

Hijacking the cell machinery to tether neurotoxins near their surface receptors (zombie scans) reveals sites of interaction.
Abstract
We show here that membrane-tethered toxins facilitate the biophysical study of the roles of toxin residues in K+ channel blockade to reveal two blocking mechanisms in the K+ channel pore. The structure of the sea anemone type I (SAK1) toxin HmK is determined by NMR. T-HmK residues are scanned by point mutation to map the toxin surface, and seven residues are identified to be critical to occlusion of the KcsA channel pore. T-HmK–Lys22 is shown to interact with K+ ions traversing the KcsA pore from the cytoplasm conferring voltage dependence on the toxin off rate, a classic mechanism that we observe as well with HmK in solution and for Kv1.3 channels. In contrast, two related SAK1 toxins, Hui1 and ShK, block KcsA and Kv1.3, respectively, via an arginine rather than the canonical lysine, when tethered and as free peptides.
INTRODUCTION
Protein toxins in animal venoms that act on ion channels are used in medical research, diagnosis, and therapy. Tethered toxins (T-toxins) were created on the basis of the discovery of lynx1, an endogenous modulator of nicotinic acetylcholine receptors tethered at the cell surface of mouse neurons via a glycosylphosphatidylinositol (GPI) anchor (1). Subsequently, μ-conotoxins and bungarotoxins were encoded with the GPI signal and processing sequences of lynx1; shown to gain surface expression, to diffuse freely on the cell surface to allow modulation of receptor function (2), and to silence voltage-gated calcium channel neurotransmission in vivo (3); and used to identify a peptide inhibitor of transient receptor potential ankyrin 1 (TRPA1) channels (4). Here, we extend the use of T-toxins to screen the role of each residue in three toxins and show that the approach can be used to characterize the biophysical parameters of toxin action, including binding affinity and kinetic parameters, to define the mechanism of action.
K+ channels that control a wide variety of cellular functions and peptide toxins that block channels by occluding the ion-conducting pore from the external side have been used to understand the structural and functional properties of the channels (5–7) and are in clinical development to treat neurodegenerative (8) and autoimmune diseases (9). Sea anemone type I (SAK1) toxins are potent K+ channel blockers (10) that we consider here.
Previously, we identified two KcsA K+ channel blockers by screening a phage display library of 1.5 million SAK1 peptides (11), a native peptide (HmK) and a combinatorial chimera of three native toxins (Hui1). HmK was found to be promiscuous, blocking the bacterial KcsA pore, human Kv1.3 (hKv1.3) and Kv1.2 channels, and Drosophila Shaker channels with half-maximal equilibrium inhibition constants (Ki) of 1 to 4 nM. In contrast, the chimera Hui1 blocked only KcsA and did so in an unexpected manner such that K+ ions traversing the pore from the cytosol side interacted with Hui1-Arg23 (rather than Hui1-Lys21), conferring voltage dependence to the toxin off rate from its external site, a “knock-off” effect classically due to the snug fit of a conserved toxin lysine in the outer K+ binding site in the conduction pathway (12–14). Here, we study the role of HmK residues in blockade by expanding the use of T-toxins as follows.
To determine binding affinity, T-toxin surface expression is quantified with single-molecule total internal reflection fluorescence (smTIRF) microscopy and an enzyme-linked immunosorbent assay (ELISA); T-HmK association rates are measured via release from tetraethylammonium (TEA) blockade, and dissociation rates are calculated from measured on rate and equilibrium affinity. As before, the KcsA pore is carried in the Shaker channel for ease of study (11), and scanning mutagenesis identifies seven T-HmK residues that alter free energy of blockade (ΔΔG) by more than 2 kcal/mol.
T-HmK and HmK peptide are shown to block KcsA with similar affinity (although the T-toxin has faster on and off rates) and to manifest the classical mechanism of inhibition with conserved Lys22, conferring voltage sensitivity to toxin dissociation. Next, we determine the structure of HmK by nuclear magnetic resonance (NMR) and mapping the seven sites including Lys22 to cluster, suggesting a toxin interaction surface with the channel.
T-HmK and HmK peptide also block hKv1.3 with similar affinity and voltage dependence via Lys22. In contrast, ShK (stichodactyla toxin) peptide (and T-ShK) reveal the Arg-dependent mechanism, in disagreement with prior mechanistic assertions (15) and similar to the chimeric toxin Hui1 (11). The fact that the two natural SAK1 toxins, HmK and ShK, block hKv1.3 with Lys and Arg mechanisms shows that both blocking strategies appear in nature and should be expected with other toxin-channel pairs.
RESULTS
T-HmK inhibition of KcsA-Shaker channels
HmK is a natural peptide in the sea anemone Heteractis magnifica (Fig. 1A) (16). It was one of the 150 SAK1 toxins that we used to design a phage display library comprising 1.5 million combinatorial SAK1 variants and was subsequently isolated by sorting the library on purified KcsA protein. HmK peptide inhibited KcsA-Shaker with a Ki ~1 nM (11).
Fig. 1. T-HmK and HmK peptide in solution block KcsA-Shaker channels similarly.
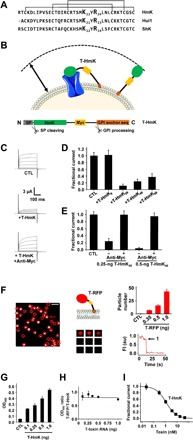
KcsA-Shaker was expressed in oocytes and studied by two-electrode voltage clamp (TEVC) to assess inhibition at equilibrium from a holding voltage of −80 mV with 300-ms test pulses to various voltages and a 5-s interpulse interval (n = 16 to 18 cells for each condition). Live oocytes were studied by ELISA and smTIRF microscopy as described in Materials and Methods. n = 12 oocytes for each condition. Values are means ± SEM. (A) Three SAK1 toxins: HmK, Hui1, and ShK peptide sequences aligned on their three disulfide bonds. (B) T-HmK was constructed as chimeric fusion protein with an N-terminal secretory signal sequence (SP; gray), the HmK sequence (green), a hydrophilic flexible linker with a c-Myc epitope tag (yellow), and a C-terminal GPI membrane anchor targeting sequence (orange). Same colors are used for marking T-HmK components in the above schematic. T-HmK is drawn free and binding to a potassium channel (blue) coexpressed in the same oocyte. The reaction volume that a T-HmK can visit (arrow) is determined by the lengths of the flexible peptide linker, GPI anchor, and the diameter of HmK toxin (Materials and Methods). (C) Representative raw current traces (steps from −80 to 60 mV) for KcsA-Shaker channels without (CTL) or with 0.25-ng T-HmK cRNA coinjection and after incubation with 2 μM anti-Myc to reverse the T-HmK blockade. (D) T-HmK linker variants (0.25 ng of cRNA) show different average KcsA-Shaker currents at 0 mV normalized to control, unblocked (CTL). T-HmK6 has 6 linker residues; T-HmK26 has 26 residues; T-HmK46 has 46 residues; and T-HmK66 has 66 residues. (E) Anti-Myc reverses blockade; KcsA-Shaker currents at 0 mV normalized to control, unblocked (CTL) with 0.25 or 0.5 ng of T-HmK46 cRNA. (F) Left: Single frame from a representative movie showing T-RFP (red fluorescent protein) on an oocyte membrane surface after 1-ng cRNA injection. Red circles mark single fluorescent spots. The white bar indicates 2 μm. Middle top: Schematic representation of T-RFP. Middle bottom: Montage of photobleaching time course of the single fluorescent particle indicated by the arrow in the left panel during continuous excitation to bleach the fluorophores. Every 12th frame is shown. Right bottom: Time course for photobleaching of RFP in the indicated particle, showing one stepwise change in fluorescence intensity (FI) (arrow). au, arbitrary units. Right top: The average number of single particles in 100-μm2 regions are 7 ± 1, 16 ± 2, or 43 ± 5 with 0.35, 0.5, or 1 ng of T-RFP cRNAs injected (n = 12 regions from three oocytes and four areas per oocytes for each condition). No fluorescent particles are observed in oocytes without T-RFP injection. (G) Increasing the amount of T-HmK cRNA injected increases surface expression on Xenopus oocytes. After 2 days of culture, surface expression of T-HmK was determined by ELISA using anti–Myc-HRP (horseradish peroxidase). The tethered control was without the toxin or c-Myc sequences (CTL). OD450, optical density at 450 nm. (H) The ELISA signals for T-RFP and T-HmK shows a stable relationship when the amount of injected cRNA increases. Two separate trials with six oocytes and five concentrations of T-RFP or T-HmK cRNA. (I) Concentration-response relationship for T-HmK (■) inhibition of KcsA-Shaker studied by TEVC and fit to the Hill relationship (Eq. 1). The determined Ki was 1.09 ± 0.01 nM with a Hill coefficient of 1.00 ± 0.01. n = 16 to 18 oocytes for each level of injected T-HmK cRNA. Coinjection of 0.35 ng of cRNA of T-HmK inhibited ~87% of the current, consistent with an effective T-HmK concentration of 7.4 ± 0.8 nM according to Eq. 1, similar to the ~8-nM surface concentration estimated by ELISA and smTIRF (table S1).
