

Abstract
Chronic diseases account for over 60% of all deaths worldwide according to the World Health Organization reports. Majority of cases are triggered by environmental exposures that lead to aberrant changes in the epigenome, specifically, the DNA methylation patterns. These changes result in altered expression of gene networks and activity of signalling pathways. Dietary antioxidants, including catechins, flavonoids, anthocyanins, stilbenes and carotenoids, demonstrate benefits in the prevention and/or support of therapy in chronic diseases. This review provides a comprehensive discussion of potential epigenetic mechanisms of antioxidant compounds in reversing altered patterns of DNA methylation in chronic disease. Antioxidants remodel the DNA methylation patterns through multiple mechanisms, including regulation of epigenetic enzymes and chromatin remodelling complexes. These effects can further contribute to antioxidant properties of the compounds. On the other hand, decrease in oxidative stress itself can impact DNA methylation delivering additional link between antioxidant mechanisms and epigenetic effects of the compounds.
Linked Articles
This article is part of a themed section on The Pharmacology of Nutraceuticals. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v177.6/issuetoc
Abbreviations
- 5‐caC
5‐carboxylcytosine
- 5‐fC
5‐formylcytosine
- 5‐hmC
5‐hydroxymethylcytosine
- 5‐mC
5‐methylcytosine
- AD
Alzheimer's disease
- ADMA
asymmetric dimethylarginine
- AMPK
AMP‐activated protein kinase
- AP‐1
activator protein‐1
- APC
adenomatous polyposis coli
- Apo(a)
apolipoprotein(a)
- APOE
apolipoprotein E
- APP
amyloid β precursor protein
- ARE
antioxidant response element
- ATRA
all‐trans‐retinoic acid
- AZA
azacytidine
- Aβ
amyloid β
- BACE1
β‐secretase 1
- BER
base excision repair
- CAT
catalase
- CDH1
cadherin 1
- CDKN2A
cyclin dependent kinase inhibitor 2A
- CHRM1
cholinergic receptor muscarinic 1
- COMT
catechol‐O‐methyltransferase
- COPD
chronic obstructive pulmonary disease
- CSR1
Phosphatidylinositol transfer protein CSR1
- CVD
cardiovascular disease
- DAC
decitabine
- DAPK1
death‐associated protein kinase‐1
- DDAH2
dimethylarginine dimethylaminohydrolase 2
- DMRs
differentially methylated regions
- DNMTi
DNMT inhibitors
- DNMTs
DNA methyltransferases
- DTX1
deltex E3 ubiquitin ligase 1
- EGC
epigallocatechin
- EGCG
epigallocatechin‐3‐gallate
- eNOS
endothelial NOS
- EPHB2
EPH receptor B2
- ER
endoplasmic reticulum
- ERα
oestrogen receptor α
- EZH2
enhancer of zeste homologue 2
- FFA
free fatty acid
- FOXP3
forkhead box P3
- GLP1R
glucagon‐like peptide 1 receptor
- GLT1D1
glycosyltransferase 1 domain containing 1
- GPx
GSH peroxidase
- GSSG
GSH disulfide
- GST
GSH‐S‐transferase
- GSTP1
GSH‐S‐transferase pi 1
- H2O2
hydrogen peroxide
- HbA1c
haemoglobin A1c
- HCC
hepatocellular carcinoma
- HERV‐w
human endogenous retrovirus‐W
- hMLH1
human mutL homologue 1
- HO‐1
hemeoxygenase‐1
- IDD
iodothyronine deiodinases
- INS
insulin
- Keap1
Kelch‐like ECH associated protein 1
- LOO•
lipid peroxyl radical
- MAPT
microtubule associated protein tau
- MBD
methyl‐CpG‐binding domain
- MGMT
O(6)‐methylguanine methyltransferase
- Na2SeO3
sodium selenite
- NEP
neprilysin
- NQO1
NAD(P)H quinone dehydrogenase 1
- Nrf2
Nuclear factor erythroid 2‐related factor 2
- O2•‐
superoxide radical
- OCM
one‐carbon metabolism
- OGG1
8‐oxoguanine DNA glycosylase
- OH•
hydroxyl radical
- ONOO−
peroxynitrite anion
- oxLDL
oxidized LDL
- PDX1
pancreatic and duodenal homeobox 1
- PSEN1
presenilin 1
- PTEN
phosphatase and tensin homologue
- PUFAs
polyunsaturated fatty acids
- RA
retinoic acid
- RARE
retinoic acid response element
- RARβ
retinoic acid receptor β
- RECK
reversion‐inducing cysteine‐rich protein with Kazal motifs
- RNS
reactive nitrogen species
- RXRα
retinoic X receptor α
- SAH
S‐adenosyl homocysteine
- SAM
S‐adenosyl‐l‐methionine
- SFN
sulforaphane
- SIRT1
sirtuin‐1
- SMC
smooth muscle cell
- Srebf1
sterol regulatory element binding transcription factor 1
- T2D
type II diabetes
- TDG
thymine DNA Glycosylase
- TET
Ten‐eleven translocation
- TNFR1
TNF receptor 1
- TRAMP
transgenic adenocarcinoma of the mouse prostate
- TRXR
thioredoxin reductase
- VHL
von Hippel‐Lindau
- WIF
WNT inhibitory factor
1. INTRODUCTION
The leading causes of death in North America and globally include five chronic diseases, cancer, cardiovascular disease (CVD), chronic obstructive pulmonary disease (COPD), type II diabetes (T2D) and Alzheimer's disease (AD; World Health Organization data, 2018). Chronic diseases affect approximately 133 million Americans and 6 million Canadians and account for over 60% of all deaths globally (World Health Organization data, 2018). Oxidative stress is believed to be a contributor to the development of these diseases, thus understanding underlying mechanisms associated with oxidative stress that are central to the prevention and/or development of therapeutic strategies will be useful to lower the prevalence of these diseases and related comorbidities. Advances in this area will translate to improved health and quality of life of millions of people.
Cancer is defined as uncontrolled cell growth with the ability to invade through the extracellular matrix and to metastasize to other parts of the human body. Many forms and sites of cancer contribute to over 600,000 deaths in the United States per year (National Cancer Institute data, 2018). CVD, including heart disease and stroke, is inextricably linked to atherosclerosis, the former being characterized by narrowing of arteries as a result of plaque build‐up in the artery wall, initiated by the modification or oxidation of LDL (Yang et al., 2017). COPD, on the other hand, is a disease occurring in the lung, defined by irreversible airflow obstruction and a progressive loss of lung function (Rahman, 2008). This is often caused by exposure to noxious particles or gases that are predominantly introduced into the lung by cigarette smoking and air pollution. T2D is a systemic metabolic disorder characterized by https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5012 resistance, decreased insulin secretion, carbonyl stress and significant apoptosis of functional pancreatic β cells, which can consequently affect almost every organ in the body (Fonseca, Gromada, & Urano, 2011; Rehman & Akash, 2017). As for the aetiology of AD, insoluble polymorphous https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4865 deposits, so‐called senile plaques, induce oxidative stress which lead to formation of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2418 adducts and massive neuronal loss in the brain (Tonnies & Trushina, 2017). Interestingly, disturbances in insulin secretion and action have been demonstrated to play a role in the formation of amyloid plaques and neurofibrillary tangles in the brain and thereb it has been proposed that AD should be classified as “type 3 diabetes” (Kandimalla, Thirumala, & Reddy, 2017). Only a fraction of the cases of chronic diseases is attributed to genetic predisposition. For instance, approximately 5–10% of cancer cases are considered hereditary. The remaining majority is associated with environmental exposures and lifestyle factors, a common feature of which, as mentioned, is the oxidative stress at the initiation stages of disease. Exposures and inflammation can induce epigenetic aberrations which are important players in the development and progression of chronic disease. Epigenetics refers to the control of gene expression without changes to the DNA sequence and comprises the following components: DNA methylation, covalent histone modifications, non‐coding RNA mechanisms, and chromatin remodelling complexes (Jones & Takai, 2001; Figure 1). Although all the components of the epigenome appear to regulate gene expression, mostly in a concerted way, DNA methylation is believed to be crucial for stabilizing the signalled changes in gene expression (Jones & Takai, 2001). A connection between oxidative stress and DNA methylation has been suggested and is under investigation (Ding et al., 2016; Ding, Miller, Savant, & O'Hagan, 2019). It is still unclear whether oxidative stress leads to aberrant DNA methylation and gene expression or whether oxidative stress is a consequence of DNA methylation changes.
Figure 1.
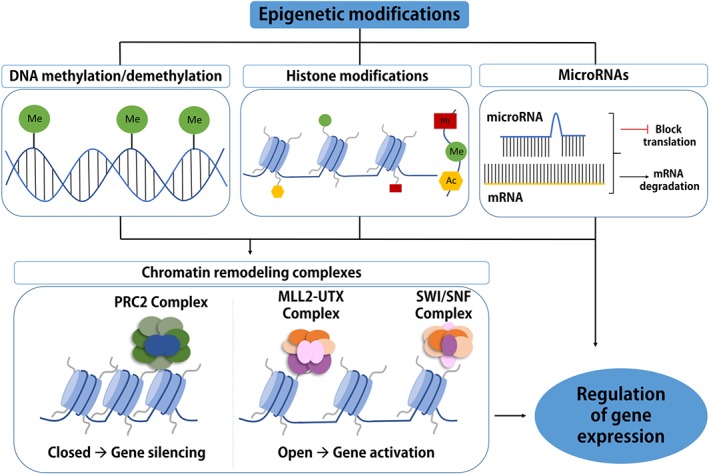
Epigenetic mechanisms of regulation of gene expression. Epigenetic regulation results in changes in gene expression without changes in the underlying DNA sequence. Epigenetic modifications include DNA methylation, histone modifications, non‐coding RNA‐related mechanisms, such as micro‐RNAs, and chromatin remodelling complexes. DNA methylation involves the covalent addition of a methyl group to the 5′ position of the cytosine base in DNA by DNA methyltransferases (DNMTs; DNMT1, DNMT3A and DNMT3B), and this reaction can be reversed by ten‐eleven translocation (TET) proteins and other proteins. Histone tails protruding from histone proteins can undergo acetylation (Ac), methylation (Me), phosphorylation (Ph), amongst other chemical modifications. Histone modifications are carried out by histone acetyltransferases (HATs) and histone deacetylases (HDACs), histone methyltransferases (HMTs) and histone demethylases (HDMs), Aurora B, and protein phosphatase 1 (PP1). miRNAs can epigenetically regulate gene expression at the post‐transcriptional level by binding to mRNA, which leads to targeted mRNA degradation and inhibition of gene expression. All these epigenetic mechanisms can influence chromatin remodelling (a condensed state or a transcriptionally accessible state), in addition to chromatin remodelling complexes such as polycomb repressive complex 2 (PRC2), MLL2‐UTX, and Swi/SNF. All of these mechanisms ultimately regulate gene expression
In the present review, we will discuss how consumption of dietary antioxidant compounds present in many commonly consumed fruits, vegetables or beverages represent an effective approach to reverse aberrant patterns of DNA methylation and cease oxidative stress, which consequently prevent and/or attenuate chronic disease. The main focus will be on potential mechanisms linking the antioxidant function of dietary compounds with DNA methylation‐remodelling activity.
2. DNA METHYLATION ALTERATIONS IN CHRONIC DISEASE
DNA methylation is thought to provide stable, long‐term regulation of gene expression over time but is also considered to be dynamic and responsive to environmental influences (Cedar & Bergman, 2009). Actively transcribed genes have non‐methylated regulatory regions, like promoters or enhancers, where RNA polymerase II and other members of the transcriptional machinery bind and drive gene transcription (Figure 2). On the other hand, methylation of promoters or enhancers in transcriptionally inactive genes results in binding of methyl‐CpG‐binding domain (MBD) proteins, which then recruit repressor complexes to further repress gene expression (Figure 2). These patterns of DNA methylation are shaped by methylating and demethylating enzymes.
Figure 2.
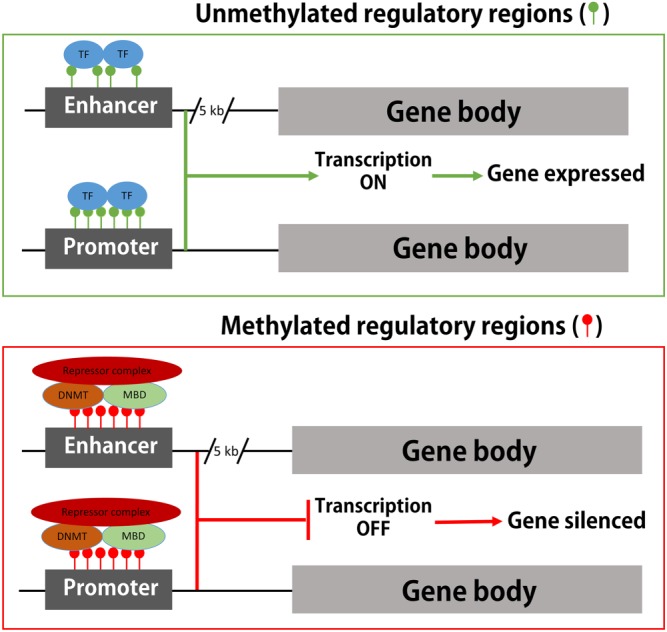
DNA methylation in the regulation of gene expression. For gene transcription to occur, transcription factors, RNA polymerase II, and other elements of the transcriptional machinery need to be able to access and bind to the gene promoter or enhancer regions. Unmethylated CpG sites within these regulatory regions enable binding of the transcriptional machinery, and gene is actively transcribed. On the other hand, methylation of the CpG sites in promoter or enhancer regions leads to recruitment of repressor complexes that prevent binding of the elements of the transcriptional machinery which results in gene silencing
DNA methylation is a covalent modification of the cytosine ring catalysed by DNA methyltransferases (DNMTs) predominantly within CpG dinucleotides in mammalian cells. Mammals have three enzymatically active DNMTs that are classified into two categories: maintenance (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2605) and de novo DNMTs (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2750 and DNMT3B). DNMTs transfer a methyl group donated by universal methyl donor https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4786 (SAM) to produce a methylated cytosine termed 5‐mC and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5265 (SAH; Cedar & Bergman, 2009; Figure 3). Two additional DNMTs, which do not possess catalytic activity towards DNA, have been described in mammalian cells, namely, DNMT2 and DNMT3L. The latter interacts with DNMT3A and presumably DNMT3B, thereby regulating their stability (Veland et al., 2019). Mechanistically, DNMT3L binds DNMT3A and consequently protects DNMT3A from degradation, contributing to DNMT3A‐dependent DNA methylation (Veland et al., 2019). Interestingly, DNMT2, the action of which was believed to be unknown for decades, can methylate RNA instead of DNA, targeting specifically cytosine 38 in the anticodon loop of aspartic acid tRNA (tRNAAsp; Goll et al., 2006).
Figure 3.
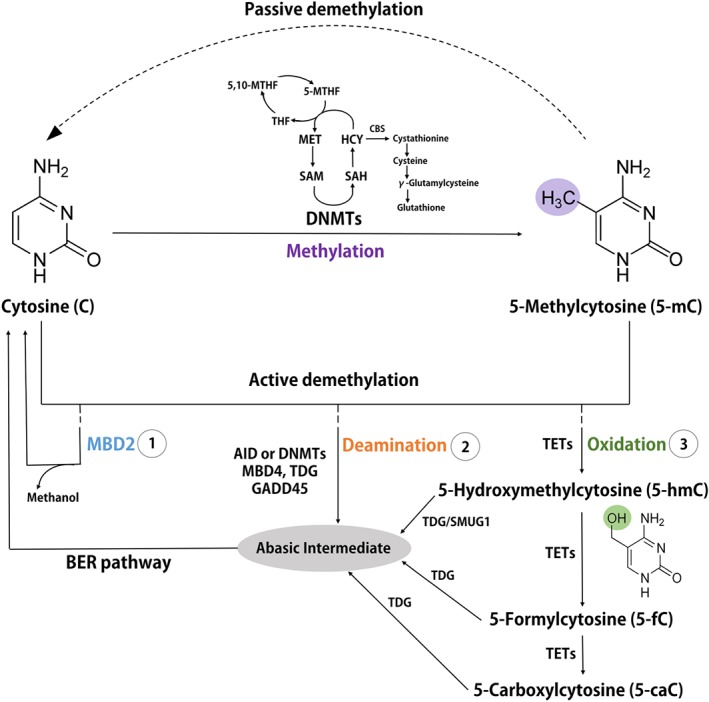
DNA methylation and demethylation occur through passive and active mechanisms. DNA methylation occurs when a methyl group is transferred to the fifth position of the cytosine ring (C ➔ 5mC) by DNA methyltransferases (DNMTs). S‐adenosyl‐l‐methionine (SAM) is the universal methyl donor for all methylation reactions, resulting in formation of S‐adenosyl homocysteine (SAH). SAM and SAH are part of the methionine cycle which converts methionine (MET) to SAM and SAH to homocysteine (HCY). The folate cycle (tetrahydrofolate [THF] ➔ 5,10‐methylene‐tetrahydrofolate [5,10‐MTHF] ➔ 5‐methyl‐tetrahydrofolate [5‐MTHF]) is then necessary to regenerate MET from HCY. HCY can also be shuttled into the trans‐sulfuration cycle by cystathionine β synthase (CBS), consisting of cysteine intermediates and results in formation of GSH. Passive DNA demethylation occurs by a reduction in activity or by absence of DNA methyltransferases (DNMTs) by dilution of 5‐mC through replication (dashed line). Active DNA demethylation can occur in one of three ways: (1) Demethylase activity of methyl binding domain 2 (MBD2) to convert 5‐mC to C and resulting in methanol as a by product. (2) Deamination of 5‐mC by activation‐induced cytidine deaminase (AID) family members or DNMTs. DNA glycosylases methyl binding domain 4 (MBD4) and thymine DNA glycosylase (TDG) yield an abasic intermediate which is converted to C through further base excision repair (BER). GADD45 repair proteins were also shown to be associated with DNA demethylation at this step. (3) Oxidation by ten‐eleven translocation methylcytosine dioxygenases (TETs) to form oxidized products 5‐hydroxymethylcytosine (5‐hmC), 5‐formylcytosine (5‐fC) and 5‐carboxylcytosine (5‐caC). DNA glycosylases such as TDG or SMUG1 yield an abasic intermediate which is converted to C through further BER
As with many chemical reactions, DNA methylation is regarded as being reversible; however, the search for DNA demethylating enzyme(s) can be challenging. Dividing cells lose DNA methylation passively when DNA replication takes place without a methylating enzyme present, which prevents the pattern of DNA methylation from being copied from a parental strand to the newly synthesized DNA strand. In terms of active demethylation, several proteins have been proposed to play a role, such as methyl‐CpG‐binding domain protein 4 (MBD4), growth arrest and DNA damage‐inducible protein GADD45 (GADD45), methyl‐CpG‐binding domain protein 2 (MBD2) and recently ten‐eleven translocation proteins (TETs; Figure 3). MBD4 and GADD45 are hypothesized to be involved in DNA demethylation by DNA repair‐based mechanisms where methylated cytosine is recognized, excised and replaced with unmethylated cytosine (Bhattacharya, Ramchandani, Cervoni, & Szyf, 1999). In 1999, a non‐DNA‐repair‐based mechanism for reversal of DNA methylation was proposed for the first time—with MBD2 as the major player (Bhattacharya et al., 1999). Although the exact mechanism of MBD2‐mediated demethylation remains unknown, the products of the reaction were reported to be non‐methylated cytosine and methanol (Bhattacharya et al., 1999). Despite the fact that several groups failed to confirm the demethylase activity of MBD2, the protein has been shown to bind to a significant fraction of hypomethylated genes and to determine both RNA polymerase II binding and the DNA methylation state in cancer cells (Stefanska et al., 2011; Stefanska et al., 2013). In addition, MBD2 depletion was shown to increase promoter methylation and silence several genes hypomethylated in tumours (Stefanska et al., 2011). Almost 10 years later, active enzymatic reversal of DNA methylation has been shown to be catalysed by proteins from the TET family (Hamm et al., 2008). The mechanism has been well described and involves oxidation of 5‐mC to 5‐hydroxymethylcytosine (5‐hmC) followed by the release of a methyl group in formaldehyde (Hamm et al., 2008). 5‐hmC can be further converted to 5‐formylcytosine (5‐fC) and 5‐carboxylcytosine (5‐caC) through consecutive oxidation reactions catalysed by TETs (Hamm et al., 2008; Figure 3). 5‐fC and 5‐caC can be excised by thymine DNA glycosylase (TDG) and replaced by unmodified cytosine by base excision repair (BER). The modified bases are also believed to be intermediates in the process of DNA demethylation by, for example, impairing maintenance of DNA methylation during DNA replication (Hamm et al., 2008).
