

Abstract
Background and Purpose
Cerebral ischaemia/reperfusion causes exacerbated neuronal damage involving excessive autophagy and neuronal loss. The present study was designed to investigate the effect of icariside II, one of main active ingredients of Herba Epimedii on this loss and whether this is related to its PDE 5 inhibitory action.
Experimental Approach
Focal cerebral ischaemia was induced in the rat by transient middle cerebral artery occlusion over 2 hr, followed by reperfusion with icariside II, 3‐methylamphetamine or rapamycin. The effect of icariside II was determined measuring behaviour changes and the size of the infarction. The expressions of PDE 5, autophagy‐related proteins and the level of phosphorylation of glycogen synthase kinase‐3β (GSK‐3β) were determined. Cultured primary cortical neurons were subjected to oxygen and glucose deprivation followed by reoxygenation in the presence and absence of icariside II. A surface plasmon resonance assay and molecular docking were used to explore the interactions of icariside II with PDE 5 or GSK‐3β.
Key Results
Icariside II not only protected against induced ischaemic reperfusion injury in rats but also attenuated such injury in primary cortical neurons. The neuroprotective effects of icariside II on such injury were attributed to interfering with the PKG/GSK‐3β/autophagy axis by directly bounding to PDE 5 and GSK‐3β.
Conclusions and Implications
These findings indicate that icariside II attenuates cerebral I/R‐induced injury via interfering with PKG/GSK‐3β/autophagy axis. This study raises the possibility that icariside II and other PDE 5 inhibitors maybe effective in the treatment ischaemia stroke.
Abbreviations
- 3‐MA
3‐methylamphetamine
- Ad‐tf‐LC3
adenovirus harbouring tf‐LC3
- ATG5
autophagy‐related gene 5
- ATG7
autophagy‐related gene 7
- GSK‐3β
glycogen synthase kinase‐3β
- I/R
ischaemia/reperfusion
- ICS II
icariside II
- MAP 1LC3/LC3
microtubule‐associated protein 1 light chain 3
- MCA
middle cerebral artery
- MCAO
middle cerebral artery occlusion
- MTT
3‐4,5‐dimethylthiazol‐2,5‐diphenyltetrazolium bromide
- OGD/R
oxygen–glucose deprivation and reoxygenation
- Rap
rapamycin
- rCBF
relative cerebral blood flow
- S9A
constitutively active mutant of GSK‐3β
- tf‐LC3
tandem fluorescent mRFP‐GFP‐LC3
- TTC
2,3,5‐triphenyl‐tetrazolium chloride solution
What is already known
Existing PDE 5 inhibitors cause common adverse effects including elevating melanoma risk.
Icariside II (ICS II) is a PDE 5 inhibitor with a broad‐spectrum anti‐cancer agent.
What this study adds
Icariside II (ICS II) protects against cerebral ischaemia/reperfusion‐induced ischaemic stroke.
Benefit of icariside II on ischaemic stroke involves interference with PKG/glycogen synthase kinase‐3β/autophagy axis.
What is the clinical significance
Iicariside II may be a promising and effective PDE 5 inhibitor against ischaemic stroke.
1. INTRODUCTION
Ischaemic stroke, one of the most severe neurological disorders, causes high disability rate and high mortality, along with a high socio‐economic burden to the patients and the society (Flippo et al., 2018; C. Wu et al., 2019). One prevailing theory for the efficient treatment of ischaemic stroke is to timely restore the cerebral blood flow (reperfusion; Parada et al., 2019). However, cerebral ischaemia and reperfusion further exacerbate neuronal demise, a phenomenon called “cerebral ischaemia/reperfusion (I/R) injury” (Granger & Kvietys, 2015). Currently, the only approved to treat ischaemic stroke is the thrombolytic agent recombinant tissue plasminogen activator, which has a narrow therapeutic window and leads to severe side effects including cerebral I/R injury and haemorrhage (Diaz‐Canestro et al., 2019). The other strategies to treat ischaemic stroke include neuroprotection, but effective neuroprotectants are still unavailable. Hence, the development of an effective neuroprotectant is a dire clinical need.
Emerging evidence suggests that autophagy is a critical neuroprotective response against neuronal injury (Chen, Sun, Liu, & Sun, 2014; Sheng et al., 2010); however, excessive autophagy is widely recognized as a culprit of neuronal injury, even cell death (Uchiyama, Koike, & Shibata, 2008). Thus, there is an ongoing debate about whether activation of autophagy during cerebral I/R is beneficial or not. A better understanding of the role of autophagy in cerebral I/R injury is needed. In this respect, glycogen synthase kinase‐3 (GSK‐3) not only plays an important role in the neuronal cell death caused by cerebral I/R injury but also regulates autophagic cell death (Venna, Benashski, Chauhan, & McCullough, 2015). Thus inhibition of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2030 may provide neuroprotective effects against cerebral I/R‐induced injury (Chuang, Wang, & Chiu, 2011).
Now https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1304 is an enzyme that specifically hydrolyses https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2347, which can trigger the activation of https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=287 (L. Zhang, Seo, Li, Nam, & Yang, 2018). Recently, PDE 5 has been shown to be present in human neurons and affect the survival of neurons in ischaemic stroke, suggesting that it may be a potential drug target for treating cerebral I/R injury (Teich et al., 2016). As such, PDE 5 inhibitors could be potential therapeutic agents for cerebral I/R injury. Icariside II (ICS II) is one of the main active ingredients of Epimedii (extracted from Epimedium grandiflorum), which is a traditional Chinese medicine used for the treatment of a range of clinical disorders including dementia, erectile dysfunction and cardiovascular disorders (Liu et al., 2015). One of the notabley ingredients is icariside II (ICS II) which has antioxidant activity and exhibits a neuroprotective effect (Deng et al., 2017; L. Wang, Xu, et al., 2015; Yan et al., 2017). However, most recent clinical studies have reported that PDE 5 inhibitors cause common adverse effects, including promoting melanin synthesis which may exacerbate melanoma development (Dhayade et al., 2016; W. Q. Li, Qureshi, Robinson, & Han, 2014). In contrast, icariside II is a PDE 5 inhibitor with broad‐spectrum anti‐cancer activity and suppresses the proliferation of melanoma (J. Wu et al., 2013; J. Wu et al., 2015), suggesting that icariside II may be an unique PDE 5 inhibitor. Furthermore, a previous study has demonstrated that pretreatment with icariside II improves cerebral microcirculatory disturbances and alleviates hippocampal injury in gerbils after I/R (Yan et al., 2014). In this respect, our previous studies had demonstrated that pretreatment with icariside II exerted a neuroprotective effect at the acute stage of stroke in rats (Deng et al., 2016), as well as alleviating oxygen–glucose deprivation and reoxygenation (OGD/R)‐induced PC12 cell death (Feng, Gao, Liu, Shi, & Gong, 2018). Intriguingly, pretreatment with icariside II also inhibits hydrogen peroxide‐excessive autophagy in PC12 cells through regulating the reactive oxygen species (ROS)/GSK‐3β signalling pathway (Gao et al., 2017). However, whether post‐treatment with icariside II protects against cerebral I/R injury via GSK‐3β‐mediated autophagy remains unknown.
Hence, the current study is designed to investigate the effects of icariside II against cerebral I/R‐induced injury after 2 hr of focal cerebral ischaemia followed by 1‐, 3‐, and 7‐day reperfusion in vivo and OGD/R‐induced injury in rat primary cultured cortical neurons in vitro and whether these beneficial effects are associated with actions on the GSK‐3β/autophagy signalling pathway.
2. METHODS
2.1. Animals
All animal experimental procedures in this study were performed in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (National Institutes of Health Publication 85‐23, revised 1996) and were approved by the Experimental Animal Ethics Committee of the Zunyi Medical University (Guizhou, China). Adult male Sprague–Dawley (SD) rats were purchased from the Experimental Animal Center of Daping Hospital (Certificate No. SCXK 2012‐0011). The rats were housed five to six per cage in the animal care facility with a 12‐hr light–dark cycles and fed on standard rodent diet and tap water ad libitum, and kept under constant ambient temperature (23 ± 1°C) and humidity (55 ± 5%). Randomization was applied to the assigning of animals to different experimental groups and the data analysis was carried out by a blinded investigator. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath, Drummond, McLachlan, Kilkenny, & Wainwright, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology.
2.2. Focal cerebral ischaemia model
Adult male SD rats (250–280 g of body weight, specific pathogen free) were anaesthetized with 1% sodium pentobarbital (45 mg·kg−1, i.p.) and focal cerebral ischaemia was induced by intraluminal occlusion of the right middle cerebral artery (MCA) as described previously (Kanemitsu et al., 2002; Zhang et al., 2018). The total number of rats used in our experiments was 760. In brief, the left common carotid artery, external carotid artery and internal carotid artery were discreetly isolated. Thereafter, a piece of 5/0 monofilament nylon suture (0.36‐mm diameter) with a round tip was inserted into the internal carotid artery via the external carotid artery stump to occlude the origin of MCA, advanced to the appropriate depth of 18–22 mm, according to the animal's body weight. Rats were subjected to 2‐hr occlusion of MCA, and then the filament was withdrawn for blood reperfusion. The body temperature was maintained at 37 ± 0.5°C with a heating blanket and monitored using a rectal probe throughout surgery (Figure S1). Laser Doppler flowmetry (Moor Instruments, UK) was used to monitor the level of middle cerebral artery occlusion (MCAO) and the severity of the relative cerebral blood flow (rCBF) above the core of the MCA territory (AP +2.0 mm, ML −4.0 mm) during the surgical procedure and at the reperfusion to guarantee the reproducibility of cerebral I/R as described previously (Jiang et al., 2014). A successful MCAO model was accepted when rCBF reduced to ≤20% and restored to >80% of baseline level (Jiang et al., 2017). Sham group underwent the same procedure as described above without MCAO. To prevent false positive outcomes, the rats without deficits after MCAO were thus excluded as described in previous study (W. Wu et al., 2011). After exclusion of these rats along with those that did not survive MCAO during the surgery, the recovery rate of this procedure under anaesthesia was more than 80%.
2.3. Drug treatments
Animals were treated with icariside II (purity ≥98%, Nanjing Zelang Medical Technology Corporation Ltd., Nanjing, China) by gavage at different doses of 4, 8, and 16 mg·kg−1 2 hr of cerebral ischaemia followed by reperfusion twice a day from Day 1 to Day 7, while rats of sham and model groups were administered volume‐matched saline. Moreover, intracerebroventricular injections were given at the onset of reperfusion in the right lateral ventricle with 5 μl of 3‐methylamphetamine (3‐MA; 600 nM) or https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6031 (Rap; 35 pM) solution prepared in saline (Figure S2A). In addition, to determine whether icariside II post‐treatment affects physiological parameters during MCAO, the physiological variables PaCO2, PaO2, mean arterial BP and glucose level were measured before, during and after MCAO at Day 1, Day 3, and Day 7. Sham group underwent the same procedure as described above without occlusion of MCA. The mortality rate was about 15% within 1 day. After 3 days of reperfusion there were no further loss. The number in each group was 28–30 per time points and they were included in the following experiments.
2.4. Behavioural assessment
Neurological deficit scores were evaluated after 1, 3 and 7 days as previously described (Xiong et al., 2016). Briefly, a 5‐point scoring scale was used:‐ grade 0, exhibiting no observable neurological deficit; grade 1, failing to completely stretch left forepaw; grade 2, circling to the left; grade 3, falling to the left and grade 4, failing to walk spontaneously and showing lethargy. These tests were performed by an experimenter who was blind to the treatment groups. Moreover, rats also were trained in the sensorimotor tests for 3 days before ischaemia as previously reported and measured at the same time points after MCAO. Rotorod and adhesive tape removal tests were done as previously reported (Bouet et al., 2009; G. Zhang et al., 2016). Prior to surgery, all rats were trained for 3 days to establish a pre‐injury baseline to achieve 2‐min duration on the rotarod. The rotarod test was used to evaluate general fitness and motor co‐ordination. Briefly, rats were placed on an accelerating rotating beam (accelerated from 4 to 40 rpm within 5 min) and a stop‐clock was started. The duration for each rat to stay on the rotarod was measured as the time it took the rats fall onto the sensing platform below. Each rat performed three trials and the mean duration on the device was recorded. In addition, the sensorimotor (motor co‐ordination and sensory neglect) deficits after MCAO were evaluated using adhesive tape removal tests. Briefly, adhesive‐backed papers (3 × 1 cm piece of yellow tape used as bilateral tactile stimuli) were placed on the distal‐radial region on the wrist of each forelimb. Thereafter, the rats were returned to their cages and time to remove adhesive tapes from the forepaws was recorded. This was repeated 3 times with a maximum observation period of 120 s and the mean time was calculated.
2.5. Novel object recognition task
Novel object recognition tasks were used to evaluate learning and memory after MCAO as previously described (Camarasa, Rodrigo, Pubill, & Escubedo, 2010). Briefly, rats were habituated to an open‐field box (50 × 50 × 50 cm) at 3 and 7 days after MCAO. During habituation phase (10 min), two of the same objects, green cubes (A1 and A2), were located in same place from the wall. During the familiarization period (10 min), two dissimilar objects (object A2 was replaced with a novel object B1, a brown box) were presented in the same box 1 hr after the first trial. The amount of time a rat spent exploring each object during two phases was recorded. The objects and the box were cleaned with 75% ethanol after each individual trial to eliminate olfactory cues. The objects discrimination index was evaluated by a discrimination index as mentioned before (Zhang et al., 2018).
