

Abstract
Multiple myeloma (MM) is a plasma cell malignancy that thrives in the bone marrow (BM), with frequent progression to new local and distant bone sites. Our previous studies demonstrated that MM cells at primary sites secrete soluble factors and suppress osteoblastogenesis via the inhibition of Runt-related transcription factor 2 (Runx2) in pre- and immature osteoblasts (OBs) in new bone sites, prior to the arrival of metastatic tumor cells. However, it is unknown whether OB-Runx2 suppression in new bone sites feeds back to promote MM dissemination to and progression in these areas. Hence, we developed a syngeneic mouse model of MM in which Runx2 is specifically deleted in the immature OBs of C57BL6/KaLwRij mice (OB-Runx2−/− mice) to study the effect of OB-Runx2 deficiency on MM progression in new bone sites. In vivo studies with this model demonstrated that OB-Runx2 deficiency attracts MM cells and promotes MM tumor growth in bone. Mechanistic studies further revealed that OB-Runx2 deficiency induces an immunosuppressive microenvironment in BM that is marked by an increase in the concentration and activation of myeloid-derived suppressor cells (MDSCs) and the suppression and exhaustion of cytotoxic CD8+ T cells. In contrast, MDSC depletion by either gemcitabine or 5-fluorouracil treatment in OB-Runx2−/− mice prevented these effects and inhibited MM tumor growth in BM. These novel discoveries demonstrate that OB-Runx2 deficiency in new bone sites promotes MM dissemination and progression by increasing metastatic cytokines and MDSCs in BM and inhibiting BM immunity. Importantly, MDSC depletion can block MM progression promoted by OB-Runx2 deficiency.
Precis
This study demonstrates that Runx2 deficiency in immature osteoblasts at distant bone sites attracts myeloma cells and allows myeloma progression in new bone sites via OB-secreted metastatic cytokines and MDSC-mediated suppression of bone marrow immunity.
Introduction
A hallmark of multiple myeloma (MM) is predominant localization of MM cells in the bone marrow (BM) and the propensity to progress from primary bone sites to new local and distant bone sites (referred to herein as “new bone sites”) (1,2). MM dissemination is devastating for patients and contributes substantially to patient mortality (3). However, the pathomechanisms involved in MM dissemination are not well defined and, as a result, MM remains incurable. Our previous studies demonstrated that MM cells at primary sites secrete soluble factors that systemically orchestrate changes in new bone sites prior to the arrival of metastatic tumor cells (4,5). One such alteration is the simultaneous suppression of osteoblastogenesis and bone formation via suppression of the critical Runt-related transcription factor 2 (Runx2) in osteoblasts (OBs) (OB-Runx2)(4). While the mechanisms regulating MM-induced OB-Runx2 suppression have been extensively studied and described (4,6–9), no studies have determined the reciprocal effect of this suppression on MM dissemination and progression.
Runx2 is a key transcription factor highly expressed in pre-OBs and immature OBs. In these cells, Runx2 induces the expression of stage-specific OB genes and drives the transition from the immature to the mature OB phenotype, thereby promoting bone formation (10). Runx2 is also required for the expression of several molecules produced by OBs at various stages of maturation, such as osteopontin (OPN), dickkopf1 (DKK1), Wnt10, transforming growth factor β1 (TGF-β1), bone morphogenetic protein 4 (BMP-4), receptor activator of nuclear factor kappa-B ligand (RANKL), and osteoprotegerin (OPG) (10,11), that in turn regulate a variety of OB and osteoclast functions. However, the impact of OB-Runx2 suppression on other types of BM cells (e.g., immune cells) and the consequent effects on MM cell dissemination to these new sites is unclear.
In this study, we used a syngeneic mouse model of MM in which Runx2 is specifically deleted in immature OBs to determine the effect of OB-Runx2 deficiency on the BM microenvironment in new bone sites and the consequent effects thereof on MM dissemination and progression.
