

Abstract
Objective.
Mucinous ovarian carcinoma (MOC) is an uncommon ovarian cancer histotype that responds poorly to conventional chemotherapy regimens. Although long overall survival outcomes can occur with early detection and optimal surgical resection, recurrent and advanced disease are associated with extremely poor survival. There are no current guidelines specifically for the systemic management of recurrent MOC. We analyzed data from a large cohort of women with MOC to evaluate the potential for clinical utility from a range of systemic agents.
Methods.
We analyzed gene copy number (n = 191) and DNA sequencing data (n = 184) from primary MOC to evaluate signatures of mismatch repair deficiency and homologous recombination deficiency, and other genetic events. Immunohistochemistry data were collated for ER, CK7, CK20, CDX2, HER2, PAX8 and p16 (n = 117–166).
Results.
Molecular aberrations noted in MOC that suggest a match with current targeted therapies include amplification of ERBB2 (26.7%) and BRAF mutation (9%). Observed genetic events that suggest potential efficacy for agents currently in clinical trials include: KRAS/NRAS mutations (66%), TP53 missense mutation (49%), RNF43 mutation (11%), ARID1A mutation (10%), and PIK3CA/PTEN mutation (9%). Therapies exploiting homologous recombination deficiency (HRD) may not be effective in MOC, as only 1/191 had a high HRD score. Mismatch repair deficiency was similarly rare (1/184).
Conclusions.
Although genetically diverse, MOC has several potential therapeutic targets. Importantly, the lack of response to platinum-based therapy observed clinically corresponds to the lack of a genomic signature associated with HRD, and MOC are thus also unlikely to respond to PARP inhibition.
Keywords: Ovarian cancer, Precision oncology, Molecular targeted therapy, Therapy, Sequencing, Genomic
1. Introduction
Mucinous ovarian carcinoma (MOC) is a rare histotype of epithelial ovarian cancer (EOC), comprising 3–5% of EOC cases [1] with distinct epidemiological differences including weaker associations with reproductive and hormonal factors such as parity and oral contraceptive use [2]. Diagnosis is often at an early stage, with surgical resection of large unilateral tumors resulting in curative outcomes in many cases. However, recurrent and in particular high-grade MOC are classically resistant to conventional platinum/taxane therapy [1,3]. Median survival for Stage III/IV disease is <15 months compared to 41 months for serous and 51 months for endometrioid histologies [4]. Nonetheless, there are no alternative approved treatment guidelines due to a lack of evidence for efficacy. Internationally coordinated attempts to develop MOC clinical trials to assess regimens designed for mucinous gastrointestinal cancers unfortunately met with little success, with recruitment of these rare cases proving to be highly challenging and overt clinical efficacy not observed with the number of primary MOC cases included [5].
As a strategic and much needed step toward the development of novel and targeted therapies for MOC, an international collaborative effort was formed to amass the largest known cohort of patients with annotated primary MOCs. With investigation including extensive genomic, transcriptomic, immunohistochemical and associated clinical data, we aimed to evaluate current and alternate therapeutic options for this rare and chemotherapy resistant tumor type.
2. Materials and methods
2.1. Cohort and pathology review
The GAMuT cohort has been described previously (Supplementary Table 1) [6]. Centers and investigators with specialized ovarian cancer interest were approached to contribute to an international collaboration to advance the knowledge and potential for mucinous ovarian cancers. We obtained primary mucinous tumor samples, and metastases to the ovary from other sites, from 12 different sources in four countries: Australia: Royal Women’s Hospital, the Victorian Cancer Biobank, The Hudson Institute of Medical Research (all Victoria); Garvan Institute, The Gynaecological Oncology Biobank at Westmead (New South Wales); Queensland Institute for Medical Research (Queensland); Australian Ovarian Cancer Study (Australia-wide); United Kingdom: Southampton, and Edinburgh Cancer Research Centre; United States: the Mayo Clinic (MN); Canadian Ovarian Experimental Unified Resource (COEUR, Quebec, Canada) and OVCARE (British Columbia, Canada). All women provided informed consent for the use of their tissue for research and this study was conducted in accordance with the Declaration of Helsinki. All cases underwent expert pathology review of at least one diagnostic slide by PEA, MC,JP, CBG or MK, as well as clinical review to confirm primary mucinous ovarian status. Clinical data was collected by medical record review or tissue bank databases. Metastatic cases were mucinous tumors excluded as being primary ovarian, mostly from the lower gastro-intestinal tract (n = 12), upper gastro-intestinal tract (n = 9) or uterus (n = 4), 1 unknown.
2.2. Genetic data
DNA was extracted from microdissected tumor cells as described [6]. DNA sequencing was performed using either whole genome (n = 5), exome (n = 46) or targeted sequencing (n = 133) using an Agilent SureSelect panel of 507 genes [6]. Four new cases analyzed with the targeted panel are included that were not part of Cheasley et al. Detected variants were filtered as previously described to obtain a high-confidence set of somatic variants. Data are available through the European Genome Archive, submission EGAS00001003545. Mutation figures generated by Mutation mapper in the cBioPortal [46].
Existing SNP array data were used when available for copy number data. Otherwise copy number data were obtained from exome sequencing using AdTex [7], whole genome sequencing using FACETs (v0.5.6) [8], and from the targeted sequencing panel using CopywriteR [9]. Thresholds were log2 ratios of ±0.2 for gains and losses, >0.6 for high level gains and <–1 for homozygous deletions.
We evaluated HRD by mutational signatures (exome/whole genome only) [10] as well as copy number profiles (HRD score). HRD score was calculated as described [11], except that calls were made manually due to the diverse platforms used and the relative simplicity of most genomes.
2.3. Immunohistochemistry
Immunohistochemical staining was performed by the Peter MacCallum Cancer Centre Anatomical Pathology department using standard clinical assays. For ER, we considered any staining in >1% of cells to be positive. In 18/132 cases, ER staining status was obtained from the pathology report. HER2 was scored according to international guidelines for breast cancer, with 0 and 1+ considered negative, 2+ equivocal, and 3+ positive [12]. Final amplification status for equivocal cases was determined by copy number analysis using sequencing or SNP array data (Supplementary Table 2).