Thereafter, rats were anaesthetized using pentobarbital sodium (45 mg·kg−1, i.v.) and decapitated under anaesthesia at 1, 3 and 7 days after reperfusion being subjected to 2 hr of MCAO and the brains or ischaemic penumbra were rapidly removed as previously described (Wang et al., 2014).
2.6. Measurement of infarct size
Infarct size after MCAO was measured using 2,3,5‐triphenyl‐tetrazolium chloride solution (TTC, Sigma‐Aldrich, 17779) and the areas of infarction were quantified using ImageJ (https://scicrunch.org/resources/Any/search?q=SCR_003070&l=SCR_003070) as described previously (Zhang et al., 2018). Brain tissues were cut into five coronal slices of 2 mm thick. Then the brain slices were stained with TTC at 37°C for 15 min in the dark and fixed in 4% paraformaldehyde. The normal tissue was stained deep red, while the infarct area was stained pale grey colour. Then slices were photographed and the infarct volume was measured as a percentage of the contralateral hemisphere after correcting for oedema as described before (Deng et al., 2016).
2.7. Determination of brain water content
The brains were rapidly removed and weighed as the wet weight. Then the brains were dried at 100°C for another 24 hr and then weighed again to calculate the brain water content. The formula used to calculate water content was as follows: water content (%) = 100 × (wet weight − dry weight) / wet weight.
2.8. Transmission electron microscope observation
We also detected autophagosomes in the ischaemic penumbra areas using transmission electron microscope (TEM). In brief, tissues were fixed with 2% paraformaldehyde and 1% glutaraldehyde for 8 hr; thereafter, the samples were fixed with 1.5% osmium tetraoxide for another 2 hr after being washed with 0.1‐M phosphate buffer (pH 7.4). The samples were dehydrated and embedded in araldite and then sectioned (80 nm), counterstained with uranylacetate and lead citrate and autophagosomes were detected with a TEM. Images were acquired digitally from a randomly selected pool of 10 to 15 fields under each condition (Eskelinen, Reggiori, Baba, Kovacs, & Seglen, 2011).
2.9. Primary cultured rat cortical neurons culture
Primary rat cortical neurons were prepared from newborn SD rats as described previously (Zhang et al., 2015). In brief, the primary cortical neurons were seeded at 1 × 105·ml−1 on 96‐well plates or 24‐well plates with poly l‐lysine‐coated and resuspended in DMEM/F12 which contains 10% FBS and 2% B27 supplement and then maintained in a humidified incubator (37°C, 5% CO2) for 10 days. Cultures contain >95% neurons as routinely identified by neuron‐specific enolase.
2.10. Oxygen–glucose deprivation and reoxygenation (OGD/R) and drug treatment
On the 10th day of the cell culture, neurons were subjected to OGD for 2 hr to mimic ischaemic injury in vitro as previous study (Wang, Gong, Wu, & Shi, 2010). In brief, neurons were incubated for 2 hr in oxygen‐free N2/CO2 (95%/5%) gas. The control group was incubated in EBSS with 10‐mM glucose. Thereafter, the medium of OGD/R group was replaced by standard culture medium or treated with different concentrations of icariside II (12.5, 25, and 50 μM) for another 24 hr. 3‐methylamphetamine (1 mM), rapamycin, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8015 (20 μM) or KT‐5823 (5 μM) were added to the medium 1 hr before the icariside II treatment.
2.11. Determination of neuronal viability and damage
The primary rat cortical neurons were treated as described above. Briefly, before the end of the treatment, 3‐4,5‐dimethylthiazol‐2,5‐diphenyltetrazolium bromide (MTT) was added to each well at an ultimate concentration of 5 mg·ml−1 for another 4 hr. Thereafter, the medium was then removed and 150‐ml DMSO was added to dissolve the formazan. Then the absorbance of formazan formation was detected at 490 nm using a microplate reader. Neuronal viability was expressed in percentage of the value of the control group. In addition, neuronal damage was also determined using an intracellular lactate dehydrogenase kit as described previously (Gao et al., 2017). Briefly, at the end of the treatments as mentioned above, the supernatants were collected after centrifuged at 400× g for 5 min. The amount of intracellular lactate dehydrogenase released from neurons was lysed in 1% Triton X‐100 according to the manufacturer's protocol.
2.12. Tandem mRFP‐GFP fluorescence microscopy
To detect the autophagic flux in neurons in vitro, we generated adenovirus and transgenic neuron harbouring tandem fluorescent mRFP‐GFP‐LC3 (tf‐LC3; Hanbio Biotechnology Co., Ltd., Shanghai, China) at 300 MOI for 24 hr. Primary rat cortical neurons were treated as mentioned above. Then the cells were fixed in 4% paraformaldehyde for 20 min and cell images were recorded by a fluorescence microscope. GFP or RFP puncta number were counted in cells as described previously (Hariharan, Zhai, & Sadoshima, 2011). Briefly, the GFP‐LC3 or RFP‐LC3 puncta in each neuronal cell were manually counted and at least 50 cells were randomly selected for counting in each group. The data presented were from one representative experiment of at least five independent experiments.
2.13. Measurement of PDE 5 plus cGMP levels and PKG and PDE 5 activities
Briefly, the rats or the primary rat cortical neurons were treated as described above. The peri‐infarct penumbra tissues or cells were collected and homogenized utilizing 0.1‐M PBS (pH 7.4). Then the tissue homogenates were centrifuged at 12000× g for 20 min at 4°C, and the supernatants were collected and stored at −80°C. Protein concentrations of the supernatants were analysed using BCA protein assay kit. The levels of PDE 5, cGMP along with the activities of PDE 5 and PKG were determined using the ELISA kits under the manufacturer's instructions.
2.14. Western blot
The peri‐infarct penumbra tissues and primary rat cortical neurons were homogenized in RIPA buffer. The lysates were normalized to equal amounts of protein, and 30‐μg protein from tissue lysates or 20‐μg protein from cell lysates were separated using SDS‐PAGE (10% and 12%) and transferred to a nitrocellulose membrane, which was blocked using 5% non‐fat milk in TBST (pH 7.4) as previous study. Membranes were probed with primary antibodies against microtubule‐associated protein 1 light chain 3 (MAP 1LC3/LC3; 1:1,000), Beclin 1 (1:1,000), p‐ser9‐GSK‐3β (1:1,000), p‐tyr216‐GSK‐3β (1:1,000), SQSTM1/p62 (1:1,000), APG5L/autophagy‐related gene 5 (ATG5; 1:1,000), PDE 5A (1:1,000), anti‐autophagy‐related gene 7 (ATG7; 1:1,000), anti‐GAPDH (1:1,000) and anti‐β‐actin (1:1,000). Thereafter, proteins were evaluated using incubation with species‐specific HRP‐conjugated secondary antibodies. Immunoreactive bands then were visualized using ECL Western blot (Alexander SPH, Roberts RE, Broughton BRS, Sobey, SG, George CH, Stanford SC…Ahluwalia, A. , 2018) detection reagents and ImageJ software was applied to quantify the band optical intensity. Intensity for each protein was normalized to that evaluated for β‐actin or GAPDH and was expressed as the relative fold to the sham or control group
2.15. Transient silencing by small interfering RNAs
Transfection was performed when the primary rat cortical neurons were cultured to 70–80% confluence in 96‐well or six‐well plates. The SQSTM1‐targeted small interfering RNA (siRNA; Invitrogen, 4390771) was diluted in Opti‐MEM and then equilibrated at room temperature for 15 min. Thereafter, neurons were transfected with SQSTM1 siRNA or scrambled siRNA by lipofectamine™ RNAiMAX transfection reagent (Invitrogen, https://www.thermofisher.com/order/catalog/product/13778075) according to the manufacturer's instructions. The knockdown of endogenous SQSTM1 by siRNA was confirmed by real‐time PCR (qRT‐PCR) and Western blot. After transfected 24 hr, the transfected cell was subjected to OGD/R and treated with icariside II as mentioned above. Then cell viability, intracellular lactate dehydrogenase release, expression of LC3 and PDE 5, and phosphorylation level of GSK‐3β were determined.
2.16. qRT‐PCR analysis
Total RNA was extracted from primary cortical neurons with the Trizol Reagent according to the manufacturer's protocol. The cDNA was reverse transcribed with the PrimeScript™ RT Reagent Kit. Gene expression was evaluated by real‐time PCR on the CFX96 real‐time PCR detection system (Bio‐Rad Laboratories Ltd, Hertfordshire, UK), and specific primers and their sequences were as follows: β‐actin forward 5′‐TCTTCCAGCCTTCCTTCCTG‐3′, reverse 5′‐CACACAGAGTACTTGCGCTC‐3′; SQSTM1 forward 5′‐AGCTCTCTAGACCCCTCACA‐3′, reverse 5′‐GTCTGTAGGAGCCTGGTGAG‐3′. Results were normalized to levels of β‐actin mRNA and expressed as the fold change (2−ΔΔCt).
2.17. Molecular docking protocol
Molecular docking studies between icariside II and GSK‐3β were performed by Autodock 4.2 (https://scicrunch.org/resources/Any/search?q=SCR_012746&l=SCR_012746) and Autodock Tools (ADT). The X‐ray crystal structure of human PDE 5 (Protein Data Bank [PDB] ID: 2H40) and GSK‐3β (PDB ID: 1H8F) were obtained from the PDB archives and used as target for molecular docking. To prepare compounds for the molecular docking, 2D structure of icariside II was obtained from the PubChem (https://scicrunch.org/resources/Any/search?q=SCR_004284&l=SCR_004284) and then converted to 3D structure. The docking results were written as a pose viewer file and the protein–ligand complex interactions were studied using the PyMOL molecular graphics system (https://scicrunch.org/resources/Any/search?q=SCR_000305&l=SCR_000305; Dajani et al., 2001). The interactions between PKG (PDB ID: 3OD0) and GSK‐3β were determined by ZDOCK and RDOCK (https://scicrunch.org/resources/Any/search?q=SCR_002838&l=SCR_002838), which has been proved as an extensive approbatory method to perform exact prediction of protein–protein docking (H. L. Li et al., 2017). Thereafter, the predicted protein poses were obtained from ZDOCK and then refined and re‐ranked by RDOCK. Then E_RDOCK (Energy RDOCK) was applied as the default scoring function to predict the binding affinity of PKG and GSK‐3β. Hence, the 12 poses of docked conformations which had lower E_RDOCK were selected for further analysis.
2.18. Surface plasmon resonance
Analysis was conducted to further verify the interaction between GSK‐3β and PKG using Biacore X100 instrument (GE Healthcare, Uppsala, Sweden) with Biacore X100 Evaluation Software (version 2.0) and sensor chip CM5 (GE, BR‐1003‐99; Papareddy et al., 2018). GSK‐3β was immobilized using Amine method with a target level of 2000 RU. HBS‐EP buffer used as the running buffer in the whole process; EDC and NHS were adopted to activate the chip. GSK‐3β (Sino Biological Inc., 10044‐H07B) at a concentration of 40 μg·ml−1 dissolved in 10‐mM sodium acetate (PH 5.0) was then absorbed for immobilization. PKG (Abcam, ab80375) was diluted at 2, 4, 8, 16, 32, and 64 μM with HBS‐EP buffer. The contact time of proteins was set to 120 s, and 300 s for dissociation. Glycine‐HCl (PH 2.0; GE Healthcare, BR‐1003‐55) was used for regeneration. Data were analysed with the Biacore evaluation 3.1 analysis software (GE Healthcare). Equilibrium dissociation constants (KD) was evaluated using global fitting of the kinetic data from different concentrations of PKG with a 1:1 Langmuir binding model.
2.19. Transient transfection
The haemagglutinin (HA) tagged constitutively active mutant (Human pcDNA3‐HA‐GSK‐3β‐S9A; S9A) was gift from Dr. Jim Woodgett (Toronto, Canada; Addgene plasmid #14754) as previously reported (Z. Wang, Bao et al., 2015). The pcDNA3 empty vector was used as a negative control. S9A was transfected into cells using Lipofectamine 2000, according to the manufacturer's protocol. After transfection with equal amounts of expression plasmid or empty vector, neurons were subjected to various treatments as indicated.
2.20. Statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Data are presented as mean ± SEM, where n represents the number of independent experiments and not replicates. All statistical analyses were analysed by GraphPad Prism 5 (https://scicrunch.org/resources/Any/search?q=SCR_002798&l=SCR_002798). Some data were normalized to control for unwanted sources of variation as follows. First, the data were normalized to bring all of the variation into proportion with one another after removing any outliers in various groups. Then the coefficients associated with each variable will scale appropriately to adjust for the disparity in the variable sizes. For the MTT assay, the viability of control group was considered to be 100%, and the neuronal viability of ICS II‐treated group was represented as a percentage of the control. For Western blot, protein expressions (e.g. PDE 5A) were normalized to that of β‐actin. The levels of protein modification (e.g. phosphorylation of GSK‐3β at ser9 and tyr216 sites) were normalized to that of un‐modified protein (e.g. GSK‐3β). The status of protein expressions or levels in ICS II‐treated group were expressed as fold changes over that of the sham or control group, which expression was set to 1. Two or multiple groups were compared using Student's unpaired t‐test or one‐way ANOVA followed by Bonferroni post hoc test with F at P < .05 and no significant variance inhomogeneity. P < .05 was considered statistically significant.