T-HmK was constructed by cloning the sequence of HmK into a T-toxin plasmid between a signal peptide sequence and a flexible linker with an embedded c-Myc epitope that was followed by a GPI targeting sequence (Fig. 1B and Materials and Methods). Complementary RNA (cRNA) for T-HmK was synthesized in vitro and coinjected with cRNA encoding KcsA-Shaker into Xenopus oocytes. The result was T-HmK expression on the extracellular leaflet of the oocyte membrane via the GPI anchor after natural cleavage of the signal peptide (Fig. 1B), as reported for other peptides by others (2, 4).
The inhibition of KcsA-Shaker currents was measured using two-electrode voltage clamp (TEVC) 2 days after injecting 1 ng of channel cRNA into oocytes, as before (11). Coinjection of 0.25 ng of T-HmK cRNA inhibited 76% of the KcsA-Shaker current, and 0.5 ng of T-HmK cRNA inhibited over 94% (Fig. 1, C and E). To assess the impact of the length of linker between HmK and the GPI anchor, we studied constructs with 6, 26, 46, and 66 intervening residues. Whereas, T-HmK with a six-residue linker showed no inhibition, suggesting that the toxin was unable to reach the pore, the other constructs were able to block effectively (Fig. 1D); the 46-residue linker was used for further studies. Demonstrating that T-HmK inhibition was reversible, application of 2 μM anti-Myc antibody was able to restore outward currents to the levels measured when no T-HmK was expressed by binding to the tether, presumably by reducing linker flexibility or creating a T-toxin–antibody complex too large to enter to the pore vestibule (Fig. 1, C and E).
Measuring the surface expression of T-HmK
To measure the affinity of a channel blocker at equilibrium (Ki), the concentration of blocker must be known (13). To estimate the concentration of T-HmK on the oocyte surface, we replaced the sequence encoding HmK in the T-HmK plasmid with the gene for a monomeric red fluorescent protein (RFP) to produce T-RFP; this allowed counting of individual T-RFP peptides on the cell surface with smTIRF and subsequent comparison of T-RFP and T-HmK expression by ELISA using the c-Myc epitope in each tethered peptide.
The number of T-RFP molecules on the oocyte surface was counted directly by smTIRF microscopy, as before (17). The average number of single particles per 100 μm2 was 43 ± 5 when cells were injected with 1 ng of T-RFP cRNA (Fig. 1F). Fluorescent particles were confirmed to be single T-RFP proteins by demonstration that continuous photobleaching yielded a single step decrease in fluorescence intensity for all 170 T-RFP particles studied (Fig. 1F). Furthermore, the distribution of T-RFP particles was shown to be uniform over the oocyte surface with the same average number of particles in four separate 100-μm2 areas of the animal and vegetal poles of three oocytes.
Next, ELISA signals at 450 nm for T-HmK (Fig. 1G) and T-RFP (Fig. 1H) using anti-Myc conjugated to horseradish peroxidase (anti-Myc-HRP) were shown to increase reproducibly as more cRNA was injected into the cells. Surface expression of the two T-peptides was related such that with equimolar cRNA injection, the optical signal with T-RFP was ~77% of that for T-HmK (Fig. 1H).
To calculate surface density, we estimated the surface area under study as follows. First, the average capacitance of oocytes (n = 5) was determined to be 198 ± 15 nF, consistent with prior studies (18). Using the reported specific membrane capacitance of 4 μF/cm2 for stage VI Xenopus oocytes (19), an average total oocyte surface area (S) of 4.95 × 106 ± 0.37 × 106 μm2 was calculated, a value similar to that reported by others (20). The reaction volume (V) available to T-HmK was then estimated as the areas between two spheres, determined by the average oocyte radius (r) and r + the length of toxin on the flexible peptide linker including the sugar chain of the GPI anchor (Materials and Methods). Thus, 0.5-ng T-RFP cRNA injection yields an average number of peptides on the oocyte surface of 8 × 105 ± 1 × 105, yielding an estimate of the effective T-HmK concentration of 17 ± 2 nM for injection of 0.5 ng of T-HmK cRNA (table S1). The same approach with injections of 0.35 and 1.0 ng of T-HmK cRNA yields an effective toxin concentration of 8 ± 1 and 47 ± 5 nM, respectively (table S1). To rule out a confounding effect of ion channel expression on surface level and localization of the T-toxin, we injected 1 ng of T-RFP or 1 ng of T-HmK with or without 1 ng of KcsA-Shaker, and neither the surface expression nor the observed distribution of tethered peptides was observed to be altered (table S2).
T-HmK equilibrium binding affinity
Since SAK1 toxin blockade of the K+ channel pore is a bimolecular interaction (11), the apparent Ki of T-HmK block of KcsA-Shaker channels can be calculated after 0.5-ng cRNA injection to be 1.1 ± 0.1 nM using the unblocked fraction of the current at equilibrium (Fun) and the effective toxin concentration [Tx] (Fig. 1E) according to Eq. 1
| (1) |
Since each point on a concentration-response curve for a bimolecular interaction follows the Hill relationship (Eq. 1) and can be used to describe the whole curve (21), we used various injected levels of T-HmK to rederive the apparent Ki and to validate the surface density estimate. Thus, when T-HmK cRNA was injected in amounts from 0.01 to 1 ng, inhibition increased from 2 to 98% (Fig. 1I). Fitting the concentration-response relationship yielded a Ki of 1.09 ± 0.01 nM with a Hill coefficient of 1.00 ± 0.01, as for the single dose of 0.5 ng.
The concentration-response curve for HmK peptide in solution was determined by measuring inhibition by 0.05 to 15 nM toxin applied in the bath solution to oocytes expressing KcsA-Shaker. Fitting the concentration-response relationship with Eq. 1 yielded a Ki of 0.94 ± 0.08 nM and a Hill coefficient of 1.03 ± 0.09 (fig. S1). Although, the free peptide and T-toxin were observed to block the pore with similar equilibrium affinity, we anticipated that the kinetics of blockade by toxin in solution and T-toxin might differ.
Rapid TEA unblock permits study of the kinetics of T-HmK inhibition
HmK applied in solution to oocytes expressing KcsA-Shaker allows direct measurement of the kinetics of current block and unblock through study of toxin wash in and wash off (11, 13). Toxin block for a bimolecular reaction can be described by the ratio of the first-order dissociation rate constant (koff) and the second-order association rate constant (kon) according to Eq. 2, and this is related to Fun according to Eq. 3 (13, 21)
| (2) |
| (3) |
Furthermore, kon and koff are related to the association constant (τon) and dissociation constant (τoff) derived from single exponential fits of the time courses for block and unblock by Eqs. 4 and 5 (13)
| (4) |
| (5) |
Thus, acute application and washout of 5 nM HmK gave τon = 126 ± 13 s, τoff = 714 ± 82 s, kon = 1.3 × 106 ± 0.1 × 106/Ms, and koff = 1.4 × 10−3 ± 0.2 × 10−3/s (Fig. 2A).
Fig. 2. Measuring the kinetics of T-HmK blockade.
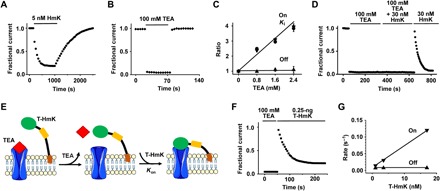
KcsA-Shaker was expressed in oocytes and studied by TEVC as in Fig. 1 with a test potential of 0 mV with 16 to 18 cells for each condition. Values are means ± SEM. (A) The time course for block and unblock of KcsA-Shaker on acute application (bar) of 5 nM peptide HmK and washout. Every 10th data point is shown normalized to the unblocked current magnitude. (B) The time course for block and unblock of KcsA-Shaker on acute application (bar) of 100 mM TEA and washout. (C) The blocking kinetics of 5 nM HmK peptide was studied at various concentrations of TEA in bath solution. Each measured parameter Ki (●), reciprocal on rate association constant (▼), and off rate dissociation constant (▲) were normalized to the value without TEA. Each point represents the mean ± SEM from six oocytes. (D) Unblock of KcsA-Shaker on washout of 100 mM TEA allows subsequent block by 30 nM free HmK peptide. (E) Schematic representation of TEA competition and T-HmK block after TEA wash off. TEA blocks KcsA-Shaker (100 mM) from the outside like HmK and prevents T-HmK binding (left). Unblock by TEA is rapid (τ ~1.3 s), so full current is recorded on washout before significant association of T-HmK due to its slow on rate (τ ~ 33 s with 0.25 ng of T-HmK cRNA) (middle). Thereafter, T-HmK relaxes to equilibrium blockade (right). (F) With T-HmK, TEA wash-off KcsA-Shaker shows full current recovery and slow binding of T-HmK, similar to that seen with free peptide. The T-HmK association constant determined by a single exponential fit of the block time course was τon = 33 ± 4 s. (G) Effect of T-HmK concentration on blocking kinetics of KcsA-Shaker. Three T-HmK concentrations were studied after coinjection of 0.1, 0.25, and 0.5 ng of T-HmK cRNA estimated to represent an effective surface concentration of ~1, 3.5, and 17 nM (table S1). The apparent first-order rate constants for association (on rate, ▼) and dissociation (off rate, ▲) are plotted as a function of effective T-HmK concentration. Increasing the concentration of T-HmK from 1 to 17 nM increased the on rate of T-HmK from ~ 0.014 to 0.119 s−1, whereas the off rate was maintained at ~7.3 × 10−3 ± 1.1 × 10−3 s−1.