The pattern of DNA methylation, established through dynamic DNA methylation and DNA demethylation, changes over the course of development and aberrant DNA methylation patterns have been linked to numerous human diseases, including chronic diseases (Barajas‐Olmos et al., 2018; Fernandez‐Sanles, Sayols‐Baixeras, Subirana, Degano, & Elosua, 2017; Lashley et al., 2015; Vucic et al., 2014).
2.1. Cancer and DNA methylation
Alterations in DNA methylation have been shown to underlie initiation, promotion and progression of cancer by providing a transcriptional environment susceptible to changing expression of key genes, clusters of genes, functional gene networks and signalling pathways (Baylin & Jones, 2011). Regulatory regions of tumour suppressor genes (e.g., MLH1, BRCA1, FANCF, and CHFR) are commonly hypermethylated leading to subsequent gene silencing, whereas oncogenes and pro‐metastatic genes (e.g., MMP2, PLAU, S100A5, MYCN, BCL2L10 and CTNNB1) lose methylation within promoters or enhancers, which results in gene activation in cancer (Saghafinia, Mina, Riggi, Hanahan, & Ciriello, 2018; Stefanska et al., 2011). In addition to aberrant silencing or activation of genes, global loss of DNA methylation, mainly within transposons and other repetitive sequences (e.g. LINE‐1 and Alu), contributes to chromosomal rearrangements and genome instability (Baylin & Jones, 2011). With the development of new genome‐wide technologies in the last decade, numerous studies in various types of cancers confirmed the altered DNA methylation patterns present in cancer, as described above. Tremendous progress in the field led to the launching of several international projects, such as the Human Epigenome Project, Encyclopedia of DNA Elements (ENCODE) Project, The Cancer Genome Atlas, and Epigenomics Consortium, amongst others, many of which work today together under The International Human Epigenome Consortium. The aim of these initiatives continues to be to sequence the entire human epigenome and establish epigenetic signatures in different tissues and different diseases (Roadmap Epigenomics et al., 2015).
Several studies have also identified changes in the pattern of genome‐wide DNA hydroxymethylation in cancer versus normal tissue, however the role of these aberrations in shaping the landscapes of DNA methylation/demethylation and consequently gene expression, is not yet fully understood (Rasmussen & Helin, 2016). It is generally observed that 5‐hmC levels are globally decreased in cancer cells (Rasmussen & Helin, 2016). Cancer‐specific mutations in TET2 and metabolic enzymes that inhibit TET enzymatic activity further implicate the loss of 5‐hmC and consequent alterations in DNA methylation in carcinogenesis (Rasmussen & Helin, 2016). However, differential changes at loci‐specific 5‐hmC levels add a new layer of complexity to the role of 5‐hmC in regulation of gene expression. Some studies report that 5‐hmC is specifically enriched at open chromatin, including gene enhancers, and that the gain of 5‐hmC is correlated with increased transcription in cancer (Bhattacharyya et al., 2017). Other investigations report that increased 5‐hmC density in gene bodies rather than regulatory regions is associated with actively transcribed genes (Bhattacharyya et al., 2017). The relationship between the distribution of 5‐hmC peaks and gene expression seems to depend on tissue type and appears to be directed by histone marks (Bhattacharyya et al., 2017). Although 5‐hmC is believed to be an intermediate of DNA demethylation and thereby expected to lead to gene activation, its role in the regulation of gene transcription may go beyond this function. Several studies indicate that 5‐hmC may attract or repel DNA‐binding proteins and consequently regulate chromatin structure and gene expression (Gao et al., 2019). During cell differentiation, 5‐hmC may regulate alternative splicing by marking exons for inclusion (Gao et al., 2019). These reports could explain the presence of 5‐hmC not only at active sites of transcription but also at sites of condensed chromatin (Gao et al., 2019).
2.2. Atherosclerosis and DNA methylation
Increasing evidence has revealed that DNA methylation has distinct roles in controlling gene expression of key players in atherosclerotic vascular tissues and consequently in CVD (Fernandez‐Sanles et al., 2017). The CpG island in the promoter region of ohttps://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=620 (ERα), a gene that protects against atherosclerosis through governing smooth muscle cell (SMC) proliferation, has consistently been found to be differentially methylated in atherosclerosis patients as compared to healthy controls (Huang, Zhi, & Wang, 2009). In healthy tissue, ERα increases the expression of https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=253, resulting in reduced SMC proliferation and platelet formation (Fernandez‐Sanles et al., 2017). During early atherogenesis, DNA hypermethylation of the ERα gene promoter contributes to gene silencing and as a consequence to the abnormally high proliferation of SMCs within atherosclerotic lesions (Fernandez‐Sanles et al., 2017). On the other hand, reduced DNA methylation in the promoter region of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998, a major player in the inflammation cascade, has been associated with greater production of cytokines from macrophages during atherogenesis (Yang, Zhao, Zhang, & Chen, 2016).
These are just a few examples amongst many others which suggest that DNA methylation plays an important function in regulating the chronic inflammatory response in arteries during the development and progression of atherosclerosis. Considering alterations in DNA methylation may address some of the current gaps in knowledge regarding the development of atherosclerosis and consequent CVD.
2.3. COPD and DNA methylation
Genome‐wide DNA methylation profiles and gene expression analyses in parenchymal cells, small airways and lung tissue suggest that aberrant DNA methylation patterns occur in numerous genes linked to functions and pathways important to COPD (Clifford et al., 2018; Morrow et al., 2018; Vucic et al., 2014). In a study examining homogenized lung tissue samples from patients with COPD, the top differentially methylated sites included https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=13 (M1), which is related to the PI3K‐Akt signalling pathway and bronchoconstriction of the airways, as well as glycosyltransferase 1 domain containing 1 (GLT1D1) and deltex E3 ubiquitin ligase 1 (DTX1), implicated respectively in reduced responses to LT modifiers in the treatment of asthma and in the development of natural killer T cells through regulating Notch signalling (Morrow et al., 2018).
Another study found multiple genes involved in key stress response pathways such as https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2497ue (PTEN) signalling, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=3057 (Nrf2)/antioxidant response element (ARE), and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5873 inflammatory response pathways, to be hypermethylated and down‐regulated in small airways of patients with COPD (Vucic et al., 2014). Interestingly, studies indicate that DNA methylation profiles differ between cell types, suggesting that DNA methylation contributes differently to the disease depending on tissue type and cell function in the lung (Clifford et al., 2018). For example, Clifford et al. (2018) found 887 differentially methylated regions (DMRs) in the airway fibroblasts of COPD individuals and 44 DMRs in parenchymal fibroblasts. Different genes were associated with the DMRs of each of these tissue types. Based on these reports, DNA methylation likely plays an essential role in contributing to the modulation of important genes implicated in COPD pathology.
2.4. T2D and DNA methylation
Recent discoveries surrounding the aetiology of T2D have shown that epigenetic modifications, in particular DNA methylation, have a significant role in pathogenesis of the disease. Studies have identified altered DNA methylation patterns in tissues of importance for T2D, including the pancreas, liver and adipose tissue (Barajas‐Olmos et al., 2018). Impaired insulin secretion from pancreatic β cells is central to the development and progression of T2D. High levels of DNA methylation in key genes important for proper functioning of pancreatic cells have been detected in cell and animal models of T2D. One of the master regulators of β‐cell maturation and survival, namely, pancreatic and duodenal homeobox 1 (PDX1) transcription factor, was found to be hypermethylated and silenced in patients with T2D compared to non‐diabetic donors (Hall et al., 2013). In addition, INS, which encodes for insulin and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2497 (GLP1R), which enhances insulin secretion, were both hypermethylated in the pancreatic islets of hyperglycaemic rats (Hall et al., 2013). In human pancreatic islets, Hall et al. (2013) further revealed that hypermethylation of GLP1R is positively associated with increased levels of glycated haemoglobin (HbA1c), an indicator of high glucose concentrations in the blood (Hall et al., 2013). These aberrations in functionally important genes may be linked to altered insulin secretion and sensitivity in T2D patients.
It is also well documented that stress exposure in utero is associated with developmental origins of T2D in accordance with the Barker hypothesis of the developmental origins of disease (Barker et al., 1993) and this link is mediated through changes in DNA methylation patterns in the fetus (Bansal & Simmons, 2018). For instance, the link between epigenetic silencing of a gene encoding for leptin in placental tissues of the offspring exposed to gestational diabetes and higher risk for T2D in adulthood has been described (Bansal & Simmons, 2018). Furthermore, low or high birth weight as a result of malnutrition during pregnancy has been strongly associated with later‐life risk of T2D (U‐shaped relation; Barker et al., 1993). A recent study of blood DNA methylation profiles in adolescents, born to mothers who lived through the Great Ice Storm in Quebec province of Canada in 1998, confirms that adverse maternal exposures (i.e. high‐level stress) predispose the offspring to metabolic disturbances later in life, including T2D, through alterations in DNA methylation patterns (Cao‐Lei et al., 2018).
2.5. AD and DNA methylation
The pathogenesis of AD is still not fully understood, but alterations in DNA methylation have been shown to be associated with AD. Interestingly, global, genome‐wide, and candidate gene DNA methylation studies conducted to date have yielded inconclusive results. Some studies have shown that there is a significant decrease in global DNA methylation in the temporal cortex, frontal cortex and hippocampus of individuals with AD compared to individuals without AD (Lashley et al., 2015). Other studies using similar techniques have obtained the opposite results (Lashley et al., 2015). Studies that analysed the DNA methylation status of promoters of genes involved in the pathophysiology of AD in human postmortem brain tissue, such as amyloid β precursor protein (APP), https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2402 (PSEN1), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9275 (MAPT), apolipoprotein E (APOE) and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2330 (BACE1), have also produced results that contradict with other published reports or show no significant differences between AD and control individuals (Iwata et al., 2014). The discrepancy between findings could be attributed to differences in methodology, lack of standardized techniques, variations across different brain regions and the use of a mixture of brain cells of different types. A recent study using the genome‐wide Illumina 450K methylation microarray platform revealed a high variability of DNA methylation profiles between AD patients that may additionally contribute to challenges in studying DNA methylation in AD (Huo et al., 2019). Moreover, they found that most of the identified variably methylated probes do not overlap with differentially methylated probes, indicating that they may be linked to different sets of genes associated with AD pathology (Huo et al., 2019). Although more studies are required to overcome the challenges, the possible contribution of DNA methylation in the pathophysiology of AD is highly supported.
3. OXIDATIVE STRESS UNDERLIES CHRONIC DISEASE
Oxidative stress is an imbalance between the production of reactive oxygen species (ROS) or reactive nitrogen species (RNS) and the elimination by antioxidant defences, which will inevitably lead to oxidative damage. ROS are oxygen‐derived molecules, including the hydroxyl radical (OH•), superoxide radical (O•) and lipid peroxyl radical (LOO•). RNS include https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2509; the reaction of NO with superoxide generates highly reactive peroxynitrate (ONOO•) radicals. Antioxidant and detoxification defence systems are critical in maintaining the balance by employing pro‐oxidant and antioxidant enzymes (Huang, Li, Su, & Kong, 2015; Vomund, Schafer, Parnham, Brune, & von Knethen, 2017). If the antioxidant system is ineffective, oxidative stress can lead to damage of lipid, DNA, RNA and proteins. Main regulators of the antioxidant response are the master transcription factor Nrf2 and the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2757 (Keap1), which were shown to be affected by dietary molecules (Huang et al., 2015; please see Section 4 for details; Figure 4). Under normal conditions, Nrf2 localizes in the cytoplasm where it interacts with Keap1 and is subsequently degraded by the ubiquitin‐proteasome pathway (Vomund et al., 2017). However, under oxidative stress conditions, Nrf2 dissociates from Keap1 and translocates to the nucleus to transactivate several cytoprotective genes that combat oxidative stress, such as SOD, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2979 (CAT), https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1441 (HO‐1), and NAD(P)H:quinone oxidoreductase (NQO1;Huang et al., 2015 ; Vomund et al., 2017). As Nrf2 plays a central role in protecting cells from damage, it has also been implicated in the prevention of major diseases. Many studies have demonstrated that inducing activation of Nrf2 is a promising approach for preventing carcinogenesis and other diseases, such as CVDs, neurodegenerative diseases, and pulmonary injury (Huang et al., 2015). Below, we discuss the role of oxidative stress in chronic disease and throughout this review highlight the ability of dietary compounds to scavenge ROS/RNS and/or activate Nrf2 as potential mechanisms to combat oxidative stress.
Figure 4.
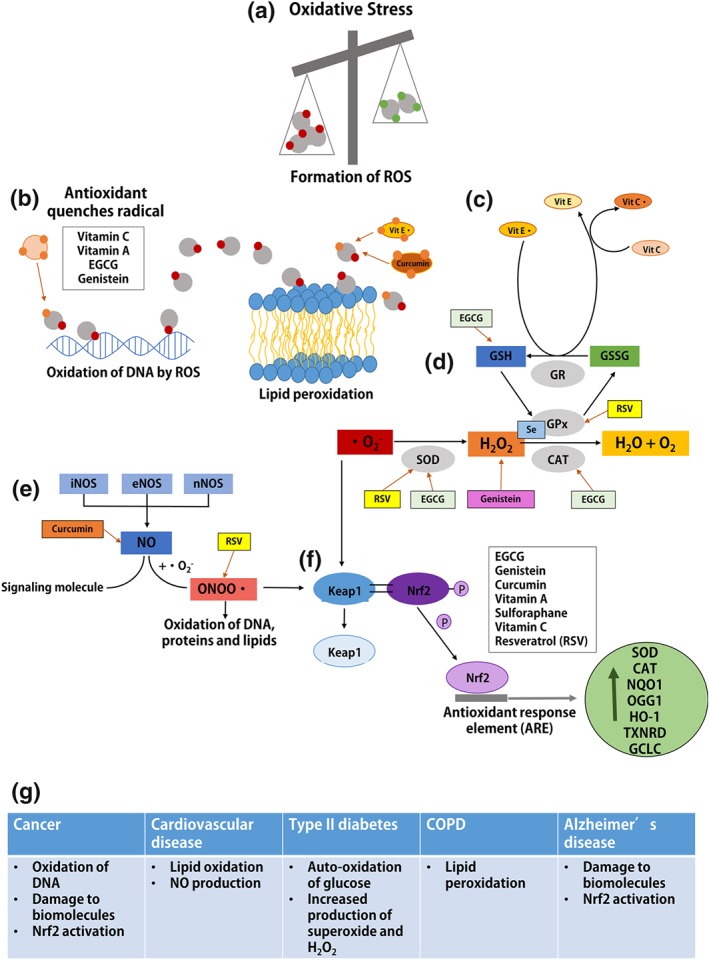
Mechanisms of oxidative stress are targets of antioxidant dietary compounds. (a) Oxidative stress occurs when there is an imbalance between ROS and antioxidant defence mechanisms. ROS target biomolecules such as DNA, proteins and lipids and impart oxidative damage which underlies many diseases. (b) Natural antioxidants such as vitamin C, vitamin A, EGCG, and genistein can neutralize free radicals by donating an electron, thus inhibiting oxidative DNA damage. Lipid soluble antioxidants, vitamin E, and curcumin prevent lipid peroxidation by quenching free radicals and protecting polyunsaturated fatty acids (PUFAs) from peroxidation. (c) The interconnection of vitamin E, vitamin C, and GSH reductase (GR) is shown, demonstrating the function of regeneration of antioxidants when oxidized. Vitamin C regenerates the reduced form of vitamin E which utilizes GR to convert GSSG to its reduced form, GSH. EGCG promotes cellular redox balance through increasing the activity of GSH. (d) SOD catalyses the reduction of superoxide anion (O2 •−) to hydrogen peroxide (H2O2), which is subsequently neutralized into water and oxygen by GSH peroxidase (GPx) and/or catalase (CAT). Antioxidants including resveratrol (RSV), EGCG, and selenium (Se) enhance the activity of specific antioxidant enzymes (SOD, CAT, and GPx) to contribute to alleviation of oxidative stress. Additionally, genistein scavenges H2O2 and free radicals leading to inhibition of H2O2‐induced oxidative damage. (e) Three isoforms of NOS, iNOS, eNOS, and nNOS (inducible, endothelial, and neuronal) can generate NO. NO can act as a signalling molecule or it can react with superoxide anion (O2 •−) generating peroxynitrite anion (ONOO−), which is a highly reactive nitrogen species. Thus, ONOO− can trigger oxidation of DNA, proteins and lipids. Antioxidants such as curcumin and resveratrol promote redox balance by targeting NO and ONOO− concentration respectively. (F) Cellular oxidative stress caused by generation of ONOO− and O2 •− stimulates nuclear factor erythroid 2‐related factor 2 (Nrf2) to dissociate from its repressor protein (Keap1) and translocate from cytoplasm into the nucleus to bind to antioxidant response elements (AREs) and activate transcription of antioxidant‐related genes. Natural antioxidants shown in the grey box have been shown to activate Keap1‐Nrf2 pathway. (g) Table of major contributors to oxidative stress in each chronic disease
3.1. Cancer and oxidative stress
Chronic inflammation has been associated with the development of numerous cancer types, thus implicating oxidative stress as a driver of carcinogenesis (Ding et al., 2016; Ding et al., 2019; Maiuri & O'Hagan, 2016; O'Hagan, 2014; O'Hagan et al., 2011; O'Hagan, Mohammad, & Baylin, 2008). Cancer cells are exposed to ROS through intrinsic and extrinsic sources. Intrinsic sources include ROS produced by mitochondria because of the increased metabolic activity of cancer cells and ROS produced by https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=993 that are activated by cancer‐related signalling pathways. Extrinsic sources include ROS released by neutrophils and macrophages present in the tumour micro‐environment. Studies have shown that oxidative stress affects all stages of carcinogenesis (Huang et al., 2015; Vomund et al., 2017). Most notably, oxidative stress has been reported to activate several transcription factors involved in inflammation and immune response, such as NFκB, AP‐1, p53, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=595, and Nrf2 (Huang et al., 2015). As mentioned previously, Nrf2 is considered to be a master transcription factor that controls expression of many antioxidant enzymes and it has been suggested that activating Nrf2 may reduce cancer formation (Vomund et al., 2017). However, persistent Nrf2 activation in cancer cells can also contribute to carcinogenesis by increasing the detoxification capacity of the cell leading to therapeutic resistance.