2.21. Materials
3‐methylamphetamine (M9281), rapamycin (37094), SB216763 (S3442), and KT‐5823 (K1388) were purchased from Sigma‐Aldrich (St. Louis, MO, USA); antibodies were used against light‐chain 3B (LC3B; Aviva Systems Biology Cat# ARP51335_P050, https://scicrunch.org/resources/Any/search?q=AB_2044956&l=AB_2044956), Beclin 1 (Novus Cat# NB500‐249B, https://scicrunch.org/resources/Any/search?q=AB_1216268&l=AB_1216268), GSK‐3β (Cell Signaling Technology Cat# 12456, https://scicrunch.org/resources/Any/search?q=AB_2636978&l=AB_2636978), phospho‐GSK‐3β (Ser9; Cell Signaling Technology Cat# 14310, https://scicrunch.org/resources/Any/search?q=AB_2798445&l=AB_2798445), phospho‐GSK‐3β (Tyr216; Abcam, Cat# 75745), SQSTM1/p62 (Cell Signaling Technology Cat# 5114, RRID:AB_10624872), APG5L/ATG5 (Abcam Cat# ab109490, https://scicrunch.org/resources/Antibodies/search?q=AB_10861245&l=AB_10861245), https://scicrunch.org/resources/Antibodies/search?q=AB_1063421&l=AB_1063421 (LifeSpan Cat# LS‐C42463‐50, RRID:AB_1063421), https://scicrunch.org/resources/Antibodies/search?q=AB_10800793&l=AB_10800793 (LifeSpan Cat# LS‐C121392‐50, RRID:AB_10800793), https://scicrunch.org/resources/Antibodies/search?q=AB_2263076&l=AB_2263076 (Proteintech Cat# 10494‐1‐AP, RRID:AB_2263076), and β‐actin (Proteintech, Cat# 60008‐1‐Ig).
2.22. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (https://scicrunch.org/resources/Any/search?q=SCR_013077&l=SCR_013077; Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Icariside ii attenuates cerebral I/R‐induced injury in rats
To determine whether icariside II could protect against cerebral I/R injury, the neurological scores, infarct volume and cerebral water content were detected after Day 1, Day 3, and Day 7 of middle cerebral artery occlusion (MCAO; Figure 1a). The results showed that cortical blood flow was successfully reduced to 20% and restored to >80% of baseline level, suggesting the successful establishment of the MCAO model (Figure S2B,C). However, icariside II dose‐dependently decreased the neurological scores of MCAO rats at Days 3 and 7 (Figure 1a). Moreover, consistent with the amelioration of neurological scores, icariside II also significantly improved rotarod test performance (Figure 1b) and adhesive removal test (Figure 1c) from 3 and 7 days post‐ischaemia compared with occlusion of MCA alone. Of note, MCAO markedly reduced the time rats spent exploring the novel environment as shown in the reduction discrimination index compared with the sham group or icariside II alone group. While, icariside II treated rats displayed better performance of recognition memory in a novel object recognition test at Days 3 and 7 after MCAO (Figure 1d–f), further indicating that icariside II improved the functional outcome of stroke rats. Furthermore, icariside II also dose‐dependently reduced the infarct volume at Day 3 (Figure 1j,k) and Day 7 (Figure 1m,n) as well as brain water contents (Figure 1l,o). It is worth noting that icariside II did not affect the motor functions (Figure 1a–c) and learning and memory (Figure 1d–f), infarct volume (Figure 1g,h) and brain water content (Figure 1i) at Day 1. These results indicated that icariside II exerted beneficial effects on cerebral I/R‐induced injury during sub‐acute stage. It is worthy to note that icariside II did not alter PaCO2, PaO2, mean arterial BP and glucose level before, during and after MCAO at Day 1, Day 3, and Day 7 (Table S1), indicating that icariside II does not affect physiological variables during the experiments.
Figure 1.
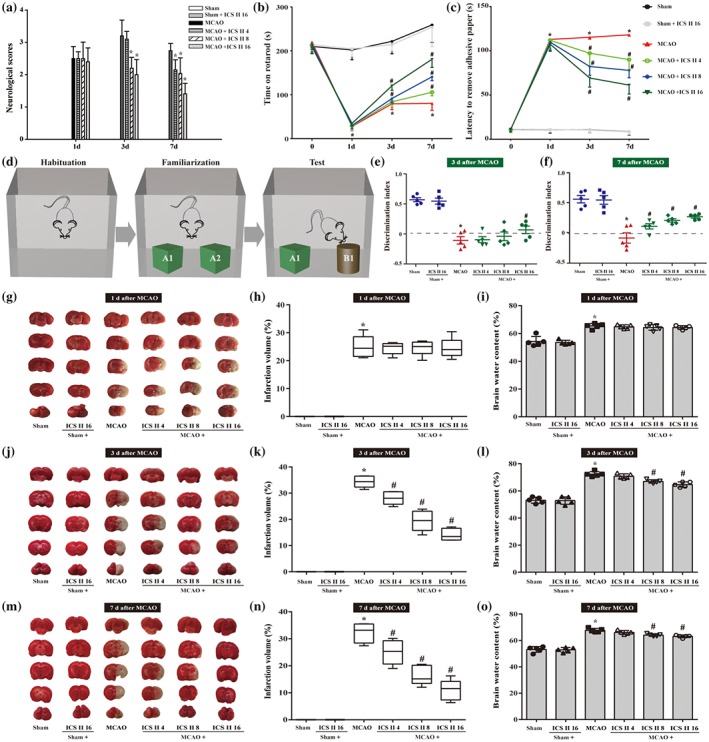
Icariside II (ICS II) protects against cerebral I/R‐induced injury in rats. Showing combinerd data from rats with or without icariside II on neurobehavioural assessments along with infarct volume and cerebral water content at Days 1, 3 and 7 after middle cerebral artery occlusion (MCAO). Neurological deficits (a) were detected by a 5‐point scale (n = 10 per group). (b) Rotarod performance (n = 10 per group) (c) Latency to remove adhesive paper (n = 10 per group, (d) Schematic diagram of novel object recognition (NOR). (e) Objects discriminated memory was evaluated by a discrimination index (DI = Time spent on familiar objects − Time spent on novel objects/Time spent on both novel and familiar objects) at Day 3. (f) Objects discriminated memory was evaluated by a discrimination index (DI = Time spent on familiar objects − Time spent on novel objects/Time spent on both novel and familiar objects) at Day 7. (g) Representative images of TTC‐stained brain sections that showed viable (red) and dead (white) tissue at Day 1. (h) Quantification of infarct size at Day 1 (n = 5 per group). (i) Brain water content was evaluated (n = 5 per group). (j) Representative images of TTC‐stained brain sections that showed viable (red) and dead (white) tissue at Day 3. (k) quantification of infarct size at Day 3 (n = 5 per group). (l) Brain water content was evaluated at Day 3 (n = 5 per group). (m) Representative images of TTC‐stained brain sections that showed viable (red) and dead (white) tissue at Day 7. (n) Quantification of infarct size at Day 7 (n = 5 per group). (o) Brain water content was evaluated at Day 7 (n = 5 per group). One‐way ANOVA with Bonferroni post hoc test (a, e–o) and two‐way ANOVA with Bonferroni post hoc test (b, c). The data were presented as the mean ± SEM. *P < .05 versus sham group; # P < .05 versus MCAO group
3.2. Icariside II inhibits neuronal autophagy after cerebral I/R‐induced injury in rats
Neurons that existed in the core of focal cerebral ischaemia are mostly and irreversibly injured. Whereas, the function of neurons located in the ischaemic penumbra, a territory of injured tissue that encircled the core of the focal cerebral ischaemia can be rescued (Arcangeli et al., 2013). Thus to investigate whether autophagic processes were involved in cerebral I/R‐induced injury in rats, autophagosomes and autophagy‐related proteins in the ischaemic penumbra (Figure S3) were detected using TEM and Western blot. The findings showed that, at Day 3 after MCAO‐induced injury, cell shrinkage, loss of cellular organelles and formation of autophagosomes are found in the ischaemic penumbra of cerebral cortex. However, most of the neurons and their organelles appeared to be normal in the icariside II ‐treated group, with less neuronal damage (Figure 2a). Furthermore, we examined the extent of conversion of LC3‐I to LC3‐II, an important marker of autophagy that is responsible for the formation of autophagosome (Dai et al., 2017), as well as Beclin 1 and SQSTM1 expression. The results showed that the accumulation of LC3‐II/LC3‐I ratio and Beclin 1 were significantly increased in penumbra tissue of rat cerebral cortex upon MCAO, but SQSTM1 was decreased. However, icariside II reversed these effects at Day 3 and Day 7. Icariside II also decreased ATG5 and ATG7 at Day 3 (Figure 2b,c) and Day 7 (Figure 2d,e). Furthermore, p‐ser9‐GSK‐3β was decreased, whereas p‐tyr216‐GSK‐3β was increased after MCAO. However, icariside II reversed these effects at Day 3 (Figure 2f,g) and Day 7 (Figure 2h,i).
Figure 2.
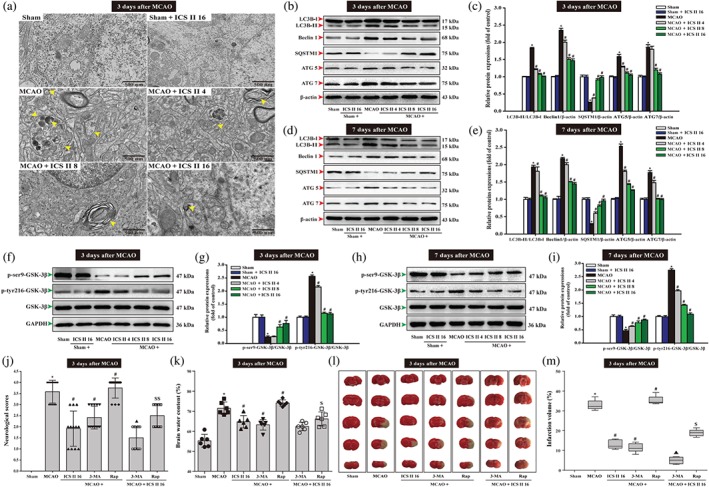
Icariside II (ICS II) inhibits neuronal excessive autophagy in penumbra after middle cerebral artery occlusion (MCAO). Showing rats treated with icariside II after MCAO. (a) Ultrastructural changes in brain tissues of penumbra from rat at Day 3 after MCAO. Yellow arrows are pointing at autophagosome. (b) Representative Western blots of LC3‐I, LC3‐II, Beclin 1, SQSTM1, ATG5 and ATG7 at Day 3. (c) Quantitation of LC3‐II/LC3‐I ratio, Beclin 1, SQSTM1, ATG5 and ATG7 at Day 3 (n = 5 per group). (d) Representative Western blots of LC3‐I, LC3‐II, Beclin 1, SQSTM1, ATG5 and ATG7 at Day 7. (e) Quantitation of LC3‐II/LC3‐I ratio, Beclin 1, SQSTM1, ATG5 and ATG7 at Day 7 (n = 5 per group). (f) Representative Western blots of p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β at Day 3. (g) Quantitation of p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β at Day 3 (n = 5 per group). (h) Representative Western blots of p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β at Day 7. (i) Quantitation of p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β at Day 7 (n = 5 per group). Treated with 3‐methylamphetamine (3‐MA) or rapamycin (Rap) or icariside II after MCAO. (j) Neurological deficits were detected by a 5‐point scale (n = 12 per group). (k) Brain water content was evaluated (n = 6 per group). (l) Representative images of TTC‐stained brain sections that show viable (red) and dead (white) tissue. (m) Quantification of infarct size (n = 6 per group). The data were presented as the mean ± SEM. *P < .05 versus sham group; # P < .05 versus MCAO group; ▲ P < .05 versus OGD/R + 3‐MA group; $ P < .05 versus OGD/R + Rap group
Thereafter, 3‐methylamphetamine and rapamycin were used to further confirm whether autophagy contributed to the beneficial effects of icariside II I after MCAO. The results showed that the neurological scores (Figure 2j), brain water content (Figure 2k) and infarct volume (Figure 2l,m) were significantly decreased after treatment with the autophagy inhibitor 3‐methylamphetamine. In contrast, treatment with the autophagy inducer rapamycin significantly aggravated the increase in the neurological scores, brain water content and infarct volume at Day 3. However, in fact, the two drugs 3‐methylamphetamine and icariside II together exerted synergism, while rapamycin together with icariside II suppressed the injury compared with that of rapamycin alone after cerebral I/R (Figure 2j–m). These results indicated that icariside II protected against MCAO‐induced injury through inhibiting excessive autophagy via inactivation of GSK‐3β.
