

Abstract
Patients with chronic enteropathy associated with SLCO2A1 (CEAS) develop multiple circular, longitudinal, or eccentric ulcers in the ileum. It is sometimes difficult to distinguish CEAS from Crohn's disease. CEAS and primary hypertrophic osteoarthropathy (PHO) are together known to be caused by a mutation of SLCO2A1 gene. The case of a 65-year-old man whose characteristic appearance due to pachydermia of the forehead folds led to the diagnosis of CEAS with PHO is presented.
Keywords: chronic enteropathy associated with SLCO2A1, primary hypertrophic osteoarthropathy, pachydermia of the forehead folds
Introduction
Chronic enteropathy associated with SLCO2A1 (CEAS) is a rare disease in which multiple circular, longitudinal, or eccentric ulcers develop in the ileum. Most CEAS patients present with anemia and hypoalbuminemia (1). However, some patients with advanced disease develop abdominal pain with intestinal stenosis and require repeated surgeries (2). Because the appearance of ileal ulcers in patients with CEAS sometimes resembles that of Crohn's disease (CD) or intestinal tuberculosis, it is sometimes difficult to distinguish CEAS from them (3-6). Umeno et al. reported that CEAS is a hereditary disease caused by mutations in the SLCO2A1 gene (1, 7). The SLCO2A1 gene is also a causal gene of primary hypertrophic osteoarthropathy (PHO), which is an extremely rare disease in which patients present digital clubbing, periostosis, and pachydermia (8). In particular, cutis verticis gyrata (CVG) and thickening and furrowing of the skin on the forehead due to pachydermia are highly characteristic signs of this condition. We herein present the case of a patient whose characteristic facial appearance led to a diagnosis of CEAS associated with PHO.
Case Report
The patient was a man who had repeated presented with anemia and positive fecal occult blood since his 30s. At 65 years of age, severe anemia was seen on a laboratory examination, and multiple gastric ulcers (Fig. 1) and a longitudinal ileal ulcer were seen on esophagogastroduodenoscopy (EGD) and ileo-colonoscopy. The patient was therefore referred to our hospital due to suspected Crohn's disease.
Figure 1.
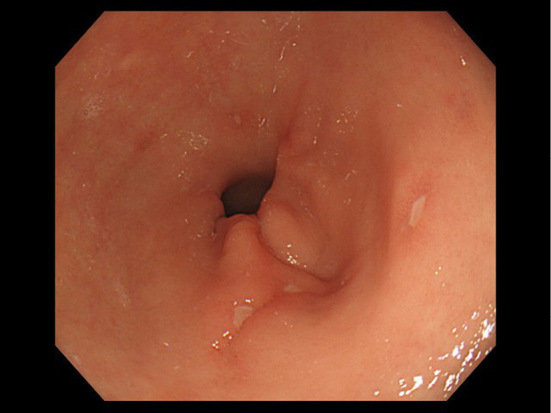
Esophagogastroduodenoscopy shows multiple ulcers in the antrum of the stomach.
Gitelman syndrome was suspected based on the presence of hypokalemia, hypomagnesemia, and a low urine potassium level when he was 58 years of age (details unknown). His other past history included appendectomy and uveitis, but not tuberculosis. He was treated with oral famotidine, potassium, magnesium oxide, and iron, but he was not taking any non-steroidal anti-inflammatory drugs (NSAIDs). He had no family history of enteritis or tuberculosis, and his parents were non-consanguineous. Vital signs were normal, and his palpebral conjunctivae looked anemic. The patient' s laboratory data were as follows: WBC count, 10,520 /μL; Hb, 8.5 mg/dL; ALB, 3.07 g/dL; K, 3.86 mEq/L; and C-reactive protein (CRP), 0.71 mg/dL. Video capsule endoscopy (VCE) was performed with a PillcamⓇ SB3 (Covidien Japan Inc., Tokyo, Japan) after assessing the patency of the small intestinal tract with a PillcamⓇ patency capsule (Covidien Japan Inc.). VCE showed multiple circular and longitudinal ulcers throughout the entire small intestine, especially in the lower ileum (except for the terminal ileum) (Fig. 2). Gastric involvement showed scarring on EGD. Double-balloon endoscopy (DBE) was performed via the anal route and showed a longitudinal shallow ulcer in the lower ileum (Fig. 3). Granulomas were not detected, and non-specific enteritis was found on a pathological analysis of biopsy specimens from the edge of the ulcer. Biopsy specimens from the ileum were subjected to culture, and polymerase chain reaction (PCR) in order to identify tuberculosis, however, all results were negative. In addition, the findings of interferon gamma release assays were also negative. The patient was diagnosed with Crohn's disease and was treated with oral mesalazine. However, it was ineffective and was discontinued after 22 months. A trial of anti-tuberculosis agents as a diagnostic therapy was considered, but the patient refused.
Figure 2.