3. Results
The current cohort includes collected tissues and associated clinical data from 12 archival sources from four different countries. Tissue and/or data was collected from 202 MOC cases diagnosed between 1993 and 2017 (Table 1, Supplementary Table 1). Available clinical data varied widely between sources, but in brief includes 157 early stage (FIGO I-II) and 25 late stage (III-IV) cases. Sequencing data was obtained by whole exome or whole genome sequencing (WES/WGS, n = 51) or using a targeted panel (n = 133). For comparison, we include data for mucinous borderline tumors (n = 28) and mucinous tumors that are metastases from extra-ovarian sites (n = 26).
Table 1.
Cohort for genomic and IHC analysis.
| Discovery sequencing | Validation sequencing | Copy number | IHC (ER) | |
|---|---|---|---|---|
| Mucinous borderline (n = 109) | 9 | 18 | 39 | 80 |
| Mucinous carcinoma (n = 198) | 51 | 133 | 191 | 132 |
| Grade | ||||
| G1 | 21 (41%) | 62 (47%) | 86 (45%) | 61 (46%) |
| G2 | 22 (43%) | 55 (42%) | 79 (41%) | 51 (39%) |
| G3 | 8 (16%) | 16 (12%) | 26 (14%) | 20 (15%) |
| FIGO stage | ||||
| I | 37 (73%) | 102 (77%) | 147 (77%) | 98 (74%) |
| II | 2 (4%) | 8 (6%) | 10 (5%) | 9 (7%) |
| III-IV | 10 (20%) | 17 (13%) | 25 (13%) | 22 (17%) |
| Stage missing | 2 (4%) | 6 (4%) | 9 (5%) | 3 (2%) |
| Extra-ovarian metastases (n = 36) | 2 | 24 | 29 | 23 |
3.1. Clinical management
A review of management was possible in 167 of the 201 cases. A total of 89 women (53%) were treated with surgery alone. For the cases with chemotherapy details available, 72/74 (97%) women received a platinum-based regimen, usually with a taxane in the first line (61/74, 82%) (Supplementary Fig. 1). Stage was strongly correlated with chemotherapy provision, increasing from 33% of patients diagnosed with Stage I MOC (41/124) to 83% for Stage II-IV (35/42). Data for second and subsequent line therapy was limited to 23 cases, across which it was notable that 16 different therapeutic regimens were prescribed, illustrating the lack of treatment guidelines or consensus for the management of this rare subset of ovarian cancer.
3.2. Homologous recombination deficiency
Homologous recombination deficiency (HRD) is an important clinical correlate in ovarian cancer, strongly predicting response to platinum-based therapies and PARP inhibition. Of 51 cases analyzed by WES/WGS, no cases showed a point mutational signature of HRD [10]. A single case carried a pathogenic BRCA2 mutation (8923; Asn1784HisfsTer2), however, the allele frequency in tumor tissue was only 0.23 and this case had a low copy-number derived HRD score [11]. A co-existing TP53 mutation had an allele frequency of 0.75, suggesting moderate tumor purity. Copy number loss and allelic imbalance across chromosome 13 indicates the loss-of-function BRCA2 allele, likely germline, may have been lost in the tumor, with detection of the allele attributable to the presence of non-tumor cells in the DNA extraction. Therefore, while occurring in a BRCA2 carrier, the MOC appeared unlikely to be an HRD-driven tumor. Copy number analysis of 191 cases found just one with a profile complex enough to be classified as HRD-high (Fig. 1A, score > 55; case WB87/8583). This HRD-high case had a co-existing TP53 mutation but no mutations in HRD-related genes. Pathology review of diagnostic slides confirmed a high-grade mucinous histology, including areas of borderline differentiation. Seven other cases (3.8%) showed an HRD score of >42, placing them in a category of possible responders [13].
Fig. 1.
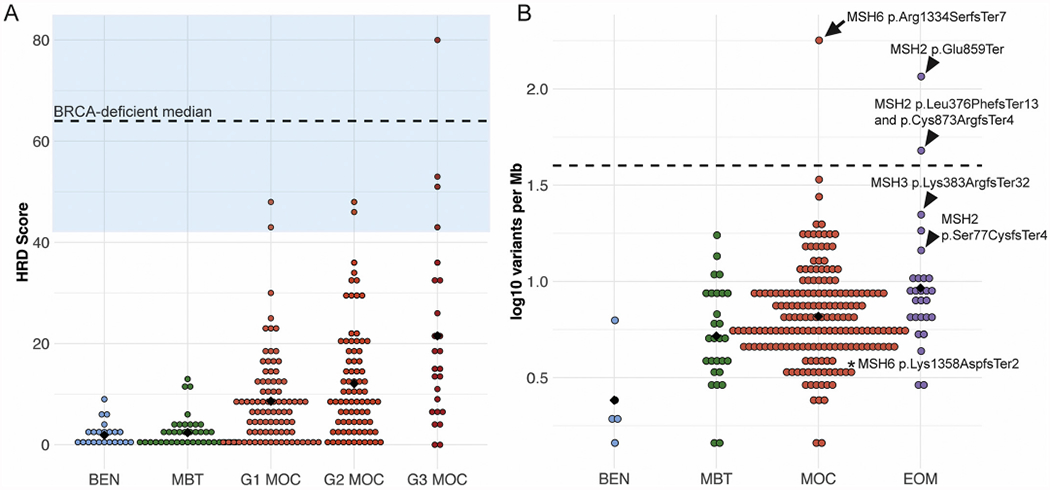
Prevalence of DNA damage repair pathways. A. Homologous recombination deficiency was measured as in Marquard et al. [11] (HRD Score) and shows that MOC rarely have a score above that used to predict response to platinum or PARP inhibitors (BRCA-deficient median and blue zone indicating HRD Score >42 that may respond to platinum from Telli et al. [13]). Black diamond is the mean HRD Score. B. Mismatch repair deficiency is also rare, as indicated by the mutation burden: Mutations per Mb, log10 transformed, black diamond is the mean, dashed line is a suggested threshold for mismatch-repair deficiency detection from Nowak et al. [14]. Arrow, MOC with MSH6 mutation, arrowheads, extra-ovarian metastases (EOM) with MMR gene mutations, *, MOC with MSH6 mutation that is likely non-pathogenic. BEN, benign mucinous tumor, MBT, mucinous borderline tumor. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Mismatch repair deficiency (MMR)
We evaluated the presence of MMR deficiency by mutation burden analysis and identifying cases with mutations in any of the genes in the pathway. One case of the 184 sequenced MOC (C1981) carried a high mutation burden on targeted sequencing (>40 variants per Mb [14], Fig. 1B) and a mutation in MSH6 (p.Arg1334SerfsTer7), although no germline DNA was available to determine if this was somatic or not. A second MOC case (IC381) diagnosed at age 73 carried a germline MSH6 variant (c.4065_4066insTTGA, p.Lys1358AspfsTer2); the tumor did not carry an elevated number of mutations, nor LOH of the wild-type allele. This variant occurred just 5 amino acids before the natural termination codon, and thus its impact on MSH6 function may be minimal, with variants in a similar region classified as class 1 or 2 (benign) in ClinVar [15], for example c.4068_4071dup (p.Lys1358Aspfs) is classed “likely benign”. In contrast, 4/23 (17%) mucinous tumors metastatic to the ovary (two colorectal, two endometrial) had a high mutation burden and/or truncating mutation in one of MSH2 or MSH3. Thus, as MOC cases harboring a high mutational burden are rare, single agent therapy with immune checkpoint inhibitors is unlikely to be helpful for the majority.