Oxidative stress associated with cancer can lead to alterations in intracellular homeostasis and damage of biomolecules such as DNA, RNA, lipids and proteins and to alterations in antioxidant defence and DNA repair mechanisms (Vomund et al., 2017; Figure 4).
3.2. Atherosclerosis and oxidative stress
Atherosclerosis is a multifactorial disease involving the interplay of endothelial dysfunction, matrix degradation, inflammation and oxidative stress (Yang et al., 2017). Increased mitochondrial oxidative stress and generation of excessive ROS are associated with atherosclerosis in humans and correlate with the severity and progression of atherosclerosis (Yang et al., 2017). In addition, the production of free oxygen radicals has been shown to initiate oxidation of lipids and proteins that impact vascular SMC migration and expression of adhesion molecules in the endothelium (Yang et al., 2017). Oxidation of polyunsaturated fatty acids (PUFAs), for example, n‐3 and n‐6, generates highly reactive unsaturated hydroxyalkenals which form protein adducts with lipoproteins. Malonaldehyde and acrolein are secondary products of lipid peroxidation and also react with lysine residues that lead to LDL modification. High levels of oxidized lipids damage blood vessels leading to macrophage activation and foam cell formation, indicating a crucial role of oxidative stress in atherogenesis (Kattoor, Pothineni, Palagiri, & Mehta, 2017). For instance, under normal physiological conditions, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1249 (eNOS) synthesizes NO, which has a vasoprotective role and studies have shown that endothelial NO prevents the uptake of LDL and reduces oxidized LDL (oxLDL) accumulation in the vascular wall (Kattoor et al., 2017). However, factors predisposing to atherosclerotic CVD, such as hyperlipidaemia and hypertension, induce vascular oxidative stress and reduce endothelial NO production. Hence, underproduction of NO leads to lipid oxidation and ROS generation in the blood vessel wall (Kattoor et al., 2017; Figure 4). Furthermore, studies have shown that several NADPH oxidases (Nox1, Nox2, Nox4, and Nox5) are important ROS generating systems and play crucial roles in atherogenesis (Kattoor et al., 2017). In animal models of atherosclerosis, Nox2 deficiency in apolipoprotein E‐null (ApoE−/−) mice resulted in significantly less atherosclerosis compared to ApoE−/− mice with elevated levels of Nox2 (Kattoor et al., 2017). This observed reduction in atherosclerosis is correlated with decreased ROS generation and increased NO bioavailability.
Overall, inflammation and oxidative stress are driving mechanisms involved in the pathogenesis of atherosclerosis. Understanding mechanisms affected by oxidative stress and its specific targets in atherosclerosis is essential for therapeutic strategies for the prevention and treatment of CVD.
3.3. COPD and oxidative stress
Chronic inflammation and oxidative stress are important features in the pathogenesis of COPD. Increased oxidative stress in COPD patients is caused by the increased burden of inhaled oxidants that lead to increased amounts of ROS produced in the epithelial cells of the airways (Rahman, 2008). Studies have demonstrated increased ROS in the bronchoalveolar lavage fluid of patients with COPD and increased footprints of oxidative stress both locally (lung) and systemically (blood and urine) in COPD patients (Rahman, 2008). Oxidative stress contributes to the pathogenesis of COPD through oxidative inactivation of antiproteases and surfactants, mucus hypersecretion, membrane lipid peroxidation, mitochondrial respiration, alveolar epithelial injury, remodelling of extracellular matrix and apoptosis (Rahman, 2008; Figure 4).
3.4. T2D and oxidative stress
Chronic inflammation and generation of ROS are amongst the most important factors for the pathogenesis of T2D (Rehman & Akash, 2017). Insulin resistance, hyperglycaemia, and excess release of fatty acids from adipose tissue induce oxidative stress, leading to inflammation and vascular complications in patients with T2D (Rehman & Akash, 2017). Studies suggest that insulin resistance occurs prior to hyperglycaemia development and factors such as TNF‐α, free fatty acids (FFAs), and leptin are known mediators of oxidative stress‐induced insulin resistance (Rehman & Akash, 2017). Hyperglycaemia also appears to enhance continuous production of free radicals via auto‐oxidation of glucose and stimulation of hexosamine pathway flux (Rehman & Akash, 2017). In addition, hyperglycaemia seems to be an important factor in PKC‐dependent activation of NAD(P)H oxidase in vascular tissues and kidney leading to increased oxidative stress in diabetic animals (Fonseca et al., 2011). Studies have shown that one of the key features of T2D is elevated circulating FFAs, which leads to increased superoxide production (Rehman & Akash, 2017). This causes mitochondrial dysfunction and reduces endogenous antioxidant defence required for restoring redox balance in T2D patients. It has been suggested that increased FFA concentration also stimulates NFκB activation, indicative of its function as a pro‐inflammatory agent in the progression of T2D (Fonseca et al., 2011). In addition, hyperglycaemia has been shown to disrupt endoplasmic reticulum (ER) homeostasis, a key player in insulin biosynthesis (Fonseca et al., 2011). ER stress leads to the progressive loss of pancreatic β cells and generation of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2448, an important source of ROS during the development of diabetes (Fonseca et al., 2011; Figure 4). Considering the damaging effects of cellular ROS in the pathogenesis of T2D and its related complications, it is anticipated that combating oxidative stress and inflammation can provide effective therapeutic strategies for the management of this chronic disease.
3.5. AD and oxidative stress
It has been suggested that oxidative stress also has a significant role in the aetiology and pathogenesis of AD (Tonnies & Trushina, 2017). ROS levels increase with age, which, together with other hallmarks of AD, such as loss of mitochondrial function, altered metal homeostasis and reduced antioxidant defence, results in the impairment in synaptic activity and neurotransmission in the brain and consequently to cognitive dysfunction (Tonnies & Trushina, 2017). One of the major antioxidant pathways, namely, the Nrf2 pathway has been reported to be down‐regulated in the hippocampal neurons and astrocytes of AD patients (Tonnies & Trushina, 2017). ROS can also contribute to abnormal cellular metabolism by oxidizing molecular targets such as nuclear and mitochondrial DNA, lipids and proteins (Tonnies & Trushina, 2017; Figure 4). This consequently results in the development of AD because it promotes the production and aggregation of amyloid‐β (Aβ) and hyperphosphorylated tau protein. The latter two elements worsen mitochondrial dysfunction and ROS production, thereby contributing to the vicious cycle that exacerbates the disease process and which eventually leads to neuronal death (Tonnies & Trushina, 2017).
4. ANTIOXIDANT DIETARY COMPOUNDS AND THEIR CAPACITY TO COMBAT OXIDATIVE STRESS
Compounds that have an antioxidant capacity can alleviate oxidative stress in environments of inflammation and immunotoxicity by inhibiting the formation of free radicals or by interrupting propagation of the free radical (Mu, Yu, & Kitts, 2019). Antioxidants function as radical scavengers, hydrogen donors, electron donors, peroxide decomposers, singlet oxygen quenchers, enzyme inhibitors, synergists and/or as metal‐chelating agents (Kurutas, 2016). Antioxidants may also work indirectly through interactions with other oxidative enzymes and cell signalling pathways important to control oxidative stress (Kurutas, 2016).
https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4781 (ascorbate), vitamin E (tocopherols and tocotrienols), and vitamin A (β‐carotene with other carotenoids, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4053) are common dietary antioxidants that function to react with free radicals and subsequently decrease oxidative damage (Traber & Stevens, 2011; Table 1). Selenium is also classified as an antioxidant because of its cofactor function in selenoproteins that possess antioxidant activities (Lipinski, 2019).
Table 1.
A list of antioxidant compounds with the structure, food source, mechanisms of antioxidant capacity, and mechanisms of modulation of DNA methylation
| Compound | Structure | Food source | Antioxidant capacity | DNA methylation mechanism | References |
|---|---|---|---|---|---|
|
Vitamin C |
|
Guava, peppers, papaya, kiwi, orange, strawberries, pineapple, grapefruit, broccoli |
• Free radical scavenger (O− 2, H2O2, ClO−, •OH, HO2, O2) • Regeneration of vitamin E • Nrf2 activator (through TET‐mediated DNA demethylation) |
• Provider of reduced iron which is required for TET function and DNA demethylation | Kurutas, 2016; Traber & Stevens, 2011 |
|
Vitamin E |
|
Wheat germ oil, sunflower oil, hazelnut, almonds, lettuce, camembert cheese, apples, carrot, wheat bread, bananas, cow's milk |
• Inhibition of lipid peroxidation (lipid soluble) • Reactivity with organic peroxyl radicals (ROO•) • O2 quencher • Nrf2 activator |
• Associated with decreased DNMT expression | Huang et al., 2012; Niki, 2014; Zappe et al., 2018 |
|
Vitamin A |
Dairy products, fish, meat (especially liver), sweet potatoes, carrots, swiss chard, kale |
• Singlet oxygen (O2.) quencher • Traps peroxyl radicals (HO2) • Nrf2 pathway activator • NF‐kB and MAPK pathway inhibitor |
• Associated with decreased expression of DNMT1 and DNMT3B (microRNA‐mediated |
Ben‐Dor et al., 2005; Bohn, 2019; Das et al., 2010; |
|
|
Selenium |
Brazil nuts, oysters, liver (lamb, chicken, turkey, pork), canned tuna | • Involvement in selenoproteins (GSH peroxidase, selenoprotein P, thioredoxin reductase) → GSH: neutralizes hydrogen peroxide and other peroxides | • Associated with decreased activity of DNMTs through regulation of one‐carbon metabolism |
Bellinger et al., 2009; Jablonska & Reszka, 2017; Speckmann et al., 2017 |
|
|
Stilbenoids |
Pterostilbene
|
Blueberries, peanuts, almonds |
• O2 radical scavenger • Inhibits generation of superoxide anion (O2•−), hydroxyl radical (OH•) and hydrogen peroxide (H2O2) • Nrf2 activator • Activates AMPK and induces NOS |
• Reduced expression of DNMT1 (PTEN, AP‐1) • Regulation of the occupancy of DNMT3B at gene loci • Indirect enzymatic inhibition of DNMT1 (p21 and PCNA competition) |
Erasalo et al., 2018; Beetch et al., 2018; Hwang et al., 2008; Iida et al., 2002; Kosuru et al., 2018; Lubecka et al., 2016; Ren et al., 2011; |
| Resveratrol
|
Grapes (red wine), mulberries, cocoa, peanuts | ||||
|
Epigallocatechin gallate (EGCG) |
|
Black and green tea, chocolate, apricots, apples, grapes (red wine), legumes |
• Free radical scavenger • Decreases lipid peroxidation • Decreases ROS and NADPH production • Decreases H2O2‐induced apoptosis • Nrf2 activator • Activates MAPKs |
• Direct enzymatic inhibition of DNMT1 and DNMT3B (binding) • Indirect enzymatic inhibition of DNMTs (COMT, p21) |
Fang et al., 2003; Khan et al., 2015; Lee et al., 2005; Lee et al., 2006; Mao et al., 2017; Mirza et al., 2013; Sun et al., 2017 |
|
Genistein |
|
Soy and soy products, peanuts, beans |
• Hydrogen atom donation from the hydroxyl group attached to the benzene ring • Effective scavenger of H2O2 and free radicals • Activation of MnSOD • Inhibits lipid peroxidation and NADH oxidase • Nrf2 activator |
• Direct enzymatic inhibition of DNMT1 (binding) • Reduced expression of DNMT1 (PTEN, AP‐1) |
Bai et al., 2019; Borras et al., 2006; Dave et al., 2005; Rahman Mazumder & Hongsprabhas, 2016; Xie et al., 2014 |
|
Curcumin |
|
Turmeric |
• Scavenges superoxide and hydroxyl radicals • Inhibits lipid peroxidation (lipid soluble) • Chelate with lead and cadmium • Activator of Nrf2 pathways |
• Direct enzymatic inhibition of DNMT1 (binding) |
Daniel et al., 2004; Lin et al., 2019; Liu et al., 2009; Manikandan et al., 2004 |
|
Sulforaphane |
|
Cauliflower, cabbage, broccoli, broccoli sprouts | • Nrf2 activator |
• Associated with decreased DNMT1 and DNMT3A expression • Direct putative binding to DNMT3B catalytic pocket • Up‐regulation of TET1 and increase in 5‐hmC |
Ali Khan et al., 2015; Kensler et al., 2013; Thaler et al., 2016; Zhang, Su, et al., 2013 |
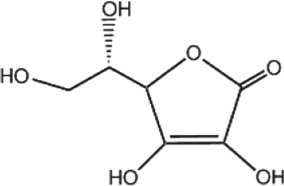
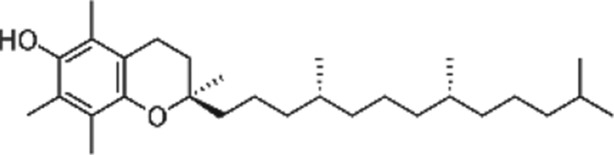
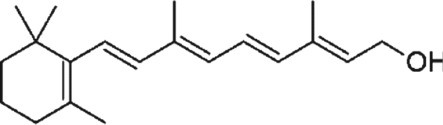
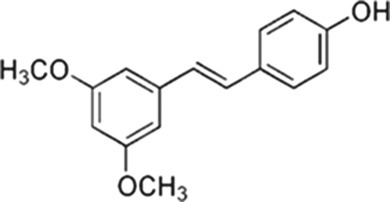
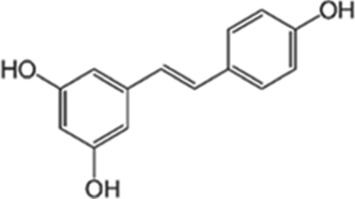
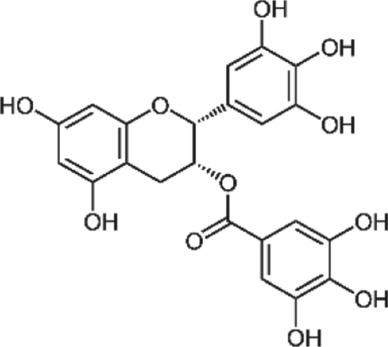
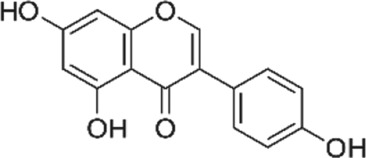
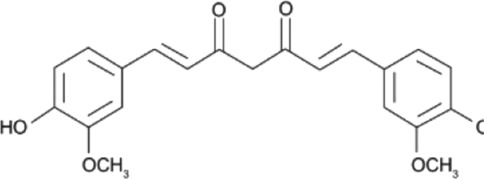
There are several phytochemicals that have potentially important biochemical properties and can be derived from the diet (Table 1). They have been shown to be involved in modulating several biological processes that include antioxidant, detoxification, apoptosis, anti‐inflammatory and anti‐carcinogenic activities. Majority of well‐studied phytochemicals belong to a large class called polyphenols. Polyphenols are a group of highly diverse natural products, chemically characterized as having phenolic structural features and possessing antioxidant activity. They are subdivided into different categories such as flavonoids, stilbenes, lignans, phenolic acids and others, according to the chemical structures of the aglycones. Phenolic acids represent a subgroup of acids that are non‐flavonoid polyphenolic compounds, which are divided into benzoic acid and cinnamic acid derivatives. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5155 and chlorogenic acids are examples of naturally occurring cinnamic acids found in many fruits and vegetables, as well as coffee. They are free radical and metal scavengers and have a role in modulating cell signalling pathways that regulate antioxidant and anti‐inflammatory reactions. Flavonoids, another category of phytochemicals, have mostly been studied from soy and citrus sources. Isoflavonoids derived from soy, namely https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2826 and daidzein, have reported antioxidant, anti‐carcinogenic activities that have been linked to epidemiologic evidence of lower risk of cancer and heart disease (Setchell & Cassidy, 1999). Soy isoflavones also have been shown to attenuate oxidative damage related to aging and AD (Hsieh, Wu, & Hu, 2009). Citrus sources of flavonoids include https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5346 and kaempferol, with well documented antioxidant, anti‐glycation, and anti‐inflammatory activities (Suantawee, Cheng, & Adisakwattana, 2016). Flavanols, such as catechin, found in tea form polymers that are referred to as proanthocyanidins and are potent antioxidants (Hu & Kitts, 2001). As much as 80% of green tea contains a mixture of catechins, namely, epigallocatechin (EGC), epicatechin‐3‐gallate (ECG) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7002 (EGCG), with the majority being the sum of EGC and EGCG.
Stilbenoids (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8741 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2681), genistein, epigallocatechin gallate (EGCG), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6569 (SFN), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7000, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4136 and coffee phenolic acids, for example, chlorogenic acid and caffeic acid are effective modulators of enzymatic antioxidants that originate from cellular regulation of redox balance. An example of this includes the capacity to activate the Nrf2 master transcription factor that modulates hundreds of genes involved in antioxidant processes and phase II enzyme detoxification (Kensler et al., 2013; Liang, Dupuis, Yada, & Kitts, 2019; Liang & Kitts, 2018). Interestingly, EGCG, genistein, and curcumin have also been shown to scavenge free radicals directly and inhibit lipid peroxidation (Hodaei, Adibian, Nikpayam, Hedayati, & Sohrab, 2019; Mao, Gu, Chen, Yu, & He, 2017; Saraf‐Bank, Ahmadi, Paknahad, Maracy, & Nourian, 2019). The antioxidant capacity of the aforementioned dietary compounds to alleviate oxidative stress are summarized in Figure 4 and Table 1.
4.1. Vitamin C
Vitamin C exists in the reduced form, called https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4781, and in the oxidized form, called dehydro‐ascorbic acid. It can accept or donate electrons and thus works directly as an antioxidant by scavenging superoxide, hydrogen peroxide, hypochlorite, the hydroxyl radical, peroxyl radicals, and the singlet oxygen (Traber & Stevens, 2011; Figure 4 and Table 1).
Another crucial antioxidant mechanism of vitamin C is to restore the antioxidant properties of vitamin E and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6737. The hydroxyl group of α‐tocopherol, an active reduced form of vitamin E, reacts with peroxyl radicals and is converted to tocopheroxyl radicals, which becomes an inactive form of vitamin E (Traber & Stevens, 2011). Vitamin C can react with tocopheroxyl radical and return it to a reduced, regenerated state. It is through this action with vitamin E that the hydrophilic vitamin C molecule protects membrane lipids and in turn triggers antioxidant defence mechanisms (Elisia & Kitts, 2013b; Niki, 2014; Traber & Stevens, 2011). Upon reduction of tocopheroxyl radicals, the formed ascorbyl radical enters the GSH‐ascorbate cycle, which detoxifies hydrogen peroxide, and is reduced by GSH‐dependent enzymes (Traber & Stevens, 2011). During this reaction, an oxidized form of GSH is formed, namely, GSH disulfide (GSSG), which is subsequently reduced by GSH reductase using NADPH as the electron donor (Traber & Stevens, 2011; Niki, 2014; Figure 4).
