). This work is also supported by the National Science Foundation of China (81702998 and 81701766). Author contributions: L.Z. conceived and designed the work, undertook the synthesis and characterization of the nanosystem, and wrote the manuscript. J.L. performed the cell experiments, established the mouse models, and performed the animal experiments. Y.S., L.Y., L.C., H.X., and H.P.T. also took part in the experiments. L.Q. and Y.W. helped and provided advice for the experimental details. J.P. assisted in the experiments, provided the mice and apparatus for the experiments, and supervised the in vivo experiments. Y.Z. supervised the project, administrated all participants, and wrote and revised the manuscript. All authors reviewed the manuscript and discussed the results. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaaz0575/DC1
Supplementary Materials and Methods
Fig. S1. Characterizations for the silica shell of calcined SCLMs.
Fig. S2. Determination of the 1O2 production by the bleaching of DPBF.
Fig. S3. Cytotoxicity assessment of C@SCLMs in the dark.
Fig. S4. Cellular apoptosis of A431 cells treated by different approaches.
Fig. S5. In vitro cell viability and the calculated therapeutic effect for different cancerous cell lines.
Fig. S6. Cell viability assay for human dermal fibroblasts.
Fig. S7. Effect of the mitochondrial protector to the combined treatment.
Fig. S8. Results of flow cytometric apoptosis analysis for A431 cells subjected to the treatment after the protection of MitoQ.
Fig. S9. Cell cycle analysis of A431 cells treated by TH588.
Fig. S10. Supplementary data for the in vivo animal experiments.
REFERENCES AND NOTES
- 1.van Straten D., Mashayekhi V., de Bruijn H. S., Oliveira S., Robinson D. J., Oncologic photodynamic therapy: Basic principles, current clinical status and future directions. Cancer 9, 54 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yun S. H., Kwok S. J. J., Light in diagnosis, therapy and surgery. Nat. Biomed. Eng. 1, 16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan W. P., Huang P., Chen X. Y., Overcoming the Achilles’ heel of photodynamic therapy. Chem. Soc. Rev. 45, 6488–6519 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Zhu X. J., Su Q. Q., Feng W., Li F. Y., Anti-stokes shift luminescent materials for bio-applications. Chem. Soc. Rev. 46, 1025–1039 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Abrahamse H., Hamblin M. R., New photosensitizers for photodynamic therapy. Biochem. J. 473, 347–364 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hague A., Faizi M. S. H., Rather J. A., Khan M. S., Next generation NIR fluorophores for tumor imaging and fluorescence-guided surgery: A review. Bioorg. Med. Chem. 25, 2017–2034 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Idris N. M., Gnanasammandhan M. K., Zhang J., Ho P. C., Mahendran R., Zhang Y., In vivo photodynamic therapy using upconversion nanoparticles as remote-controlled nanotransducers. Nat. Med. 18, 1580–1585 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Cheng L., Wang C., Liu Z., Upconversion nanoparticles and their composite nanostructures for biomedical imaging and cancer therapy. Nanoscale 5, 23–37 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Yuan H. X., Chong H., Wang B., Zhu C. L., Liu L. B., Yang Q., Lv F. T., Wang S., Chemical molecule-induced light-activated system for anticancer and antifungal activities. J. Am. Chem. Soc. 134, 13184–13187 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y. R., Pang L., Ma C., Tu Q., Zhang R., Saeed E., Mahmoud A., Wang J. Y., Small molecule-initiated light-activated semiconducting polymer dots: An integrated nanoplatform for targeted photodynamic therapy and imaging of cancer cells. Anal. Chem. 86, 3092–3099 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Xu X. Q., An H. J., Zhang D. L., Tao H., Dou Y., Li X. H., Huang J., Zhang J. X., A self-illuminating nanoparticle for inflammation imaging and cancer therapy. Sci. Adv. 5, eaat2953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Z. Z., Sun Q. Q., Pan W., Li N., Tang B., A near-infrared triggered nanophotosensitizer inducing domino effect on mitochondrial reactive oxygen species burst for cancer therapy. ACS Nano 9, 11064–11074 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Vankayala R., Kuo C.-L., Nuthalapati K., Chiang C.-S., Hwang K. C., Nucleus-targeting gold nanoclusters for simultaneous in vivo fluorescence imaging, gene delivery, and NIR-light activated photodynamic therapy. Adv. Funct. Mater. 25, 5934–5945 (2015). [Google Scholar]