3.3. Icariside II increases cerebral I/R‐induced cGMP level, PKG activity and mitigated expression and activity of PDE 5
To investigate the effects of icariside II on cGMP, PDE 5 and PKG activities in cerebral I/R injury in vivo, the level of cGMP and the activities of PDE 5 and PKG were determined using according kits in the penumbra area. The results showed that icariside II mitigated the MCAO‐induced increase in the expression of PDE 5 at Day 3 (Figure 3a,b) and Day 7 (Figure 3c,d) and PDE 5 activity at Day 3 (Figure 3e) and Day 7 (Figure 3h). Moreover, icariside II suppressed the decrease in cGMP level at Day 3 (Figure 3f) and Day 7 (Figure 3i) and PKG activity at Day 3 (Figure 3g) and Day 7 (Figure 3j), suggesting that cGMP/PKG pathway was involved in the inhibition of PDE 5 by icariside II.
Figure 3.
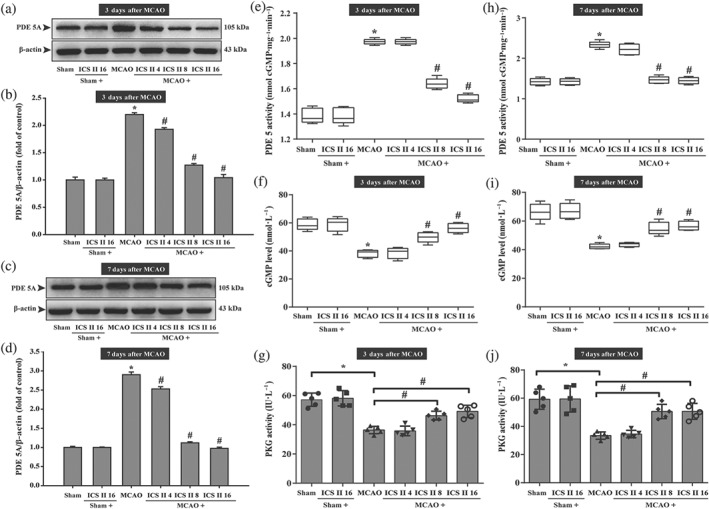
Icariside II (ICS II) mitigates expression and activity of PDE 5 and increased cerebral I/R‐induced cGMP level and PKG activity in penumbra. (a) PDE 5A expression at Day 3. (b) Quantitation of PDE 5A at Day 3 (n = 5 per group). (c) PDE 5A expression at Day 7. (d) Quantitation of PDE 5A at Day 7 (n = 5 per group). (e) PDE 5 activity at Day 3. (f) cGMP level at Day 3 (n = 5 per group). (g) PKG activity at Day 3 (n = 5 per group). (h) PDE 5 activity at Day 7 (n = 5 per group). (i) cGMP level at Day 7 (n = 5 per group). (j) PKG activity at Day 7 (n = 5 per group). The data were presented as the mean ± SEM. *P < .05 versus sham group; # P < .05 versus MCAO group
3.4. Icariside II attenuates oxygen–glucose deprivation and reoxygenation (OGD/R)‐induced primary rat cortical neuronal injury by inhibiting PDE 5 and autophagy
Next, in vitro, the effect of icariside II on OGD/R‐induced primary rat cortical neuronal injury was first investigated. Icariside II (6.25–25 μM) exerted no effect on neurons at lower concentrations, but higher concentrations of icariside II (50–100 μM) dramatically suppressed cell survival than that of control group within 48 hr (Figure 4a). Thus, icariside II (6.25–25 μM) was used in the subsequent experiments because these concentration levels did not induce cytotoxity. When primary cortical neurons were subject to OGD/R and were returned to normal medium for 24 hr after OGD 2 hr, icariside II (6.25, 12.5, and 25 μM) concentration‐dependently reduced the OGD/R‐induced decrease of neuronal cell viability (Figure 4b). Parallel experiment of intracellular lactate dehydrogenase release showed that icariside II (6.25, 12.5, and 25 μM) also concentration‐dependently suppressed OGD/R‐induced cytotoxicity (Figure 4c). In addition, OGD/R reduced the number of neurons, whereas icariside II reversed these changes, as evidenced by morphologic observations (Figure 4d). These results demonstrate that icariside II exerts beneficial effect on OGD/R‐induced neuronal injury, consistent with the results in vivo.
Figure 4.
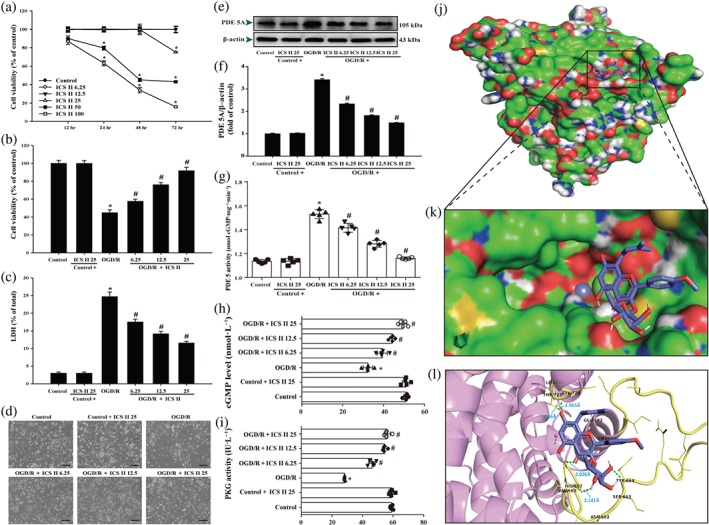
Icariside II (ICS II) attenuates oxygen–glucose deprivation and reoxygenation (OGD/R)‐induced cytotoxicity and inhibited PDE 5. (a) Primary cortical neurons were treated with different concentrations of icariside II and cell viability was determined by MTT (n = 5). Neuronal cells were treated with or without icariside II for 24 hr after OGD/R. (b) Cell viability was measured using the MTT assay (n = 5). (c) Cytotoxicity was detected using the intracellular lactate dehydrogenase (LDH) release assay (n = 5). (d) The morphology of neuronal cells was observed using reverse‐phase microscope. (e) PDE 5A expression. (f) Quantitation of PDE 5A (n = 5). (g) PDE 5 activity. (h) cGMP level (n = 5). (i) PKG activity (n = 5). The docking results of icariside II with the PDE 5 complex. (j) The whole view of the icariside II dimer and PDE 5 displaying the molecular binding pocket. (k) A close‐up amplification of the molecule binding pocket from the side. (l) Crystal structure of icariside II (blue) displaying PDE 5 (yellow and pink) bound to the docking pocket. Hydrogen bonds were shown as green dotted lines. Data were expressed as mean ± SEM of five independent experiments. *P < .05 versus control group; # P < .05 versus OGD/R group
Furthermore, OGD/R enhanced the expression of PDE 5 and PDE 5 activity and decreased the cGMP level and PKG activity. In contrast, icariside II suppressed OGD/R‐induced increase both in the expression of PDE 5 (Figure 4e,f) and PDE 5 activity (Figure 4g). Moreover, icariside II suppressed the decrease in cGMP level (Figure 4h) and PKG activity (Figure 4i) after OGD/R, which were consistent with the findings in vivo. Intriguingly, the results also showed a strong binding affinity between icariside II and PDE 5, with binding energy of −6.52 kcal·mol−1 (Figure 4j,k). We thereafter tested the suppositive binding modes and interaction within the amino acid pocket, including VAL660, HIS675, LEU725, and THR723 as evidenced by a molecular docking simulation (Figure 4l), further confirming that icariside II bound to the hydrophobic pocket of PDE 5, which is in line with the findings in vivo and in vitro as well as our previous study.
3.5. Icariside II suppresses autophagy‐related proteins during OGD/R‐induced neuronal injury
We then investigated the possible molecular mechanism in the beneficial effect of icariside II on OGD/R exposure. The results showed that icariside II significantly suppressed the increase of LC3‐II/LC3‐I ratio, Beclin 1 expression, and the decrease of SQSTM1 expression after OGD/R. Intriguingly, icariside II also mitigated the increase of ATG5 and ATG7 expressions after OGD/R, consistent with the in vivo findings (Figure 5a,b). Moreover, icariside II attenuated the increase in PDE 5A, p‐tyr216‐GSK‐3β and the decrease in p‐ser9‐GSK‐3β after OGD/R (Figure 5c,d),again are consistent with in vivo the results. Furthermore, under the OGD/R condition, silencing of SQSTM1 (Figure 5e) apparently suppressed the increase in cell viability (Figure 5f) and the decrease in intracellular lactate dehydrogenase release (Figure 5g) after treatment with icariside II upon OGD/R than those of the scrambled siRNA group. Moreover, silencing of SQSTM1 reversed the effects of icariside II on LC3‐II/LC3‐I ratio but did not alter the effects of icariside II on the PDE 5A expression and phosphorylation level of p‐tyr216‐GSK‐3β and p‐ser9‐GSK‐3β than those of the scrambled siRNA group (Figure 5h–j). These findings further confirm that icariside II suppresses OGD/R‐induced neuronal injury mediated by autophagy.
Figure 5.
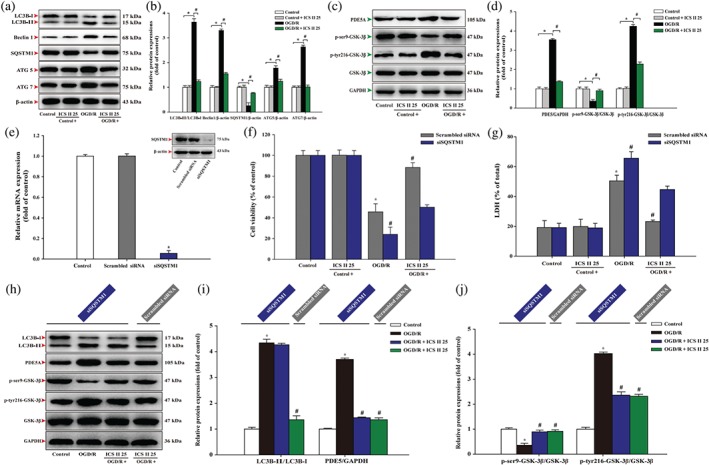
Icariside II (ICS II) suppresses oxygen–glucose deprivation and reoxygenation (OGD/R)‐induced increase in autophagy‐related proteins in neurons. Neuronal cells were treated with or without icariside II 25 μM for 24 hr after OGD/R. (a) Representative Western blots of LC3‐I, LC3‐II, Beclin 1, SQSTM1, ATG5 and ATG7. (b) Quantitation of LC3‐II/LC3‐I ratio, Beclin 1, SQSTM1, ATG5 and ATG7 (n = 5). (c) Representative Western blots of PDE 5A, p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β. (d) Quantitation of PDE 5A, p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β (n = 5). (e) Quantitation of qRT‐PCR and representative Western blot were shown for SQSTM1 siRNA. (f) Cell viability was measured in neurons transfected with or without SQSTM1 siRNA. (g) Intracellular lactate dehydrogenase release was detected in neurons transfected with or without SQSTM1 siRNA. (h) Representative Western blots of LC3‐I, LC3‐II, PDE 5A, p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β. (i) Quantitation of the ratio of LC3‐II/LC3‐I and PDE 5A. (j) Quantitation of p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β. Data were expressed as mean ± SEM of five independent experiments. *P < .05 versus control group; # P < .05 versus OGD/R group
3.6. Icariside II inhibits autophagic flux during OGD/R‐induced neuronal injury
In the OGD/R group, 3‐methylamphetamine increased but rapamycin decreased neuronal cell viability. Parallel experiment on intracellular lactate dehydrogenase release showed the suppression and aggravation of OGD/R‐induced cytotoxicity by 3‐methylamphetamine and rapamycin, respectively. These results indicated that autophagy plays an important role in OGD/R‐induced cell death. However, 3‐methylamphetamine and icariside II together exerted synergism, while rapamycin together with icariside II mitigated the increase in neuronal injury caused by rapamycin alone after OGD/R (Figure 6a–c). The results demonstrated that icariside II protects against OGD/R‐induced injury in neurons through inhibiting excessive autophagy, which were consistent with the findings in vivo.
Figure 6.