Materials and Methods
Cell line and cell culture
Wild-type 5TGM1 murine MM cell line was a gift from Dr. Ralph Sanderson (University of Alabama at Birmingham, UAB). 5TGM1 cells expressing GFP (5TGM1-GFP) or firefly luciferase (5TGM1-Luc) were from Dr. Fenghuang Zhan (Iowa University). Cell authentication was conducted by assessing the following features: (1) the expression of IgG2bκ and CD138 (two markers of 5TGM1 cells) by flow cytometry (FACS); (2) in vitro growth curves by MTT and migration rates by cell migration assay; (3) in vivo growth by injecting cells into C57BL/KaLwRij mice via tail vein and measuring levels of IgG2bκ (a soluble marker of 5TGM1 cells) in murine serum by enzyme-linked immunosorbent assay (ELISA). We confirmed that 5TGM1-Luc and 5TGM1-GFP cells maintain the same characteristics as wild-type 5TGM1 cells and the 5TGM1 cells used in the publications of other researchers. We also confirmed that these 5TGM1 cells are mycoplasma negative, using Immu-Mark Myco-Test Kit (MP Biomedicals). 5TGM1 cells were grown in RPMI 1640 medium (Corning) supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a 37°C incubation chamber at 5% CO2 for 7–10 days before used in experiments (medium changed every 3 days).
Development of a syngeneic model of murine MM with specific deletion of Runx2 in immature OBs
We previously generated a mouse model with conditional deletion of Runx2 in immature OBs on a C57BL6 background using Cre-recombinase driven by the Col1a promoter (Runx2−/− mice). The detailed characteristics of this model have been published (12). To define the effect of OB-Runx2 deficiency on MM progression, we backcrossed the original Runx2+/− mice with C57BL/KaLwRij mice, an immunocompetent syngeneic model of murine MM (13,14) (Harlan Laboratories Inc.), and intercrossed the resulting Runx2+/− C57BL/KaLwRij mice to generate litters for the experiments described herein (supplementary methods and Supplementary Fig. S1A). We used OB-Runx2+/+ and OB-Runx2−/− to denote wild-type Runx2+/+ C57BL6//KaLwRij mice and homozygous Runx2−/− C57BL6//KaLwRij mice, respectively. We confirmed that the genotyping and skeletal phenotypes of OB-Runx2−/− mice phenocopy those of the original Runx2−/− mice (12): Col1a-Cre activity is limited to OBs in cortical and trabecular bone. There is no significant alteration in the number of OBs, osteoclasts, or osteocytes or an obvious skeletal phenotype in 5-week-old OB-Runx2−/− mice (Supplementary Fig. S1B–S1E and S2). In this study, we used 5-week-old mice to observe the effect of OB-Runx2 deficiency on the BM microenvironment and MM dissemination to bone. Detailed descriptions of the generation and characteristics of OB-Runx2+/+ and OB-Runx2−/− mice are provided in the supplementary methods. All animal studies were approved by UAB Institutional Animal Care and Use Committee. Equal numbers of male and female mice were used in each experiment.
Evaluation of MM bone-homing and growth in OB-Runx2+/+ and OB-Runx2−/− mice
Equal numbers of 5TGM1-GFP or 5TGM1-Luc cells were injected into 5-week-old syngeneic OB-Runx2+/+ (wild-type control) and OB-Runx2−/− mice via lateral tail vein. To evaluate MM bone-homing rates, the BM cells were harvested from both legs of each mouse 1, 2, and 6 hours after tumor cell injection, and the numbers of GFP+ MM cells in BM were counted by FACS. Tumor growth was monitored biweekly by bioluminescence imaging and by measuring levels of IgG2bκ in murine serum by ELISA.
Isolation of BM supernatants and BM cells for cytokine arrays and FACS
BM was flushed from the femurs/tibias of 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice in 1 ml of PBS and centrifuged to collect BM cells and supernatants. FACS was used to analyze and sort BM cells. A complete list of antibodies used for FACS is provided in Supplementary Table S1. BM supernatants were used for cytokine arrays (15,16) and other in vitro experiments (detailed in supplementary methods).
Isolation and culture of pre-OBs and OBs from mouse calvaria and long bones
Tibias and femurs were harvested from 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice. The BM was flushed out and bone cavities were washed several times with PBS. Bones were then cut into small chips and incubated in 4 ml of α-MEM containing 2 mg/ml collagenase A (Sigma) for 90 minutes. The bone pieces were then washed three times with medium and incubated in α-MEM containing 10% FBS at 37°C in a humidified 5% CO2/95% air atmosphere. After 5–8 days, OBs migrated from the bone pieces were harvested and plated at 2.5−5.0×103 cells/cm2 in T25 flasks. The conditioned medium (CM) was collected after 3 days of culture and stored at −80°C for cytokine arrays and MM migration assays.