4.2. Vitamin E
Vitamin E is an important antioxidant, and its most common and biologically active form is α‐tocopherol, albeit different isoforms have varying capacities to scavenge free radicals (Elisia & Kitts, 2013a; Figure 4 and Table 1). Tocopherol is a singlet oxygen quencher and also donates an electron to the peroxyl radicals produced in the lipid peroxidation chain reaction (Niki, 2014). Peroxyl radicals react 1,000 times faster with α‐tocopherol than with PUFA, and thus, vitamin E prevents the propagation of the free radical chain reaction in cell membranes and plasma lipoproteins (Niki, 2014). Of note, vitamin E at high doses has pro‐oxidant properties by inducing microsomal lipid peroxidation upon oxidation to phenoxyl radicals (Tafazoli, Wright, & O'Brien, 2005). In a colorectal cancer cell model, Zappe et al. (2018) reported that vitamin E at lower 10‐μM concentration exerted a more potent effects in reducing oxidative stress compared to higher 50‐μM concentration (Zappe et al., 2018).
4.3. Vitamin A
Vitamin A consists of a group of organic compounds that include retinol, retinal, retinoic acid (RA) and several provitamin A carotenoids (most notably β‐carotene). Carotenoids are strong scavengers of O2 and peroxyl radicals (Bohn, 2019). Singlet oxygen scavenging by carotenoids is largely imparted to a physical quenching mechanism, however can also act via electron acceptance, donation, or via hydrogen abstraction/acceptance (Bohn, 2019; Kurutas, 2016). Carotenoids also have an important role in free radical reaction inhibition and protecting membranes from lipid peroxidation (Kurutas, 2016). They also act through interactions with cellular signalling cascades, such as NFκB, MAPK, or Nrf2 (Ben‐Dor et al., 2005; Figure 4 and Table 1).
4.4. Selenium
Selenium is an essential trace element and a major component of selenoproteins, exhibiting both antioxidant and anti‐inflammatory properties depending on concentration (Lipinski, 2019). There are approximately 25 selenoproteins that have been identified in humans, but the three major classes that are the most extensively studied are GSH peroxidase (GPx), thioredoxin reductase (TRXR), and iodothyronine deiodinases (IDD). Selenoproteins have critical roles in both the GSH‐dependent and TRX‐dependent antioxidant systems. GPX1, the most abundant selenoprotein, is a sensitive parameter to measure active selenium in the body (Bellinger, Raman, Reeves, & Berry, 2009). GPX1 is a cofactor for GSH peroxidase and catalyses the breakdown of peroxides into water (Figure 4 and Table 1). This process ultimately helps detoxify oxidized proteins and lipids (Bellinger et al., 2009).
4.5. Stilbenoids
Stilbenoid compounds, such as resveratrol and pterostilbene—the most abundant respectively in grapes and blueberries, are a class of polyphenols that have antioxidant, anti‐inflammatory and anti‐cancer properties (Erasalo et al., 2018; Soufi, Sheervalilou, Vardiani, Khalili, & Alipour, 2012). These compounds lead to scavenging of O2 radicals (e.g., superoxide anion, hydroxyl radical and hydrogen peroxide), inhibition of lipid peroxidation, and reduction of oxidative stress markers, such as TNF‐α, mainly via enhancing the activity of the antioxidant cellular network (Erasalo et al., 2018; Soufi et al., 2012). https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8741 has been shown to increase expression and translocation of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=3057 to the nucleus where Nrf2 acts as a transcription factor, increasing expression of ROS scavenging enzymes and antioxidant signalling molecules, such as SOD, CAT, HO‐1, NQO1, https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=988, γ‐glutamylcysteine synthetase and GSH‐S‐transferase (GST; Ren, Fan, Chen, Huang, & Yang, 2011). Similarly, activation of the Nrf2 antioxidant pathway is observed upon exposure to https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2681 in many disease models (Kosuru et al., 2018). Stilbenoids also have an important antioxidant role via activation of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1540 (AMPK) and induction of NOS (Hwang, Kwon, Park, & Kim, 2008; Figure 4 and Table 1). As a consequence, higher production of NO by NOS contributes to NO‐mediated inhibition of oxidation of lipids and proteins (Mu et al., 2019).
4.6. Epigallocatechin gallate
EGCG is the most active polyphenol found in green tea with antioxidant activities that are partially related to the oxidation of phenolic groups in EGCG structure (Mao et al., 2017). The compound acts as a radical scavenger and an electron donor, balancing cellular redox status and decreasing lipid peroxidation, ROS, and NADPH (Mao et al., 2017; Figure 4 and Table 1). For instance, EGCG at low concentration (<10 μM) prevented DNA damage caused by hydrogen peroxide in lymphoblastoid cells (Sugisawa, Kimura, Fenech, & Umegaki, 2004). Antioxidant treatment with EGCG has been suggested to prevent the progression of diabetes‐induced oxidative damage and diabetic neuropathy in animal models (Sun et al., 2017). Apart from ROS scavenging ability, activation of Nrf2 signalling has been proposed to be a major mechanism through which EGCG alleviates oxidative stress (Han, Han, Toborek, & Hennig, 2012; Shin, Jeon, Park, & Chang, 2016; Figure 4 and Table 1). A study by Sun et al. (2017) showed that EGCG prevented diabetes‐driven molecular changes associated with diabetic neuropathy such as fibrosis, oxidative stress and inflammation in a mouse model of streptozotocin‐induced diabetes (Sun et al., 2017). However, in Nrf2 knockout animals in the same study, the protective roles of EGCG were diminished indicating that the antioxidant action of EGCG is mediated through the Nrf2 pathway (Sun et al., 2017). Activation of Nrf2 consequently leads to up‐regulation of Nrf2 responsive genes, which can explain increased expression of antioxidant enzymes, such as SOD, GST, GSH and CAT, upon treatment with EGCG (Han et al., 2012; Shin et al., 2016). Another proposed mechanism of EGCG‐mediated up‐regulation of phase II antioxidant enzymes is via the MAPK signalling pathway (Han et al., 2012; Shin et al., 2016). Interestingly, the polyphenols in green tea can instead act as pro‐oxidants when used at high dose and in a certain disease state (Mao et al., 2017). In a disease like cancer, induction of additional oxidative stress by the pro‐oxidant effects of EGCG can lead to ROS‐mediated cell death of cancer cells providing a beneficial therapeutic outcome (Mao et al., 2017).
4.7. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2826
Genistein, a soy isoflavone, has been shown to have powerful scavenging capacity towards hydrogen peroxide (H2O2) and free radicals and protect cells against oxidative stress (Rahman Mazumder & Hongsprabhas, 2016; Figure 4 and Table 1). This potent action is associated with genistein's capacity to donate a hydrogen atom from the hydroxyl group attached to the benzene ring in its structure (Rahman Mazumder & Hongsprabhas, 2016). As a phytooestrogen, genistein can also mimic the action of oestrogen in regulating antioxidant genes (Borras et al., 2006). Briefly, genistein binds to oestrogen receptors and induces signal transduction leading to phosphorylation of ERK1/2 kinase, which activates NFκB and subsequently increases expression of the antioxidant enzyme manganese SOD (MnSOD; Borras et al., 2006). Furthermore, genistein was reported to inhibit microsomal lipid peroxidation induced by an Fe2+‐ADP‐complex and NADPH, suppress NADH oxidase and respiratory chain in rat liver mitochondria (Rahman Mazumder & Hongsprabhas, 2016). Similar to other natural antioxidant compounds, genistein was shown to maintain redox balance by activating the Nrf2 pathway and genes associated with ARE (Bai & Wang, 2019).
4.8. Curcumin
Curcumin, the most active constituent of turmeric, has been shown to exhibit potent antioxidant properties (Hodaei et al., 2019; Saraf‐Bank et al., 2019). It has been reported that the phenolic hydroxyl group present in curcumin plays an important role in the singlet oxygen, superoxide, and hydroxyl radical scavenger capacity of this compound (Figure 4 and Table 1; Manikandan et al., 2004). Curcumin was shown to indirectly reduce NO concentration in cells by trapping nitrogen dioxide, an important reaction intermediate, which then leads to redox balance (Lin et al., 2019). Interestingly, curcumin was reported to effectively chelate lead and cadmium, thereby blocking the free radical chain reaction induced by these heavy metals and subsequently protecting the rat brain from lipid peroxidation damage (Daniel, Limson, Dairam, Watkins, & Daya, 2004). Furthermore, curcumin induces the activity of phase II antioxidant enzymes through activating the Keap1/Nrf2/ARE pathway (Lin et al., 2019). Treatment of rats with myocardial ischaemia with curcumin has been shown to reduce levels of superoxide anion, xanthine oxidase and lipid peroxides with simultaneous increase in antioxidant enzymes, SOD, GPx, and GST (Manikandan et al., 2004).
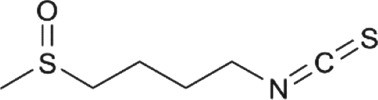
4.9. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6569
SFN is a phytochemical commonly found in cruciferous vegetables such as broccoli, brussels sprouts and cabbage. It has gained interest for its function as an indirect antioxidant since it can induce several ARE‐dependent antioxidant enzymes (e.g. NQO1, GCLC, GSRR and TXNRD) that help maintain cellular redox homeostasis in the Keap1/Nrf2 signalling pathways (Kensler et al., 2013; Figure 4 and Table 1).
5. INTERRELATIONSHIP BETWEEN ANTIOXIDANT PROPERTIES AND DNA METHYLATION MECHANISMS OF DIETARY COMPOUNDS
Studies show that bioactive compounds may exert antioxidant capacity, at least in part, via epigenetic mechanisms, specifically, remodelling of DNA methylation patterns. Examples of such mechanisms of action are discussed in the following paragraphs.
5.1. Vitamin C
5.1.1. Mechanisms behind changes in DNA methylation in response to vitamin C
There are several lines of evidence to show vitamin C is a required factor in the DNA demethylating process involving TET proteins. Vitamin C was observed to enhance 5‐hmC levels in embryonic cells in a dose‐ and time‐dependent manner. This increase in 5‐hmC levels was confirmed to be mediated by TET proteins in embryonic cells as well as cancer cells (Minor, Court, Young, & Wang, 2013). Accordingly, vitamin C has been investigated in disease models, specifically cancer, as a TET‐activating compound increasing 5‐hmC levels (Peng et al., 2018). For instance, in bladder cancer, where genome‐wide mapping of 5‐hmC revealed loss of 5‐hmC in cancer‐related genes, vitamin C treatment led to increased 5‐hmC levels at gene‐specific loci, a shift in the transcriptome profile and inhibition of the malignant phenotypes (Peng et al., 2018). A positive correlation between plasma concentration of vitamin C and hydroxymethylation of leukocyte DNA was further observed in patients with inflammatory bowel disease, adenomatous polyps and colorectal cancer (Starczak et al., 2018). Additionally, vitamin C was found to increase the efficacy of DNA demethylating agents, such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6805 (DAC) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6796 (AZA), that work through inhibition of DNMTs (Gerecke et al., 2018). Combination treatment of vitamin C with these DNMT inhibitors (DNMTi) reactivated tumour suppressor genes, for example, p21 in colon cancer and hepatocellular carcinoma (HCC) cells, through targeting both DNA methylation and demethylation mechanisms (Gerecke et al., 2018).
5.1.2. A link between antioxidant properties of vitamin C and vitamin C‐mediated changes in DNA methylation
Due to the antioxidant activity of vitamin C and the established role of the nutrient in DNA demethylation, studies in different chronic disease models have sought to investigate the relationship between vitamin C‐mediated changes in DNA methylation/demethylation and oxidative stress‐related pathways. An excellent connection between vitamin C and DNA methylation is a direct regulation of TET enzyme activity (Yin et al., 2013). As an antioxidant, vitamin C reduces ferric iron Fe3+ to ferrous iron Fe2+, making it available for the TET enzyme catalytic centre. TET activity, in turn, converts a methylated cytosine to 5‐hmC and further oxidized forms (Figure 3), which eventually leads to DNA demethylation and gene activation, including genes from the Nrf2 antioxidant pathway (Yin et al., 2013; Peng et al., 2018; Figure 4). In the study by Peng et al. (2018), the Nrf2‐mediated oxidative stress response pathway was the most abundant pathway associated with enhancers that overlapped with vitamin C‐increased 5‐hmC peaks in bladder cancer (Peng et al., 2018). This finding provides evidence that DNA demethylation induction in response to vitamin C plays a vital role in the antioxidant effects of vitamin C (Peng et al., 2018).
Furthermore, an epigenetic mechanism has been established for vitamin C‐mediated suppression of the LPA gene encoding for lipoprotein(a) [LP(a)] (Qu et al., 2017). This effect may provide an explanation for an inverse correlation between dietary intake of vitamin C and LP(a) plasma concentration observed in a transgenic mice model (Cha, Niedzwiecki, & Rath, 2015). LP(a) is a type of LDL, consisting of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2718 and apolipoprotein(a) [apo(a)], high levels of which are considered a major risk factor for atherosclerosis and CVD (Orso & Schmitz, 2017). In response to vitamin C in HepG2 HCC cells, increased TET2‐induced hydroxymethylation at the ELK1 promoter was accompanied by ELK1 up‐regulation and higher occupancy of ELK1 at the LPA promoter (Qu et al., 2017). ELK1 acts as a transcriptional inhibitor of LPA and down‐regulates apo(a) expression. Taking into account that LP(a) is susceptible to oxidative modifications followed by the formation of pro‐inflammatory oxidized lipids and lipid‐protein adducts, a decrease in LP(a) expression has been linked to lower cellular oxidative damage and decreased pro‐inflammatory responses (Orso & Schmitz, 2017).
5.2. Vitamin E
5.2.1. Mechanisms behind changes in DNA methylation in response to vitamin E
Limited studies have investigated the effects of vitamin E on DNA methylation in chronic disease. However, the few studies that have examined global changes in DNA methylation were done with cancer models. For example, Huang et al. (2012) found that DNMT1, DNMT3A and DNMT3B were suppressed by a γ‐tocopherol‐rich mixture of tocopherols administered to transgenic adenocarcinoma of the mouse prostate (TRAMP) mice (Huang et al., 2012). Interestingly, only DNMT3B was suppressed by the tocopherol mixture in an in vitro model, namely, a TRAMP cell line (Huang et al., 2012). Other studies have observed changes in DNA methylation at LINE‐1, which is used as an indicator of global DNA methylation (Zappe et al., 2018). Upon vitamin E treatment, LINE‐1 methylation was increased, indicating a global increase in DNA methylation in response to vitamin E in cancer cells. Specific gene targets unrelated to antioxidant pathways as well as other mechanisms contributing to changes in global DNA methylation in response to vitamin E have not been identified, nor has a mechanism as to how vitamin E might directly or indirectly influence DNMTs been proposed.
5.2.2. A link between antioxidant properties of vitamin E and vitamin E‐mediated changes in DNA methylation
The most compelling study to date that assessed the antioxidant effect of vitamin E in relation to DNA methylation was performed in a cancer model, where a γ‐tocopherol‐rich mixture of tocopherols re‐activated the expression of the methylation‐silenced Nrf2 gene in TRAMP mice and prostate cancer cells (Huang et al., 2012; Figure 4).
Another mode of action of vitamin E that is key for prevention of atherosclerosis/CVD, involves up‐regulation of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=595 a transcription factor with protective properties against oxidative stress (Tang et al., 2014). Interestingly, an animal study with transgenic mice with macrophage‐specific overexpression of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2605 demonstrates that DNA hypermethylation of the proximal PPARγ promoter was mediated by DNMT1 (Yu et al., 2016). Additionally, the luciferase activity of the unmethylated PPARγ promoter was significantly greater than that of the fully methylated promoter, indicating that PPARγ promoter activity is regulated by DNA methylation (Yu et al., 2016). Hence, since vitamin E down‐regulates DNMT1 expression (Huang et al., 2012), there is a possibility that vitamin E also mediates PPARγ up‐regulation indirectly by altering epigenetic regulation of PPARγ.
5.3. Vitamin A
5.3.1. Mechanisms behind changes in DNA methylation in response to vitamin A
RA, an active metabolite of vitamin A, has profound effects on cellular proliferation and differentiation. This effect is mainly through its role as a transcription factor in a complex with a receptor, which is able to recognize and bind the retinoic acid response element (RARE) in DNA followed by regulation of transcriptional activity of numerous target genes (Stefanska, Rudnicka, Bednarek, & Fabianowska‐Majewska, 2010; Stefanska, Salame, Bednarek, & Fabianowska‐Majewska, 2012; Sun et al., 2018).
Moreover, several studies reported changes in expression of genes, which were related to changes in DNA methylation of regulatory regions of those genes rather than binding of RA to RAREs (Stefanska et al., 2010; Stefanska et al., 2012; Sun et al., 2018). For instance, exposure of breast cancer cells to https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2644 (ATRA) led to DNA hypomethylation within promoters of PTEN and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=591 (RARβ2), which was accompanied by increased expression of these tumour suppressor genes (Stefanska et al., 2010; Stefanska et al., 2012). A recent study on T cells derived from systemic sclerosis patients demonstrated that ATRA increases expression of FOXP3, a molecular marker crucial for development and function of T regulatory immune cells, through decrease in DNA methylation of the FOXP3 promoter (Sun et al., 2018). Using a genome‐wide technology based on methylated DNA immunoprecipitation, a network of differentially methylated genes functionally related to differentiation of neuroblastoma cells was identified in response to treatment with ATRA (Das et al., 2010). In search for potential mechanisms, Das et al. (2010) assessed the expression of the DNMTs. DNMT1 and DNMT3B were down‐regulated in response to ATRA likely explaining ATRA‐mediated remodelling of DNA methylation patterns (Das et al., 2010). Mechanistic studies further showed that expression of the DNMTs decreases upon treatment with ATRA because their mRNAs are targeted by micro‐RNAs, specifically miR‐152 targeting DNMT1 and miR‐26a/b and miR‐125a/b targeting DNMT3B (Das et al., 2010).
DNA methylation mechanisms regarding other forms of vitamin A, such as β‐carotene and other carotenoids which are classically considered antioxidants, have not yet been elucidated but have been studied in diseases with underlying inflammation.
5.3.2. A link between antioxidant properties of vitamin A and vitamin A‐mediated changes in DNA methylation
As mentioned above, carotenoids are classical antioxidant forms of vitamin A working as free radical scavengers (Bohn, 2019; Figure 4). Intake of β‐carotene and other carotenoids in overweight and obese individuals has been associated with changes in the DNA methylation patterns in white blood cells (Bollati et al., 2014). Higher intake was linked to higher methylation of the endogenous retrovirus gene HERV‐w, in contrast to TNF‐α, whose methylation was lower with increasing intake (Bollati et al., 2014). Interestingly, these changes in the methylation state of HERV‐w and TNF‐α may contribute to antioxidant effects of carotenoids. HERV‐w, which was hypermethylated in the high intake group and thereby potentially silenced, is part of a superfamily of repetitive and transposable elements and acts as a pro‐inflammatory molecule activating pathways that lead to ER stress and production of free radicals (Bollati et al., 2014). TNF‐α, which was hypomethylated and thereby potentially expressed at higher levels in the high intake group, is a master regulator of the immune system and takes part in the propagation of inflammation. However, TNF‐α's action may vary depending on the receptor it interacts with (Fischer & Maier, 2015). When TNF‐α binds to its receptor https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1870, it acts as a pro‐inflammatory molecule leading to tissue degeneration (Fischer & Maier, 2015). On the other hand, the interaction with another receptor, TNFR2, was shown to have protective effects against tissue degeneration (Fischer & Maier, 2015). Hence, hypomethylation of TNF‐α in response to high intake of carotenoids could be considered as part of the antioxidant mechanism of action of carotenoids.