In contrast, when KcsA-Shaker channels are expressed with T-HmK, they are blocked at baseline because the toxin is fixed to the membrane by its tether. A strategy was required to study unblocked channels and, to this end, we used TEA, an organic cation blocker of K+ channel pores to displace the T-HmK from the pores and to study T-toxin rebinding, as follows.
TEA block of KcsA-Shaker has a Ki of ~0.8 mM (22). At 100 mM, TEA block is complete and wash-in and wash-off kinetics are rapid (Fig. 2B). Association and dissociation of TEA are complete in less than one recording sweep at the 5-s interval used to study HmK block of KcsA-Shaker. This is because the dissociation of TEA (τoff ~1.3 s) is two orders of magnitude faster than the association of 5 nM HmK (Fig. 2, A and B). Supporting the expectation that TEA and HmK bind at overlapping pore sites, the Ki of free HmK increases linearly as TEA concentration increases, entirely as a result of a decrease in τon while τoff is unaffected (Fig. 2C). Further evidence that TEA and HmK both bind in the external vestibule, mutation of the KcsA-Shaker pore residue Tyr82 to Val, decreased their equilibrium affinities by ~200- and ~550-fold, respectively (fig. S2, A and B). These findings are reminiscent of competition studies with TEA and scorpion α-KTx peptides such as CTX (13, 23) and the effects of homologous mutations (24).
Incubation with 100 mM TEA suppresses block of KcsA-Shaker by free HmK peptide because TEA moves into the pore rapidly whenever HmK is unbound. Thus, no HmK inhibition is apparent when cells are incubated with 100 mM TEA and HmK, as reflected by the rapid, full current recovery on TEA wash off and subsequent slow association of HmK (Fig. 2D). Association of 30 nM HmK after TEA wash off showed τon = 27 ± 3 s and a kon = 1.2 × 106 ± 0.1 × 106/Ms, the same as determined in the absence of prior TEA blockade (Fig. 2, A and D, and Table 1).
Table 1. Blocking parameters of KcsA-Shaker by T-HmK and HmK peptide variants.
KcsA-Shaker inhibition at equilibrium (Ki ± SEM) was determined as described in Figs. 1 and 2; the kinetic parameters were assessed at 0 mV. n = 16 to 18 oocytes for T-toxins, and n = 6 oocytes for peptides.
| Toxins | Ki (nM) | kon (1/Ms) | koff (1/s) |
| T-HmK | 1.09 ± 0.01 | 6.6 × 106 ± 0.8 × 106 | 7.3 × 10−3 ± 0.9 × 10−3 |
| T-HmK–Lys22Asn | 35 ± 4 | 3.1 × 106 ± 0.3 × 106 | 1.1 × 10−1 ± 0.1 × 10−1 |
| T-HmK–Tyr23Ala | 28 ± 4 | 6.1 × 106 ± 0.8 × 106 | 1.7 × 10−1 ± 0.2 × 10−1 |
| T-HmK–Arg24Gln | 0.91 ± 0.11 | 6.9 × 106 ± 0.7 × 106 | 6.3 × 10−3 ± 0.8 × 10−3 |
| Peptide HmK | 0.94 ± 0.08 | 1.3 × 106 ± 0.2 × 106 | 1.4 × 10−3 ± 0.3 × 10−3 |
| Peptide HmK-Lys22Asn | 225 ± 37 | 3.8 × 105 ± 0.5 × 105 | 1.0 × 10−1 ± 0.2 × 10−1 |
| Peptide HmK-Tyr23Ala | 162 ± 13 | 9.3 × 105 ± 1.2 × 105 | 1.5 × 10−1 ± 0.1 × 10−1 |
Block by TEA was also used to determine the association rate of T-HmK (Fig. 2E). With 100 mM TEA, T-HmK was displaced from channel pore, and when TEA was washed off, the association of T-HmK could be observed (Fig. 2F). The association of T-HmK determined from single exponential fits with 0.25 ng of T-HmK cRNA was τon = 33 ± 4 s. Using measured Fun and estimated surface concentration (table S1), we calculated the binding kinetics of T-HmK to be kon = 6.6 × 106 ± 0.8 × 106/Ms and koff = 7.3 × 10−3 ± 0.9 × 10−3/s (Eqs. 2 to 5). Blocking kinetics of T-HmK at lower and higher levels of expression showed that the association of T-HmK was dependent on its effective surface concentration in a linear manner, whereas dissociation was insensitive to concentration, findings consistent with a simple bimolecular interaction for the T-toxin, as observed for HmK in solution (Fig. 2G); while the results demonstrate the utility of Eqs. 1 to 3, it is important to recognize that the values are approximations using relationships derived for evaluation of mass action and random diffusion.
Comparing the blocking kinetics of KcsA-Shaker by the tethered and free toxin revealed that the T-HmK on rate was ~5.1-fold faster than for HmK peptide in solution when they were studied at the same estimated concentration (Table 1); the tether also increased the T-HmK off rate so that it was ~5.2-fold faster than free peptide. Thus, the observation that free HmK and T-HmK have similar apparent equilibrium affinity can now be explained by the tether increasing both association and dissociation rates in an offsetting manner.
Ready identification of important toxin residues using T-HmK variants
HmK has 35 residues, 6 in disulfide bonds that maintain the toxin backbone fold (Fig. 1A). To identify the functional residues of HmK that physically interact with the KcsA-Shaker pore, we produced 29 T-HmK variants by individual mutation of the non-Cys residues (Fig. 3A). Injection of 0.5 ng of wild-type (WT) T-HmK cRNA inhibited ~94% of KcsA-Shaker current, while seven of the T-HmK variants blocked less than 38% (Fig. 3A, top). None of the 29 mutants showed a significant change in surface expression using ELISA, ruling out decreased expression as a cause for altered activity (Fig. 3A, bottom). Thus, seven HmK residues change the free energy of toxin blocking (ΔΔG) by more than 2 kcal/mol (Kvariant/KWT, ~26-fold), using Eqs. 6 and 7, where R is the gas constant and T is the temperature in Kelvin
| (6) |
| (7) |
Fig. 3. T-HmK screening mutagenesis and demonstration that Lys22 mediates voltage-dependent block of KcsA-Shaker channels.
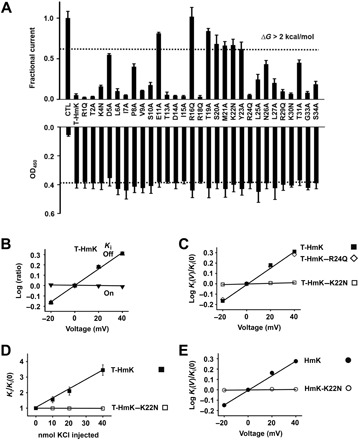
KcsA-Shaker was expressed in oocytes, and inhibition and surface expression of T-HmK and point variants were studied by TEVC or ELISA after coinjection of 0.5 ng of cRNAs. The equilibrium inhibition and kinetic blocking parameters of T-HmK after 0.25-ng cRNA coinjection and peptide HmK were studied by TEVC as in Fig. 2. n = 18 oocytes for each T-HmK concentration, and n = 6 for each level of HmK peptide. Values are means ± SEM. Some error bars are smaller than symbols. (A) Top: KcsA-Shaker currents at 0 mV measured as in Fig. 1E with T-HmK variants normalized to the unblocked condition (CTL). n = 16 to 18 cells for each variant. Bottom: ELISA performed as in Fig. 1G with T-HmK variants. Control was without the toxin or c-Myc sequences (CTL). n = 12 cells for each condition. (B) Effect of voltage on T-HmK blocking kinetics. The on rate is insensitive to voltage, whereas the off rate and, thus, the Ki are responsive, consistent with trans-ion knockoff. Each parameter was measured with test steps from −20 to 40 mV and normalized to its value at 0 mV, ▼, kon; ▲, koff; ■, Ki. Ki was determined from the fraction of unblocked current at equilibrium (Fun) according to Eq. 1. The association constant was determined by single exponential fits to the time course for T-HmK inhibition after TEA instant wash off. The dissociation rate constant was determined from Ki and kon according to Eq. 2. (C) Effect of voltage on blockade by T-HmK mutants. Neutralizing Lys22 (K22N) but not Arg24 (R24Q) removes the voltage dependence of toxin blockade. Ki for each variant was determined from −20 to 40 mV on the basis of the fraction of unblocked current at equilibrium (Fun) and plotted as a ratio to the value at 0 mV. (D) Raising intracellular KCl concentration speeds up the off rate of WT T-HmK but not when HmK-Lys22 is neutralized by mutation. Microinjection of 40 nl of KCl (0.25 to 1 M) increased the dissociation rate of T-HmK, whereas T-HmK–K22N was insensitive. Oocyte solute space has been estimated to be ~400 nl, and intracellular potassium concentration has been estimated to be ~90 mM; therefore, a 20-nmol KCl microinjection should raise intracellular potassium by ~50 to 140 mM, leading to an 11-mV shift in EK and a 50% increase in outward current, close to the observed values here of 10 ± 2 mV and a 45 ± 9% (n = 6 oocytes), as described before (11, 38). The new Ki were stable during the period of recording, that is, from 30 to 120 min after microinjection. (E) HmK peptides show the same Lys22 dependence of voltage blockade of KcsA-Shaker as T-HmK. Ki were determined from −20 to 40 mV on the basis of the fraction of unblocked current at equilibrium (Fun) and plotted as a ratio to the value at 0 mV. Association and dissociation time constants were determined by single exponential fits to the time course for block or unblock on acute application or washout of 5 nM HmK or 500 nM HmK-K22N.