3.4. HER2 and HER3
Copy number data were assessed in 191 cases, with ERBB2 amplification found in 51 cases (26.7%). Amplifications were all high level and focal, similar to those seen in breast and gastric cancer (Fig. 2, Supplementary Fig. 2). IHC for HER2 was carried out in 61 of 191 cases. We compared our copy number results derived from SNP arrays and sequencing to IHC and observed strong concordance of 3+ staining with amplified and 0/1+ with non-amplified status in MOC (56/61, 91.8%). Two cases were amplified but IHC negative (10673, 8981), and three cases were either 2+ for IHC but not amplified (8488, VOA1675) or 2+ and amplified (IC392). The reasons for the 2+ interpretation varied. In two MOC (IC392, VOA1675) this was likely due to a subclonal amplification whereby intense membranous IHC staining was only focally present (Fig. 2C). Amplified/IHC positive cells may have been sampled (IC392) or not (VOA1675) from the frozen tumor piece used for DNA extraction. In contrast, for 8488 sequencing data indicated a low-level copy number gain across all of 17q associated with 2+ staining.
Fig. 2.
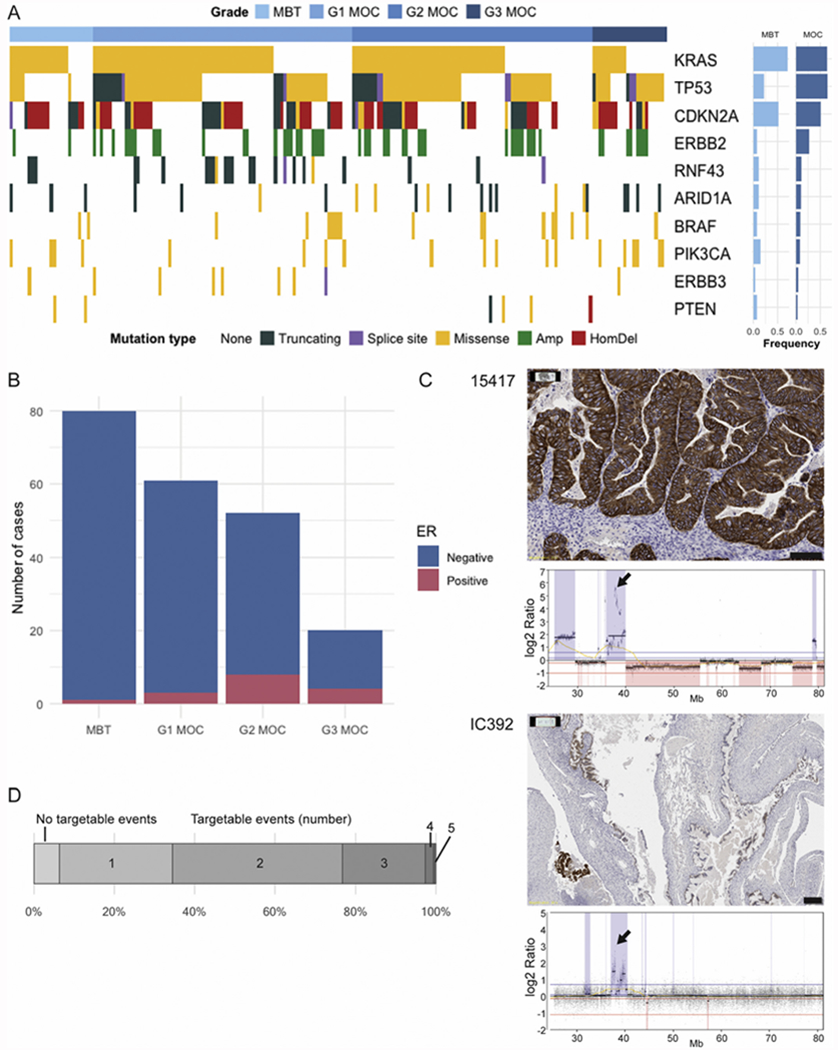
Targetable events in mucinous ovarian cancer. A. Genetic events potentially targetable in MOC. MBT = mucinous borderline tumor n = 27, G1 MOC n = 83, G2 MOC n = 78, G3 MOC n = 24. Mutation type: Amp = Amplification (ERBB2 only); HomDel = Homozygous deletion (CDKN2A only). B. ER immunohistochemistry status by MOC grade. C. ERBB2 amplification and HER2 immunohistochemistry. Case 15417 showing concordance of 3+ staining and high-level amplification (Scale bar 100 μm). Case IC392 showing HER2 heterogeneity (Scale bar 200 μm). Arrows indicate location of ERBB2 gene. D. Percentage of cases with the number of genes (from A) affected by mutation or copy number.