Calvarial pre-OBs were isolated from newborn OB-Runx2+/+ and OB-Runx2−/− mice and seeded in cell culture dishes at a density of 3×103 cells/cm2 in proliferation or osteogenic differentiation medium, as we previously described (4). The CM was collected after 3 days of culture for cytokine arrays and replaced with fresh medium. Medium was replaced every 3 days thereafter. After 14 days, alkaline phosphatase (ALP) staining was performed for evaluation of differentiated OBs (4).
Detailed methods are provided in supplementary methods.
Depletion of MDSCs in vivo
MDSCs in BM were depleted in 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice by intraperitoneal (i.p.) injection of gemcitabine (GEM; Selleckchem), an inhibitor of MDSCs that does not directly affect lymphocytes (17,18), or 5-fluorouracil (5-FU, Millipore) to validate the effects of GEM (19,20,21). GEM was injected once per week for 4 weeks (60 mg/kg body weight/injection, equal to 180 mg/m2 body surface area/week) (19). 5-FU was injected once every 3 days for 2 or 4 weeks (30 mg/kg body weight/injection, equal to 180 mg/m2 body surface area/week).
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Differences in means were analyzed with Student’s t-test to evaluate continuous variables of two groups and with one-way ANOVA to evaluate continuous variables of more than two groups. Survival patterns in tumor-bearing mice were evaluated via the Kaplan–Meier method. A p< 0.05 was considered statistically different.
Results
OB-Runx2 deficiency promotes MM cell homing to and progression in bone in vivo
To determine whether OB-Runx2 deficiency contributes to MM cell homing to new bone sites, we injected 5TGM1-GFP MM cells into 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice via tail vein and collected BM cells 1, 2, and 6 hours after tumor cell injection. FACS analysis showed significantly more GFP+ MM cells homed to the BM in OB-Runx2−/− mice than in OB-Runx2+/+ control mice (Fig. 1A). To then determine the impact of OB-Runx2 deficiency on MM progression in bone, we injected 5TGM1-Luc cells into 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice via tail vein and monitored tumor growth by bioluminescence imaging and measurement of IgG2bκ (a soluble marker of 5TGM1 MM cells) in murine serum by ELISA. Compared with OB-Runx2+/+ mice, OB-Runx2−/− mice had substantially larger tumors within bone (Fig. 1B), more bone sites with a tumor (Fig. 1C), and a higher total tumor burden (Fig. 1D) 4 weeks after tumor cell injection. In