Video capsule endoscopy on admission shows multiple circular and longitudinal ulcers throughout the entire small intestine.
Figure 3.

Double balloon endoscopy on admission shows a longitudinal shallow ulcer in the lower ileum.
Symptomatic treatment was then continued, and 46 months after his first visit to our hospital, we noticed his characteristic facial appearance due to pachydermia of the forehead folds (Fig. 4a). The patient had been aware of thickening and furrowing of the skin on the forehead and CVG since his 30s. Digital clubbing was also present (Fig. 4b), and an X-ray examination revealed periostosis of the long bones (Fig. 4c). A diagnosis of PHO was made, and CEAS was strongly suspected. A genetic analysis was performed, and compound heterozygous mutations of the SLCO2A1 gene [c.940+1 G>A (splice site variant) and c.1475 G>A (p.Cys792Tyr)] were detected (Fig. 5). CEAS was definitely diagnosed, and symptomatic treatment was continued.
Figure 4.

a: Characteristic facial appearance due to pachydermia of the forehead folds. b: Digital clubbing. c: Periostosis of the tibia and fibula.
Figure 5.

Sequencing the SLCO2A1 mutations. Compound heterozygous mutations of SLCO2A1 gene, c.940+1G>A (splice site variant) and c.1475G>A (p.Cys792Tyr), were detected. Upper: Wild-type. Lower: The present patient. Left: Exon7. Right: Exon11.
Discussion
CEAS, which was previously called chronic nonspecific multiple ulcers of the small intestine (CNSU), is rare in the Japanese population (2). CEAS is a hereditary disease and it has been reported that approximately one-quarter of patients have consanguinity or siblings with enteropathy (1, 9). Recently, mutations in the SLCO2A1 gene were identified in patients with CNSU, and a more appropriate name, CEAS, was suggested (1, 7). Among SLCO2A1 mutations, a splice-site mutation at intron 7 (c.940+1 G>A; rs765249238), which was detected in the present patient, was the most frequently observed in CEAS patients (1). This mutant allele frequency is 0.091% in the Japanese population (1), but it is rarely found in Caucasian populations (1, 7). SLCO2A1 gene encodes OATP2A1, which is a prostaglandin (PG) transporter that may be involved in mediating both the influx and efflux of prostaglandins in numerous tissues (10). It is interesting that the circular ulcers in the small intestine observed by VCE in the present patient are very unique and resemble those of NSAID-induced enteropathy. NSAIDs are known to block cyclooxygenase-derived PG synthesis (11). Nakanishi et al. hypothesized that the local PGE2 concentration is also decreased in patients with CEAS due to impairment of the PG efflux function, but not the influx function, of OATP2A1 (10). Umeno et al. proposed a new entity of gastrointestinal disorders together with NSAID-induced enteropathy called “prostaglandin-associated enteropathy” due to impaired prostaglandin use (1).
CEAS is characterized by multiple small intestinal ulcers of nonspecific histology and chronic persistent gastrointestinal bleeding. However, most patients do not experience hematochezia (1). The hematochezia in the present patient confused us when CEAS was diagnosed. Some patients with advanced disease develop intestinal stenosis and require repeated surgeries. Although total parenteral nutrition (TPN) is the only effective therapy (9), it was not tried because it had the potential to worsen the patient' s intestinal stenosis. We consider symptomatic treatment to be necessary and sufficient for patients with mild symptoms.
The endoscopic findings of CEAS are characterized by multiple circular, longitudinal, or oblique shallow ulcers with discrete margins in the ileum (except for the terminal ileum), which resemble Crohn's disease or intestinal tuberculosis (3, 6).
We think that it is difficult to diagnose this rare disease, especially in a patient without consanguinity or a family history. The key to the diagnosis in the present case was the characteristic facial appearance of patients with PHO. PHO is an extremely rare disease that causes digital clubbing, periostosis, and pachydermia (8). In particular, increased furrowing of the forehead folds due to pachydermia is a highly characteristic sign of the condition. PHO is also known to be caused by SLCO2A1 gene mutation (12). Umeno et al. reported that digital clubbing or periostosis was found in 28% of patients with CEAS, with 10.8% fulfilling the major diagnostic criteria of PHO. It is interesting that all of these patients were male, like the present patient, even though CEAS predominantly affects females (1). The symptoms of PHO are especially important for the diagnosis of CEAS in males. Some patients with CD and PHO have been previously reported (13, 14); they seem to have had CEAS rather than CD. The present patient had a history of suspected Gitelman's syndrome. Jiang et al. also reported a case of PHO with Bartter's-like syndrome (15). An association between PHO and hypokalemia has been suggested.