In one MOC case (8981) with negative HER2 IHC from a tissue microarray (TMA), but positive copy number amplification by sequencing, we repeated in situ analyses using whole sections. The results provide evidence for intratumoral heterogeneity (Supplementary Fig. 2). The sensitive sequencing assay was able to detect an amplification, but the original IHC by TMA did not contain the part of the tumor that was amplified. Interestingly, this case was primarily a borderline tumor, and a small group of cells showing abnormal morphology within the full section was the only area positive by CISH but showed only modest protein staining (Supplementary Fig. 2).
We observed no ERBB2 mutations, but a small number of ERBB3 mutated tumors were detected in our cohort (n = 8, 4.3% MOC, Supplementary Fig. 3). The mutation distribution was similar to that seen in COSMIC [16], with the majority of variants being observed in the receptor and furin domains.
3.5. Other targetable genes
We assessed 184 cases for mutations in other potentially targetable genes (Fig. 2). We observed 16 (8.7%) cases with BRAF mutations in MOC, however, 7 of these cases did not carry the typical V600E mutation (Supplementary Fig. 3), and four of these seven also carried a KRAS mutation (although three of these were atypical but likely still oncogenic: p.Lys5Asn, p.Gly13dup and p.Gln61His). KRAS mutations were observed in 118/184 MOC (64%) and NRAS in 3/184 cases (1 also with a KRAS mutation).
Fifteen of 184 MOC and 4/39 mucinous borderline tumors were found to harbor mutations in PIK3CA. However, the mutation spectrum was very different to that in COSMIC, with 8 variants in the PI3-kinase domain, just one at the p.H1047 hotspot and two nearby, and the remaining 8 at other unusual locations in the first 500 amino acids (Supplementary Fig. 3). In COSMIC, <20% of the mutations are outside of the hotspots in exons 9 and 20 (+/− 5 amino acids), but these comprised 10/19 (53%) of the mutations we observed. This 2.5-fold higher rate of non-hotspot mutations is significantly different from expected based on COSMIC (p = 0.003, binomial exact two-tailed test). Two of 185 MOC had inactivating events in PTEN (1 truncating mutation, 1 homozygous deletion) that were exclusive of PIK3CA mutations.
Other molecularly targeted agents that are not currently in the clinic, but are in clinical trials include Wnt-pathway inhibitors (RNF43 mutations), mutant p53 reactivating drugs (TP53 missense mutations) and epigenetic modifiers (ARID1A mutations). Mutations in these genes are detected in our cohort at varying frequencies (Table 2).
Table 2.
Potentially targetable events in MOC.
| Genetic event | Na in MOC (%) | Potential therapy |
|---|---|---|
| Currently in the clinic for other cancer types | 79 (42.9%) | |
| ERBB2 amplification | 51 (26.7%) | Anti Her2 monoclonal antibody therapy, Anti Her2 Mab conjugate therapies, Anti-Her2 tyrosine kinase inhibitors |
| BRAF mutation (any) | 16 (8.7%) | BRAF inhibitors |
| BRAF mutation (V600E) | 9 (4.9%) | |
| High HRD score (>55) | 1 (0.5%) | Platinum salts, PARP inhibitors |
| Moderate HRD score (42-55) | 7 (3.8%) | |
| ER positivity | 14 (10.6%) | Anti-estrogens, CDK4/6 inhibition |
| Mismatch repair deficiency | 1 (0.5%) | Immune checkpoint inhibitors |
| Currently in trials for other cancer types | 167 (90.8%) | |
| KRAS, NRAS mutation | 118, 3 (65.8%) | Broad spectrum RAS/RAF inhibition, MEK inhibition |
| TP53 missense mutation | 90 (48.9%) | Mutant p53 reactivators |
| RNF43 mutation | 21 (11.4%) | FZD inhibition, PORCN inhibitors |
| ARID1A mutation | 19 (10.3%) | Epigenetic modifiers (e.g. BET inhibitors, EZH2 inhibitors), ATR inhibitors |
| PIK3CA mutation, PTEN inactivating | 15,2 (9.2%) | PI3-kinase inhibitors, AKT inhibitors |
| ERBB3 mutation | 8 (4.3%) | Anti-Her2 tyrosine kinase inhibitors, anti-Her3 Mab conjugate therapies |
| Not yet targetable | ||
| CDKN2A inactivating | 82 (44.6%) | |
| TP53 truncating/ESS mutation | 30 (16.3%) |
Mutation data denominator = 184; copy number denominator = 191.
We evaluated whether any mutations were enriched in grade 3 tumors, as a proxy for the infiltrative subtype of MOC that is at higher risk of recurrence [17], and found that mutations in PIK3CA were more common in grade 3 than other grades (grade 3 6/24 versus grade 1, 4/83 and grade 2, 5/77, p = 0.049, Fishers exact test with Benjamini-Hochberg correction for multiple testing). Conversely, mutations in RNF43 were absent in grade 3 cases (corrected p value = 0.003).
3.6. Estrogen receptor (ER) status
We evaluated ER expression in 132 MOC cases, with positive staining seen in 14 cases (10.6%, Supplementary Fig. 4). There was a trend for ER positivity to increase with grade from 5% of grade 1 cases up to 20% of grade 3 cases (p = 0.035, Cochran-Armitage test for trend, Z = −2.10). In contrast, just 1/80 borderline cases (1.25%) were ER+ (Fig. 2C). ER staining in MOC was positively associated with tumor PAX8 staining, with 8/12 ER+ cases also PAX8+ (Supplementary Fig. 4). The genetic events in this group were distinct, with significantly fewer TP53 mutations and CDKN2A losses in ER+ MOC and significantly more CTNNB1 mutations (Supplementary Fig. 4).