5.4. Selenium
5.4.1. Mechanisms behind changes in DNA methylation in response to selenium
Multiple studies performed on the associations between selenium status or selenium supplementation and DNA methylation report alterations in both global and gene‐specific DNA methylation patterns (Jablonska & Reszka, 2017; Speckmann et al., 2017). For instance, in the study by Speckmann et al. (2017), physiological doses of selenium led to increased global methylation levels as measured by LINE‐1 methylation state and increased SAM/SAH ratio in mice liver as compared with non‐supplemented group (Speckmann et al., 2017). However, loci‐specific DNA methylation levels were reduced, including the Srebf1 regulatory region, which was linked to Srebf1 up‐regulation. These findings clearly indicate complex modulation of the methylation machinery by selenium levels rather than one mode of action. In a disease model, namely, human LNCaP prostate cancer cells, non‐toxic doses of selenium also led to demethylation and activation of genes such as GSH‐S‐transferase pi 1 (GSTP1), APC and CSR1 tumour suppressor genes (Xiang, Zhao, Song, & Zhong, 2008). On the other hand, severe selenium‐deficiency increased promoter methylation and down‐regulated the VHL tumour suppressor genes in Caco‐2 colon cancer cells and rat colon mucosa, sensitizing the epithelium to cancer development upon exposure to a carcinogen (Uthus, Begaye, Ross, & Zeng, 2011). A similar correlation between low plasma selenium levels and high promoter methylation of the WIF1 tumour suppressor gene was reported in human studies (Tapp et al., 2013). In the same study, the effect on another tumour suppressor gene, APC, was sex dependent, indicating possible sex differences in selenium metabolism (Tapp et al., 2013).
The mechanisms explaining these changes in DNA methylation in response to selenium have been elucidated. Selenium intake impacts the activity of the one‐carbon metabolism (OCM) pathway and thus DNA methylation rate (Jablonska & Reszka, 2017; Speckmann et al., 2017). Specifically, one of the pathways of selenium metabolism requires methylation by methyltransferases and utilizes SAM as a methyl donor. As a consequence, an excess of selenium leads to competition for SAM resulting in less SAM available for DNMTs to methylate DNA. On the other hand, selenium deficiency redirects https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5198 towards GSH synthesis in the trans‐sulfuration pathway. It consequently decreases levels of homocysteine that are available for restoration of SAM in the re‐methylation pathway (Jablonska & Reszka, 2017; Speckmann et al., 2017). Hence, both selenium excess and selenium deficiency result in inhibition of DNMT activity/expression and dysregulation of DNA methylation machinery.
Despite well‐established mechanistic involvement of selenium in the regulation of DNA methylation machinery, some studies have yielded mixed results (Jablonska & Reszka, 2017; Speckmann et al., 2017). This inconsistency may result from using different experimental models, methods to measure DNA methylation, doses of selenium, times of exposure, forms of selenium and even nutritional context in the animal experiments.
5.4.2. A link between antioxidant properties of selenium and selenium‐mediated changes in DNA methylation
Selenium is an essential trace element and a major component of selenoproteins, exhibiting a wide range of antioxidant and anti‐inflammatory properties (Lipinski, 2019). In addition, a study by Xiang et al. (2008) demonstrated that the epigenetic activity of selenium may contribute to its antioxidant properties (Xiang et al., 2008). Treatment of prostate cancer cells with sodium selenite (Na2SeO3) resulted in partial promoter demethylation of GSTP1, an important player in detoxification and antioxidant defence maintaining the cellular redox state (Xiang et al., 2008). Demethylation in response to selenium was accompanied by decreased DNMT1 and DNMT3A expression levels which may explain decrease in loci‐specific DNA methylation (Figure 4 and Table 1). Further studies should be focused on altered DNA methylation upon selenium treatment and to investigate the antioxidant role of selenium in these processes.
5.5. Stilbenoids
5.5.1. Mechanisms behind changes in DNA methylation in response to stilbenoids
Stilbenoid compounds such as resveratrol and pterostilbene alter DNA methylation patterns at gene‐specific loci (Beetch, Lubecka, Kristofzski, Suderman, & Stefanska, 2018; Kala & Tollefsbol, 2016; Lubecka et al., 2016; Medina‐Aguilar et al., 2016; Papoutsis, Lamore, Wondrak, Selmin, & Romagnolo, 2010; Stefanska et al., 2010; Stefanska et al., 2012). Our group (Beetch et al., 2018; Beetch et al., 2019; Lubecka et al., 2016) and others (Medina‐Aguilar et al., 2016) using genome‐wide technologies have demonstrated that stilbenoids remodel patterns of DNA methylation in human cells, including cancer and normal cells, leading to CpG‐loci specific hypermethylation or hypomethylation. In cancer cells, CpG sites, which were hypomethylated in response to stilbenoids, are associated with genes falling into a category of tumour suppressor genes, whereas genes containing hypermethylated CpG sites upon exposure to stilbenoids are enriched with oncogenic and pro‐metastatic functions. The latter group of genes included many active regulators of oncogenic signalling pathways, such as NOTCH, WNT, MAPK, and JAK/STAT (Lubecka et al., 2016; Medina‐Aguilar et al., 2016).
Several mechanisms attributed to stilbenoid‐mediated changes in DNA methylation have been suggested. First of all, DNMT3B may be the key enzyme catalysing hypermethylation that occurs in regulatory regions of genes with oncogenic functions, for example, MAML2, in response to stilbenoids in cancer cells (Lubecka et al., 2016). It has been suggested that stilbenoids increase the occupancy of DNMT3B at these genes which coincides with de‐localization of transcription factors, hypermethylation, and gene silencing (Lubecka et al., 2016). Secondly, a decrease in expression and/or activity of DNMT1 has been proposed as a possible link to loci‐specific decrease in methylation during cell division and consequent activation of methylation‐silenced tumour suppressor genes in response to stilbenoids (Kala &Tollefsbol, 2016; Mirza et al., 2013; Papoutsis et al., 2010; Stefanska et al., 2010; Stefanska et al., 2012). The compounds may decrease DNMT1 expression by affecting proteins that control DNMT1 transcription, such as the AP‐1 transcription factor (Bigey, Ramchandani, Theberge, Araujo, & Szyf, 2000). The cascade of events starts with PTEN, which was shown to be up‐regulated by stilbenoids (Stefanska et al., 2010; Stefanska et al., 2012). PTEN is a tumour suppressor gene exerting an inhibitory effect on Ras/Raf/MAPK signalling (Chung et al., 2006). The up‐regulation of PTEN can consequently decrease AP‐1‐dependent transcription (Karin, 1995; Figure 5). In addition, resveratrol was reported to decrease AP‐1 activity through PTEN‐independent pathways (Manna, Mukhopadhyay, & Aggarwal, 2000).
Figure 5.
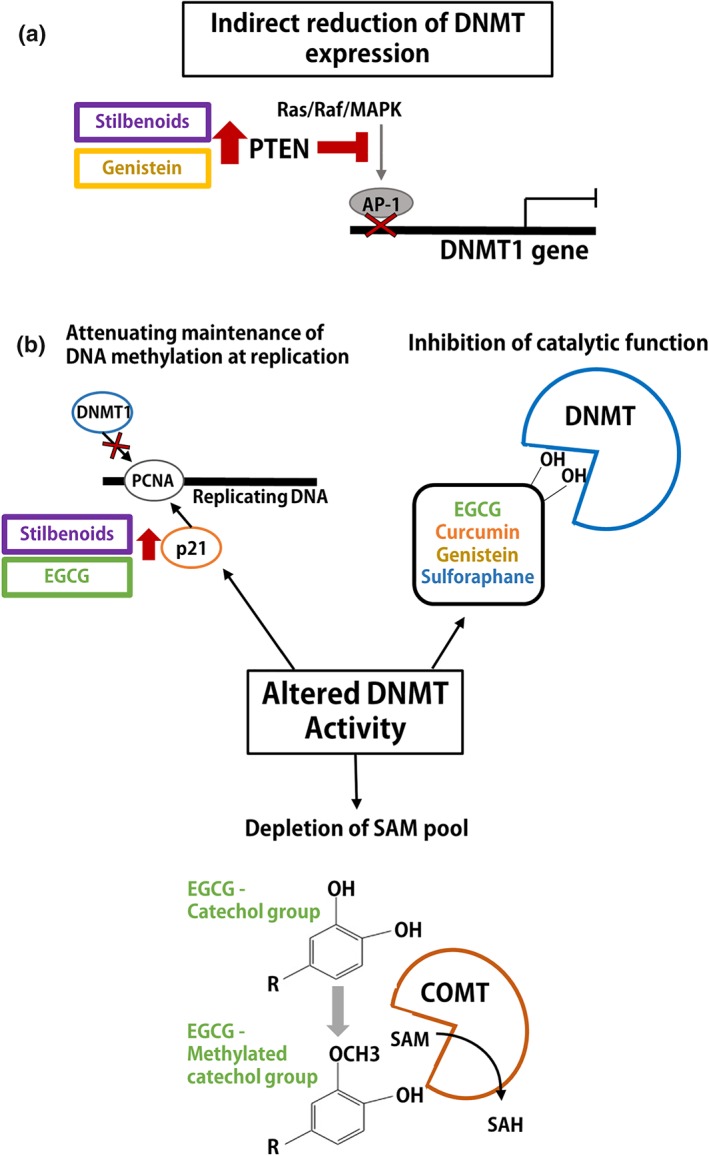
Mechanisms of polyphenol‐mediated effects on expression and activity of DNA methyltransferases (DNMTs). (a) Indirect reduction in expression of DNMT1 by polyphenol‐mediated increase in phosphatase and tensin homologue (PTEN) to inhibit AP‐1 transcription factor binding at the DNMT1 promoter. (b) DNMT activity is altered through mechanisms associated with attenuation of DNMT1 binding at replicating DNA, inhibition of catalytic activity of DNMTs, and depletion of the S‐adenosyl‐l‐methionine (SAM) pool
Apart from regulating expression of DNMT1, stilbenoids may indirectly affect DNMT1 activity through increasing levels of p21, which was reported in response to resveratrol (Stefanska et al., 2010; Stefanska et al., 2012). Because DNMT1 is targeted to replicating DNA by PCNA, this nuclear antigen is necessary for DNMT1 activity (Iida et al., 2002). Of note, the interaction of DNMT1 and PCNA at sites of DNA replication is disrupted in the presence of high p21 levels, since p21 competes with DNMT1 for a binding site on PCNA (Chuang et al., 1997; Figure 5).
5.5.2. A link between antioxidant properties of stilbenoids and stilbenoid‐mediated changes in DNA methylation
Resveratrol has been reported to regulate one of the major antioxidant pathways, namely, the Nrf2 pathway through an epigenetic mechanism(s) (Singh et al., 2014; Figure 4 and Table 1). In female rats treated with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1013 (E2), often used as an inducer of the oxidative stress affecting breast cancer, Nrf2 was significantly up‐regulated in response to resveratrol. Expression of Nrf2 downstream targets, such as NQO1, SOD, and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=3060, was also increased, indicating activation of this antioxidant pathway. Using methylation‐specific PCR, it was determined that the up‐regulation of Nrf2 upon resveratrol treatment was accompanied by DNA hypomethylation of the Nrf2 promoter in the E2‐treated rat model and in oestrogen‐receptor‐negative MCF10A breast cells treated with E2 (Singh et al., 2014). These epigenetic changes were concomitant with lower levels of 7,8‐dihydroxy‐8‐oxo‐deoxyguaninosine (8‐oxo‐dG), an indicator of DNA damage, in response to resveratrol, which clearly indicates direct epigenetic mechanisms of the antioxidant action of stilbenoids (Singh et al., 2014).
5.6. Epigallocatechin gallate
5.6.1. Mechanisms behind changes in DNA methylation in response to EGCG
Many studies conducted in cancer models have reported EGCG‐mediated transcriptional activation of methylation‐silenced tumour suppressor genes through a decrease in promoter methylation (Fang et al., 2003; Kato et al., 2008; Khan et al., 2015; Lee, Shim, & Zhu, 2005; Morris et al., 2016). For instance, RARβ, cadherin 1 (CDH1), and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2002 (DAPK1) were demethylated and re‐activated in EGCG‐treated HeLa cells (Khan et al., 2015). Similar changes in methylation and expression of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=610 (RXRα) in colon cancer (Morris et al., 2016), p16(INK4a), RARβ, O(6)‐methylguanine methyltransferase (MGMT) and human mutL homologue 1 (hMLH1) in oesophageal cancer cells (Fang et al., 2003), and RECK in oral squamous cell carcinoma (Kato et al., 2008) were observed.
These effects were often accompanied by attenuation of expression and/or activity of DNMTs (Fang et al., 2003; Khan et al., 2015; Lee et al., 2005; Mirza et al., 2013). Indeed, EGCG is one of the few bioactive compounds that can directly bind to a catalytic pocket of DNMT1, thus inhibiting the enzyme's activity (Fang et al., 2003; Lee et al., 2005). Molecular modelling and docking site data support DNMT3B as another direct target of EGCG (Khan et al., 2015; Figure 5).
Another mechanism which indirectly contributes to a decrease in loci‐specific DNA methylation in response to EGCG is linked to https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2472 (COMT; Table 1). EGCG as a catechol compound is an excellent substrate for COMT which, similarly to DNMTs, utilizes methyl donor SAM to methylate catechol groups. This methylation reaction results in less methyl donor available for DNMTs to methylate DNA and additionally leads to formation of SAH, a potent non‐competitive inhibitor of DNMTs (Lee & Zhu, 2006; Figure 5).
Moreover, EGCG treatment up‐regulated p21 expression in breast cancer cells (Mirza et al., 2013). As described in Section 5.5, high levels of p21 may decrease DNMT1 activity due to p21 competition for binding PCNA (Chuang et al., 1997; Table 1).
5.6.2. A link between antioxidant properties of EGCG and EGCG‐mediated changes in DNA methylation
EGCG has been shown to epigenetically regulate dimethylargininase 2 (DDAH2) in HUVEC, which initiates a cascade of events leading to a decrease in oxidation of lipids and proteins and consequently to protective cardiovascular effects (Zhang, Lai, Niu, Zhao, & Jia, 2013). Specifically, EGCG decreases methylation within the DDAH2 promoter which results in DDAH2 up‐regulation in HUVEC cells (Zhang, Lai, et al., 2013). Mechanistic experiments showed lower expression of DNMT1 and impaired binding of DNMT1 to the DDAH2 promoter, which may explain demethylation of the promoter in response to EGCG (Zhang, Lai, et al., 2013). Increase in DDAH2 activity, in turn, is known to contribute to a decrease in levels of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5229 (ADMA), the endogenous NOS inhibitor (Tang, Hu, Chen, Deng, & Li, 2006). This drop in ADMA allows for higher production of NO by NOS and NO‐mediated inhibition of oxidation of lipids and proteins (Mu et al., 2019).
5.7. Genistein
5.7.1. Mechanisms behind changes in DNA methylation in response to genistein
Genistein can target aberrant changes in DNA methylation and cause associated changes in gene expression in chronic diseases, particularly in cancer (Adjakly et al., 2011; Fang et al., 2005; Majid et al., 2010; Xie et al., 2014). Anti‐proliferative and pro‐apoptotic effects of this isoflavone have been linked in many studies to re‐activation of expression of tumour suppressor genes that were silenced by promoter DNA methylation during carcinogenesis (Adjakly et al., 2011; Fang et al., 2005; Majid et al., 2010; Xie et al., 2014). For instance, genistein reversed hypermethylation which led to re‐activation of RARβ2, p16, and MGMT tumour suppressor genes in KYSE 510 esophageal squamous carcinoma cells (Fang et al., 2005). A similar mode of action was detected for B‐cell translocation gene 3 (BTG3/ANA/APRO4), a tumour suppressor gene in LNCaP and PC3 prostate cancer cells (Majid et al., 2010), for BRCA1, GSTP1, and EPHB2 promoters in DU‐145 and PC‐3 prostate cancer cells (Adjakly et al., 2011), and for ATM, APC, and PTEN tumour suppressor genes in MCF‐7 and MDA‐MB‐231 breast cancer cells (Xie et al., 2014).
In addition to in vitro disease model studies, genistein has also been shown to modify loci‐specific DNA methylation status in offspring in transgenerational Agouti mice (Dolinoy, Weidman, Waterland, & Jirtle, 2006). Supplementing the maternal diet with genistein led to methylation of a retrotransposon in the 5′ end of the Agouti gene, which decreased ectopic Agouti expression, shifting the fur colour towards a more brown wild type shade and moreover, protected the offspring from obesity later in life (Dolinoy et al., 2006).
Studies conducted in cell culture and rodent models defined mechanisms driving genistein‐mediated changes in DNA methylation. It appears that similarly to other bioactive compounds described above, genistein acts differentially to impact DNA methylation states, depending on the gene‐loci or gene function. An additional level of complexity for the epigenetic effects of genistein concerns the dose‐dependent effects. A study in healthy pre‐menopausal women demonstrated that circulating levels of genistein impacted the direction of changes in loci‐specific DNA methylation levels, which suggests different mechanisms of action for high versus low doses of genistein (Qin et al., 2009). This clearly indicates a complex change in the DNA methylation machinery in response to genistein. Studies have shown that genistein exhibits dose‐dependent inhibition of DNMT activity within nuclear extracts from KYSE 510 cells (Fang et al., 2005). Molecular modelling and docking of genistein into the DNMT1 enzymatic pocket indicates that the compound may bind to and form bonds with the catalytic domain of DNMT1, resulting in inhibition of the enzymatic activity of DNMT1 (Xie et al., 2014). Moreover, a decrease in expression of DNMT1 was detected upon exposure to genistein in cancer cell models indicating additional levels of genistein‐mediated regulation of DNMT1(Xie et al., 2014). Since genistein was shown to up‐regulate PTEN (Dave et al., 2005), it has been speculated that transcriptional regulation of DNMT1 in response to genistein may be driven by inhibition of AP‐1 transcription factor via PTEN action on MAPK signalling, as we described for stilbenoids in Section 5.5 (Karin, 1995; Chung et al., 2006; Figure 5).
5.7.2. A link between antioxidant properties of genistein and genistein‐mediated changes in DNA methylation
While numerous studies have explored mechanisms of remodelling DNA methylation patterns at gene‐specific targets in response to genistein, investigations on the link between these epigenetic mechanisms and antioxidant action of the compound are lacking. There is only one line of evidence demonstrating that genistein changes the DNA methylation status of Keap1 from the Keap1‐Nrf2 signalling pathway. However, this alteration actually contributes to an increase in oxidative stress and oxidative damage sensitizing cancer cells to radiotherapy (Liu et al., 2016; Figure 4 and Table 1). Liu et al. (2016) demonstrated that genistein leads to demethylation and activation of Keap1 in human A549 non‐small‐cell lung cancer cells, which lead to inhibition of Nrf2 translocation to the nucleus and prevented Nrf2‐mediated activation of antioxidant networks (Liu et al., 2016). Interestingly, such effects of genistein were not observed in the human MRC‐5 lung fibroblast cell line (a normal cell model), where the compound instead increased nuclear localization of Nrf2 and similarly activated antioxidant pathways (Liu et al., 2016). However, mechanisms behind these distinct actions of genistein in normal versus cancer cells remain to be elucidated.