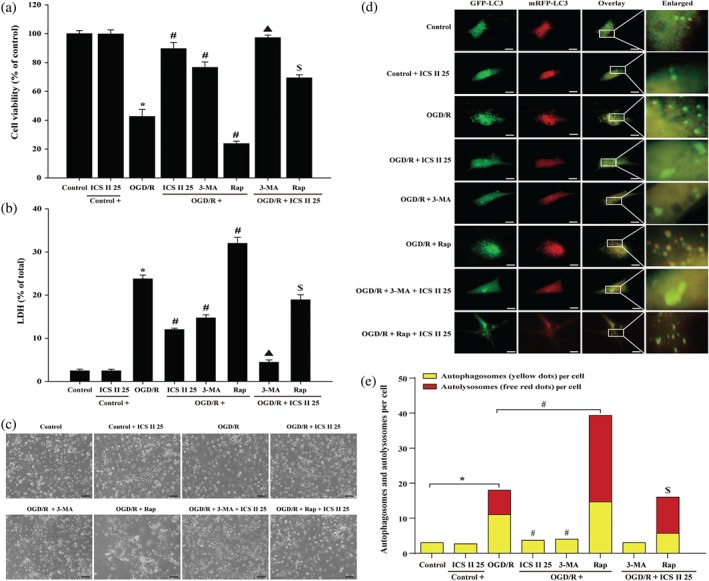
Icariside II (ICS II)suppresses oxygen–glucose deprivation and reoxygenation (OGD/R)‐induced increase in autophagy‐related proteins and autophagic flux in neuron OGD models. Neuronal cells were treated with or without 1‐mM 3‐methylamphetamine (3‐MA) or 20‐μM rapamycin (Rap) together with icariside II 25 μM for 24 hr after OGD/R. (a) Cell viability was measured using the MTT assay (n = 5). (b) Cytotoxicity was detected using the intracellular lactate dehydrogenase release assay (n = 5). (c) The morphology of neuronal cells was observed using reverse‐phase microscope. (d) Neurons were transduced with 300 MOI of Ad‐tf‐LC3 for 24 hr; representative images of fluorescent LC3 puncta were shown. Scale bar represented 50 μm. (e) Mean number of autophagosomes represented by yellow dots in overlaid images and autolysosomes represented by red dots in overlaid images per cell (n = 5).*P < .05 versus control group; # P < .05 versus OGD/R group; ▲ P < .05 versus OGD/R + 3‐MA group; $ P < .05 versus OGD/R + Rap group
Moreover, to better determine the inhibitory effect of icariside II on OGD/R‐induced autophagic flux in neurons, we generated an adenovirus harbouring tf‐LC3 (Ad‐tf‐LC3), which is a very useful tool for evaluating the extent of autophagic flux, as a marker to distinguish between autophagosomes and autolysosomes (Hariharan, Zhai, & Sadoshima, 2011). Neurons were transduced with Ad‐tf‐LC3 for 24 hr. By applying Ad‐tf‐LC3 and determining the number of red dots that overlay green dots and merged yellow dots in overlaid images, the number of autophagosomes can be counted. The red dots that do not overlay green dots appeared in merged images, which indicates autolysosome formation (Kimura, Noda, & Yoshimori, 2007; Klionsky et al., 2008). The results showed that the numbers of GFP and mRFP dots per cell were both increased by OGD/R, suggesting that OGD/R triggered autophagic flux. However, icariside II significantly attenuated the increase in yellow dots and free red dots, suggesting that icariside II suppressed OGD/R‐induced autophagic flux. Furthermore, 3‐methylamphetamine also inhibited autophagic flux, whereas rapamycin promoted the procession of autophagic flux, further confirming that autophagy was involved in the inhibitory action of icariside II on OGD/R‐induced neuronal injury. Whereas, the two drugs 3‐methylamphetamine or rapamycin and icariside II together mitigated the autophagic flux after OGD/R (Figure 6d,e).
3.7. Icariside II suppresses OGD/R‐induced neuronal injury by mediating PKG/GSK‐3β/autophagy axis
The binding affinity of icariside II and GSK‐3β was determined using molecular docking method. The results showed the strong binding affinity between icariside II and GSK‐3β, with a binding energy of −8.23 kcal·mol−1. We thereafter tested the suppositive binding modes and the interaction within the amino acid pocket, including Phe293, Ala292, Phe93, Lys94, Asn95, Ala92, Arg96, Leu88, Gly90, Gln295, Pro294, and Gln89 (Figure 7a). Overall, in molecular docking, analysis revealed that icariside II might be directly affecting GSK‐3β.
Figure 7.
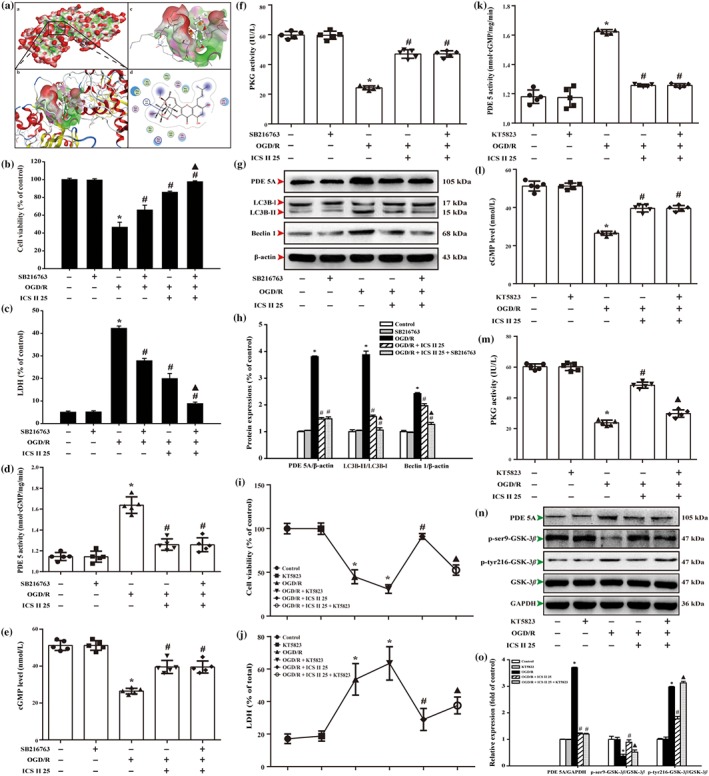
Icariside II (CS II) suppresses oxygen–glucose deprivation and reoxygenation (OGD/R)‐induced neuronal injury by mediating PKG/GSK‐3β/autophagy axis. (a) The binding energy has been surfaced and key residues of icariside II with GSK‐3β were displayed using in silico molecular docking. (A) The substrate binding surface. (B) The substrate binding sites. (C) The substrate binding pocket. (D) Amino acid residues. Neurons were treated with or without icariside II and GSK‐3β inhibitor SB216763 after OGD/R for 24 hr. (b) Cell viability was detected using the MTT assay. (c) Cell toxicity was detected using the intracellular lactate dehydrogenase (LDH) release assay. (d) PD5 activity (n = 5). (e) cGMP level (n = 5). (f) PKG activity (n = 5). (g) Representative Western blots were shown for PDE 5A, LC3B, and Beclin 1 expression. (h) Quantitation of PDE 5A expression, LC3‐II/LC3‐I ratio, and Beclin 1 expression (n = 3). Neurons were treated with or without icariside II and PKG inhibitor KT5823 after OGD/R for 24 hr. (i) Cell viability was detected using the MTT assay. (j) Cell toxicity was detected using the LDH release assay. (k) PD5 activity (n = 5). (l) cGMP level (n = 5). (m) PKG activity (n = 5). (n) Representative Western blots were shown for PDE 5A expression and levels of p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β. (o) Quantitation of PDE 5A expression and levels of p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β. The data are presented as the mean ± SEM. *P < .05 versus control group; # P < .05 versus OGD/R group; ▲ P < .05 versus OGD/R + ICS II 25 μM group
Furthermore, GSK‐3β inhibitor was applied to further confirm the role of GSK‐3β in mediating the inhibition effect of icariside II on OGD/R‐induced autophagic cell death in neurons. The findings showed that GSK‐3β inhibitor alone did not affect neuronal cell survival and cytotoxicity, consistent with the previous study (Lin, Chen, Yang, & Shih, 2014). However, the neuronal cell survival and cytotoxicity of GSK‐3β inhibitor‐treated cells were increased and decreased after OGD/R. Notably, the suppression effect of icariside II was enhanced together with GSK‐3β inhibitor after OGD/R than that of icariside II group, as evidenced by MTT assay (Figure 7b) and intracellular lactate dehydrogenase release (Figure 7c), respectively. Furthermore, SB216763 significantly enhanced the effect of icariside II on OGD/R‐induced changes of LC3‐II/LC3‐I ratio and Beclin 1, but did not alter the effects of icariside II on the expression of PDE 5A (Figure 7g,h) and activity of PDE 5 (Figure 7d), cGMP level (Figure 7e) and PKG activity on OGD/R (Figure 7f). Of note, to further verify the role of GSK‐3β in the protective effects of icariside II on OGD/R, overexpression of GSK‐3β in cultured primary rat cortical neurons was manipulated by transient transfection of the S9A. The results showed that overexpression due to the transfected S9A mutant in neurons resulted in dephosphorylation of ser9 and activation of GSK‐3β (decreased p‐ser9‐GSK‐3β level; Figure S4A). In neurons expressing S9A, which is an uninhibitable mutant of GSK‐3β as the serine 9 site was replaced by alanine, protective effects of icariside II on OGD/R were blunted (Figure S4B,C). Moreover, these changes were also altered in other endpoints, including LC3‐II/LC3‐I ratio (Figure S4D,E), Beclin 1 expression (Figure S3D,F) and PDE 5A expression (Figure S4D,G). These results imply that the suppressive phosphorylation of GSK‐3β is necessary for the protective effect of icariside II on OGD/R‐induced neuron injury.
Moreover, KT‐5823, a PKG inhibitor, alone did not affect neuronal cell survival and cytotoxicity. Whereas, KT‐5823 not only decreased the cell viability and increased intracellular lactate dehydrogenase release on OGD/R but also significantly abolished icariside II‐induced increase in cell viability and decreased in intracellular lactate dehydrogenase release after OGD/R injury, as evidenced by the MTT and intracellular lactate dehydrogenase assay, respectively (Figure 7i,j). Furthermore, KT‐5823 did have any effect on the levels of PDE 5, cGMP and PKG activity per se in control conditions. However, KT‐5823 markedly attenuated the effects of icariside II on OGD/R‐induced changes of PKG activity (Figure 7m) and p‐ser9‐GSK‐3β and p‐tyr216‐GSK‐3β levels (Figure 7n,o), but did not alter the effects of icariside II on PDE 5 activity (Figure 7k) and PDE 5A expression (Figure 7n,o), as well as cGMP level upon OGD/R (Figure 7l). These findings suggest that PKG‐dependent phosphorylation of GSK‐3β is involved in the neuroprotective effect of icariside II on cerebral I/R‐induced excessive autophagy.
3.8. The interaction between PKG and GSK‐3β
The docking of PKG and GSK‐3β was performed using ZDOCK and RDOCK method. ZDOCK is a docking module to display protein structure and the output results of RDOCK contain ZRANK and the E_RDOCK (Energy RDOCK) scores. The E_RDOCK score is often preferred to predict the better docking pose to ZRANK score. Moreover, the clashes of the selected poses were zero demonstrated better docking positions. Higher negative score of E_RDOCK predicted the stronger interactions between PKG and GSK‐3β. The output values of PKG with GSK‐3β docking were shown in Table S2. Notably, the results indicated that hydrogen bond, charge interactions were formed by the residues such as THR38, TYR56, THR39, SER55, VAL40, VAL54, ALA42, GLN52, THR43 and PHE115 and Pi interactions with the residues such as PHE291 and PHE293, TYR171 and HIS106, PHE181 and TYR156, PHE257 and TRP241, PHE181 and TYR156 (Figure 8a). Of note, we further confirmed the direct interaction between PKG and GSK‐3β using surface plasmon resonance. The results showed that PKG directly bound to GSK‐3β in a concentration‐dependent manner with a KD value of 1.31e−5 M (Figure 8b). These findings indicate that there is strong affinity between PKG and GSK‐3β.
Figure 8.