4. Discussion
In molecular profiling of MOC, we have identified current therapies that are unlikely to be effective (cisplatin, PARPi, immune checkpoint inhibitors), and in contrast, we highlight a range of targeted therapies worthy of focus, which could have an impact in the near future on individual cases of MOC (Table 2). The low rate of HRD in MOC is consistent with previous studies showing a generally stable genomic profile and a low rate of germline BRCA1/2 mutations [18]. It is also consistent with the reported lack of response to platinum-based therapies, although we were unable to directly correlate HRD scores with response. It is important to highlight that we should avoid prescribing therapy for patients with a rare cancer if there is evidence for a high probability that patients are highly unlikely to receive benefit. Instead, such patients would be better off having investigation for molecular matching of therapy, if evidence suggests the likelihood of targetable aberrations in their rare tumor type [19].
Early reports suggested an association of MMR deficiency with non-serous ovarian cancer histotypes, including mucinous carcinoma [20]. This indication, together with the potential for MMR deficiency in gastrointestinal/endometrial origin tumors with adnexal metastasis [21,22], means it is not uncommon for clinicians to request immunohistochemistry of MMR proteins in MOC. More recent studies have suggested that MMR-deficient ovarian cancer is only common in the endometrioid histotype [23,24]. The difference in the rates of MMR deficiency observed in our study compared to older cohorts may be explained by our careful selection of primary MOC, whereas older studies will include cases of endometrial and colorectal origin. The detection of MMR deficiency in a fairly high proportion of our non-ovarian mucinous cohort supports this contention. Our findings suggest little benefit from reflex MMR IHC testing for MOC unless there was a strong suspicion of extra-ovarian origin. The low rate of MMR-deficiency suggests that single agent immune checkpoint inhibition may have limited potential benefit, however the immune microenvironment remains unexplored in MOC, and it is necessary to assess whether these tumors are immunologically “hot” by other mechanisms and could therefore benefit [25].
Previous data in MOC found that ERBB2 amplification was present in ~20% of cases [26], and our data found a similar proportion. Targeting HER2 in MOC has been attempted anecdotally with mixed results [26] but a clinical trial failed to recruit enough patients. The high-level and focal nature of the amplifications and the strong over-expression in most MOC suggests this could be a viable target, however, response cannot be guaranteed. We observed substantial heterogeneity in the presence of amplification, and in the strength of HER2 positive staining by IHC (e.g. 10405, Supplementary Fig. 2). ERBB2 amplification appears to be a relatively late event in MOC development, as it occurs significantly less frequently in borderline tumors. Heterogeneity within the carcinoma component may indicate firstly, that treatment with anti-HER2 therapies may not be effective on all tumor cells and secondly, that recurrence testing may be required for metastatic disease. Additionally, anti-HER2 antibody monotherapy (e.g. trastuzumab) is most effective in the presence of a strong immune infiltrate [27] and a combination with a small molecule tyrosine kinase inhibitor may be required in tumors with low immune response. Alternatively, a conjugate therapy that exploits an antibody to the overexpressed HER2 to deliver a toxic payload may be of greater efficacy (e.g. Trastuzumab-DM1 or Trastuzumab-deruxtecan). In addition, the strong co-existence of TP53 mutation with ERBB2 amplification provides a strong rationale for attempting p53 normalizing therapies [28] in combination with anti-HER2 therapy.
It is unclear at present whether mutations in ERBB3 lead to longterm responses by small molecular inhibitors. A recent basket trial did not see any responses to neratinib across several tumor types [29]. In addition, the same trial saw responses in ERBB2 mutated tumors, most often in breast cancer but not colorectal cancer. Differences in response were also observed depending on the mutation type and location. In the MOSCATO trial, the best response of an ERBB3 mutated tumor was a partial response to combined trastuzumab and lapatinib, while stable disease was recorded in 7/9 patients treated with afatinib (n = 2), combined trastuzumab and lapatinib (n = 2), lapatinib alone (n = 1) or other combinations (n = 2) [30]. The best responses to lapatinib were in tumors with mutations in the tyrosine kinase domain [30], however only one of the mutations detected in our MOC cohort was located in this domain. The small percentage of MOC with ERBB3 mutations could present an alternative therapeutic opportunity but will likely require combination therapy.
BRAF inhibitors are in current clinical practice for melanoma and show promise for low-grade serous ovarian carcinoma [31], but have shown little efficacy in colorectal carcinoma [32]. Whether MOC would respond to this class of agent remains unclear but again may be worthy of exploration. The high rate of non-V600E mutations in MOC suggests a lower potential response to existing BRAF inhibitors such as vemurafinib, but MOC may be responsive to new combined RAS-RAF inhibitors. BRAF, NRAS and KRAS mutant cases together comprise over 70% of MOC and these women could now be included in combination clinical trials of dual RAS/RAF inhibitors such as BGB-283 [33] (e.g. ). Similarly, while we observed an overall mutation rate of ~9% in PIK3CA/PTEN, suggesting that targeting the PI3-kinase pathway is worth considering during therapy selection [34], the clinical significance of the frequent PIK3CA mutations outside of the usual hotspots is unclear. Inhibitors targeting the PI3-kinase pathway are in clinical development for the management of advanced endometrial, breast and other cancer types (for example alpelisib (PIK3CA-mutated) and buparlisib (pan-PI3K) [35]), with some inhibitors not limited specifically to the PI3kinase domains, but others more broadly targeting the AKT/PTEN/PI3K pathways. One of the most common genetic events in MOC is CDKN2A inactivation, however, whether this gene is a biomarker for response to CDK4/6 inhibition remains an open question that may be tumor-type specific [36].