5.8. Curcumin
5.8.1. Mechanisms behind changes in DNA methylation in response to curcumin
A mechanism of direct inhibition of DNA methylation has been described for curcumin. Molecular docking studies have suggested that curcumin binds a cysteine residue to covalently inhibit catalytic activity of DNMT1 (Liu et al., 2009; Figure 5). Accordingly, curcumin has been shown to increase expression of several tumour suppressor genes via hypomethylation of regulatory regions in various cancer types (Jiang et al., 2015; Liu, Zhou, Hu, Wang, & Yuan, 2017; Yu et al., 2013). Liu et al. (2017) reported that the deleted in liver cancer 1 (DLC1) gene was up‐regulated upon treatment with curcumin in breast cancer cells with concomitant reduction of DNA methylation in the DLC1 promoter (Liu et al., 2017). Similar results were obtained for p15 in acute myeloid leukaemia cells (Yu et al., 2013). Both studies observed that curcumin decreased DNMT1 expression (Liu et al., 2017; Yu et al., 2013), which agrees with the proposed direct inhibition of DNMT1 by curcumin (Liu et al., 2009). Furthermore, a study using lung cancer cells and a xenograft mouse model found that RARβ mRNA and protein expression was increased in response to curcumin with an accompanied decrease in promoter DNA methylation (Jiang et al., 2015). In this study, DNMT3B levels were decreased upon curcumin treatment in both cancer cells and xenografts, suggesting that curcumin may have a role in altering activity of other DNMTs as well. Whether direct or indirect, mechanisms of DNA methylation alterations via DNMT3A and DNMT3B in response to curcumin are needed to be investigated in future studies. Interestingly, a genome‐wide DNA methylation study in colorectal cancer cells exposed to curcumin shows that curcumin caused loci‐specific both hypermethylation and hypomethylation, predominantly in partially methylated CpG sites (Link et al., 2013). Thus, curcumin effects on the DNA methylation patterns are differential and likely depend of CpG location and gene function as suggested for stilbenoids (Beetch et al., 2018; Lubecka et al., 2016).
Curcumin‐mediated epigenetic regulation of gene expression was also described in non‐cancer models, namely, in AD. Deng et al. (2014) found that curcumin was able to induce demethylation of the neprilysin (NEP) gene, which was followed by transcriptional NEP reactivation and inhibition of AKT/NFκB signalling (Deng et al., 2014).
5.8.2. A link between antioxidant properties of curcumin and curcumin‐mediated changes in DNA methylation
A genome‐wide study testing changes of DNA methylation and expression patterns in response to curcumin in a mouse model of colitis‐induced colon cancer (AOM/DSS) demonstrated that curcumin increases DNA methylation and causes silencing of a set of genes functionally linked to oxidative stress and inflammation (Guo et al., 2018). For instance, Tnf, one of the major mediators of ROS production and inflammation (Fischer & Maier, 2015), was epigenetically activated via demethylation of its regulatory region in AOM/DSS group. Curcumin treatment restored the high methylation profile of Tnf, which coincided with a decrease in Tnf expression (Guo et al., 2018). Earlier gene candidate studies performed in murine RAW 264.7 macrophage cells and TRAMP C1 prostate cancer cells delivered evidence for curcumin‐mediated demethylation and activation of Nrf2, a potent activator of antioxidant pathways (Khor et al., 2011). Another member of the antioxidant network, GSTP1, was also found to be demethylated and activated upon exposure of breast cancer cells to curcumin (Kumar, Sharma, & Rathi, 2017). Furthermore, Maugeri et al. (2018) reported up‐regulation of DNMT1 expression and activity along with oxidative stress induced by chronic high glucose levels in a cellular model of diabetic retinopathy (Maugeri et al., 2018). Interestingly, curcumin restored normal levels of DNMT1 and inhibited the overproduction of ROS (Maugeri et al., 2018).
5.9. Sulforaphane
5.9.1. Mechanisms behind changes in DNA methylation in response to SFN
SFN has been shown to impact the DNA methylation status of multiple candidate genes (Ali Khan et al., 2015; Lubecka, Kaufman‐Szymczyk, & Fabianowska‐Majewska, 2018; Lubecka‐Pietruszewska et al., 2015). Demethylation and re‐activation of tumour suppressor genes in response to SFN have been reported, for example, for CDKN2A at a non‐invasive breast cancer stage (Lubecka et al., 2018), PTEN and RARβ2 in breast cancer cells (Lubecka‐Pietruszewska et al., 2015), and RARβ, CDH1, DAPK1, and GSTP1 in cervical cancer cells (Ali Khan et al., 2015). As with stilbenoids, SFN may exert differential effects on loci‐specific DNA methylation depending on a gene location and/or gene function. Although a genome‐wide study is missing, the CD14 gene that is up‐regulated by bacterial LPS and aggravates the inflammatory response was shown to be down‐regulated by SFN via hypermethylation of its promoter region (Yang et al., 2016).
Importantly, the ability of SFN to alter DNA methylation patterns can have a much broader spectrum of effects, not only limited to a given gene. The compound can affect a network of genes, as it has been shown to alter the DNA methylation status of microRNAs that further impact post‐transcriptional regulation of multiple target genes (Gao et al., 2018).
Several mechanisms by which SFN may impact DNA methylation machinery have been proposed. Firstly, a decrease in protein levels of DNMT1 and DNMT3A, as detected in prostate cancer cells, may explain the loss of methylation in response to SFN (Zhang, Su, Khor, Shu, & Kong, 2013). Secondly, Ali Khan et al. (2015) demonstrated that SFN reduces DNMT3B expression and DNMTs enzymatic activity, and predicted, using molecular modelling, a putative direct binding of SFN to DNMT3B (Ali Khan et al., 2015; Figure 5). Finally, changes in DNA methylation patterns in response to SFN may also be closely linked to TET demethylating enzymes. In a bone cell model, SFN was shown to increase global levels of 5‐hmC, which was accompanied by up‐regulation of TET1 and was dependent on TET1 action (Thaler et al., 2016).
5.9.2. A link between antioxidant properties of SFN and SFN‐mediated changes in DNA methylation
One of the antioxidant mechanisms of SFN is the activation of the Nrf2 pathway, which may be linked to epigenetic effects of SFN (Figure 4 and Table 1). A study by Zhang et al. (2013) reported that, like many other dietary antioxidants, SFN restores Nrf2 expression via demethylation of the Nrf2 promoter and up‐regulates downstream targets of Nrf2 such as NQO1 in prostate cancer cells (Zhang, Su, et al., 2013). Epigenetic activation of Nrf2 was also shown to be involved in the anti‐cancer effect of SFN against 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA)‐induced neoplastic transformation of mouse skin cells (Su et al., 2014). In a cellular model of AD, similar findings were described (Zhao, Zhang, & Chang, 2018). The study showed that SFN‐mediated demethylation and activation of Nrf2 was associated with induction of Nrf2‐dependent genes such as HO‐1 and NQO1. Additionally, the altered pattern of Nrf2 methylation in response to SFN was accompanied by down‐regulation of DNMT1, DNMT3A, and DNMT3B (Zhao et al., 2018). The latter result could provide a possible mechanism behind loci‐specific demethylation upon treatment with SFN.
6. DECREASING OXIDATIVE STRESS—AN INDUCER OF ABERRANT DNA METHYLATION—MAY MEDIATE EFFECTS OF ANTIOXIDANTS ON THE DNA METHYLATION PATTERNS
From this review, we have reported considerable evidence to show how antioxidant effects of many dietary compounds are linked to their ability to alter DNA methylation patterns at specific genes, for example, Nrf2. Studies on oxidative stress and inflammation in the last decade suggest that oxidative stress can induce alterations in DNA methylation through activation of repair mechanisms (O'Hagan et al., 2008; O'Hagan et al., 2011). Thus, compounds that decrease oxidative stress could consequently impact DNA methylation landscapes.
As shown in Figure 4, oxidative stress causes damage to DNA, proteins, and lipids. ROS‐induced DNA damage includes base damage, with 8‐oxo‐dG being the most common, as well as single‐ and double‐strand DNA breaks (SSBs and DSBs; O'Hagan, 2014; Maiuri & O'Hagan, 2016). It has been demonstrated that in response to ROS, DNMT1, along with the histone modifying proteins, NAD‐dependent protein deacetylase https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2707 (SIRT1) and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2654 (EZH2), is recruited to sites of oxidative DNA damage through interaction with mismatch repair proteins MSH2 and MSH6 (Ding et al., 2016; Ding et al., 2019; O'Hagan et al., 2008). This localization to sites of DNA damage is proposed to be part of the normal repair process resulting in local inhibition of transcription to allow for DNA repair to occur (Ding et al., 2016; O'Hagan, 2014). Indeed, repressive histone modifications, DNA methylation of promoter CpG islands, and transcriptional repression of associated genes were observed upon oxidative damage, which was dependent on the occupancy of the region by DNMT1 and mismatch repair proteins (Ding et al., 2016).
During the normal repair process once repair is completed, the chromatin should be restored to its original state, and transcription should resume. However, with repetitive damage that occurs during chronic exposure to stressors or inflammation, such transient chromatin modifications occurring during the repair process may lead to stable epigenetic alterations that contribute to disease development. As an example, recruitment of DNMT1 and/or DNMT3B to sites of DSBs can result in DNA hypermethylation and heritable silencing of associated genes (Ding et al., 2016; O'Hagan et al., 2008). Furthermore, in an in vivo model of inflammation‐driven colon tumorigenesis mismatch repair protein‐dependent recruitment of DNMT1 to sites of oxidative damage was required for DNA hypermethylation and gene silencing of tumour suppressor genes during tumorigenesis (Maiuri et al., 2017). On the other hand, the localization of DNMT1 to sites of DNA damage may decrease the pool of DNMT1 available for binding to other sites in the genome and consequently lead to DNA hypomethylation and gene activation at those sites. This hypothesis is consistent with the global loss of DNA methylation observed in cancer cells. By decreasing oxidative stress, antioxidant compounds could potentially prevent ROS‐induced aberrant changes in the DNA methylation patterns and protect against disease development. It is likely that dietary antioxidant compounds alter DNA methylation through multiple mechanisms, including by directly altering the activity/function of DNMTs, as detailed in Section 5, and by decreasing oxidative stress and subsequent methylation changes.
7. CONCLUSIONS
Antioxidant dietary compounds are emerging as vital nutraceuticals due to their anti‐inflammatory, cardioprotective and antitumor properties. The research on epigenetic mechanisms of action of dietary antioxidants in chronic disease is relatively new and requires more comprehensive studies in different models and human populations. There is a need for integrated approaches, including epigenome, transcriptome, and metabolome profiling, to systematically assess the mechanisms of action of antioxidant nutraceuticals in different diseases. The available experimental data have revealed that bioactive antioxidants exert potent health effects by remodelling the epigenome, specifically, reversing aberrant patterns of DNA methylation and thus ceasing oxidative stress. Interestingly, scientific evidence in last few years provides support for oxidative stress as a driver of alterations in DNA methylation. Hence, dietary compounds that decrease oxidative stress may protect from aberrant changes in the epigenome. Future research should reveal mechanistic targets and answer a question whether oxidative stress leads to aberrant DNA methylation and gene expression or whether oxidative stress is a consequence of DNA methylation changes. As such, understanding mechanistic underpinning of these targets could provide opportunities for clinical utilization whereby antioxidant compounds and DNA methylation modifying drugs could be strategically administered to combat chronic disease and related comorbidities. Due to the widespread negative impact of chronic diseases, advances in this area will translate to improved health and quality of life of millions of people.
7.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
This publication was made possible with support from (a) the University of British Columbia VP Academic (10R76632) Award granted to B.S.; (b) the Canadian Foundation for Innovation (CFI) John R. Evans Leaders Fund and the British Columbia Knowledge Development Fund (37105) granted to B.S.; (c) the Indiana Clinical and Translational Sciences Institute and the Indiana CTSI Collaboration in Biomedical/Translational Research (CBR/CTR) Pilot Program Grants; and (d) Grant UL1TR001108 from the National Institutes of Health, National Center for Advancing Translational Science, Clinical and Translational Sciences‐Award granted to H.M.O. and B.S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Beetch M, Harandi‐Zadeh S, Shen K, et al. Dietary antioxidants remodel DNA methylation patterns in chronic disease. Br J Pharmacol. 2020;177:1382–1408. 10.1111/bph.14888
REFERENCES
- Adjakly, M. , Bosviel, R. , Rabiau, N. , Boiteux, J. P. , Bignon, Y. J. , Guy, L. , & Bernard‐Gallon, D. (2011). DNA methylation and soy phytoestrogens: Quantitative study in DU‐145 and PC‐3 human prostate cancer cell lines. Epigenomics, 3(6), 795–803. 10.2217/epi.11.103 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, J. F. , … CGTP Collaborators (2019). The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S398. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali Khan, M. , Kedhari Sundaram, M. , Hamza, A. , Quraishi, U. , Gunasekera, D. , Ramesh, L. , … Hussain, A. (2015). Sulforaphane reverses the expression of various tumor suppressor genes by targeting DNMT3B and HDAC1 in human cervical cancer cells. Evidence‐Based Complementary and Alternative Medicine: Ecam, 2015(412149), 1–12. 10.1155/2015/412149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, Z. , & Wang, Z. (2019). Genistein protects against doxorubicin‐induced cardiotoxicity through Nrf‐2/HO‐1 signaling in mice model. Environmental Toxicology, 34(5), 645–651. [DOI] [PubMed] [Google Scholar]
- Bansal, A. , & Simmons, R. A. (2018). Epigenetics and developmental origins of diabetes: Correlation or causation? American Journal of Physiology. Endocrinology and Metabolism, 315(1), E15–E28. 10.1152/ajpendo.00424.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barajas‐Olmos, F. , Centeno‐Cruz, F. , Zerrweck, C. , Imaz‐Rosshandler, I. , Martinez‐Hernandez, A. , Cordova, E. J. , … Campos, F. (2018). Altered DNA methylation in liver and adipose tissues derived from individuals with obesity and type 2 diabetes. BMC Medical Genetics, 19(1), 28‐36. 10.1186/s12881-018-0542-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker, D. J. , Hales, C. N. , Fall, C. H. , Osmond, C. , Phipps, K. , & Clark, P. M. (1993). Type 2 (non‐insulin‐dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): Relation to reduced fetal growth. Diabetologia, 36(1), 62–67. [DOI] [PubMed] [Google Scholar]
- Baylin, S. B. , & Jones, P. A. (2011). A decade of exploring the cancer epigenome—Biological and translational implications. Nature Reviews. Cancer, 11(10), 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beetch, M. , Lubecka, K. , Kristofzski, H. , Suderman, M. , & Stefanska, B. (2018). Subtle alterations in DNA methylation patterns in normal cells in response to dietary stilbenoids. Molecular Nutrition & Food Research, 62(14), 1‐13, e1800193. [DOI] [PubMed] [Google Scholar]
- Beetch, M. , Lubecka, K. , Shen, K. , Flower, K. , Harandi‐Zadeh, S. , Suderman, M. , … Stefanska, B. (2019). Stilbenoid‐mediated epigenetic activation of semaphorin 3A in breast cancer cells involves changes in dynamic interactions of DNA with DNMT3A and NF1C transcription factor. Molecular Nutrition & Food Research, 63(19), 1‐15, e1801386. [DOI] [PubMed] [Google Scholar]
- Bellinger, F. P. , Raman, A. V. , Reeves, M. A. , & Berry, M. J. (2009). Regulation and function of selenoproteins in human disease. The Biochemical Journal, 422(1), 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Dor, A. , Steiner, M. , Gheber, L. , Danilenko, M. , Dubi, N. , Linnewiel, K. , … Levy, J. (2005). Carotenoids activate the antioxidant response element transcription system. Molecular Cancer Therapeutics, 4(1), 177–186. [PubMed] [Google Scholar]
- Bhattacharya, S. K. , Ramchandani, S. , Cervoni, N. , & Szyf, M. (1999). A mammalian protein with specific demethylase activity for mCpG DNA. Nature, 397(6720), 579–583. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya, S. , Pradhan, K. , Campbell, N. , Mazdo, J. , Vasantkumar, A. , Maqbool, S. , … Verma, A. (2017). Altered hydroxymethylation is seen at regulatory regions in pancreatic cancer and regulates oncogenic pathways. Genome Research, 27(11), 1830–1842. 10.1101/gr.222794.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigey, P. , Ramchandani, S. , Theberge, J. , Araujo, F. D. , & Szyf, M. (2000). Transcriptional regulation of the human DNA Methyltransferase (dnmt1) gene. Gene, 242(1‐2), 407–418. [DOI] [PubMed] [Google Scholar]
- Bohn, T. (2019). Carotenoids and markers of oxidative stress in human observational studies and intervention trials: Implications for chronic diseases. Antioxidants, 8(6), 179‐223. 10.3390/antiox8060179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollati, V. , Favero, C. , Albetti, B. , Tarantini, L. , Moroni, A. , Byun, H. M. , … Pesatori, A. (2014). Nutrients intake is associated with DNA methylation of candidate inflammatory genes in a population of obese subjects. Nutrients, 6(10), 4625–4639. 10.3390/nu6104625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borras, C. , Gambini, J. , Gomez‐Cabrera, M. C. , Sastre, J. , Pallardo, F. V. , Mann, G. E. , … Sastre, J. (2006). Genistein, a soy isoflavone, up‐regulates expression of antioxidant genes: Involvement of estrogen receptors, ERK1/2, and NFκB. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 20(12), 2136–2138. 10.1096/fj.05-5522fje [DOI] [PubMed] [Google Scholar]
- Cao‐Lei, L. , Dancause, K. N. , Elgbeili, G. , Laplante, D. P. , Szyf, M. , & King, S. (2018). DNA methylation mediates the effect of maternal cognitive appraisal of a disaster in pregnancy on the child's C‐peptide secretion in adolescence: Project Ice Storm. PLoS ONE, 13(2), 1‐17, e0192199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar, H. , & Bergman, Y. (2009). Linking DNA methylation and histone modification: Patterns and paradigms. Nature Reviews. Genetics, 10(5), 295–304. 10.1038/nrg2540 [DOI] [PubMed] [Google Scholar]
- Cha, J. , Niedzwiecki, A. , & Rath, M. (2015). Hypoascorbemia induces atherosclerosis and vascular deposition of lipoprotein(a) in transgenic mice. American Journal of Cardiovascular Disease, 5(1), 53–62. [PMC free article] [PubMed] [Google Scholar]
- Chuang, L. S. , Ian, H. I. , Koh, T. W. , Ng, H. H. , Xu, G. , & Li, B. F. (1997). Human DNA‐(cytosine‐5) methyltransferase‐PCNA complex as a target for p21WAF1. Science, 277(5334), 1996–2000. [DOI] [PubMed] [Google Scholar]
- Chung, J. H. , Ostrowski, M. C. , Romigh, T. , Minaguchi, T. , Waite, K. A. , & Eng, C. (2006). The ERK1/2 pathway modulates nuclear PTEN‐mediated cell cycle arrest by cyclin D1 transcriptional regulation. Human Molecular Genetics, 15(17), 2553–2559. [DOI] [PubMed] [Google Scholar]
- Clifford, R. L. , Fishbane, N. , Patel, J. , MacIsaac, J. L. , McEwen, L. M. , Fisher, A. J. , … Knox, A. J. (2018). Altered DNA methylation is associated with aberrant gene expression in parenchymal but not airway fibroblasts isolated from individuals with COPD. Clinical Epigenetics, 10, 32‐46. 10.1186/s13148-018-0464-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel, S. , Limson, J. L. , Dairam, A. , Watkins, G. M. , & Daya, S. (2004). Through metal binding, curcumin protects against lead‐ and cadmium‐induced lipid peroxidation in rat brain homogenates and against lead‐induced tissue damage in rat brain. Journal of Inorganic Biochemistry, 98(2), 266–275. [DOI] [PubMed] [Google Scholar]