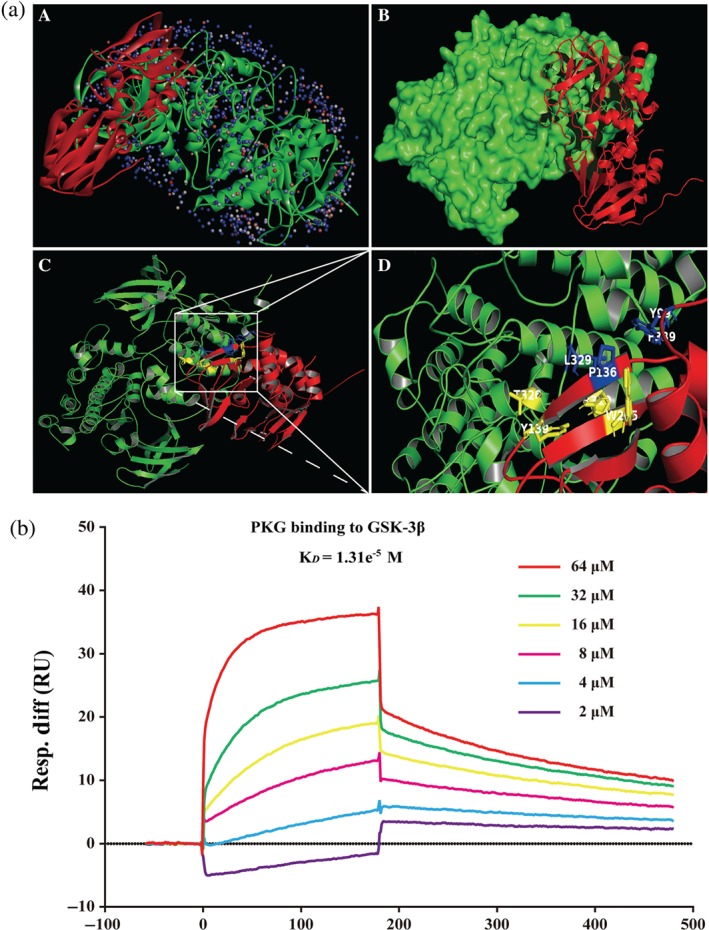
The interaction between PKG and GSK‐3β. (a) The interaction between PKG and GSK‐3β was displayed using ZDOCK. (A) The substrate binding sites. (B) The substrate binding surface. (C) PKG bound to GSK‐3β. (D) Interactions of PKG with active site amino acids and Pi of GSK‐3β. The PKG protein was shown in red colour solid ribbon, while GSK‐3β protein was in green. (b) The binding affinity of PKG with GSK‐3β was evaluated by a surface plasmon resonance assay. Various concentrations of PKG were mixed with GSK‐3β, and binding was detected
4. DISCUSSION
The present study revealed that:‐ (i) ICS II, a naturally ‐occurring PDE 5 inhibitor derived from herbal Epimedii, exerted beneficial effects on cerebral I/R‐induced injury both in vivo and in vitro; (ii) the attenuation of icariside II on cerebral I/R‐induced injury was due to inhibition of excessive autophagy; (iii) icariside II not only inactivated GSK‐3β but also restored cGMP/PKG pathway and (iv) most intriguingly, the present findings also demonstrated that GSK‐3β is downstream of PKG and upstream of autophagy‐related proteins. Therefore PKG/GSK‐3β/autophagy axis plays an extremely important role in mediating the beneficial effect of icariside II on cerebral I/R‐induced excessive autophagic neuronal death (Figure 9).
Figure 9.
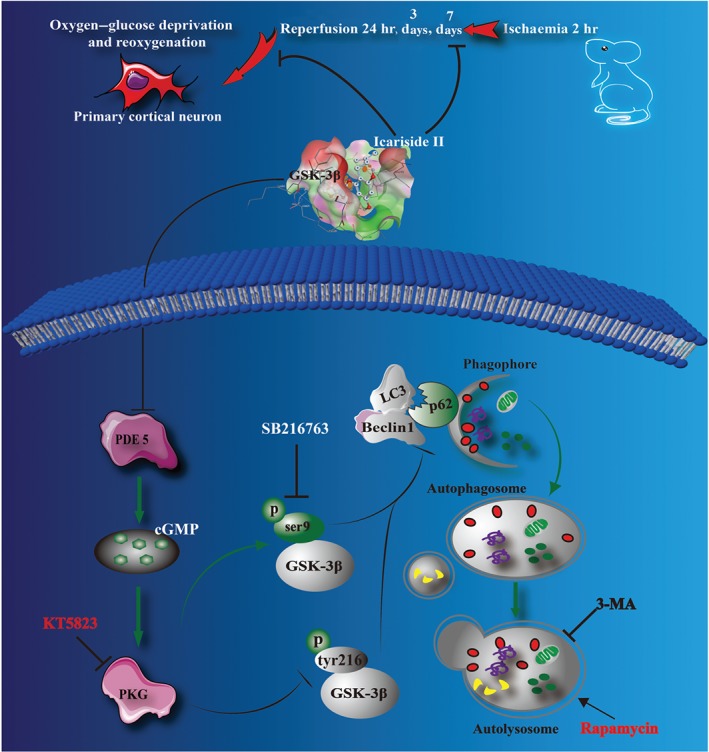
The schematic presentation elucidating proposed mechanisms that icariside II protects neurons against cerebral I/R‐induced injury. Cerebral I/R induces excessive autophagy, resulting in neuronal death. ICS II, a PDE 5 inhibitor, reduces autophagic influx after OGD/R insults via mediating the PKG/GSK‐3β/autophagy axis
Thrombolysis for ischaemic stroke is an accepted treatment, although fewer than 5% of stroke patients are treated with recombinant tissue plasminogen activator (rt‐PA) due to its narrow therapeutic window (Kinouchi et al., 2018). Prophylactic treatment may be a promising strategy for prevention of ischaemic stroke recurrence or long‐term complications; however, there is difficult to implement clinically during the treatment of ischaemic stroke in subacute periods, of which penumbra can be restored with drug treatment. Therefore, although previous studies have been proved that pretreatment with icariside II exerted neuroprotective effect on cerebral I/R injury, whether post‐treatment with icariside II can suppress cerebral I/R injury needs to be further investigated. Interestingly, the present study indicates that post‐treatment with icariside II exerted significant neuroprotective effect on cerebral I/R‐induced injury at Days 3 and 7, whereas it almost has no effect at Day 1, which suggests that post‐treatment with icariside II protects against cerebral I/R injury in subacute periods. These findings indicate that icariside II will be a potential agent to translate into the clinic for treating ischaemic stroke. However, to elucidate its exact mechanism, more detailed studies are urgent.
Autophagy is the cellular process that mediates degradation of cellular proteins and organelles and maintains homeostasis (Cao et al., 2019). Excessive activation of autophagic pathways is shown to be detrimental in cerebral I/R, although the role of autophagy is controversial with some reports demonstrating that autophagy exhibited neuroprotective effects in cerebral I/R. Our findings clearly demonstrated that 3‐methylamphetamine, an inhibitor of autophagy, strongly reduced the lesion volume. While, rapamycin, an inducer of autophagy, reinforced the increase in infarct volume after MCAO at Day 3, which is the peak for cerebral I/R. The data indicated that autophagy is required for neuronal death during cerebral I/R. Notably, neuronal cell death after OGD/R was attenuated and aggravated with 3‐methylamphetamine and rapamycin, respectively. These results were consistent with the hypothesis that activation of autophagy in cerebral I/R does not provide neuroprotection but triggers cell death (Luo et al., 2017). Most importantly, icariside II significantly inhibited autophagosome formation (a special double‐membrane structure) in neurons upon cerebral I/R stress, which was observed by ultrastructural analysis. Additionally, as we expected, icariside II also protected against OGD/R‐induced neuronal death in vitro, which further suggested that the attenuation of icariside II on cerebral I/R‐induced injury is due to the inhibition of excessive autophagy.
Autophagy is a complex dynamic process which is mediated by imperative molecular mechanisms and numerous autophagy‐related (ATG) proteins (Y. Li, Zhou, et al., 2019). MAP 1LC3/LC3 proteins, mammalian subfamily of orthologs of yeast Atg8, are ubiquitin‐like proteins involving the process of autophagy. Since unconjugated LC3 (LC3‐I) protein can be converted to phosphatidylethanolamine‐conjugated LC3 (LC3‐II), which is located at the membrane of the autophagosome. LC3 is an appropriate marker protein of autophagy (He et al., 2017). Beclin 1 is a key subunit of the phosphatidylinositol‐3‐kinase complex, which plays a vital role in the process of autophagosome formation and could also induce autophagy in cerebral ischaemia (X. Li, Li, et al., 2019). Moreover, ATG5 is an important protein associated with phagophore formation and ATG7 is an essential protein involved in the ubiquitin‐like conjugation systems of LC3. Both ATG5 and ATG7 are pivotal components in the process of autophagosomes formation. The results showed that icariside II not only inhibited LC3‐I shift to LC3‐II but also decreased ATG expressions including Beclin 1, ATG5 and ATG7 after stimulation both in vivo and in vitro. Functionally, SQSTM1, an autophagy substrate, binds directly to LC3 and is degraded through autophagic‐lysosomal pathway, monitoring autophagic flux. The present findings also demonstrated that icariside II suppressed the decrease in SQSTM1 expression after MCAO or OGD/R insult indicating autophagic flux, which refers to the entire process of autophagy. Therefore, we further introduced mRFP‐GFP‐LC3 reporter into neurons to evaluate the effect of icariside II on autophagic flux through measuring both autophagosomes and autolysosomes formation simultaneously in neurons. The results obtained from the in vitro experiment with mRFP‐GFP‐LC3 demonstrated that OGD/R increased both autophagosomes and autolysosomes, triggering autophagic flux in neurons. Furthermore, rapamycin significantly promoted autophagic flux, especially autolysosomes formation. On the contrary, 3‐methylamphetamine treatment markedly induce autophagic flux. Intriguingly, silencing of SQSTM1 partially abolished the beneficial effects of icariside II, further confirming that autophagic flux is involved OGD/R. Notably, icariside II inhibited OGD/R‐triggered autophagic flux, suggesting that the suppression of icariside II on cerebral I/R‐induced injury is due to repressing autophagic flux, combined with the decrease of ATG and the increase of the SQSTM1 accumulation.
There is mounting evidence that GSK‐3 plays an important role in regulating autophagic cell death. Phosphorylation of ser9 decreases GSK‐3β activity, whereas phosphorylation of tyr216 increases GSK‐3β activity (Duan et al., 2019; Peixoto, Nunes, & Garcia‐Osta, 2015). In the present study, icariside II robustly augmented GSK‐3β phosphorylation at Ser9 and reduced GSK‐3β phosphorylation at tyr216 in a dose‐dependent manner, suggesting that icariside II can inactivate GSK‐3β in neuronal cells, consistent with our previous study (Gao et al., 2017). Based on the in vivo findings we speculated that icariside II can directly bind to GSK‐3β. To evaluate this hypothesis, molecular docking was used. As expected, icariside II exhibited high affinity for GSK‐3β, which further confirmed that GSK‐3β plays an imperative role in the effect of icariside II on cerebral I/R‐induced injury. Thereafter, GSK‐3β inhibitor significantly promoted the beneficial effect of icariside II, further confirming that the neuroprotective mechanism of icariside II is through inactivating GSK‐3β, at least partly. Of note, there exists a link between GSK‐3β and cGMP, which is regulated by its highly mediated hydrolytic degradation by PDE 5 (Inserte & Garcia‐Dorado, 2015). Interestingly, PDE 5 plays a pivotal role in the cGMP/PKG axis of cellular signalling in neurons (de Witte et al., 2018) and blockade of PDE 5 has been demonstrated to limit infarct size in different experimental animal models (Royl et al., 2009). The results demonstrated that icariside II decreased both the expression and activity of PDE 5, thereby enhancing cGMP level and activating PKG in vivo and in vitro. Furthermore, neuroprotection of icariside II against OGD/R was significantly muted by the PKG inhibitor, which further suggested that icariside II was a potential PDE 5 inhibitor. This data indicates that the cGMP/PKG signalling pathway was involved in the inhibitory effect of icariside II on cerebral I/R‐induced excessive autophagic neuronal death. Collectively, these results suggested that there exists a strong link between GMP/PKG signalling and GSK‐3β. Accordingly, it would be of interest to determine whether the PKG/GSK‐3β/autophagy axis was involved in the suppressive effect of icariside II on cerebral I/R insults. Intriguingly, we found that the cGMP/PKG pathway acts upstream on GSK‐3β inactivation in the inhibitory effect of icariside II on OGD/R‐induced autophagic neuronal death, as evidenced by GSK‐3β inhibitor, PKG inhibitor, silencing SQSTM1 or overexpression of GSK‐3β. As a result, it seems that icariside II protected against cerebral I/R‐induced injury, at least in partly, via the PKG/GSK‐3β/autophagy axis. In view of these findings, there indeed seem to exist a crosstalk between cGMP/PKG, GSK‐3β and autophagy.
Intriguingly, our findings suggested a direct interaction between icariside II and GSK‐3β as evidenced by the docking model, whereas the results also showed the indirect effect of icariside II on GSK‐3β via PKG activation that would seem to be contradictory. Thus, we supposed that there might exist a direct interaction between PKG and GSK‐3β. As we expected, PKG could bind to GSK‐3β directly, as evidenced by ZDOCK and RDOCK as well as surface plasmon resonance. These findings revealed that icariside II inactivated GSK‐3β via, not only by directly affecting GSK‐3β but also by activating PKG, due to the reciprocal action between PKG and GSK‐3β or the PDE 5/cGMP/PKG axis which are involved as icariside II is a PDE 5 inhibitor.
Of note, there still remains limitations in this study. First, although icariside II has been proved to cross the blood brain barrier (Xu et al., 2017), whereas under the I/R condition what percentage of icariside II can cross the blood brain barrier is unknown, as its pharmacokinetic, distribution and metabolism in the brain are unclear. Secondly, icariside II has been suggested to directly bind to PDE 5 and GSK‐3β; whether icariside II can be defined as a dual PDE 5/GSK‐3β inhibitor is worth investigating further. Finally, more robust data would be needed to be provided to substantiate these findings using loss‐of‐function animal models and the long‐term effects of icariside II on cerebral ischaemia.