The mutation that had the most marked effect was HmK-Arg16Gln, which fully eliminates the toxin block. The other six mutations that change the ΔΔG of block by >2 kcal/mol were E11A, T19A, S20A, M21A, K22N, and Y23A (Fig. 3A, top), suggesting that these residues may mediate direct interaction with KcsA-Shaker pore.
T-HmK and HmK peptide occlude the KcsA-Shaker pore via Lys22
To classify the blocking mechanism of T-HmK, we studied the basis for affinity changes at the “dyad residues” (Lys and Tyr/Phe) that have been proposed to be important for high-affinity binding of α-KTx toxins and SAK1 toxins (25), HmK-Lys22 and HmK-Tyr23 (Fig. 3A, top). Thus, compared to WT T-HmK, T-HmK–Lys22Asn showed a ~30-fold decrease in affinity due to a ~2-fold slower association rate and ~15-fold faster dissociation rate, and T-HmK-Tyr23Ala showed an ~25-fold decrease in affinity primarily due to an ~23-fold faster dissociation rate (Table 1). In contrast, neutralization of Arg24 to produce T-HmK–Arg24Gln did not significantly alter toxin inhibition (Fig. 3A, top), surface expression (Fig. 3A, bottom), or kinetics of inhibition compared to WT T-HmK (Table 1). The fact that the changes in affinity were largely due to changes in dissociation suggested that the mutations did not modify toxin folding.
The blocking mechanism of SAK1 toxins on K+ channels had been assumed to be uniformly based on association of the conserved dyad Lys in the pore to occlude the ion conduction pathway (15), like α-KTx toxins (12–14), endowing toxin dissociation rate with dependence on membrane voltage due to electrostatic repulsion between K+ in the conduction pathway and the pore occluding residue. Thus, we were surprised when Arg23 in the chimeric SAK1 toxin Hui1 mediated voltage dependence to the toxin off rate (11). To assess the basis for voltage dependence of inhibition by T-HmK and HmK peptide, we compared WT toxin and two neutralization variants, Lys22Asn and Arg24Gln.
As expected, the dissociation rate of T-HmK from KcsA-Shaker was sensitive to voltage, showing an effective valence (zδ) of ∼0.44, whereas the toxin on rate was insensitive to voltage (Fig. 3B). Similarly, the voltage dependence of inhibition of T-HmK–Arg24Gln showed a zδ of ~0.42 based on changes in off rate (Fig. 3C). In contrast, the T-HmK–Lys22Asn variant eliminated the voltage dependence of T-HmK (Fig. 3C).
Consistent with the interaction of HmK-Lys22 with K+ ions traversing the pore, increasing the level of intracellular K+ increased the off rate of WT T-HmK, a trans-ion effect, whereas T-HmK–Lys22Asn was insensitive to changes in the cytoplasmic K+ concentration (Fig. 3D). Similar to T-HmK, the dissociation rate of HmK was sensitive to voltage, showing an zδ of ∼0.40 (Fig. 3E), and neutralization of Lys22 decreased toxin inhibition (~240-fold; Table 1) and eliminated the voltage dependence of HmK dissociation rate (Fig. 3E).
Native SAK1 toxins block hKv1.3 channel by two mechanisms
hKv1.3 channels are required for human T lymphocyte function and a validated target for therapeutic modulation of the immune system (26). We therefore sought to validate the T-toxin strategy as a means to study the mechanism of voltage dependence of inhibition with the human channel with two natural SAK1 toxins, HmK and ShK.
Injection of 0.5 ng of cRNA for T-HmK inhibited ~85% of hKv1.3 current, consistent with an apparent Ki of 3.0 ± 0.4 nM, while T-HmK–Lys22Asn and T-HmK–Arg24Gln blocked ~2 and ~68%, decreasing the apparent Ki by ~230- and 3-fold, respectively (Fig. 4A and table S3). The off rate of T-HmK from hKv1.3 was sensitive to voltage with an zδ of ∼0.61 (Fig. 4B), and T-HmK–Arg24Gln showed the same zδ as the WT toxin (Fig. 4B). In contrast, neutralizing Lys22 in T-HmK–Lys22Asn eliminated the voltage dependence of toxin off rate consistent with Lys22-dependent pore blockade. Similarly, HmK peptide inhibited hKv1.3 with a Ki of 3.1 ± 0.2 nM, the dissociation rate of the peptide was sensitive to voltage with zδ of ∼0.6, and neutralization of Lys22 decreased toxin inhibition ~730-fold, and it eliminated the voltage dependence of HmK dissociation (Fig. 4D and table S3).
Fig. 4. Voltage-dependent block of hKv1.3 channels is mediated by Lys22 of HmK but by Arg24 of ShK.
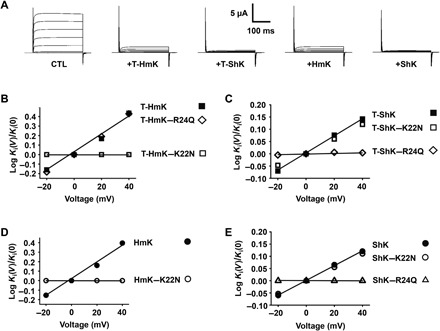
hKv1.3 was expressed in oocytes and studied by TEVC to assess equilibrium inhibition and kinetic blocking parameters of T-HmK after 0.5-ng cRNA coinjection, T-ShK after 0.1-ng cRNA coinjection, and peptide of ShK by the protocol described in Fig. 2. The Ki for T-HmK, T-ShK, peptide ShK, and their variants were determined from −20 to 40 mV on the basis of the fraction of unblocked current at equilibrium (Fun) and plotted as a ratio to the value at 0 mV. n = 18 oocytes for each condition of T-toxins, and n = 6 oocytes for each condition of peptide toxins. Values are means ± SEM. Some error bars are smaller than symbols. (A) Representative raw current traces for hKv1.3 (steps from −80 to 60 mV) without blocker (CTL), with 0.5-ng T-HmK cRNA coinjection, with 0.1-ng T-ShK cRNA coinjection, and after application of HmK or ShK peptides at 20 or 1 nM, respectively. (B) Neutralizing Lys22 (K22N) but not Arg24 (R24Q) removes the voltage dependence of T-HmK blockade of hKv1.3 channel. (C) Neutralizing Arg24 (R24Q) but not Lys22 (K22N) removes the voltage dependence of T-ShK blockade of hKv1.3 channel. (D) HmK peptides show the same Lys22 dependence of voltage blockade of hKv1.3 as T-HmK, parameters calculated as in Fig. 3E on acute application and washout of 20 nM HmK and 5 μM HmK-K22N. (E) ShK peptides show the same Arg24 dependence of voltage blockade of hKv1.3 as T-ShK, parameters calculated as in Fig. 3E on acute application and washout of 1 nM ShK, 5 nM ShK-K22N, and 1 nM ShK-R24Q.
The chimeric SAK1 toxin Hui1 blocks KcsA-Shaker but not hKv1.3, and therefore, we chose to study ShK, a potent toxin blocker of hKv1.3 that uses the same disulfide bond scaffold as HmK and Hui1 (11). While it has been long assumed that ShK uses Lys22 to occlude the hKv1.3 pore (15), this has been a matter of controversy (27).
Injection of 0.1 ng of cRNA for T-ShK inhibited ~91% of hKv1.3 current, consistent with an apparent Ki of 0.098 ± 0.012 nM, while T-ShK–Lys22Asn and T-ShK–Arg24Gln blocked ~70 and ~93% of the current, with apparent Ki of 0.42 ± 0.05 and 0.075 ± 0.010 nM, respectively (Fig. 4A and table S3). The off rate of T-ShK was sensitive to voltage with an zδ of ∼0.21 (Fig. 4C), and T-ShK–Lys22Asn showed the same zδ (Fig. 4C). In contrast, T-ShK–Arg24Gln eliminated the voltage dependence of toxin off rate consistent with Arg24-dependent pore blockade (Fig. 4C).
Similarly, ShK peptide inhibited hKv1.3 with a Ki of 0.05 ± 0.01 nM; the dissociation rate of the peptide was sensitive to voltage with zδ of ∼0.20 (Fig. 4E), and while neutralization of Lys22 decreased toxin inhibition ~24-fold (table S3), the voltage dependence was the same as WT (Fig. 4E). In contrast, ShK-Arg24Gln eliminated the voltage dependence of ShK dissociation rate (Fig. 4E).