Although no effective treatment other than TPN has been established, the diagnosis of CEAS and the exclusion of other diseases are important to avoid unnecessary treatment. The number of patients diagnosed with CEAS will increase with progress in modalities for examining the small intestine and improved recognition of the disease. The characteristic facial appearance that is seen in some patients can be very helpful for making the diagnosis.
Conclusion
The characteristic facial appearance of pachydermia of the forehead was useful for diagnosing CEAS.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
The authors would like to thank all of the staff of Oita University Hospital who took care of the patient.
References
- 1. Umeno J, Esaki M, Hirano A, et al. Clinical features of chronic enteropathy associated with SLCO2A1 gene: a new entity clinically distinct from Crohn's disease. J Gastroenterol 53: 907-915, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matsumoto T, Iida M, Matsui T, Yao T, Watanabe H, Okabe H. Non-specific multiple ulcers of the small intestine unrelated to non-steroidal anti-inflammatory drugs. J Clin Pathol 57: 1145-1150, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hosoe N, Ohmiya N, Hirai F, et al. Chronic enteropathy associated with SLCO2A1 gene [CEAS]-characterisation of an enteric disorder to be considered in the differential diagnosis of Crohn's disease. J Crohns Colitis 11: 1277-1281, 2017. [DOI] [PubMed] [Google Scholar]
- 4. Matsumoto T, Nakamura S, Esaki M, et al. Endoscopic features of chronic nonspecific multiple ulcers of the small intestine: comparison with nonsteroidal anti-inflammatory drug-induced enteropathy. Dig Dis Sci 51: 1357-1363, 2006. [DOI] [PubMed] [Google Scholar]
- 5. Yanai S, Yamaguchi S, Nakamura S, et al. Distinction between chronic enteropathy associated with the SLCO2A1 gene and Crohn's disease. Gut Liver 13: 62-66, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uchida K, Nakajima A, Ushijima K, et al. Pediatric-onset chronic nonspecific multiple ulcers of small intestine: a nationwide survey and genetic study in Japan. J Pediatr Gastroenterol Nutr 64: 565-568, 2017. [DOI] [PubMed] [Google Scholar]
- 7. Umeno J, Hisamatsu T, Esaki M, et al. A hereditary enteropathy caused by mutations in the SLCO2A1 gene, encoding a prostaglandin transporter. PLoS Genet 11: e1005581, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Z, Zhang C. Primary hypertrophic osteoarthropathy: an update. Front Med 7: 60-64, 2013. [DOI] [PubMed] [Google Scholar]
- 9. Matsumoto T, Kubokura N, Matsui T, Iida M, Yao T. Chronic nonspecific multiple ulcer of the small intestine segregates in offspring from consanguinity. J Crohns Colitis 5: 559-565, 2011. [DOI] [PubMed] [Google Scholar]
- 10. Nakanishi T, Tamai I. Roles of organic anion transporting polypeptide 2A1 (OATP2A1/SLCO2A1) in regulating the pathophysiological actions of prostaglandins. AAPS J 20: 13, 2017. [DOI] [PubMed] [Google Scholar]
- 11. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294: 1871-1875, 2001. [DOI] [PubMed] [Google Scholar]
- 12. Zhang Z, Xia W, He J, et al. Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am J Hum Genet 90: 125-132, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Compton RF, Sandborn WJ, Yang H, et al. A new syndrome of Crohn's disease and pachydermoperiostosis in a family. Gastroenterology 112: 241-249, 1997. [DOI] [PubMed] [Google Scholar]
- 14. Ohata K, Niinami C, Ohya T, et al. Education and imaging: gastrointestinal: pachydermoperiostosis associated with Crohn's disease. J Gastroenterol Hepatol 24: 1576, 2009. [DOI] [PubMed] [Google Scholar]
- 15. Jiang Y, Du J, Song YW, et al. Novel SLCO2A1compound heterozygous mutation causing primary hypertrophic osteoarthropathy with Bartter-like hypokalemia in a Chinese family. J Endocrinol Invest 42: 1245-1252, 2019. [DOI] [PubMed] [Google Scholar]