In conclusion, the present study demonstrates that inhibition of PDE 5 by icariside II exerts beneficial effect on cerebral I/R‐induced autophagic neuronal death through action on the cGMP/PKG/GSK‐3β signalling pathway. These findings raise the possibility that icariside II I may be a potent and effective PDE 5 inhibitor against ischaemic stroke.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Q.H.G. and J.S.S. designed the experiments. J.M.G., L.L., F.X., L.Y.F., and Y.G.L. performed the experiments in vivo and in vitro. J.M.G. and Q.H.G. prepared the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Table S1 Icariside II (ICS II) does not affect physiological parameters. Physiological parameters are for vehicle (saline)‐ administrated (Vehicle) rats and ICS II 16 mg/kg ‐treated (Treated) rats by transient MCAO before, during, or after ischemia. GI, glucose level; MABP, mean arterial blood pressure; All values are expressed as mean ± SEM. n = 5 per group. There are no significant differences between the groups.
Table S2 ZDOCK and RDOCK score of PKG and GSK‐3β. aLower values of ZRANK score and E_RDOCK and higher ZDOCK score indicate top docking of PKG and GSK‐3β. bClash ‘0’ indicates no stearic clash between the proteins after refined using RDOCK.
Figure S1 All rats were maintained at 37 ± 0.5°C rectal temperature. The body temperature was maintained with a heating blanket and monitored using a rectal probe throughout surgery.
Figure S2 The experimental workflow and cerebral blood flow change in rats before, during and after ischemia. (A) The timeline of experiment. The nylon monofilament was withdrawn after 2 h of ischemia. Rats were initially treated with icariside II (ICS II) when the nylon monofilament was withdrawn and then once daily until sacrifice; 3‐MA or Rap was administrated by intracerebroventricular injection at the onset of reperfusion (B) In the rat MCAO model, laser‐doppler flowmeter showed that cortical blood flow reduced to ≤20% and restored to >80% of baseline level (n = 10 per group). (C) Quantitation of cortical blood flow. * P < 0.05 versus before occlusion; # P < 0.05 versus after occlusion.
Figure S3 A schematic diagram of the selected penumbra area for experimental analysis. The dotted line in the brain tissue is considered to be cerebral ischemic penumbra.
Figure S4 Forced expression of a mutant S9A‐GSK3β‐HA/pcDNA3 blocks the beneficial effect of icariside II (ICS II) on OGD/R‐induced injury in neurons. Neurons were transiently transfected with a constitutively active GSK3β mutant S9A (S9A) and an empty vector control (EV) for 24 h. Neurons were then treated with or without ICS II (25 μM) for another 24 h upon OGD/R. (A) Western blot analysis was used to detect the expression of GSK3β and level of p‐Ser9‐GSK3β (n = 5). (B) Cell viability was assessed by MTT assay (n = 5). (C) cell death was determined by intracellular lactate dehydrogenase (LDH) assay (n = 5). (D) Western blot analysis of LC3‐I, LC3‐II, Beclin 1 and PDE 5A expressions in the primary cultured rat cortical neurons. (E) Quantitation of LC3‐II/LC3‐I ratio (n = 5). (F) Quantitation of Beclin 1 expression (n = 5). (G) Quantitation of PDE 5A expression (n = 5). All values are expressed as mean ± SEM. * P < 0.05 versus control group; # P < 0.05 versus OGD/R group (EV).
ACKNOWLEDGEMENTS
This work was supported by Natural Science Foundation of China (81560666), National Key R&D Program “Research on Modernization of Traditional Chinese Medicine” (2017YFC1702005), Program for excellent young talents of Zunyi Medical University (15zy‐002), Science and Technology Innovation Talent Team of Guizhou Province (20154023), Post‐subsidy Project of State Key R&D Program in the Field of Social Development (SQ2017YFC17020405), The hundred level of high‐level innovative talents in Guizhou Province (QKHRCPT 20165684), Shijingshan's Tutor Studio of Pharmacology (GZS‐201607), and Program for Changjiang Scholars and Innovative Research Team in University, China (IRT_17R113).
Gao J, Long L, Xu F, et al. Icariside II, a phosphodiesterase 5 inhibitor, attenuates cerebral ischaemia/reperfusion injury by inhibiting glycogen synthase kinase‐3β‐mediated activation of autophagy. Br J Pharmacol. 2020;177:1434–1452. 10.1111/bph.14912
REFERENCES
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, S. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British. Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S.P.H. , Mathie, A., Peters, J.A. , Veale, E.M. , Striessnig, J. , Kelly, E. , … CGTP collaborators .(2019). The Concise Guide to PHARMACOLOGY 2019/20: ion channels. British Journal of Pharmacology 176, S142–S228 10.1111/bph.14749 [DOI] [PMC free article] [PubMed]
- Arcangeli, S. , Tozzi, A. , Tantucci, M. , Spaccatini, C. , de Iure, A. , Costa, C. , … Calabresi, P. (2013). Ischemic‐LTP in striatal spiny neurons of both direct and indirect pathway requires the activation of D1‐like receptors and NO/soluble guanylate cyclase/cGMP transmission. Journal of Cerebral Blood Flow and Metabolism, 33, 278–286. 10.1038/jcbfm.2012.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouet, V. , Boulouard, M. , Toutain, J. , Divoux, D. , Bernaudin, M. , Schumann‐Bard, P. , & Freret, T. (2009). The adhesive removal test: A sensitive method to assess sensorimotor deficits in mice. Nature Protocols 4, 1560–1564. [DOI] [PubMed] [Google Scholar]
- Camarasa, J. , Rodrigo, T. , Pubill, D. , & Escubedo, E. (2010). Memantine is a useful drug to prevent the spatial and non‐spatial memory deficits induced by methamphetamine in rats. Pharmacological Research, 62, 450–456. 10.1016/j.phrs.2010.05.004 [DOI] [PubMed] [Google Scholar]
- Cao, Y. , Li, Q. , Liu, L. , Wu, H. , Huang, F. , Wang, C. , … Wu, X . (2019). Modafinil protects hippocampal neurons by suppressing excessive autophagy and apoptosis in mice with sleep deprivation. British Journal of Pharmacology, 179, 1282–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Sun, Y. , Liu, K. , & Sun, X. (2014). Autophagy: A double‐edged sword for neuronal survival after cerebral ischemia. Neural Regeneration Research, 9, 1210–1216. 10.4103/1673-5374.135329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang, D. M. , Wang, Z. , & Chiu, C. T. (2011). GSK‐3 as a target for lithium‐induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Frontiers in Molecular Neuroscience, 4, 1–32. 10.3389/fnmol.2011.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, S. H. , Chen, T. , Li, X. , Yue, K. Y. , Luo, P. , Yang, L. K. , … Jiang, X. F. (2017). Sirt3 confers protection against neuronal ischemia by inducing autophagy: Involvement of the AMPK‐mTOR pathway. Free Radical Biology & Medicine, 108, 345–353. 10.1016/j.freeradbiomed.2017.04.005 [DOI] [PubMed] [Google Scholar]
- Dajani, R. , Fraser, E. , Roe, S. M. , Young, N. , Good, V. , Dale, T. C. , & Pearl, L. H. (2001). Crystal structure of glycogen synthase kinase 3β: Structural basis for phosphate‐primed substrate specificity and autoinhibition. Cell, 105, 721–732. 10.1016/s0092-8674(01)00374-9 [DOI] [PubMed] [Google Scholar]
- de Witte, W. E. A. , Versfelt, J. W. , Kuzikov, M. , Rolland, S. , Georgi, V. , Gribbon, P. , … de Lange, E. C. M. (2018). In vitro and in silico analysis of the effects of D2 receptor antagonist target binding kinetics on the cellular response to fluctuating dopamine concentrations. British Journal of Pharmacology, 175, 4121–4136. 10.1111/bph.14456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Y. , Long, L. , Wang, K. , Zhou, J. , Zeng, L. , He, L. & Gong, Q. . (2017). Icariside II, a broad‐spectrum anti‐cancer agent, reverses beta‐amyloid‐induced cognitive impairment through reducing inflammation and apoptosis in rats. Frontiers in Pharmacology, 8, 1–24. 10.3389/fphar.2017.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Y. , Xiong, D. , Yin, C. , Liu, B. , Shi, J. , & Gong, Q. (2016). Icariside II protects against cerebral ischemia‐reperfusion injury in rats via nuclear factor‐κB inhibition and peroxisome proliferator‐activated receptor up‐regulation. Neurochemistry International, 96, 56–61. 10.1016/j.neuint.2016.02.015 [DOI] [PubMed] [Google Scholar]
- Dhayade, S. , Kaesler, S. , Sinnberg, T. , Dobrowinski, H. , Peters, S. , Naumann, U. , … Feil, R. (2016). Sildenafil potentiates a cGMP‐dependent pathway to promote melanoma growth. Cell Reports, 14, 2599–2610. 10.1016/j.celrep.2016.02.028 [DOI] [PubMed] [Google Scholar]
- Diaz‐Canestro, C. , Reiner, M. F. , Bonetti, N. R. , Liberale, L. , Merlini, M. , Wust, P. … Camici, G.G. (2019). AP‐1 (activated protein‐1) transcription factor JunD regulates ischemia/reperfusion brain damage via IL‐1β (interleukin‐1β). Stroke, 50, 469–477. 10.1161/STROKEAHA.118.023739 [DOI] [PubMed] [Google Scholar]
- Duan, J. , Cui, J. , Yang, Z. , Guo, C. , Cao, J. , Xi, M. , … Wen, A. (2019). Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK‐3β/Nrf2 signaling. Journal of Neuroinflammation, 16, 1–16. 10.1186/s12974-019-1406-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen, E. L. , Reggiori, F. , Baba, M. , Kovacs, A. L. , & Seglen, P. O. (2011). Seeing is believing: The impact of electron microscopy on autophagy research. Autophagy, 7, 935–956. 10.4161/auto.7.9.15760 [DOI] [PubMed] [Google Scholar]
- Feng, L. , Gao, J. , Liu, Y. , Shi, J. , & Gong, Q. (2018). Icariside II alleviates oxygen‐glucose deprivation and reoxygenation‐induced PC12 cell oxidative injury by activating Nrf2/SIRT3 signaling pathway. Biomedicine & Pharmacotherapy, 103, 9–17. 10.1016/j.biopha.2018.04.005 [DOI] [PubMed] [Google Scholar]
- Flippo, K. H. , Gnanasekaran, A. , Perkins, G. A. , Ajmal, A. , Merrill, R. A. , Dickey, A. S. , … Strack, S. (2018). AKAP1 protects from cerebral ischemic stroke by inhibiting Drp1‐dependent mitochondrial fission. The Journal of Neuroscience, 38, 8233–8242. 10.1523/JNEUROSCI.0649-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, J. , Deng, Y. , Yin, C. , Liu, Y. , Zhang, W. , Shi, J. , & Gong, Q. (2017). Icariside II, a novel phosphodiesterase 5 inhibitor, protects against H2O2‐induced PC12 cells death by inhibiting mitochondria‐mediated autophagy. Journal of Cellular and Molecular Medicine, 21, 375–386. 10.1111/jcmm.12971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger, D. N. , & Kvietys, P. R. (2015). Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biology, 6, 524–551. 10.1016/j.redox.2015.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan, N. , Zhai, P. , & Sadoshima, J. (2011). Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxidants & Redox Signaling, 14, 2179–2190. 10.1089/ars.2010.3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Z. , Guo, L. , Shu, Y. , Fang, Q. , Zhou, H. , Liu, Y. , … Chai, R. (2017). Autophagy protects auditory hair cells against neomycin‐induced damage. Autophagy, 13, 1884–1904. 10.1080/15548627.2017.1359449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inserte, J. , & Garcia‐Dorado, D. (2015). The cGMP/PKG pathway as a common mediator of cardioprotection: Translatability and mechanism. British Journal of Pharmacology, 172, 1996–2009. 10.1111/bph.12959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, J. , Luo, Y. , Qin, W. , Ma, H. , Li, Q. , Zhan, J. , & Zhang, Y. (2017). Electroacupuncture suppresses the NF‐κB signaling pathway by upregulating cylindromatosis to alleviate inflammatory injury in cerebral ischemia/reperfusion rats. Frontiers in Molecular Neuroscience, 10, 1–14. 10.3389/fnmol.2017.00363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, T. , Yu, J. T. , Zhu, X. C. , Wang, H. F. , Tan, M. S. , Cao, L. , … Tan, L. (2014). Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre‐activation of AMPK‐dependent autophagy. British Journal of Pharmacology, 171, 3146–3157. 10.1111/bph.12655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemitsu, H. , Nakagomi, T. , Tamura, A. , Tsuchiya, T. , Kono, G. , & Sano, K. (2002). Differences in the extent of primary ischemic damage between middle cerebral artery coagulation and intraluminal occlusion models. Journal of Cerebral Blood Flow and Metabolism, 22, 1196–1204. 10.1097/01.wcb.0000037992.07114.95 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, S. , Noda, T. , & Yoshimori, T. (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent‐tagged LC3. Autophagy, 3, 452–460. 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- Kinouchi, T. , Kitazato, K. T. , Shimada, K. , Yagi, K. , Tada, Y. , Matsushita, N. , … Nagahiro, S. (2018). Treatment with the PPARγ agonist pioglitazone in the early post‐ischemia phase inhibits pro‐inflammatory responses and promotes neurogenesis via the activation of innate‐ and bone marrow‐derived stem cells in rats. Translational Stroke Research, 9, 306–316. 10.1007/s12975-017-0577-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D. J. , Abeliovich, H. , Agostinis, P. , Agrawal, D. K. , Aliev, G. , Askew, D. S. , … Deter, R. L. (2008). Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy, 4, 151–175. 10.4161/auto.5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. L. , Ma, Y. , Ma, Y. , Li, Y. , Chen, X. B. , Dong, W. L. , & Wang, R. L. (2017). The design of novel inhibitors for treating cancer by targeting CDC25B through disruption of CDC25B‐CDK2/Cyclin A interaction using computational approaches. Oncotarget, 8, 33225–33240. 10.18632/oncotarget.16600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. Q. , Qureshi, A. A. , Robinson, K. C. , & Han, J. (2014). Sildenafil use and increased risk of incident melanoma in US men: A prospective cohort study. JAMA Internal Medicine, 174, 964–970. 10.1001/jamainternmed.2014.594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Li, J. , Qian, J. , Zhang, D. , Shen, H. , Li, X. , … Chen, G. (2019). Loss of ribosomal RACK1 (receptor for activated protein kinase C 1) induced by phosphorylation at T50 alleviates cerebral ischemia‐reperfusion injury in rats. Stroke, 50, 162–171. 10.1161/STROKEAHA.118.022404 [DOI] [PubMed] [Google Scholar]