The unexpected role for Hui1-Arg23 was confirmed using T-toxins to study KcsA-Shaker. Thus, injection of 0.25 ng of cRNA of T-Hui1 with KcsA-Shaker inhibited ~82% of the current, consistent with an apparent Ki of 0.77 ± 0.09 nM. Whereas WT T-Hui1 block of KcsA-Shaker was sensitive to voltage with a zδ of ∼0.32, similar to the zδ of peptide Hui1 (zδ ~0.3) that we previously reported (11), and Lys neutralization in T-Hui1–Lys21Asn did not alter the zδ (~0.31), T-Hui1–Arg23Ala eliminated the voltage dependence of toxin blockade (fig. S3B). Notably, T-Hui1–Lys21Asn and T-Hui1–Arg23Ala inhibited ~32 and ~89% of the current, decreasing the apparent Ki by ~10- and 0.6-fold, respectively, demonstrating that the Lys has an important role in binding affinity even if it is no longer destabilized by ions traversing the pore (fig. S3A).
NMR structure of HmK and the toxin interaction surface
To assess the structural basis for toxin inhibition, we determined the three-dimensional (3D) solution structure of HmK using standard 2D NMR measurements (Materials and Methods). A total of 328 distance and dihedral angle constraints were used to derive an ensemble of 20 low-energy structures exhibiting a backbone (all heavy atoms) root mean square deviation of 0.31 Å (Fig. 5A and tables S4 to S6). Superposition of the structure of HmK with ShK [Protein Data Bank (PDB) 4LFQ] or with Hui1 (PDB 2N6B) showed that HmK retained a well-defined SAK1 toxin scaffold (Fig. 5B). Specifically, HmK residues 14 to 18 and 22 to 26 form two short perpendicular α helixes, adopting the typical SAK1 helix-kink-helix conformation, and the side chains of important residues (i.e., Met21, Lys22, Tyr23, and Arg24) adopted similar orientations to those of homologous residues in ShK and Hui1 (fig. S4).
Fig. 5. HmK structure, interaction surface, and binding orientations.
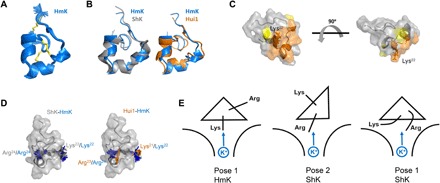
The HmK structure was determined from 328 distance and dihedral constraints derived from homo- and heteronuclear 2D NMR spectra (see Materials and Methods). (A) HmK structure (PDB accession code 6EI7). The 25 lowest-energy structures obtained from distance geometry/simulated annealing determination are shown. (B) Superposition of HmK (blue) with the crystal structure of ShK (4LFQ; gray) or the NMR structure of Hui1 (2N6B; orange) based on alignment of HmK/ShK residues 3 to 35 (Hui1, 2 to 34) shows that HmK retains an SAK1 scaffold. (C) Interaction surface of HmK mapped with T-toxin scanning as described in Fig. 3A. Orange indicates residues forming the primary toxin interaction surface (mutation caused ΔΔG > 2 kcal/mol). Yellow indicates Pro8 and Asn26 (mutation caused ΔΔG > 1.3 kcal/mol). The side chain of Lys22 is shown in dark orange. The two structures are separated by a rotation of 90o. (D) Left: Superposition of ShK and HmK highlighting ShK-Arg24 (gray)/HmK-Lys22 (blue) that mediate voltage dependence and ShK-Lys22 (gray)/HmK-Arg24 (blue) that do not. Right: Superposition of Hui1 and HmK highlighting Hui1-Arg23 (orange)/HmK-Lys22 (blue) that mediate voltage dependence and Hui1-Lys21 (orange)/HmK-Arg24 (blue) that do not. (E) Cartoon suggesting SAK1 toxin binding orientations in the hKv1.3 external vestibule with Lys or Arg near the conduction pore. Left: Pose 1 for HmK with Lys22 toward K+ in the pore. Middle: Pose 2 for ShK with Arg24 near K+ in the pore. Right: Pose 1 for ShK with Arg24 flexing to interact with K+ in the conduction pore.
The high-impact HmK residues identified by T-toxin scanning were mapped onto the HmK structure (Fig. 5C, orange). The seven residues (Glu11, Arg16, Thr19, Ser20, Met21, Lys22, and Tyr23) responsible for a change of ΔΔG > 2 kcal/mol in T-HmK blockade lie together on a continuous surface of the toxin presumed to make close contact with KcsA pore. Close to this primary surface are residues Pro8 and Asn26 (yellow); mutation of either to alanine reduced the affinity of T-HmK ~10-fold. A similar effect of two other residues, Asp5 and Thr31, upon toxin affinity should probably be attributed to their proposed contribution to the conformational stability of SAK1 toxins (28, 29). The HmK structure, similar to structures of the other toxins, appeared to confirm this proposition since both residues are marginally exposed to solvent and are oriented directly away from the interaction surface noted above. Given that the mutation at Lys30 does not affect the affinity, ruling out the formation of an Asp5-Lys30 salt bridge, these two side chains are presumably involved in one (or more) fold-stabilizing hydrogen bonding interactions.
Of note are the differing orientations of the two residues implicated in SAK1 toxin inhibition of channels by direct interaction with ions traversing the pore, Lys22 (in HmK) and Arg24/Arg23 (in ShK and Hui1, respectively). Located on opposite faces of the second helix, the side chains of these two residues are oriented offset by 120° to 160° in the various low-energy structures (Fig. 5D). This implies that the two toxin orientations suggested by our findings are separated by a significant rotation around the long axis of the toxins and therefore may involve quite different contacts with the channel pore.
DISCUSSION
Measuring the affinity and kinetics of blockade with T-toxins
Here, we use the T-toxin strategy, previously used to identify toxin-receptor partnerships, to facilitate screening of the energetic and mechanistic roles of individual toxin residues. The equilibrium affinities of T-HmK and HmK peptide for KcsA-Shaker were similar, showing Ki of 1.09 ± 0.01 and 0.94 ± 0.08 nM, respectively; because T-toxin association and dissociation were both about fivefold faster than for HmK peptide, the apparent equilibrium affinity of T-HmK was similar to that for the free peptide (Table 1).
As expected, the second-order association rate constant of peptide block depends on the toxin concentration (13, 21). Thus, just as Hoshi et al. (30) observed on shortening and lengthening the chain length separating the N-terminal inactivation ball peptide from its inner pore receptor site in Shaker K+ channels, we find that the short linker in T-HmK6 yields no blockade as if the toxin cannot reach its pore site and increasing the linker length in T-HmK26, T-HmK46, and T-HmK66 decreases blockade, consistent with a decrease in the effective concentration of T-HmK linked to the oocyte membrane surface, slowing the apparent toxin association constant from ~15 to 57 s, consistent with increased reaction volumes from ~7.4 × 10−11 to 1.3 × 10−10 liters (Fig. 1, B to D).
The tether accelerates the association of T-HmK (~5-fold), T-HmK–K22N (~8-fold), and T-HmK–Y23A (~6.5-fold) compared to the homologous peptides, perhaps, by favoring proximity of the toxin to the receptor within the reaction space (Table 1). Moreover, the effect of mutation on on rate appear to be preserved in T-toxins such that the approximately twofold slowing of T-HmK–K22N compared to T-HmK is similar when HmK-K22N and HmK peptides are compared (approximately threefold).
In contrast, the approximately fivefold increase in dissociation rate for T-HmK relative to free peptide was not observed for T-HmK–K22N and T-HmK–Y23A, perhaps because the mutant toxins had such short blocking dwell times. Tethering improved the affinity of the two weakly binding toxins by improving their on rate without speeding their off rates (Table 1). For example, T-HmK–K22N shows ~32-fold lower affinity than WT T-HmK, whereas HmK-K22N peptide shows ~240-fold affinity decrease compared to HmK peptide. This suggests that T-toxins may prove more useful than their soluble analogs for studies of low-affinity toxin-target pairs.
It is important to highlight that the block parameters that we report are estimates. Equations 1 to 3 describe mass action for molecules subject to random diffusion and the long flexible linker, and GPI anchor was observed to speed both toxin association and dissociation compared to the free peptide. Furthermore, invagination domains of the oocyte surface (31) and unrecognized toxin conformational variation beyond the encoded point change in a residue side chain may influence the behavior of free toxins and T-toxins differently. Conversely, the correlation of expression level with blockade, as well as the similar mechanism of action of free toxins and T-toxins, supports the utility of T-toxins to compare the roles of individual residues and approximate the energetics of binding.
Rapid identification of toxin high-impact residues
Although different in their binding kinetics, we found T-HmK and HmK peptide block KcsA-Shaker by the same pore occluding mechanism wherein Lys22 mediates voltage-dependent dissociation due to interaction with trans-ions in the conduction pore. Therefore, neutralization of Lys22 abolishes the voltage dependence of block in both cases, and the zδ of voltage-dependent inhibition was the same for T-HmK and HmK peptide, supporting a similar binding position for Lys22 in the two toxin forms in the KcsA-Shaker pore. Because variants were expressed on the cell surface at similar levels as WT T-HmK, scanning with alanine point mutations provided a rapid and efficient process, avoiding the time and expense of producing peptides and the risks attendant with handling, such as difficulty of peptide synthesis and folding in vitro (32), residue modification such as methionine oxidation, and peptide degradation during storage (33). The result was a facile mapping of the seven most impactful residues in T-HmK on block of KcsA-Shaker, pointing to the binding interface upon the NMR-derived HmK structure. The structure also distinguished residues contributing to structural stabilization of the toxin. Last, T-toxins conveniently highlighted the dual inhibition mode used by SAK1 toxins, with ShK and Hui1 dependent on residue Arg24 for channel blocking and HmK requiring residue Lys22 for this purpose, a clear indication that two distinct binding modes to the channel are at work.