Low-grade serous and endometrioid ovarian carcinomas have high frequencies of ER protein expression [37]. It has been suggested that these histotypes may respond to hormonal therapy [38–40]. The percentage of ER+ cases we observed (10.6%) is consistent with a recent OTTA study, which found that 16% of MOC had strong ER expression [37], but others report rates as low as 4% up to 19% [41–44]. None of the MOC cases in our cohort appear to have received hormonal therapy, hence hormonal response data within this cohort is unknown. It is worth noting that this subset of ER+ MOC in our cohort appears to be distinct from ER- MOC, based on their genetic features. We speculate that these ER+ cases could represent a subset of MOC with an origin in endometriosis, in a spectrum of tumors incorporating those previously known as “seromucinous” or “endocervical” types. Unfortunately, the clinical and histopathology data with respect to the presence or absence of endometriosis was incomplete in our cohort, and we could not assess this potential association. An alternative explanation is that some of these ER+ cases could in fact have been misdiagnosed endometrioid carcinomas, a recently recognized issue [44]. Data from Rambau et al. showed that the seromucinous type most often resembled an endometrioid ovarian carcinoma at a molecular level [45]. The ER+MOC in our cohort were intermediate on a molecular level, with more CTNNB1 mutations than ER- MOC, but more TP53 mutations and fewer PIK3CA mutations than endometrioid carcinomas (Supplementary Fig. 4). Inclusion of ER (and PR [44]) in the diagnostic work-up for MOC in the future will therefore be important, both to exclude misdiagnosed endometrioid cases and for therapeutic stratification. Inclusion of patients with ER positive MOC in trials, for example basket trials of Aromatase Inhibitors, would provide useful information.
In summary, this exploration of the molecular therapeutic landscape of MOC is the largest undertaken to date and has identified many avenues for investigation. We have summarized these avenues in Fig. 3. If we consider all variants reported in this study as potentially targetable, plus the known targets in other cancer types (BRAF and ERBB2/3), then the proportion of cases with at least one theoretically targetable event was 93.5%, with 65% of cases having two or more targetable events. However, there is currently no evidence for these therapies in MOC. Given the tissue specificity of some targeted therapies, preclinical data using genomically characterized patient-derived models in vitro and in vivo are urgently required to evaluate efficacy. Nonetheless, multiple clinical trials with specific molecular targets are in progress and women diagnosed with MOC would greatly benefit from trials that performed DNA sequencing to identify the most suitable targeted therapy (Table 2). In addition, in order to maximize benefit, such trials should be targeted to those agents with the greatest pre-clinical efficacy. Clinical response, and lack of response, to targeted therapies must be reported along with biomarker information to build an evidence base in humans. Importantly, 23% of women were found to have MOC harboring three or more targetable mutations which may increase their chance of responding to targeted therapy combinations in the future [19]. This approach contrasts with current practice of prescribing platinum-based chemotherapy, which provides little benefit but imposes considerable toxicity. While these potential targets likely over-represent the reality of tumor response, for a tumor type with such poor outcomes and no specific treatment recommendations the potential for therapeutic exploration appears bright.
Fig. 3.

Clinical pathway for therapy choice in MOC. We suggest that high-risk disease (grade 3/advanced stage/infiltrative subtype) should be pre-emptively tested for genomic events using a suitable panel method, since these are unlikely to respond completely to the adjuvant chemotherapy that will be offered while genomic testing is performed. On recurrence of non-high risk disease, genomic testing should also be performed, preferably on recurrence tumor tissue if this is available through surgical debulking or biopsy. If not, primary tumor tissue could be used. ER, estrogen receptor; HRD, homologous recombination deficiency; TMB, tumor mutational burden; “i”, inhibitor. TKi, tyrosine kinase inhibitor.
Supplementary Material
HIGHLIGHTS.
MOC often carry genetic events indicating a targeted therapy and should be entered in basket trials of such agents.
A distinct subset of MOC are estrogen receptor positive (~11%), suggesting hormonal therapy as an option.
Mismatch repair deficiency is present in <1% of MOC; the role of checkpoint inhibitors is uncertain.
Rarity of homologous recombination deficiency means MOC is unlikely to respond to platinum-based therapy/PARP inhibitors.
Acknowledgements
The authors acknowledge the Bioinformatics and Molecular Genomics Core Facilities of the Peter MacCallum Cancer Centre, which were supported by the Australian Cancer Research Foundation. We thank Margot Osinski and Estefania Vicario (Royal Women’s Hospital) for database assistance, Rhiannon Dudley and Nicole Fairweather (Hudson Institute of Medical Research), Maret Böhm (Garvan Institute of Medical Research), Gwo-Yaw Ho (WEHI) and Kimberly Kalli (Mayo Clinic) for assistance in sample collection and/or annotation.
Financial support
This study was supported by the National Health and Medical Research Council of Australia (NHMRC) Grants #APP1045783 and #628434, the Victorian Cancer Agency (Clinical Fellowships to CLS CRF10-20, CRF16014), Cancer Council Victoria (Sir Edward Dunlop Fellowship in Cancer Research to CLS); the Peter MacCallum Cancer Foundation, and the Stafford Fox Medical Research Foundation. This work was made possible through the Australian Cancer Research Foundation, the Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS. The Bioinformatics and Molecular Genomics Core Facilities of the Peter MacCallum Cancer Centre were supported by the Australian Cancer Research Foundation.
The following cohorts were supported as follows:
CASCADE: Supported by the Peter MacCallum Cancer Foundation.
AOCS: The Australian Ovarian Cancer Study Group was supported by the U.S. Army Medical Research and Materiel Command under DAMD17-01-1-0729, The Cancer Council Victoria, Queensland Cancer Fund, The Cancer Council New South Wales, The Cancer Council South Australia, The Cancer Council Tasmania and The Cancer Foundation of Western Australia (Multi-State Applications 191, 211 and 182) and the National Health and Medical Research Council of Australia (NHMRC; ID400413 and ID400281).
The Australian Ovarian Cancer Study gratefully acknowledges additional support from Ovarian Cancer Australia and the Peter MacCallum Foundation. The AOCS also acknowledges the cooperation ofthe participating institutions in Australia and acknowledges the contribution of the study nurses, research assistants and all clinical and scientific collaborators to the study. The complete AOCS Study Group can be found at www.aocstudy.org. We would like to thank all of the women who participated in these research programs.