- Das, S. , Foley, N. , Bryan, K. , Watters, K. M. , Bray, I. , Murphy, D. M. , … Stallings, R. L. (2010). MicroRNA mediates DNA demethylation events triggered by retinoic acid during neuroblastoma cell differentiation. Cancer Research, 70(20), 7874–7881. 10.1158/0008-5472.CAN-10-1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave, B. , Eason, R. R. , Till, S. R. , Geng, Y. , Velarde, M. C. , Badger, T. M. , & Simmen, R. C. M. (2005). The soy isoflavone genistein promotes apoptosis in mammary epithelial cells by inducing the tumor suppressor PTEN. Carcinogenesis, 26(10), 1793–1803. 10.1093/carcin/bgi131 [DOI] [PubMed] [Google Scholar]
- Deng, Y. , Lu, X. , Wang, L. , Li, T. , Ding, Y. , Cao, H. , … Yu, G. (2014). Curcumin inhibits the AKT/NF‐κB signaling via CpG demethylation of the promoter and restoration of NEP in the N2a cell line. The AAPS Journal, 16(4), 649–657. 10.1208/s12248-014-9605-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, N. , Bonham, E. M. , Hannon, B. E. , Amick, T. R. , Baylin, S. B. , & O'Hagan, H. M. (2016). Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. Journal of Molecular Cell Biology, 8(3), 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, N. , Miller, S. A. , Savant, S. S. , & O'Hagan, H. M. (2019). JAK2 regulates mismatch repair protein‐mediated epigenetic alterations in response to oxidative damage. Environmental and Molecular Mutagenesis, 60(4), 308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy, D. C. , Weidman, J. R. , Waterland, R. A. , & Jirtle, R. L. (2006). Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environmental Health Perspectives, 114(4), 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elisia, I. , & Kitts, D. D. (2013a). Different tocopherol isoforms vary in capacity to scavenge free radicals, prevent inflammatory response, and induce apoptosis in both adult‐ and fetal‐derived intestinal epithelial cells. BioFactors, 39(6), 663–671. [DOI] [PubMed] [Google Scholar]
- Elisia, I. , & Kitts, D. D. (2013b). Modulation of NF‐κB and Nrf2 control of inflammatory responses in FHs 74 Int cell line is tocopherol isoform‐specific. American Journal of Physiology. Gastrointestinal and Liver Physiology, 305(12), G940–G949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erasalo, H. , Hamalainen, M. , Leppanen, T. , Maki‐Opas, I. , Laavola, M. , Haavikko, R. , … Moilanen, E. (2018). Natural stilbenoids have anti‐inflammatory properties in vivo and down‐regulate the production of inflammatory mediators NO, IL6, and MCP1 possibly in a PI3K/Akt‐dependent manner. Journal of Natural Products, 81(5), 1131–1142. 10.1021/acs.jnatprod.7b00384 [DOI] [PubMed] [Google Scholar]
- Fang, M. Z. , Chen, D. , Sun, Y. , Jin, Z. , Christman, J. K. , & Yang, C. S. (2005). Reversal of hypermethylation and reactivation of p16INK4a, RARβ, and MGMT genes by genistein and other isoflavones from soy. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 11(19 Pt 1), 7033–7041. [DOI] [PubMed] [Google Scholar]
- Fang, M. Z. , Wang, Y. , Ai, N. , Hou, Z. , Sun, Y. , Lu, H. , … Yang, C. S. (2003). Tea polyphenol (‐)‐epigallocatechin‐3‐gallate inhibits DNA methyltransferase and reactivates methylation‐silenced genes in cancer cell lines. Cancer Research, 63(22), 7563–7570. [PubMed] [Google Scholar]
- Fernandez‐Sanles, A. , Sayols‐Baixeras, S. , Subirana, I. , Degano, I. R. , & Elosua, R. (2017). Association between DNA methylation and coronary heart disease or other atherosclerotic events: A systematic review. Atherosclerosis, 263, 325–333. [DOI] [PubMed] [Google Scholar]
- Fischer, R. , & Maier, O. (2015). Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxidative Medicine and Cellular Longevity, 2015, 1‐18, 610813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca, S. G. , Gromada, J. , & Urano, F. (2011). Endoplasmic reticulum stress and pancreatic β‐cell death. Trends in Endocrinology and Metabolism: TEM, 22(7), 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, D. , Pinello, N. , Nguyen, T. V. , Thoeng, A. , Nagarajah, R. , Holst, J. , … Wong, J. J. L. (2019). DNA methylation/hydroxymethylation regulate gene expression and alternative splicing during terminal granulopoiesis. Epigenomics, 11(1), 95–109. 10.2217/epi-2018-0050 [DOI] [PubMed] [Google Scholar]
- Gao, L. , Cheng, D. , Yang, J. , Wu, R. , Li, W. , & Kong, A. N. (2018). Sulforaphane epigenetically demethylates the CpG sites of the miR‐9‐3 promoter and reactivates miR‐9‐3 expression in human lung cancer A549 cells. The Journal of Nutritional Biochemistry, 56, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerecke, C. , Schumacher, F. , Edlich, A. , Wetzel, A. , Yealland, G. , Neubert, L. K. , … Kleuser, B. (2018). Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget, 9(67), 32822–32840. 10.18632/oncotarget.25999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll, M. G. , Kirpekar, F. , Maggert, K. A. , Yoder, J. A. , Hsieh, C. L. , Zhang, X. , … Bestor, T. H. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science, 311(5759), 395–398. 10.1126/science.1120976 [DOI] [PubMed] [Google Scholar]
- Guo, Y. , Wu, R. , Gaspar, J. M. , Sargsyan, D. , Su, Z. Y. , Zhang, C. , … Kong, A. N. (2018). DNA methylome and transcriptome alterations and cancer prevention by curcumin in colitis‐accelerated colon cancer in mice. Carcinogenesis, 39(5), 669–680. 10.1093/carcin/bgy043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, E. , Dayeh, T. , Kirkpatrick, C. L. , Wollheim, C. B. , Dekker Nitert, M. , & Ling, C. (2013). DNA methylation of the glucagon‐like peptide 1 receptor (GLP1R) in human pancreatic islets. BMC Medical Genetics, 14, 76‐83. 10.1186/1471-2350-14-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm, S. , Just, G. , Lacoste, N. , Moitessier, N. , Szyf, M. , & Mamer, O. (2008). On the mechanism of demethylation of 5‐methylcytosine in DNA. Bioorganic & Medicinal Chemistry Letters, 18(3), 1046–1049. 10.1016/j.bmcl.2007.12.027 [DOI] [PubMed] [Google Scholar]
- Han, S. G. , Han, S. S. , Toborek, M. , & Hennig, B. (2012). EGCG protects endothelial cells against PCB 126‐induced inflammation through inhibition of AhR and induction of Nrf2‐regulated genes. Toxicology and Applied Pharmacology, 261(2), 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodaei, H. , Adibian, M. , Nikpayam, O. , Hedayati, M. , & Sohrab, G. (2019). The effect of curcumin supplementation on anthropometric indices, insulin resistance and oxidative stress in patients with type 2 diabetes: A randomized, double‐blind clinical trial. Diabetology & Metabolic Syndrome, 11, 41‐49. 10.1186/s13098-019-0437-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, H. M. , Wu, W. M. , & Hu, M. L. (2009). Soy isoflavones attenuate oxidative stress and improve parameters related to aging and Alzheimer's disease in C57BL/6J mice treated with D‐galactose. Food and Chemical Toxicology: An International Journal Published for the British Industrial Biological Research Association, 47(3), 625–632. [DOI] [PubMed] [Google Scholar]
- Hu, C. , & Kitts, D. D. (2001). Evaluation of antioxidant activity of epigallocatechin gallate in biphasic model systems in vitro. Molecular and Cellular Biochemistry, 218(1‐2), 147–155. 10.1023/A:1007220928446 [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Khor, T. O. , Shu, L. , Saw, C. L. , Wu, T. Y. , Suh, N. , … Kong, A. N. (2012). A γ‐tocopherol‐rich mixture of tocopherols maintains Nrf2 expression in prostate tumors of TRAMP mice via epigenetic inhibition of CpG methylation. The Journal of Nutrition, 142(5), 818–823. 10.3945/jn.111.153114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. , Li, W. , Su, Z. Y. , & Kong, A. N. (2015). The complexity of the Nrf2 pathway: Beyond the antioxidant response. The Journal of Nutritional Biochemistry, 26(12), 1401–1413. 10.1016/j.jnutbio.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. S. , Zhi, Y. F. , & Wang, S. R. (2009). Hypermethylation of estrogen receptor‐α gene in atheromatosis patients and its correlation with homocysteine. Pathophysiology: The Official Journal of the International Society for Pathophysiology, 16(4), 259–265. [DOI] [PubMed] [Google Scholar]
- Huo, Z. , Zhu, Y. , Yu, L. , Yang, J. , De Jager, P. , Bennett, D. A. , & Zhao, J. (2019). DNA methylation variability in Alzheimer's disease. Neurobiology of Aging, 76, 35–44. 10.1016/j.neurobiolaging.2018.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, J. T. , Kwon, D. Y. , Park, O. J. , & Kim, M. S. (2008). Resveratrol protects ROS‐induced cell death by activating AMPK in H9c2 cardiac muscle cells. Genes & Nutrition, 2(4), 323–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida, T. , Suetake, I. , Tajima, S. , Morioka, H. , Ohta, S. , Obuse, C. , & Tsurimoto, T. (2002). PCNA clamp facilitates action of DNA cytosine methyltransferase 1 on hemimethylated DNA. Genes to Cells: Devoted to Molecular & Cellular Mechanisms, 7(10), 997–1007. 10.1046/j.1365-2443.2002.00584.x [DOI] [PubMed] [Google Scholar]
- Iwata, A. , Nagata, K. , Hatsuta, H. , Takuma, H. , Bundo, M. , Iwamoto, K. , … Tsuji, S. (2014). Altered CpG methylation in sporadic Alzheimer's disease is associated with APP and MAPT dysregulation. Human Molecular Genetics, 23(3), 648–656. 10.1093/hmg/ddt451 [DOI] [PubMed] [Google Scholar]
- Jablonska, E. , & Reszka, E. (2017). Selenium and epigenetics in cancer: Focus on DNA methylation. Advances in Cancer Research, 136, 193–234. [DOI] [PubMed] [Google Scholar]
- Jiang, A. , Wang, X. , Shan, X. , Li, Y. , Wang, P. , Jiang, P. , & Feng, Q. (2015). Curcumin reactivates silenced tumor suppressor gene RARβ by reducing DNA methylation. Phytotherapy Research: PTR, 29(8), 1237–1245. 10.1002/ptr.5373 [DOI] [PubMed] [Google Scholar]
- Jones, P. A. , & Takai, D. (2001). The role of DNA methylation in mammalian epigenetics. Science, 293(5532), 1068–1070. [DOI] [PubMed] [Google Scholar]
- Kala, R. , & Tollefsbol, T. O. (2016). A novel combinatorial epigenetic therapy using resveratrol and pterostilbene for restoring estrogen receptor‐α (ERα) expression in ERα‐negative breast cancer cells. PLoS ONE, 11(5), 1‐17, e0155057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandimalla, R. , Thirumala, V. , & Reddy, P. H. (2017). Is Alzheimer's disease a type 3 diabetes? A critical appraisal. Biochimica et Biophysica Acta, Molecular Basis of Disease, 1863(5), 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin, M. (1995). The regulation of AP‐1 activity by mitogen‐activated protein kinases. The Journal of Biological Chemistry, 270(28), 16483–16486. [DOI] [PubMed] [Google Scholar]
- Kato, K. , Long, N. K. , Makita, H. , Toida, M. , Yamashita, T. , Hatakeyama, D. , … Shibata, T. (2008). Effects of green tea polyphenol on methylation status of RECK gene and cancer cell invasion in oral squamous cell carcinoma cells. British Journal of Cancer, 99(4), 647–654. 10.1038/sj.bjc.6604521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattoor, A. J. , Pothineni, N. V. K. , Palagiri, D. , & Mehta, J. L. (2017). Oxidative stress in atherosclerosis. Current Atherosclerosis Reports, 19(11), 42‐53. [DOI] [PubMed] [Google Scholar]
- Kensler, T. W. , Egner, P. A. , Agyeman, A. S. , Visvanathan, K. , Groopman, J. D. , Chen, J. G. , … Talalay, P. (2013). Keap1‐nrf2 signaling: A target for cancer prevention by sulforaphane. Topics in Current Chemistry, 329, 163–177. 10.1007/128_2012_339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M. A. , Hussain, A. , Sundaram, M. K. , Alalami, U. , Gunasekera, D. , Ramesh, L. , … Quraishi, U. (2015). (‐)‐Epigallocatechin‐3‐gallate reverses the expression of various tumor‐suppressor genes by inhibiting DNA methyltransferases and histone deacetylases in human cervical cancer cells. Oncology Reports, 33(4), 1976–1984. 10.3892/or.2015.3802 [DOI] [PubMed] [Google Scholar]
- Khor, T. O. , Huang, Y. , Wu, T. Y. , Shu, L. , Lee, J. , & Kong, A. N. (2011). Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochemical Pharmacology, 82(9), 1073–1078. 10.1016/j.bcp.2011.07.065 [DOI] [PubMed] [Google Scholar]
- Kosuru, R. , Kandula, V. , Rai, U. , Prakash, S. , Xia, Z. , & Singh, S. (2018). Pterostilbene decreases cardiac oxidative stress and inflammation via activation of AMPK/Nrf2/HO‐1 pathway in fructose‐fed diabetic rats. Cardiovascular Drugs and Therapy, 32(2), 147–163. [DOI] [PubMed] [Google Scholar]
- Kumar, U. , Sharma, U. , & Rathi, G. (2017). Reversal of hypermethylation and reactivation of glutathione S‐transferase pi 1 gene by curcumin in breast cancer cell line. Tumour Biology: The Journal of the International Society for Oncodevelopmental Biology and Medicine, 39(2), 1‐8, 1010428317692258 10.1177/1010428317692258 [DOI] [PubMed] [Google Scholar]
- Kurutas, E. B. (2016). The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutrition Journal, 15(1), 71‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashley, T. , Gami, P. , Valizadeh, N. , Li, A. , Revesz, T. , & Balazs, R. (2015). Alterations in global DNA methylation and hydroxymethylation are not detected in Alzheimer's disease. Neuropathology and Applied Neurobiology, 41(4), 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W. J. , Shim, J. Y. , & Zhu, B. T. (2005). Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Molecular Pharmacology, 68(4), 1018–1030. [DOI] [PubMed] [Google Scholar]
- Lee, W. J. , & Zhu, B. T. (2006). Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol‐containing coffee polyphenols. Carcinogenesis, 27(2), 269–277. [DOI] [PubMed] [Google Scholar]
- Liang, N. , Dupuis, J. H. , Yada, R. Y. , & Kitts, D. D. (2019). Chlorogenic acid isomers directly interact with Keap 1‐Nrf2 signaling in Caco‐2 cells. Molecular and Cellular Biochemistry, 457(1‐2), 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, N. , & Kitts, D. D. (2018). Amelioration of oxidative stress in Caco‐2 cells treated with pro‐inflammatory proteins by chlorogenic acid isomers via activation of the Nrf2‐Keap1‐ARE‐signaling pathway. Journal of Agricultural and Food Chemistry, 66(42), 11008–11017. [DOI] [PubMed] [Google Scholar]
- Lin, X. , Bai, D. , Wei, Z. , Zhang, Y. , Huang, Y. , Deng, H. , & Huang, X. (2019). Curcumin attenuates oxidative stress in RAW264.7 cells by increasing the activity of antioxidant enzymes and activating the Nrf2‐Keap1 pathway. PLoS ONE, 14(5), 1‐13, e0216711 10.1371/journal.pone.0216711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link, A. , Balaguer, F. , Shen, Y. , Lozano, J. J. , Leung, H. C. , Boland, C. R. , & Goel, A. (2013). Curcumin modulates DNA methylation in colorectal cancer cells. PLoS ONE, 8(2), 1‐13, e57709 10.1371/journal.pone.0057709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski, B. (2019). Redox‐active selenium in health and disease: A conceptual review. Mini Reviews in Medicinal Chemistry, 19(9), 720–726. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Sun, C. , Liu, B. , Jin, X. , Li, P. , Zheng, X. , … Li, Q. (2016). Genistein mediates the selective radiosensitizing effect in NSCLC A549 cells via inhibiting methylation of the keap1 gene promoter region. Oncotarget, 7(19), 27267–27279. 10.18632/oncotarget.8403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Zhou, J. , Hu, Y. , Wang, J. , & Yuan, C. (2017). Curcumin inhibits growth of human breast cancer cells through demethylation of DLC1 promoter. Molecular and Cellular Biochemistry, 425(1‐2), 47–58. [DOI] [PubMed] [Google Scholar]
- Liu, Z. , Xie, Z. , Jones, W. , Pavlovicz, R. E. , Liu, S. , Yu, J. , … Chan, K. K. (2009). Curcumin is a potent DNA hypomethylation agent. Bioorganic & Medicinal Chemistry Letters, 19(3), 706–709. 10.1016/j.bmcl.2008.12.041 [DOI] [PubMed] [Google Scholar]
- Lubecka, K. , Kaufman‐Szymczyk, A. , & Fabianowska‐Majewska, K. (2018). Inhibition of breast cancer cell growth by the combination of clofarabine and sulforaphane involves epigenetically mediated CDKN2A upregulation. Nucleosides, Nucleotides & Nucleic Acids, 37(5), 280–289. [DOI] [PubMed] [Google Scholar]
- Lubecka, K. , Kurzava, L. , Flower, K. , Buvala, H. , Zhang, H. , Teegarden, D. , … Stefanska, B. (2016). Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis, 37(7), 656–668. 10.1093/carcin/bgw048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubecka‐Pietruszewska, K. , Kaufman‐Szymczyk, A. , Stefanska, B. , Cebula‐Obrzut, B. , Smolewski, P. , & Fabianowska‐Majewska, K. (2015). Sulforaphane alone and in combination with clofarabine epigenetically regulates the expression of DNA methylation‐silenced tumour suppressor genes in human breast cancer cells. Journal of Nutrigenetics and Nutrigenomics, 8(2), 91–101. [DOI] [PubMed] [Google Scholar]
- Maiuri, A. R. , & O'Hagan, H. M. (2016). Interplay between inflammation and epigenetic changes in cancer. Progress in Molecular Biology and Translational Science, 144, 69–117. [DOI] [PubMed] [Google Scholar]
- Maiuri, A. R. , Peng, M. , Podicheti, R. , Sriramkumar, S. , Kamplain, C. M. , Rusch, D. B. , … O'Hagan, H. M. (2017). Mismatch repair proteins initiate epigenetic alterations during inflammation‐driven tumorigenesis. Cancer Research, 77(13), 3467–3478. 10.1158/0008-5472.CAN-17-0056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majid, S. , Dar, A. A. , Shahryari, V. , Hirata, H. , Ahmad, A. , Saini, S. , … Dahiya, R. (2010). Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B‐Cell translocation gene 3 in prostate cancer. Cancer, 116(1), 66–76. 10.1002/cncr.24662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manikandan, P. , Sumitra, M. , Aishwarya, S. , Manohar, B. M. , Lokanadam, B. , & Puvanakrishnan, R. (2004). Curcumin modulates free radical quenching in myocardial ischaemia in rats. The International Journal of Biochemistry & Cell Biology, 36(10), 1967–1980. [DOI] [PubMed] [Google Scholar]
- Manna, S. K. , Mukhopadhyay, A. , & Aggarwal, B. B. (2000). Resveratrol suppresses TNF‐induced activation of nuclear transcription factors NF‐κB, activator protein‐1, and apoptosis: Potential role of reactive oxygen intermediates and lipid peroxidation. Journal of Immunology, 164(12), 6509–6519. [DOI] [PubMed] [Google Scholar]
- Mao, X. , Gu, C. , Chen, D. , Yu, B. , & He, J. (2017). Oxidative stress‐induced diseases and tea polyphenols. Oncotarget, 8(46), 81649–81661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri, A. , Mazzone, M. G. , Giuliano, F. , Vinciguerra, M. , Basile, G. , Barchitta, M. , & Agodi, A. (2018). Curcumin modulates DNA methyltransferase functions in a cellular model of diabetic retinopathy. Oxidative Medicine and Cellular Longevity, 2018, 1‐12, 5407482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina‐Aguilar, R. , Pérez‐Plasencia, C. , Marchat, L. A. , Gariglio, P. , Mena, J. G. , Cuevas, S. R. , … López‐Camarillo, C. (2016). Methylation landscape of human breast cancer cells in response to dietary compound resveratrol. PLoS ONE, 11(6), 1‐20, e0157866 10.1371/journal.pone.0157866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor, E. A. , Court, B. L. , Young, J. I. , & Wang, G. (2013). Ascorbate induces ten‐eleven translocation (Tet) methylcytosine dioxygenase‐mediated generation of 5‐hydroxymethylcytosine. The Journal of Biological Chemistry, 288(19), 13669–13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza, S. , Sharma, G. , Parshad, R. , Gupta, S. D. , Pandya, P. , & Ralhan, R. (2013). Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. Journal of Breast Cancer, 16(1), 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, J. , Moseley, V. R. , Cabang, A. B. , Coleman, K. , Wei, W. , Garrett‐Mayer, E. , & Wargovich, M. J. (2016). Reduction in promotor methylation utilizing EGCG (epigallocatechin‐3‐gallate) restores RXRα expression in human colon cancer cells. Oncotarget, 7(23), 35313–35326. 10.18632/oncotarget.9204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow, J. D. , Glass, K. , Cho, M. H. , Hersh, C. P. , Pinto‐Plata, V. , Celli, B. , … DeMeo, D. L. (2018). Human lung DNA methylation quantitative trait loci colocalize with chronic obstructive pulmonary disease genome‐wide association loci. American Journal of Respiratory and Critical Care Medicine, 197(10), 1275–1284. 10.1164/rccm.201707-1434OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, K. , Yu, S. , & Kitts, D. D. (2019). The role of nitric oxide in regulating intestinal redox status and intestinal epithelial cell functionality. International Journal of Molecular Sciences, 20(7), 1‐14. 10.3390/ijms20071755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCI (2018). National cancer statistics 2018.