We find T-toxins to be very useful for structure-function studies for at least four reasons. First, they afford experimental flexibility, especially in the iterative study of peptides at various positions where examining each successive hypothesis otherwise requires synthesis and purification of peptides rather than simple point mutations. Thus, scanning 29 HmK variants here took weeks using T-toxin point mutants, whereas peptide synthesis would have taken months at a cost of ~30,000 USD. T-toxins also circumvent the problem that some peptides degrade on storage and need to be refolded or synthesized, whereas cDNA remains stable. Last, T-toxins allow studies of very low affinity variants that are hard or impossible to apply at sufficient levels in peptide form. Thus, we now use T-toxins to scan and then confirm inferred mechanisms with a few required peptides.
Two SAK1 toxin–blocking mechanisms
The scorpion toxin CTX blocks K+ channels by positioning Lys27 in the pore to impede passage of ions so that neutralization of the residue by mutation abolishes the dependence of toxin dissociation on voltage (12, 13). This mechanism was visualized in the crystal structure of CTX and Kv1.2 showing the ε-amino group of Lys27 near the first (S1) K+ binding site in the conduction pore (14). Similarly, we observe here that the SAK1 toxin HmK interacts with trans-ions passing through either KcsA-Shaker or hKv1.3 via Lys22 in tethered and peptide form (Figs. 3 and 4). In contrast, we previously observed the chimeric SAK1 toxin peptide Hui1 to interact with trans-ions in the KcsA-Shaker via Arg23, reflecting a new blocking mechanism (11), a finding that we recapitulate here using T-Hui1 and variants (fig. S3); Hui1 does not inhibit hKv1.3 (11). Here, we find that ShK blocks hKv1.3 by a similar, Arg-dependent mechanism (Fig. 4).
ShK blocks Kv1.3 channel in T lymphocytes with picomolar affinity (10). Its synthetic analog ShK-186 is in development for treatment of autoimmune diseases (9). Weak energetic coupling of ShK-Lys22 and a Tyr residue in the selectivity filter of Kv1.3 by mutant cycle analysis was the basis for hypothesizing occlusion of the pore by the residue (34). However, weak coupling is also consistent with interaction through an intervening residue (14), and molecular dynamic simulations have modeled ShK-Lys22 in the external vestibule of Kv1.3 away from the narrow filter region of the pore (27). In addition, consistent with localization of ShK-Lys22 in the outer pore vestibule rather than in the filter of a Ca2+-activated potassium channel, mutant cycle analysis showed coupling of K+ ions in the external bath solution and the toxin residue (15).
Because neutralization of ShK-Arg24, but not ShK-Lys22, abolishes the voltage dependence of hKv1.3 blockade, we infer Arg24 to be closer to the selectivity filter than Lys22. Given that potassium channel pores are conserved (7) and, therefore, the first K+ ion binding site in the selectivity filter (S1) in hKv1.3 is likely to be similar in size and geometry to that in the Kv1.2 chimera channel studied by Banerjee et al. (14), we conclude that ShK-Arg24 is too large to fit snugly into the site, suggesting that the residue is near but not deeply within the conduction pathway. Consistent with a weaker interaction of Arg than Lys with ions in the hKv1.3 permeation pathway, the voltage dependence of ShK mediated by Arg24 (zδ of ∼0.2) was about one-third of that of HmK mediated by Lys22 (zδ of ∼0.6). Similarly, it seems reasonable to assume that a better fit of Lys in the pore than Arg rationalizes the greater loss of binding energy with changes of HmK-Lys22 compared to ShK-Arg24 and Hui1-Arg23.
HmK and ShK share a SAK1 scaffold structure, and both block hKv1.3 pore; however, the voltage dependence of channel blockade is dependent on Lys22 in HmK and Arg24 in ShK, two residues whose side chains are shown by their NMR solution structures to be offset by 120° to 160° (Fig. 5D, left). Similarly, HmK and Hui1 also share the SAK1 scaffold in our 3D NMR structures, yet it is HmK-Lys22 and Hui1-Arg23 that mediate toxin destabilization by K+ ions traversing the KcsA-Shaker pore (Fig. 5D, right). Consistent with a more important role for HmK-Lys22 in blocking hKv1.3 than ShK-Lys22, neutralization of the two residues decreases affinity ~730- and ~24-fold, respectively.
We favor the idea that despite their strong structural similarities, the toxins adopt two different binding orientations in the pores (Fig. 5E, left and middle). The most straightforward notion is that variations in toxin sequences, presumably in the N-terminal region where most differences reside (11), are sufficient to favor different toxin-specific binding orientations that place the Arg or Lys closest to the conduction pathway. Alternatively, the structures of the bound toxins may diverge from the canonical SAK1 scaffold that we observe in solution based on the local energy landscape in the channel pores, implying that the toxins are somewhat more flexible than we assume when treating them as rigid “molecular calipers” (6). Whereas significant conformational flexibility may be afforded by the two positively charged side chains, for example, allowing the Arg to flex toward the pore to interact with K+ ions (Fig. 5E, right), replacing the Lys, the distances involved suggest some change in binding orientation, or the toxin 3D structure is required as well.
Peptides that occlude K+ channel pores can fit like a key in a lock in the ion conduction pathway so that positively charged, ε-amino group of a toxin lysine competes with trans-K+ ions leaving the pore, thereby conferring voltage dependence to inhibition (6, 14). A number of recent observations are suggesting variations on this mechanism. Here, as in a prior report (11), we demonstrate that SAK1 toxins can use a second binding orientation in some channels so that an Arg confers voltage dependence rather than the nearby canonical Lys. Recently, high-speed atomic force microscopy was used to show that the affinity of the peptide AgTx2 increases in the KcsA pore over time, consistent with an induced fit binding model (35), and another group demonstrated that CTX can wobble between several bound conformations in Shaker channels so that the pore occluding Lys detaches sufficiently to allow cis-K+ ions to enter the pore and alter blockade (36). Here, we also extend the utility of T-toxins to screening and quantifying the parameters of SAK1 toxin-channel interactions to facilitate the study of variants, particularly those with low affinity, in a manner that is amenable to the study of other peptides and membrane receptors. Although, trans-ion–enhanced toxin dissociation is most simply explained by Lys or Arg near the conduction pore or directly in the selectivity filter as visualized with CTX by crystallography (14), our studies do not preclude other mechanisms for elimination of the voltage dependence of blockade on neutralization of the residues including changes in the conformation of the toxins or induced changes in the pore receptor in response to the altered toxin, as recently described (37). The limitations of this study will be best addressed by the accomplishment of direct visualization of a toxin-channel pair showing the basis for Arg-dependent inhibition.
MATERIALS AND METHODS
Molecular biology
All T-toxins and T-RFP were constructed by replacing the sequence of lynx1, a natural tethered nicotinic acetylcholine receptor peptide modulator, with the sequences of toxins or TagRFP-T (RFP) so that they are exposed at the N terminus after cleavage of the secretory signal sequence; the toxin or RFP is followed by a flexible linker of Gly-Asn repeats (various lengths as noted) bearing a c-Myc epitope tag (Fig. 1B). The sequence encoding the hydrophobic chain of lynx1 for GPI attachment was inserted after the flexible linker. The whole sequences of T-toxins and T-RFP were cloned into pCS2+ plasmid vector with SP6 promoter for in vitro transcription of cRNAs. Point mutations for T-toxins and KcsA-Shaker were introduced using QuikChange Site-Directed Mutagenesis Kit (Agilent). T-toxins with different length linkers were constructed using gBlocks gene fragments (Integrated DNA Technologies) and Gibson Assembly (New England BioLabs). The sequences of all constructs were confirmed by DNA sequencing.
Peptide toxins synthesis
HmK (O16846), HmK-K22N, HmK-Y23A, ShK (P29187), ShK-K22N, and ShK-R24Q were purchased as synthetic peptides from CSBio. Peptide toxins folding reactions were quenched by acidification and purified by reverse-phase high-performance liquid chromatography, as before (11). Peptides that were more than 95% pure were lyophilized and stored at −20°C. The identity of the synthetic peptides was confirmed by mass spectral analysis. Peptides were dissolved in recording solution for TEVC before use.