- Li, Y. , Zhou, D. , Ren, Y. , Zhang, Z. , Guo, X. , Ma, M. , … Zhang, R. (2019). Mir223 restrains autophagy and promotes CNS inflammation by targeting ATG16L1. Autophagy, 15, 478–492. 10.1080/15548627.2018.1522467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. J. , Chen, T. H. , Yang, L. Y. , & Shih, C. M. (2014). Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death & Disease, 5, 1–23. 10.1038/cddis.2014.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, R. H. , Kang, X. , Xu, L. P. , Nian, H. L. , Yang, X. W. , Shi, H. T. , & Wang, X. J. (2015). Effects of the combined extracts of Herba Epimedii and Fructus Ligustri Lucidi on bone mineral content and bone turnover in osteoporotic rats. BMC Complementary and Alternative Medicine, 15, 1–17. 10.1186/s12906-015-0641-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, C. , Ouyang, M. W. , Fang, Y. Y. , Li, S. J. , Zhou, Q. , Fan, J. , … Tao, T. (2017). Dexmedetomidine protects mouse brain from ischemia‐reperfusion injury via inhibiting neuronal autophagy through up‐regulating HIF‐1α. Frontiers in Cellular Neuroscience, 11, 1–23. 10.3389/fncel.2017.00197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , Drummond, G. B. , McLachlan, E. M. , Kilkenny, C. , & Wainwright, C. L. (2010). Guidelines for reporting experiments involving animals: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1573–1576. 10.1111/j.1476-5381.2010.00873.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papareddy, P. , Kasetty, G. , Alyafei, S. , Smeds, E. , Salo‐Ahen, O. M. H. , Hansson, S. R. , … Herwald, H. (2018). An ecoimmunological approach to study evolutionary and ancient links between coagulation, complement and Innate immunity. Virulence, 9, 724–737. 10.1080/21505594.2018.1441589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada, E. , Casas, A. I. , Palomino‐Antolin, A. , Gomez‐Rangel, V. , Rubio‐Navarro, A. , Farre‐Alins, V. , et al. (2019). Early toll‐like receptor 4 blockade reduces ROS and inflammation triggered by microglial pro‐inflammatory phenotype in rodent and human brain ischaemia models. British Journal of Pharmacology, 176, 2764–2779. 10.1111/bph.14703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto, C. A. , Nunes, A. K. , & Garcia‐Osta, A. (2015). Phosphodiesterase‐5 inhibitors: Action on the signaling pathways of neuroinflammation, neurodegeneration, and cognition. Mediators of Inflammation, 2015, 1–52. 10.1155/2015/940207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royl, G. , Balkaya, M. , Lehmann, S. , Lehnardt, S. , Stohlmann, K. , Lindauer, U. , … Meisel, A. (2009). Effects of the PDE5‐inhibitor vardenafil in a mouse stroke model. Brain Research, 1265, 148–157. 10.1016/j.brainres.2009.01.061 [DOI] [PubMed] [Google Scholar]
- Sheng, R. , Zhang, L. S. , Han, R. , Liu, X. Q. , Gao, B. , & Qin, Z. H. (2010). Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy, 6, 482–494. 10.4161/auto.6.4.11737 [DOI] [PubMed] [Google Scholar]
- Teich, A. F. , Sakurai, M. , Patel, M. , Holman, C. , Saeed, F. , Fiorito, J. , & Arancio, O. (2016). PDE5 exists in human neurons and is a viable therapeutic target for neurologic disease. Journal of Alzheimer's Disease, 52, 295–302. 10.3233/JAD-151104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama, Y. , Koike, M. , & Shibata, M. (2008). Autophagic neuron death in neonatal brain ischemia/hypoxia. Autophagy, 4, 404–408. 10.4161/auto.5598 [DOI] [PubMed] [Google Scholar]
- Venna, V. R. , Benashski, S. E. , Chauhan, A. , & McCullough, L. D. (2015). Inhibition of glycogen synthase kinase‐3β enhances cognitive recovery after stroke: The role of TAK1. Learning & Memory, 22, 336–343. 10.1101/lm.038083.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Xu, Y. , Li, H. , Lei, H. , Guan, R. , Gao, Z. , & Xin, Z. (2015). Antioxidant icariside II combined with insulin restores erectile function in streptozotocin‐induced type 1 diabetic rats. Journal of Cellular and Molecular Medicine, 19, 960–969. 10.1111/jcmm.12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P. , Xu, T. Y. , Wei, K. , Guan, Y. F. , Wang, X. , Xu, H. , … Miao, C. Y. (2014). ARRB1/β‐arrestin‐1 mediates neuroprotection through coordination of BECN1‐dependent autophagy in cerebral ischemia. Autophagy, 10, 1535–1548. 10.4161/auto.29203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Gong, Q. , Wu, Q. , & Shi, J. (2010). Neuroprotective effects of Dendrobium alkaloids on rat cortical neurons injured by oxygen‐glucose deprivation and reperfusion. Phytomedicine, 17, 108–115. 10.1016/j.phymed.2009.05.010 [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Bao, H. , Ge, Y. , Zhuang, S. , Peng, A. , & Gong, R. (2015). Pharmacological targeting of GSK3β confers protection against podocytopathy and proteinuria by desensitizing mitochondrial permeability transition. British Journal of Pharmacology, 172, 895–909. 10.1111/bph.12952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. , Chen, J. , Yang, R. , Duan, F. , Li, S. , & Chen, X. (2019). Mitochondrial protective effect of neferine through the modulation of nuclear factor erythroid 2‐related factor 2 signalling in ischaemic stroke. British Journal of Pharmacology, 176, 400–415. 10.1111/bph.14537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Song, T. , Liu, S. , Li, X. , Li, G. , & Xu, J. (2015). Icariside II inhibits cell proliferation and induces cell cycle arrest through the ROS‐p38‐p53 signaling pathway in A375 human melanoma cells. Molecular Medicine Reports, 11, 410–416. 10.3892/mmr.2014.2701 [DOI] [PubMed] [Google Scholar]
- Wu, J. , Xu, J. , Eksioglu, E. A. , Chen, X. , Zhou, J. , Fortenbery, N. , … Dong, J. (2013). Icariside II induces apoptosis of melanoma cells through the downregulation of survival pathways. Nutrition and Cancer, 65, 110–117. 10.1080/01635581.2013.741745 [DOI] [PubMed] [Google Scholar]
- Wu, W. N. , Wu, P. F. , Chen, X. L. , Zhang, Z. , Gu, J. , Yang, Y. J. , … Chen, J. G. (2011). Sinomenine protects against ischaemic brain injury: Involvement of co‐inhibition of acid‐sensing ion channel 1a and L‐type calcium channels. British Journal of Pharmacology, 164, 1445–1459. 10.1111/j.1476-5381.2011.01487.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, D. , Deng, Y. , Huang, B. , Yin, C. , Liu, B. , Shi, J. , & Gong, Q. (2016). Icariin attenuates cerebral ischemia‐reperfusion injury through inhibition of inflammatory response mediated by NF‐κB, PPARα and PPARγ in rats. International Immunopharmacology, 30, 157–162. 10.1016/j.intimp.2015.11.035 [DOI] [PubMed] [Google Scholar]
- Xu, S. , Yu, J. , Zhan, J. , Yang, L. , Guo, L. , & Xu, Y. (2017). Pharmacokinetics, tissue distribution, and metabolism study of icariin in rat. BioMed Research International, 2017, 1–20. 10.1155/2017/4684962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, B. Y. , Pan, C. S. , Mao, X. W. , Yang, L. , Liu, Y. Y. , Yan, L. , … Han, J. Y. (2014). Icariside II improves cerebral microcirculatory disturbance and alleviates hippocampal injury in gerbils after ischemia‐reperfusion. Brain Research, 1573, 63–73. 10.1016/j.brainres.2014.05.023 [DOI] [PubMed] [Google Scholar]
- Yan, L. , Deng, Y. , Gao, J. , Liu, Y. , Li, F. , Shi, J. , et al. (2017). Icariside II effectively reduces spatial learning and memory impairments in Alzheimer's disease model mice targeting beta‐amyloid production. Frontiers in Pharmacology, 8, 1–25. 10.3389/fphar.2017.00106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. , Zhang, F. , Zhang, T. , Gu, J. , Li, C. , Sun, Y. , … Wang, Y. (2016). Tetramethylpyrazine nitrone improves neurobehavioral functions and confers neuroprotection on rats with traumatic brain injury. Neurochemical Research, 41, 2948–2957. 10.1007/s11064-016-2013-y [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Seo, J. H. , Li, H. , Nam, G. , & Yang, H. O. (2018). The phosphodiesterase 5 inhibitor, KJH‐1002, reverses a mouse model of amnesia by activating a cGMP/cAMP response element binding protein pathway and decreasing oxidative damage. British Journal of Pharmacology, 175, 3347–3360. 10.1111/bph.14377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Liu, J. , Hu, X. , Li, P. , Leak, R. K. , Gao, Y. , & Chen, J. (2015). n‐3 polyunsaturated fatty acids reduce neonatal hypoxic/ischemic brain injury by promoting phosphatidylserine formation and Akt signaling. Stroke, 46, 2943–2950. 10.1161/STROKEAHA.115.010815 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Icariside II (ICS II) does not affect physiological parameters. Physiological parameters are for vehicle (saline)‐ administrated (Vehicle) rats and ICS II 16 mg/kg ‐treated (Treated) rats by transient MCAO before, during, or after ischemia. GI, glucose level; MABP, mean arterial blood pressure; All values are expressed as mean ± SEM. n = 5 per group. There are no significant differences between the groups.
Table S2 ZDOCK and RDOCK score of PKG and GSK‐3β. aLower values of ZRANK score and E_RDOCK and higher ZDOCK score indicate top docking of PKG and GSK‐3β. bClash ‘0’ indicates no stearic clash between the proteins after refined using RDOCK.
Figure S1 All rats were maintained at 37 ± 0.5°C rectal temperature. The body temperature was maintained with a heating blanket and monitored using a rectal probe throughout surgery.
Figure S2 The experimental workflow and cerebral blood flow change in rats before, during and after ischemia. (A) The timeline of experiment. The nylon monofilament was withdrawn after 2 h of ischemia. Rats were initially treated with icariside II (ICS II) when the nylon monofilament was withdrawn and then once daily until sacrifice; 3‐MA or Rap was administrated by intracerebroventricular injection at the onset of reperfusion (B) In the rat MCAO model, laser‐doppler flowmeter showed that cortical blood flow reduced to ≤20% and restored to >80% of baseline level (n = 10 per group). (C) Quantitation of cortical blood flow. * P < 0.05 versus before occlusion; # P < 0.05 versus after occlusion.
Figure S3 A schematic diagram of the selected penumbra area for experimental analysis. The dotted line in the brain tissue is considered to be cerebral ischemic penumbra.
Figure S4 Forced expression of a mutant S9A‐GSK3β‐HA/pcDNA3 blocks the beneficial effect of icariside II (ICS II) on OGD/R‐induced injury in neurons. Neurons were transiently transfected with a constitutively active GSK3β mutant S9A (S9A) and an empty vector control (EV) for 24 h. Neurons were then treated with or without ICS II (25 μM) for another 24 h upon OGD/R. (A) Western blot analysis was used to detect the expression of GSK3β and level of p‐Ser9‐GSK3β (n = 5). (B) Cell viability was assessed by MTT assay (n = 5). (C) cell death was determined by intracellular lactate dehydrogenase (LDH) assay (n = 5). (D) Western blot analysis of LC3‐I, LC3‐II, Beclin 1 and PDE 5A expressions in the primary cultured rat cortical neurons. (E) Quantitation of LC3‐II/LC3‐I ratio (n = 5). (F) Quantitation of Beclin 1 expression (n = 5). (G) Quantitation of PDE 5A expression (n = 5). All values are expressed as mean ± SEM. * P < 0.05 versus control group; # P < 0.05 versus OGD/R group (EV).