- Niki, E. (2014). Role of vitamin E as a lipid‐soluble peroxyl radical scavenger: in vitro and in vivo evidence. Free Radical Biology & Medicine, 66, 3–12. 10.1016/j.freeradbiomed.2013.03.022 [DOI] [PubMed] [Google Scholar]
- O'Hagan, H. M. (2014). Chromatin modifications during repair of environmental exposure‐induced DNA damage: A potential mechanism for stable epigenetic alterations. Environmental and Molecular Mutagenesis, 55(3), 278–291. 10.1002/em.21830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan, H. M. , Mohammad, H. P. , & Baylin, S. B. (2008). Double strand breaks can initiate gene silencing and SIRT1‐dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genetics, 4(8), 1‐16, e1000155 10.1371/journal.pgen.1000155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan, H. M. , Wang, W. , Sen, S. , Destefano Shields, C. , Lee, S. S. , Zhang, Y. W. , … Casero, R. A. (2011). Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell, 20(5), 606–619. 10.1016/j.ccr.2011.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orso, E. , & Schmitz, G. (2017). Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clinical Research in Cardiology Supplements, 12(Suppl 1), 31–37. 10.1007/s11789-017-0084-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papoutsis, A. J. , Lamore, S. D. , Wondrak, G. T. , Selmin, O. I. , & Romagnolo, D. F. (2010). Resveratrol prevents epigenetic silencing of BRCA‐1 by the aromatic hydrocarbon receptor in human breast cancer cells. The Journal of Nutrition, 140(9), 1607–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, D. , Ge, G. , Gong, Y. , Zhan, Y. , He, S. , Guan, B. , … Zhou, L. (2018). Vitamin C increases 5‐hydroxymethylcytosine level and inhibits the growth of bladder cancer. Clinical Epigenetics, 10(1), 94‐107. 10.1186/s13148-018-0527-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, W. , Zhu, W. , Shi, H. , Hewett, J. E. , Ruhlen, R. L. , MacDonald, R. S. , … Sauter, E. R. (2009). Soy isoflavones have an antiestrogenic effect and alter mammary promoter hypermethylation in healthy premenopausal women. Nutrition and Cancer, 61(2), 238–244. 10.1080/01635580802404196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu, K. , Ma, X. F. , Li, G. H. , Zhang, H. , Liu, Y. M. , Zhang, K. , … Wang, Z. (2017). Vitamin C down‐regulate apo(a) expression via Tet2‐dependent DNA demethylation in HepG2 cells. International Journal of Biological Macromolecules, 98, 637–645. 10.1016/j.ijbiomac.2017.02.025 [DOI] [PubMed] [Google Scholar]
- Rahman, I. (2008). Antioxidant therapeutic advances in COPD. Therapeutic Advances in Respiratory Disease, 2(6), 351–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman Mazumder, M. A. , & Hongsprabhas, P. (2016). Genistein as antioxidant and antibrowning agents in in vivo and in vitro: A review. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie, 82, 379–392. [DOI] [PubMed] [Google Scholar]
- Rasmussen, K. D. , & Helin, K. (2016). Role of TET enzymes in DNA methylation, development, and cancer. Genes & Development, 30(7), 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman, K. , & Akash, M. S. H. (2017). Mechanism of generation of oxidative stress and pathophysiology of type 2 diabetes mellitus: How are they interlinked? Journal of Cellular Biochemistry, 118(11), 3577–3585. [DOI] [PubMed] [Google Scholar]
- Ren, J. , Fan, C. , Chen, N. , Huang, J. , & Yang, Q. (2011). Resveratrol pretreatment attenuates cerebral ischemic injury by upregulating expression of transcription factor Nrf2 and HO‐1 in rats. Neurochemical Research, 36(12), 2352–2362. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics, C. , Kundaje, A. , Meuleman, W. , Ernst, J. , Bilenky, M. , Yen, A. , … Amin, V. (2015). Integrative analysis of 111 reference human epigenomes. Nature, 518(7539), 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghafinia, S. , Mina, M. , Riggi, N. , Hanahan, D. , & Ciriello, G. (2018). Pan‐cancer landscape of aberrant DNA methylation across human tumors. Cell Reports, 25(4), 1066, e1068–1080. [DOI] [PubMed] [Google Scholar]
- Saraf‐Bank, S. , Ahmadi, A. , Paknahad, Z. , Maracy, M. , & Nourian, M. (2019). Effects of curcumin supplementation on markers of inflammation and oxidative stress among healthy overweight and obese girl adolescents: A randomized placebo‐controlled clinical trial. Phytotherapy Research: PTR, 33, 2015–2022. 10.1002/ptr.6370 [DOI] [PubMed] [Google Scholar]
- Setchell, K. D. , & Cassidy, A. (1999). Dietary isoflavones: Biological effects and relevance to human health. The Journal of Nutrition, 129(3), 758S–767S. [DOI] [PubMed] [Google Scholar]
- Shin, J. H. , Jeon, H. J. , Park, J. , & Chang, M. S. (2016). Epigallocatechin‐3‐gallate prevents oxidative stress‐induced cellular senescence in human mesenchymal stem cells via Nrf2. International Journal of Molecular Medicine, 38(4), 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, B. , Shoulson, R. , Chatterjee, A. , Ronghe, A. , Bhat, N. K. , Dim, D. C. , & Bhat, H. K. (2014). Resveratrol inhibits estrogen‐induced breast carcinogenesis through induction of NRF2‐mediated protective pathways. Carcinogenesis, 35(8), 1872–1880. 10.1093/carcin/bgu120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi, F. G. , Sheervalilou, R. , Vardiani, M. , Khalili, M. , & Alipour, M. R. (2012). Chronic resveratrol administration has beneficial effects in experimental model of type 2 diabetic rats. Endocrine Regulations, 46(2), 83–90. [DOI] [PubMed] [Google Scholar]
- Speckmann, B. , Schulz, S. , Hiller, F. , Hesse, D. , Schumacher, F. , Kleuser, B. , … Kipp, A. P. (2017). Selenium increases hepatic DNA methylation and modulates one‐carbon metabolism in the liver of mice. The Journal of Nutritional Biochemistry, 48, 112–119. 10.1016/j.jnutbio.2017.07.002 [DOI] [PubMed] [Google Scholar]
- Starczak, M. , Zarakowska, E. , Modrzejewska, M. , Dziaman, T. , Szpila, A. , Linowiecka, K. , … Olinski, R. (2018). In vivo evidence of ascorbate involvement in the generation of epigenetic DNA modifications in leukocytes from patients with colorectal carcinoma, benign adenoma and inflammatory bowel disease. Journal of Translational Medicine, 16(1), 204‐215. 10.1186/s12967-018-1581-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanska, B. , Huang, J. , Bhattacharyya, B. , Suderman, M. , Hallett, M. , Han, Z. G. , & Szyf, M. (2011). Definition of the landscape of promoter DNA hypomethylation in liver cancer. Cancer Research, 71(17), 5891–5903. 10.1158/0008-5472.CAN-10-3823 [DOI] [PubMed] [Google Scholar]
- Stefanska, B. , Rudnicka, K. , Bednarek, A. , & Fabianowska‐Majewska, K. (2010). Hypomethylation and induction of retinoic acid receptor β2 by concurrent action of adenosine analogues and natural compounds in breast cancer cells. European Journal of Pharmacology, 638(1‐3), 47–53. [DOI] [PubMed] [Google Scholar]
- Stefanska, B. , Salame, P. , Bednarek, A. , & Fabianowska‐Majewska, K. (2012). Comparative effects of retinoic acid, vitamin D and resveratrol alone and in combination with adenosine analogues on methylation and expression of phosphatase and tensin homologue tumour suppressor gene in breast cancer cells. The British Journal of Nutrition, 107(6), 781–790. [DOI] [PubMed] [Google Scholar]
- Stefanska, B. , Suderman, M. , Machnes, Z. , Bhattacharyya, B. , Hallett, M. , & Szyf, M. (2013). Transcription onset of genes critical in liver carcinogenesis is epigenetically regulated by methylated DNA‐binding protein MBD2. Carcinogenesis, 34(12), 2738–2749. [DOI] [PubMed] [Google Scholar]
- Su, Z. Y. , Zhang, C. , Lee, J. H. , Shu, L. , Wu, T. Y. , Khor, T. O. , … Kong, A. N. T. (2014). Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA‐induced mouse skin cell transformation by sulforaphane. Cancer Prevention Research, 7(3), 319–329. 10.1158/1940-6207.CAPR-13-0313-T [DOI] [PubMed] [Google Scholar]
- Suantawee, T. , Cheng, H. , & Adisakwattana, S. (2016). Protective effect of cyanidin against glucose‐ and methylglyoxal‐induced protein glycation and oxidative DNA damage. International Journal of Biological Macromolecules, 93(Pt A), 814–821. [DOI] [PubMed] [Google Scholar]
- Sugisawa, A. , Kimura, M. , Fenech, M. , & Umegaki, K. (2004). Anti‐genotoxic effects of tea catechins against reactive oxygen species in human lymphoblastoid cells. Mutation Research, 559(1‐2), 97–103. [DOI] [PubMed] [Google Scholar]
- Sun, W. , Liu, X. , Zhang, H. , Song, Y. , Li, T. , Liu, X. , … Wu, H. (2017). Epigallocatechin gallate upregulates NRF2 to prevent diabetic nephropathy via disabling KEAP1. Free Radical Biology & Medicine, 108, 840–857. 10.1016/j.freeradbiomed.2017.04.365 [DOI] [PubMed] [Google Scholar]
- Sun, X. , Xiao, Y. , Zeng, Z. , Shi, Y. , Tang, B. , Long, H. , … Xiao, R. (2018). All‐trans retinoic acid induces CD4+CD25+FOXP3+ regulatory T cells by increasing foxp3 demethylation in systemic sclerosis CD4+ T cells. Journal of Immunology Research, 2018, 1, 8658156–7. 10.1155/2018/8658156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafazoli, S. , Wright, J. S. , & O'Brien, P. J. (2005). Prooxidant and antioxidant activity of vitamin E analogues and troglitazone. Chemical Research in Toxicology, 18(10), 1567–1574. [DOI] [PubMed] [Google Scholar]
- Tang, F. , Lu, M. , Zhang, S. , Mei, M. , Wang, T. , Liu, P. , & Wang, H. (2014). Vitamin E conditionally inhibits atherosclerosis in ApoE knockout mice by anti‐oxidation and regulation of vasculature gene expressions. Lipids, 49(12), 1215–1223. 10.1007/s11745-014-3962-z [DOI] [PubMed] [Google Scholar]
- Tang, W. J. , Hu, C. P. , Chen, M. F. , Deng, P. Y. , & Li, Y. J. (2006). Epigallocatechin gallate preserves endothelial function by reducing the endogenous nitric oxide synthase inhibitor level. Canadian Journal of Physiology and Pharmacology, 84(2), 163–171. 10.1139/y05-156 [DOI] [PubMed] [Google Scholar]
- Tapp, H. S. , Commane, D. M. , Bradburn, D. M. , Arasaradnam, R. , Mathers, J. C. , Johnson, I. T. , & Belshaw, N. J. (2013). Nutritional factors and gender influence age‐related DNA methylation in the human rectal mucosa. Aging Cell, 12(1), 148–155. 10.1111/acel.12030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler, R. , Maurizi, A. , Roschger, P. , Sturmlechner, I. , Khani, F. , Spitzer, S. , … van Wijnen, A. J. (2016). Anabolic and antiresorptive modulation of bone homeostasis by the epigenetic modulator sulforaphane, a naturally occurring isothiocyanate. The Journal of Biological Chemistry, 291(13), 6754–6771. 10.1074/jbc.M115.678235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnies, E. , & Trushina, E. (2017). Oxidative stress, synaptic dysfunction, and Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 57(4), 1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traber, M. G. , & Stevens, J. F. (2011). Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radical Biology & Medicine, 51(5), 1000–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uthus, E. , Begaye, A. , Ross, S. , & Zeng, H. (2011). The von Hippel‐Lindau (VHL) tumor‐suppressor gene is down‐regulated by selenium deficiency in Caco‐2 cells and rat colon mucosa. Biological Trace Element Research, 142(2), 223–231. [DOI] [PubMed] [Google Scholar]
- Veland, N. , Lu, Y. , Hardikar, S. , Gaddis, S. , Zeng, Y. , Liu, B. , … Chen, T. (2019). DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Research, 47(1), 152–167. 10.1093/nar/gky947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vomund, S. , Schafer, A. , Parnham, M. J. , Brune, B. , & von Knethen, A. (2017). Nrf2, the master regulator of anti‐oxidative responses. International Journal of Molecular Sciences, 18(12), 1‐19. 10.3390/ijms18122772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic, E. A. , Chari, R. , Thu, K. L. , Wilson, I. M. , Cotton, A. M. , Kennett, J. Y. , … Lam, W. L. (2014). DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. American Journal of Respiratory Cell and Molecular Biology, 50(5), 912–922. 10.1165/rcmb.2013-0304OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2018). World Health statistics 2018.
- Xiang, N. , Zhao, R. , Song, G. , & Zhong, W. (2008). Selenite reactivates silenced genes by modifying DNA methylation and histones in prostate cancer cells. Carcinogenesis, 29(11), 2175–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Q. , Bai, Q. , Zou, L. Y. , Zhang, Q. Y. , Zhou, Y. , Chang, H. , … Mi, M. T. (2014). Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes, Chromosomes & Cancer, 53(5), 422–431. 10.1002/gcc.22154 [DOI] [PubMed] [Google Scholar]
- Yang, Q. , Proll, M. J. , Salilew‐Wondim, D. , Zhang, R. , Tesfaye, D. , Fan, H. , … Hölker, M. (2016). LPS‐induced expression of CD14 in the TRIF pathway is epigenetically regulated by sulforaphane in porcine pulmonary alveolar macrophages. Innate Immunity, 22(8), 682–695. 10.1177/1753425916669418 [DOI] [PubMed] [Google Scholar]
- Yang, Q. , Zhao, Y. , Zhang, Z. , & Chen, J. (2016). Association of interleukin‐6 methylation in leukocyte DNA with serum level and the risk of ischemic heart disease. Scandinavian Journal of Clinical and Laboratory Investigation, 76(4), 291–295. 10.3109/00365513.2016.1149616 [DOI] [PubMed] [Google Scholar]
- Yang, X. , Li, Y. , Li, Y. , Ren, X. , Zhang, X. , Hu, D. , … Shang, H. (2017). Oxidative stress‐mediated atherosclerosis: Mechanisms and therapies. Frontiers in Physiology, 8, 1‐16, 600. 10.3389/fphys.2017.00600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, R. , Mao, S. Q. , Zhao, B. , Chong, Z. , Yang, Y. , Zhao, C. , … Wang, H. (2013). Ascorbic acid enhances Tet‐mediated 5‐methylcytosine oxidation and promotes DNA demethylation in mammals. Journal of the American Chemical Society, 135(28), 10396–10403. 10.1021/ja4028346 [DOI] [PubMed] [Google Scholar]
- Yu, J. , Peng, Y. , Wu, L. C. , Xie, Z. , Deng, Y. , Hughes, T. , … Liu, Z. (2013). Curcumin down‐regulates DNA methyltransferase 1 and plays an anti‐leukemic role in acute myeloid leukemia. PLoS ONE, 8(2), 1‐9, e55934 10.1371/journal.pone.0055934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J. , Qiu, Y. , Yang, J. , Bian, S. , Chen, G. , Deng, M. , … Huang, L. (2016). DNMT1‐PPARγ pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Scientific Reports, 6, 1‐11, 30053 10.1038/srep30053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappe, K. , Pointner, A. , Switzeny, O. J. , Magnet, U. , Tomeva, E. , Heller, J. , … Haslberger, A. G. (2018). Counteraction of oxidative stress by vitamin E affects epigenetic regulation by increasing global methylation and gene expression of MLH1 and DNMT1 dose dependently in Caco‐2 cells. Oxidative Medicine and Cellular Longevity, 2018, 1‐13, 3734250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B. K. , Lai, Y. Q. , Niu, P. P. , Zhao, M. , & Jia, S. J. (2013). Epigallocatechin‐3‐gallate inhibits homocysteine‐induced apoptosis of endothelial cells by demethylation of the DDAH2 gene. Planta Medica, 79(18), 1715–1719. [DOI] [PubMed] [Google Scholar]
- Zhang, C. , Su, Z. Y. , Khor, T. O. , Shu, L. , & Kong, A. N. (2013). Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochemical Pharmacology, 85(9), 1398–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, F. , Zhang, J. , & Chang, N. (2018). Epigenetic modification of Nrf2 by sulforaphane increases the antioxidative and anti‐inflammatory capacity in a cellular model of Alzheimer's disease. European Journal of Pharmacology, 824, 1–10. [DOI] [PubMed] [Google Scholar]