Two-electrode voltage clamp
Xenopus laevis oocytes consistent with stage VI were selected on the basis of their size and clear white equatorial bands and were injected with 1 ng of cRNA encoding KcsA-Shaker, KcsA-Shaker–Y82V, or hKv1.3 (NM_002232) carried in a laboratory vector with the 5′ and 3′ portions of the X. laevis β-globin gene and a T7 promoter for in vitro transcription. For accessing the blocking effect of T-toxins, cRNAs of T-toxins and channels were mixed and coinjected into the oocytes. Recording solution was 4 mM KCl, 100 mM NaCl, 1 mM MgCl2, 0.3 mM CaCl2, and 10 mM Hepes (pH 7.5). Recordings were performed with constant gravity flow of solution at 2 ml/min, yielding chamber exchange in 5 s. Currents were recorded 2 days after cRNA injection using an oocyte clamp amplifier (OC-725C; Warner Instruments) and electrodes filled with 3 M KCl with resistance of 0.3 to 1 megohm. Data were filtered at 1 kHz and digitized at 20 kHz using pCLAMP software and assessed with Clampfit version 9.0 and Origin 6.0.
The fractional blockade by T-toxins was determined from the mean Itoxin with T-toxin cRNA coinjection normalized to the mean Icontrol with only channel cRNA injection. The competition of T-HmK binding with TEA to determine the association rate of T-HmK was performed by incubation of oocytes in solutions containing 100 mM TEACl, 4 mM KCl, 1 mM MgCl2, 0.3 mM CaCl2, and 10 mM Hepes (pH 7.5) for 1 hour before measurement. The reversion of T-HmK blocking by antibody was performed by incubation of oocytes in the same TEA containing solution with additional 3% bovine serum albumin (BSA; Fraction V; Thermo Fisher Scientific) and 2 μM c-Myc antibody (9E10; Developmental Studies Hybridoma Bank). To study the effects of changing intracellular KCl concentration, oocytes were microinjected with unbuffered aqueous solutions of KCl and studied 30 to 120 min thereafter (11, 38). The capacitance of oocytes was determined using a ramp-shaped voltage protocol, as previously reported by others (18).
Equilibrium inhibition of HmK was determined by fitting the concentration-response curve, whereas it was calculated for other peptide toxins from the fraction of unblocked current achieved by one to three toxin concentrations that inhibited 20 to 80% of the current, as described before (11). kon and koff were estimated from fits of the kinetics of toxin wash in and wash off. Thus, according to a bimolecular scheme with a single toxin-bound state and changes in toxin concentration compared to the speed of block and unblock, one can assess steady-state and kinetic parameters of blockade. The unblocked fractional current at equilibrium (Fun) with a toxin concentration [Tx] relates to the concentration dependence of inhibition according to Eq. 1. Fun is related to the second-order association rate constant kon (1/Ms) and first-order dissociation rate constant koff (1/s) according to Eq. 3, and the equilibrium inhibition constant for half-maximal blockade Ki is given by Eq. 2. Time constants of block and unblock (τon and τoff) were determined by single exponential fits of the kinetics of toxin wash in and wash off, and these parameters are related kon and koff by Eqs. 4 and 5. The zδ of voltage-dependent inhibition of toxins was calculated according to log[Ki(V)/Ki(0)] = zδFV/RT, where F is the Faraday constant, R is the gas constant, and T is the temperature in Kelvin.
Enzyme-linked immunosorbent assay
Oocytes injected with cRNAs of T-HmK, T-HmK variants, and T-RFP were blocked for 1 hour in recording solution with 3% BSA and then bound with c-Myc–Tag monoclonal antibody HRP (1 μg/ml) (Invitrogen) for 1 hour. Oocytes were extensively washed using recording solution with 0.1% Tween 20 and transferred to recording solution without BSA and Tween 20. Individual oocytes were placed in V-bottom 96-well plate (Thermo Fisher Scientific) with 50 μl of 1-Step Ultra TMB-ELISA solution (Thermo Fisher Scientific) in each well and incubated for 15 min. The reaction was stopped by adding 50 μl of 2 M H2SO4. Surface ELSIA signal was quantitated at 450 nm using Epoch Microplate Reader (BioTek).
Single-molecule total internal reflection fluorescence microscopy
Two days after T-RFP cRNA injection, Xenopus oocytes were treated enzymatically with neuraminidase and hyaluronidase, devitellinized manually, and placed on glass-bottom dishes (Chemglass Life Sciences) with recording solution bath. The critical angle for TIRF was adjusted using a cellTIRF illuminator (Olympus) and a high–numerical aperture (NA) apochromat objective (150×, 1.45 NA) mounted on an automated fluorescence microscope controlled using MetaMorph software (Molecular Devices). cellTIRF software (Olympus) was used to illuminate fluorophores at a critical angle such that only 100 nm deep was illuminated. RFP was excited with the 561-nm laser line and recorded by a back-illuminated electron-multiplying charge-coupled device. Stoichiometry of T-RFP was assessed by simultaneous photobleaching with continual excitation at 4°C for reducing particles diffusion. Data were captured as movies of 300 frames acquired at 1 Hz. Data were analyzed as previously described (17). The surface particle number of T-RFP was manually counted in separate regions (10 μm by 10 μm field), and four regions (two in animal hemisphere and two in vegetal hemisphere) were counted per oocyte using ImageJ software.
Single-molecule photobleaching events are missed in practical application to biological systems because of fluorophore prebleaching, the quantum efficiency of the fluorophore, and the time resolution of smTIRF system; in this case, the error is estimated to be less than 10% (17). Protein misfolding or incomplete maturation can also decrease expression (39). Together, these challenges have been estimated to maximally decrease membrane protein signals by up to ~18% in our studies (17) and those by others (39); this could yield an error in our absolute estimate of T-HmK affinity (1.09 nM) of ~0.19 nM. As far as our assessment of the role of individual toxin residues in blockade, it is reassuring that scanning point mutations do not appear to alter T-protein expression differentially (Fig. 3A) and that free toxins with point mutations show the same relative change in potency and mechanism of action as their tethered congeners (Figs. 3 and 4).
Calculation of the membrane surface concentration for T-toxins
The average number of single T-RFP particles (n) in a 100-μm2 membrane surface area (s) was measured by smTIRF counting. The total surface area of one oocyte (S) was calculated from average capacitance of oocytes (C; 198 ± 15 nF) and specific capacitance of stage VI Xenopus oocytes (c; 4 μF/cm2). The total molecular number of T-RFP on the oocytes surface can be calculated. The total molecular number of T-HmK on the oocytes surface can be estimated by the ratio of optical signal between T-HmK and T-RFP determined by ELISA and converted to moles (mol) using Avogadro’s number (NA)
| (8) |
The reaction volume (V) available to T-HmK on the oocyte surface was estimated as the areas between two spheres (Fig. 1B). The smaller sphere has a radius equal to the average radius of oocyte (r ~0.6 mm), and the bigger sphere has a radius (R) equal to the addition of r and the full extent of the peptide linker (number of residues × 3 Å for each peptide bond, ~138 Å), the length of the sugar chain of the GPI anchor (~60 Å), plus the diameter of HmK toxin (28 Å), that is, for T-HmK, L = 46 × 3 + 60 + 28 = 226 Å. The reaction volume can be estimated using Eq. 9
| (9) |
and the concentration of T-toxins in nM ([Tx]) can be calculated from the ratio of the amount of toxin and the reaction volume (mol/V). This method for calculation of the reaction volume was similarly used by others to study the expression of membrane-tethered peptides in rat PC12 cells (40).
NMR spectroscopy and structure determination
Lyophilized HmK peptide was dissolved to a final concentration of 0.2 to 0.5 mM in 20 mM potassium phosphate buffer (pH 6.6) containing 10 mM NaCl and 0.02% NaN3 in 7 or 99% 2H2O. NMR spectra were collected on a Bruker DRX 700 spectrometer equipped with cryogenic triple-resonance TCI probe with z-axis pulsed field gradients. All experiments were conducted at 16.4 T and 293 K. Resonance assignments and structural restraints (distances and dihedral angles) were obtained from 2D homonuclear and heteronuclear spectra, and secondary chemical shift analysis were obtained using the TALOS program. Detailed experimental conditions are provided in the Supplementary Materials. Structure calculation was performed using the Crystallography and NMR System software using distance geometry and simulating annealing protocols. Calculations were performed iteratively, introducing short-range and later long-range distance constraints, followed by dihedral constraints, to eliminate erroneous assignments. Structure quality of the final ensemble was validated by the PROCHECK-NMR program. Protein structures were visualized using the PyMOL program.
Supplementary Material
Acknowledgments
Funding: We are grateful for grant support from the NIH (GM111716 to S.A.N.G.) and the United States–Israel Binational Science Foundation (BSF 2013185 and 2017243) to S.A.N.G. and J.H.C. Author contributions: R.Z. and H.D. designed and performed research and wrote the paper. N.M. performed research. J.H.C. and S.A.N.G. designed research and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaaz3439/DC1
Fig. S1. Concentration-response relationship of HmK blockade of KcsA-Shaker.
Fig. S2. Effect of mutation KcsA-Shaker–Y82V on block by TEA and HmK.
Fig. S3. Hui1-Arg23 mediates the voltage-dependent block of KcsA-Shaker.
Fig. S4. Structures of HmK, ShK, and Hui1 toxins noting some important, conserved residues.
Table S1. Single-particle counting and calculation of effective T-HmK surface concentration.
Table S2. Single-particle counting and calculation of effective T-HmK surface concentration with coexpression of KcsA-Shaker channels.