COEUR: This study uses resources provided by the Canadian Ovarian Cancer Research Consortium’s - COEUR biobank funded by the Terry Fox Research Institute and managed and supervised by the Centre hospitalier de l’Université de Montréal (CRCHUM). The Consortium acknowledges contributions to its COEUR biobank from 1nstitutions across Canada (for a full list see http://www.tfri.ca/en/research/translational-research/coeur/coeur_biobanks.aspx).
The Gynaecological Oncology Biobank at Westmead is a member of the Australasian Biospecimen Network-Oncology group, which was funded by the National Health and Medical Research Council Enabling Grants ID 310670 & ID 628903 and the Cancer Institute NSW Grants ID 12/RIG/1-17 & 15/RIG/1-16.
OVCARE receives core funding from The BC Cancer Foundation and the VGH and UBC Hospital Foundation.
Mayo Clinic: National Institutes of Health (R01-CA122443, P30-CA15083, P50-CA136393); Mayo Foundation; Minnesota Ovarian Cancer Alliance; Fred C. and Katherine B. Andersen Foundation.
Edinburgh: We extend our thanks to the Edinburgh Ovarian Cancer Database from which data were collected for this research and the Nicola Murray Foundation for supporting the Nicola Murray Centre for Ovarian Cancer Research.
Declaration of competing interest
The authors declare no conflicts of interest. ADF, NT and DDLB have received research grant funding from AstraZeneca, unrelated to the contents on this manuscript. DDLB also reports funding from Roche-Genentech and BeiGene, also unrelated. CG reports funding from AstraZeneca, Roche, Clovis, Tesaro, Foundation One, Nucana, Aprea, Novartis, Chugai, and MSD, all outside the submitted work. CLS reports non-financial support and/or other support from Clovis Oncology, Roche, Eisai Australia, Beigene, Sierra Oncology, and AstraZeneca, all outside the submitted work.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ygyno.2019.12.015.
Ethics statement
This study was approved by the Peter MacCallum Cancer Centre and Royal Melbourne Hospital Human Research Ethics Committees, ID #14/76, #01/38 and #2011-248. Informed consent was obtained for all patients in the study. All relevant ethical regulations have been complied with.
References
- [1].Shimada M, Kigawa J, Ohishi Y, et al. , Clinicopathological characteristics of mucinous adenocarcinoma of the ovary, Gynecol. Oncol. 113 (3) (2009) 331–334. [DOI] [PubMed] [Google Scholar]
- [2].Gates MA, Rosner BA, Hecht JL, Tworoger SS, Risk factors for epithelial ovarian cancer by histologic subtype, Am. J. Epidemiol. 171 (1) (2010) 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bamias A, Sotiropoulou M, Zagouri F, et al. , Prognostic evaluation of tumour type and other histopathological characteristics in advanced epithelial ovarian cancer, treated with surgery and paclitaxel/carboplatin chemotherapy: cell type is the most useful prognostic factor, Eur. J. Cancer 48 (10) (2011) 1476–1483. [DOI] [PubMed] [Google Scholar]
- [4].Mackay HJ, Brady MF, Oza AM, et al. , Prognostic relevance of uncommon ovarian histology inwomen with stage III/IV epithelial ovarian cancer, Int. J. Gynecol. Cancer 20 (6) (2010) 945–952. [DOI] [PubMed] [Google Scholar]
- [5].Gore M, Hackshaw A,Brady WE, et al. , An international, phase III randomized trial in patients with mucinous epithelial ovarian cancer (mEOC/GOG 0241) with longterm follow-up: and experience of conducting a clinical trial in a rare gynecological tumor, Gynecol. Oncol. 153 (3) (2019) 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cheasley D,Wakefield MJ, Ryland GL, et al. , The molecular origin and taxonomy of mucinous ovarian carcinoma, Nat. Commun. 10 (1) (2019) 3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Amarasinghe KC, Li J, Hunter SM, et al. , Inferring copy number and genotype in tumour exome data, BMC Genomics 15 (2014) 732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shen R, Seshan VE, FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing, Nucleic Acids Res. 44 (16) (2016) e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kuilman T, Velds A, Kemper K, et al. , CopywriteR: DNA copy number detection from off-target sequence data, Genome Biol. 16 (2015) 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Alexandrov LB, Nik-Zainal S, Wedge DC, et al. , Signatures of mutational processes in human cancer, Nature 500 (7463) (2013) 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Marquard AM, Eklund AC, Joshi T, et al. , Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs, Biomarker Res. 3 (2015) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wolff AC, Hammond ME, Hicks DG, et al. , Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update, Arch. Pathol. Lab. Med. 138 (2) (2014) 241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Telli ML, Timms KM, Reid J, et al. , Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer, Clin. Cancer Res. 22 (15) (2016) 3764–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nowak JA, Yurgelun MB, Bruce JL, et al. , Detection of mismatch repair deficiency and microsatellite instability in colorectal adenocarcinoma by targeted next-generation sequencing, J. Mol. Diagn. 19 (1) (2017) 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Landrum MJ, Lee JM, Benson M, et al. , ClinVar: public archive of interpretations of clinically relevant variants, Nucleic Acids Res. 44 (D1) (2016) D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tate JG, Bamford S, Jubb HC, et al. , COSMIC: the catalogue of somatic mutations in cancer, Nucleic Acids Res. 47 (D1) (2019)(D941–D7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tabrizi AD, Kalloger SE, Kobel M, et al. , Primary ovarian mucinous carcinoma of intestinal type: significance of pattern of invasion and immunohistochemical expression profile in a series of 31 cases, Int. J. Gynecol. Pathol. 29 (2) (2010) 99–107. [DOI] [PubMed] [Google Scholar]