- 14.Zhou Z. J., Song J. B., Nie L. M., Chen X. Y., Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy. Chem. Soc. Rev. 45, 6597–6626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J. N., Bu W. B., Shi J. L., Chemical design and synthesis of functionalized probes for imaging and treating tumor hypoxia. Chem. Rev. 117, 6160–6224 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Sheng Y., Nesbitt H., Callan B., Taylor M. A., Love M., McHale A. P., Callan J. F., Oxygen generating nanoparticles for improved photodynamic therapy of hypoxic tumours. J. Control. Release 264, 333–340 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Turan I. S., Yildiz D., Turksoy A., Gunaydin G., Akkaya E. U., A bifunctional photosensitizer for enhanced fractional photodynamic therapy: Singlet oxygen generation in the presence and absence of light. Angew. Chem. Int. Ed. 55, 2875–2878 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Tang W., Zhen Z. P., Wang M. Z., Wang H., Chuang Y. J., Zhang W. Z., Wang G. D., Todd T., Cowger T., Chen H. M., Liu L., Li Z. B., Xie J., Red blood cell-facilitated photodynamic therapy for cancer treatment. Adv. Funct. Mater. 26, 1757–1768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng Y. H., Cheng H., Jiang C. X., Qiu X. F., Wang K. K., Huan W., Yuan A., Wu J. H., Hu Y. Q., Perfluorocarbon nanoparticles enhance reactive oxygen levels and tumour growth inhibition in photodynamic therapy. Nat. Commun. 6, 8785 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allison R. R., Photodynamic therapy: Oncologic horizons. Future Oncol. 10, 123–124 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Oleinick N. L., Morris R. L., Belichenko T., The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. Sci. 1, 1–21 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Buytaert E., Dewaele M., Agostinis P., Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta Rev. Cancer 1776, 86–107 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Hirakawa K., Kawanishi S., Segawa H., Hirano T., Guanine-specific DNA oxidation photosensitized by the tetraphenylporphyrin P(V) complex. J. Porphyr. Phthalocyanines 10, 1285–1292 (2006). [Google Scholar]
- 24.Zhou X., Wang Y. P., Si J., Zhou R., Gan L., Di C. X., Xie Y., Zhang H., Laser controlled singlet oxygen generation in mitochondria to promote mitochondrial DNA replication in vitro. Sci. Rep. 5, 16925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakumi K., Furuichi M., Tsuzuki T., Kakuma T., Kawabata S., Maki H., Sekiguchi M., Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J. Biol. Chem. 268, 23524–23530 (1993). [PubMed] [Google Scholar]
- 26.Nakabeppu Y., Cellular levels of 8-oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int. J. Mol. Sci. 15, 12543–12557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gad H., Koolmeister T., Jemth A. S., Eshtad S., Jacques S. A., Strom C. E., Svensson L. M., Schultz N., Lundback T., Einarsdottir B. O., Saleh A., Gokturk C., Baranczewski P., Svensson R., Berntsson R. P. A., Gustafsson R., Stromberg K., Sanjiv K., Jacques-Cordonnier M. C., Desroses M., Gustavsson A. L., Olofsson R., Johansson F., Homan E. J., Loseva O., Brautigam L., Johansson L., Hoglund A., Hagenkort A., Pham T., Altun M., Gaugaz F. Z., Vikingsson S., Evers B., Henriksson M., Vallin K. S. A., Wallner O. A., Hammarstrom L. G. J., Wiita E., Almlof I., Kalderen C., Axelsson H., Djureinovic T., Puigvert