Table S3. Blocking parameters of hKv1.3 by T-HmK, T-ShK, and the analogous free peptide variants.
Table S4. 1H chemical shifts of HmK.
Table S5. Heteronuclear chemical shifts of HmK.
Table S6. Statistics for HmK structure calculation.
REFERENCES AND NOTES
- 1.Miwa J. M., Iban̆ez-Tallon I., Crabtree G. W., Sánchez R., S̆ali A., Role L. W., Heintz N., lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron 23, 105–114 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Ibañez-Tallon I., Wen H., Miwa J. M., Xing J., Tekinay A. B., Ono F., Brehm P., Heintz N., Tethering naturally occurring peptide toxins for cell-autonomous modulation of ion channels and receptors in vivo. Neuron 43, 305–311 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Auer S., Stürzebecher A. S., Jüttner R., Santos-Torres J., Hanack C., Frahm S., Liehl B., Ibañez-Tallon I., Silencing neurotransmission with membrane-tethered toxins. Nat. Methods 7, 229–236 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Gui J., Liu B., Cao G., Lipchik A. M., Perez M., Dekan Z., Mobli M., Daly N. L., Alewood P. F., Parker L. L., King G. F., Zhou Y., Jordt S.-E., Nitabach M. N., A tarantula-venom peptide antagonizes the TRPA1 nociceptor ion channel by binding to the S1-S4 gating domain. Curr. Biol. 24, 473–483 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalia J., Milescu M., Salvatierra J., Wagner J., Klint J. K., King G. F., Olivera B. M., Bosmans F., From foe to friend: Using animal toxins to investigate ion channel function. J. Mol. Biol. 427, 158–175 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldstein S. A. N., Pheasant D. J., Miller C., The charybdotoxin receptor of a Shaker K+ channel: Peptide and channel residues mediating molecular recognition. Neuron 12, 1377–1388 (1994). [DOI] [PubMed] [Google Scholar]
- 7.MacKinnon R., Cohen S. L., Kuo A., Lee A., Chait B. T., Structural conservation in prokaryotic and eukaryotic potassium channels. Science 280, 106–109 (1998). [DOI] [PubMed] [Google Scholar]
- 8.de Souza J. M., Goncalves B. D. C., Gomez M. V., Vieira L. B., Ribeiro F. M., Animal toxins as therapeutic tools to treat neurodegenerative diseases. Front. Pharmacol. 9, 145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chi V., Pennington M. W., Norton R. S., Tarcha E. J., Londono L. M., Sims-Fahey B., Upadhyay S. K., Lakey J. T., Iadonato S., Wulff H., Beeton C., Chandy K. G., Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 59, 529–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diochot S., Lazdunski M., Sea anemone toxins affecting potassium channels. Prog. Mol. Subcell. Biol. 46, 99–122 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Zhao R., Dai H., Mendelman N., Cuello L. G., Chill J. H., Goldstein S. A. N., Designer and natural peptide toxin blockers of the KcsA potassium channel identified by phage display. Proc. Natl. Acad. Sci. U.S.A. 112, E7013–E7021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacKinnon R., Miller C., Mechanism of charybdotoxin block of the high-conductance, Ca2+−activated K+ channel. J. Gen. Physiol. 91, 335–349 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein S. A. N., Miller C., Mechanism of charybdotoxin block of a voltage-gated K+ channel. Biophys. J. 65, 1613–1619 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banerjee A., Lee A., Campbell E., Mackinnon R., Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. eLife 2, e00594 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rauer H., Pennington M., Cahalan M., Chandy K. G., Structural conservation of the pores of calcium-activated and voltage-gated potassium channels determined by a sea anemone toxin. J. Biol. Chem. 274, 21885–21892 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Gendeh G. S., Young L. C., de Medeiros C. L. C., Jeyaseelan K., Harvey A. L., Chung M. C. M., A new potassium channel toxin from the sea anemone Heteractis magnifica: Isolation, cDNA cloning, and functional expression. Biochemistry 36, 11461–11471 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Plant L. D., Xiong D., Dai H., Goldstein S. A. N., Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proc. Natl. Acad. Sci. U.S.A. 111, E1438–E1446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitt B. M., Koepsell H., An improved method for real-time monitoring of membrane capacitance in Xenopus laevis oocytes. Biophys. J. 82, 1345–1357 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.G. Baker, S. Dunn, A. Holt, A. Lajtha, eds. Handbook of Neurochemistry and Molecular Neurobiology: Practical Neurochemistry Methods (Springer, 2006). [Google Scholar]
- 20.Preston G. M., Carroll T. P., Guggino W. B., Agre P., Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science 256, 385–387 (1992). [DOI] [PubMed] [Google Scholar]
- 21.J. Kuriyan, B. Konforti, D. Wemmer, The Molecules of Life: Physical and Chemical Principles (Garland Science, Taylor & Francis Group, 2013). [Google Scholar]
- 22.Lu Z., Klem A. M., Ramu Y., Ion conduction pore is conserved among potassium channels. Nature 413, 809–813 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Miller C., Competition for block of a Ca2+-activated K+ channel by charybdotoxin and tetraethylammonium. Neuron 1, 1003–1006 (1988). [DOI] [PubMed] [Google Scholar]
- 24.Heginbotham L., MacKinnon R., The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron 8, 483–491 (1992). [DOI] [PubMed] [Google Scholar]
- 25.Dauplais M., Lecoq A., Song J., Cotton J., Jamin N., Gilquin B., Roumestand C., Vita C., de Medeiros C. L. C., Rowan E. G., Harvey A. L., Ménez A., On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 272, 4302–4309 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Cahalan M. D., Chandy K. G., The functional network of ion channels in T lymphocytes. Immunol. Rev. 231, 59–87 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin L., Wu Y., Molecular mechanism of the sea anemone toxin ShK recognizing the Kv1.3 channel explored by docking and molecular dynamic simulations. J. Chem. Inf. Model. 47, 1967–1972 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Tudor J. E., Pallaghy P. K., Pennington M. W., Norton R. S., Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat. Struct. Biol. 3, 317–320 (1996). [DOI] [PubMed] [Google Scholar]
- 29.Dang B., Shen R., Kubota T., Mandal K., Bezanilla F., Roux B., Kent S. B. H., Inversion of the side-chain stereochemistry of indvidual Thr or Ile residues in a protein molecule: Impact on the folding, stability, and structure of the ShK toxin. Angew. Chem. Int. Ed. Engl. 56, 3324–3328 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoshi T., Zagotta W. N., Aldrich R. W., Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250, 533–538 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Massover W. H., Complex surface invaginations in frog oocytes. J. Cell Biol. 58, 485–491 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milescu M., Vobecky J., Roh S. H., Kim S. H., Jung H. J., Kim J. I., Swartz K. J., Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 130, 497–511 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren J., Li R., Ning J., Zhu X., Zhangsun D., Wu Y., Luo S., Effect of methionine oxidation and substitution of α-conotoxin TxID on α3β4 nicotinic acetylcholine receptor. Mar. Drugs 16, E215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalman K., Pennington M. W., Lanigan M. D., Nguyen A., Rauer H., Mahnir V., Paschetto K., Kem W. R., Grissmer S., Gutman G. A., Christian E. P., Cahalan M. D., Norton R. S., Chandy K. G., ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 273, 32697–32707 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Sumino A., Sumikama T., Uchihashi T., Oiki S., High-speed AFM reveals accelerated binding of agitoxin-2 to a K+ channel by induced fit. Sci. Adv. 5, eaax0495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moldenhauer H., Díaz-Franulic I., Poblete H., Naranjo D., Trans-toxin ion-sensitivity of charybdotoxin-blocked potassium-channels reveals unbinding transitional states. eLife 8, e46170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labro A. J., Cortes D. M., Tilegenova C., Cuello L. G., Inverted allosteric coupling between activation and inactivation gates in K+ channels. Proc. Natl. Acad. Sci. U.S.A. 115, 5426–5431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tai K. K., Goldstein S. A. N., The conduction pore of a cardiac potassium channel. Nature 391, 605–608 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Ulbrich M. H., Isacoff E. Y., Subunit counting in membrane-bound proteins. Nat. Methods 4, 319–321 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt D., Tillberg P. W., Chen F., Boyden E. S., A fully genetically encoded protein architecture for optical control of peptide ligand concentration. Nat. Commun. 5, 3019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaaz3439/DC1
Fig. S1. Concentration-response relationship of HmK blockade of KcsA-Shaker.
Fig. S2. Effect of mutation KcsA-Shaker–Y82V on block by TEA and HmK.
Fig. S3. Hui1-Arg23 mediates the voltage-dependent block of KcsA-Shaker.
Fig. S4. Structures of HmK, ShK, and Hui1 toxins noting some important, conserved residues.
Table S1. Single-particle counting and calculation of effective T-HmK surface concentration.
Table S2. Single-particle counting and calculation of effective T-HmK surface concentration with coexpression of KcsA-Shaker channels.
Table S3. Blocking parameters of hKv1.3 by T-HmK, T-ShK, and the analogous free peptide variants.
Table S4. 1H chemical shifts of HmK.
Table S5. Heteronuclear chemical shifts of HmK.
Table S6. Statistics for HmK structure calculation.