- [18].Alsop K, Fereday S, Meldrum C, et al. , BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group, J. Clin. Oncol. 30 (21) (2012) 2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sicklick JK, Kato S, Okamura R, et al. ,Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study, Nat. Med. 25 (5) (2019) 744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pal T, Permuth-Wey J, Kumar A, Sellers TA, Systematic review and meta-analysis of ovarian cancers: estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology, Clin. Cancer Res. 14 (21) (2008) 6847–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kakar S, Aksoy S, Burgart LJ, Smyrk TC, Mucinous carcinoma of the colon: correlation of loss of mismatch repair enzymes with clinicopathologic features and survival, Mod. Pathol. 17 (6) (2004) 696–700. [DOI] [PubMed] [Google Scholar]
- [22].Raut CP, Pawlik TM, Rodriguez-Bigas MA, Clinicopathologic features in colorectal cancer patients with microsatellite instability, Mutat. Res. 568 (2) (2004) 275–282. [DOI] [PubMed] [Google Scholar]
- [23].Chui MH, Ryan P, Radigan J, et al. , The histomorphology of Lynch syndrome associated ovarian carcinomas: toward a subtype-specific screening strategy, Am. J. Surg. Pathol. 38 (9) (2014) 1173–1181. [DOI] [PubMed] [Google Scholar]
- [24].Rambau PF, Duggan MA, Ghatage P, et al. , Significant frequency of MSH2/MSH6 abnormality in ovarian endometrioid carcinoma supports histotype-specific Lynch syndrome screening in ovarian carcinomas, Histopathology 69 (2) (2016) 288–297. [DOI] [PubMed] [Google Scholar]
- [25].Keenan TE, Burke KP, Van Allen EM, Genomic correlates of response to immune checkpoint blockade, Nat. Med. 25 (3) (2019) 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].McAlpine JN, Wiegand KC, Vang R, et al. , HER2 overexpression and amplification is present in a subset of ovarian mucinous carcinomas and can be targeted with trastuzumab therapy, BMC Cancer 9 (2009) 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bianchini G, Gianni L, The immune system and response to HER2-targeted treatment in breast cancer, Lancet Oncol. 15 (2) (2014) e58–e68. [DOI] [PubMed] [Google Scholar]
- [28].Lehmann S, Bykov VJ, Ali D, et al. , Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer, J. Clin. Oncol. 30 (29) (2012) 3633–3639. [DOI] [PubMed] [Google Scholar]
- [29].Hyman DM, Piha-Paul SA, Won H, et al. , HER kinase inhibition in patients with HER2- and HER3-mutant cancers, Nature 554 (7691) (2018) 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Verlingue L, Hollebecque A, Lacroix L, et al. , Human epidermal receptor family inhibitors in patients with ERBB3 mutated cancers: entering the back door, Eur. J. Cancer 92 (2018) 1–10. [DOI] [PubMed] [Google Scholar]
- [31].Moujaber T, Etemadmoghadam D, Kennedy CJ, et al. , BRAF mutations in low-grade serous ovarian cancer and response to BRAF inhibition, JCO Precis. Oncol. (2) (2018) 1–14. [DOI] [PubMed] [Google Scholar]
- [32].Kopetz S, Desai J, Chan E, et al. , Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer, J. Clin. Oncol. 33 (34) (2015) 4032–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tang Z, Yuan X, Du R, et al. , BGB-283, a novel RAF kinase and EGFR inhibitor, displays potent antitumor activity in BRAF-mutated colorectal cancers, Mol. Cancer Ther. 14 (10) (2015) 2187–2197. [DOI] [PubMed] [Google Scholar]
- [34].Weigelt B, Downward J, Genomic determinants of PI3K pathway inhibitor response in cancer, Front. Oncol. 2 (2012) 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Massacesi C, Di Tomaso E, Urban P, et al. , PI3K inhibitors as new cancer therapeutics: implications for clinical trial design, Onco Targets Ther. 9 (2016) 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Knudsen ES, Witkiewicz AK, The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies, Trends Cancer 3 (1) (2017) 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sieh W, Kobel M, Longacre TA, et al. , Hormone-receptor expression and ovarian cancer survival: an ovarian tumor tissue analysis consortium study, Lancet Oncol. 14 (9) (2013) 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Smyth JF, Gourley C, Walker G, et al. , Antiestrogen therapy is active in selected ovarian cancer cases: the use of letrozole in estrogen receptor-positive patients, Clin. Cancer Res. 13 (12) (2007) 3617–3622. [DOI] [PubMed] [Google Scholar]
- [39].Pan Y, Kao MS, Endometrioid ovarian carcinoma benefits from aromatase inhibitors: case report and literature review, Curr. Oncol. 17 (6) (2010) 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gershenson DM, Bodurka DC, Coleman RL, Lu KH, Malpica A, Sun CC, Hormonal maintenance therapy for women with low-grade serous cancer of the ovary or peritoneum, J. Clin. Oncol. 35 (10) (2017) 1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ajani MA, Salami A, Awolude OA, Oluwasola AO, Hormone-receptor expression status of epithelial ovarian cancer in Ibadan, South-western Nigeria, Pan Afr. Med. J. 27 (2017) 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hogdall EV, Christensen L, Hogdall CK, et al. , Prognostic value of estrogen receptor and progesterone receptor tumor expression in Danish ovarian cancer patients: from the ‘MALOVA’ ovarian cancer study, Oncol. Rep. 18 (5) (2007) 1051–1059. [PubMed] [Google Scholar]
- [43].Shen F, Zhang X, Zhang Y, Ding J, Chen Q, Hormone receptors expression in ovarian cancer taking into account menopausal status: a retrospective study in Chinese population, Oncotarget 8 (48) (2017) 84019–84027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Woodbeck R, Kelemen LE, Kobel M, Ovarian endometrioid carcinoma misdiagnosed as mucinous carcinoma: an underrecognized problem, Int. J. Gynecol. Pathol. 38 (6) (2018) 568–575. [DOI] [PubMed] [Google Scholar]
- [45].Rambau PF, McIntyre JB, Taylor J, et al. , Morphologic reproducibility, genotyping, and immunohistochemical profiling do not support a category of seromucinous carcinoma of the ovary, Am. J. Surg. Pathol. 41 (5) (2017) 685–695. [DOI] [PubMed] [Google Scholar]
- [46].Gao J, Aksoy BA, Dogrusoz U, et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci. Signal. 6 (269) (2013) l1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.