J. C., Haggblad M., Jeppsson F., Martens U., Lundin C., Lundgren B., Granelli I., Jensen A. J., Artursson P., Nilsson J. A., Stenmark P., Scobie M., Berglund U. W., Helleday T., MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 508, 215–221 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Topal M. D., Baker M. S., DNA precursor pool: A significant target for N-methyl-N-nitrosourea in C3H-10T1/2 clone 8 cells. Proc. Natl. Acad. Sci. U.S.A. 79, 2211–2215 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laberge R. M., Adler D., DeMaria M., Mechtouf N., Teachenor R., Cardin G. B., Desprez P. Y., Campisi J., Rodier F., Mitochondrial DNA damage induces apoptosis in senescent cells. Cell Death Dis. 4, e727 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oka S., Ohno M., Tsuchimoto D., Sakumi K., Furuichi M., Nakabeppu Y., Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 27, 421–432 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao L. Z., Yuan W., Ang C. Y., Qu Q. Y., Dai Y., Gao Y., Luo Z., Wang J. G., Chen H., Li M. H., Li F., Zhao Y., Silica-polymer hybrid with self-assembled PEG corona excreted rapidly via a hepatobiliary route. Adv. Funct. Mater. 26, 3036–3047 (2016). [Google Scholar]
- 32.Niu D. C., Li Y. S., Shi J. L., Silica/organosilica cross-linked block copolymer micelles: A versatile theranostic platform. Chem. Soc. Rev. 46, 569–585 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Zhao L. Z., Yuan W., Tham H. P., Chen H. Z., Xing P. Y., Xiang H. J., Yao X., Qiu X. C., Dai Y., Zhu L. L., Li F. Y., Zhao Y. L., Fast-clearable nanocarriers conducting chemo/photothermal combination therapy to inhibit recurrence of malignant tumors. Small 13, 1700963 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Cai X. J., Jia X., Gao W., Zhang K., Ma M., Wang S. G., Zheng Y. Y., Shi J. L., Chen H. R., A versatile nanotheranostic agent for efficient dual-mode imaging guided synergistic chemo-thermal tumor therapy. Adv. Funct. Mater. 25, 2520–2529 (2015). [Google Scholar]
- 35.Tsuzuki T., Egashira A., Kura S., Analysis of MTH1 gene function in mice with targeted mutagenesis. Mutat. Res. 477, 71–78 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Nakabeppu Y., Regulation of intracellular localization of human MTH1, OGG1, and MYH proteins for repair of oxidative DNA damage. Progr. Nucleic Acid Res. Mol. Biol. 68, 75–94 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Kazak L., Reyes A., Holt I. J., Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 13, 659–671 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Cline S. D., Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta 1819, 979–991 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Houten B., Hunter S. E., Meyer J. N., Mitochondrial DNA damage induced autophagy, cell death, and disease. Front. Biosci. 21, 42–54 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Georgakilas A. G., Martin O. A., Bonner W. M., p21: A two-faced genome guardian. Trends Mol. Med. 23, 310–319 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Abbas T., Dutta A., p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 9, 400–414 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaaz0575/DC1
Supplementary Materials and Methods
Fig. S1. Characterizations for the silica shell of calcined SCLMs.
Fig. S2. Determination of the 1O2 production by the bleaching of DPBF.
Fig. S3. Cytotoxicity assessment of C@SCLMs in the dark.
Fig. S4. Cellular apoptosis of A431 cells treated by different approaches.
Fig. S5. In vitro cell viability and the calculated therapeutic effect for different cancerous cell lines.
Fig. S6. Cell viability assay for human dermal fibroblasts.
Fig. S7. Effect of the mitochondrial protector to the combined treatment.
Fig. S8. Results of flow cytometric apoptosis analysis for A431 cells subjected to the treatment after the protection of MitoQ.
Fig. S9. Cell cycle analysis of A431 cells treated by TH588.
Fig. S10. Supplementary data for the in vivo animal experiments.