addition, OB-Runx2−/− mice bearing tumors had significantly shorter overall survival than OB-Runx2+/+ counterparts had (Fig. 1E). Together, these results suggest that OB-Runx2 deficiency in bone recruits MM cells, supports tumor growth therein, and increases MM-related mortality. Of note, there was no difference in tumor growth kinetics between male and female mice within the same genotype.
Figure 1.
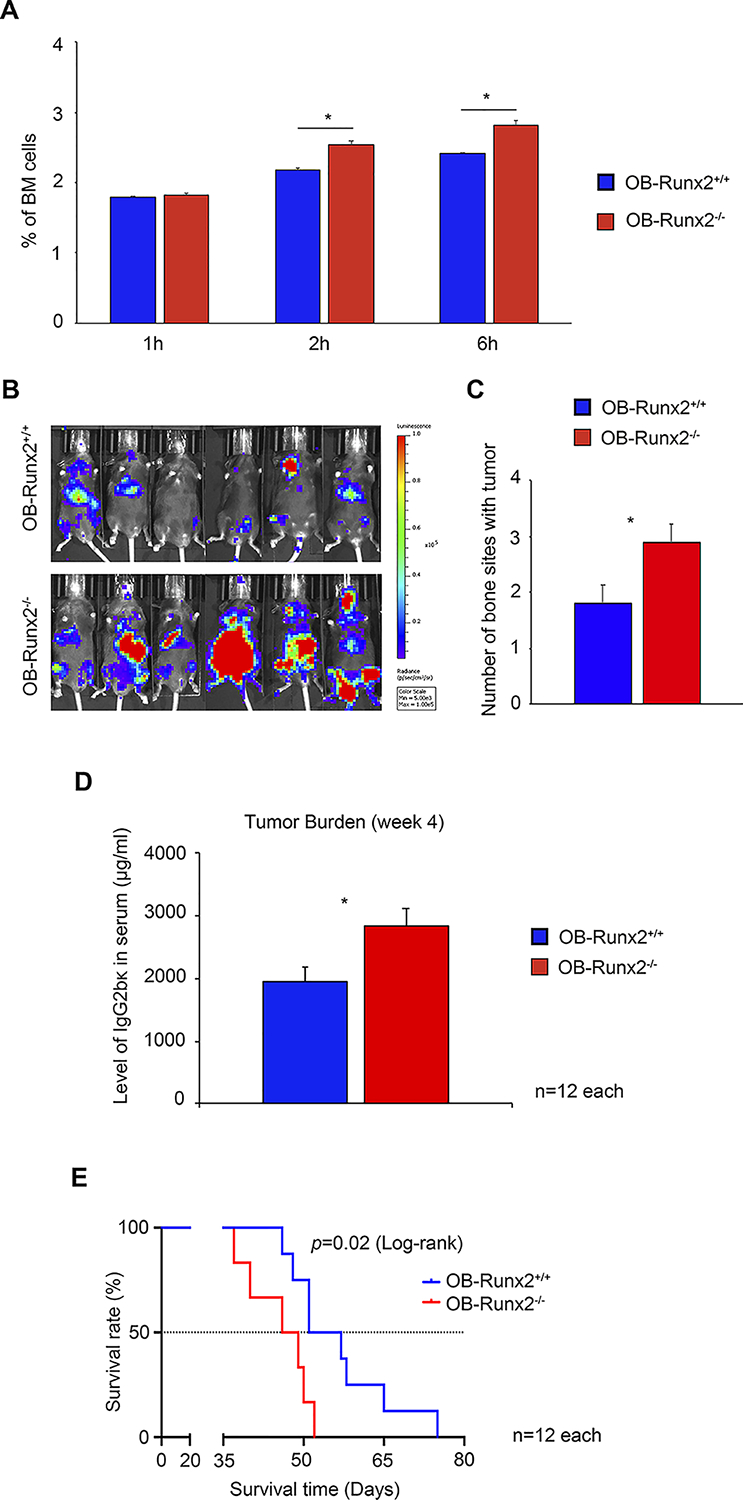
Accelerated bone-homing and growth of 5TGM1 MM cells in the OB-Runx2−/− mouse model. A, To assess bone-homing of MM cells, OB-Runx2+/+ and OB-Runx2−/− mice were i.v. injected with 5TGM1-GFP MM cells (2×106 cells per mouse, n=12 mice/group). BM cells were harvested 1, 2 and 6 hours after tumor cell injection (n=4 mice/group/time point), and GFP+ MM cells were counted by FACS. The percentage of MM cells among all BM cells is shown for each time point. B-E, To assess MM growth and mouse survival rate, OB-Runx2+/+ and OB-Runx2−/− mice were i.v. injected with 5TGM1-Luc MM cells (106 cells per mouse; n=12 mice/group), and tumor growth was analyzed 4 weeks later. Representative bioluminescence imaging of OB-Runx2+/+ (top) and OB-Runx2−/− (bottom) mice (B), the number of bone sites with tumor in each group (C), and the levels of IgG2bκ in the serum of OB-Runx2+/+ and OB-Runx2−/− mice measured by ELISA (in duplicate) (D), Kaplan-Meier survival analysis of OB-Runx2+/+ and OB-Runx2−/− mice after tumor cell injection (E). For A, C, and D, data are presented as mean ± SEM. *P < 0.05 between groups.
OB-Runx2 deficiency increases the production of protumorigenic and immunosuppressive soluble factors by immature OBs
We next elucidated whether the robust bone-homing and growth of MM cells in OB-Runx2−/− mice is due to changes in the concentration of soluble factors within the BM. In BM supernatants of 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice (without tumor cell injection), we measured the concentrations of 30 chemokines, cytokines, and growth factors important for tumor metastasis by cytokine array. Compared with OB-Runx2+/+ controls, OB-Runx2−/− mice had significantly elevated levels of many protumorigenic cytokines in BM (Fig. 2A and B). Additionally, factors important for the expansion and activation of immunosuppressive MDSCs, namely the myeloid-specific growth factors GM-CSF, G-CSF, and M-CSF as well as IL-1β and IL-6 (22), were significantly enhanced in the BM of OB-Runx2−/− mice, with levels of IL-6 and IL-1β increased the most substantially (Fig. 2B).
Figure 2.
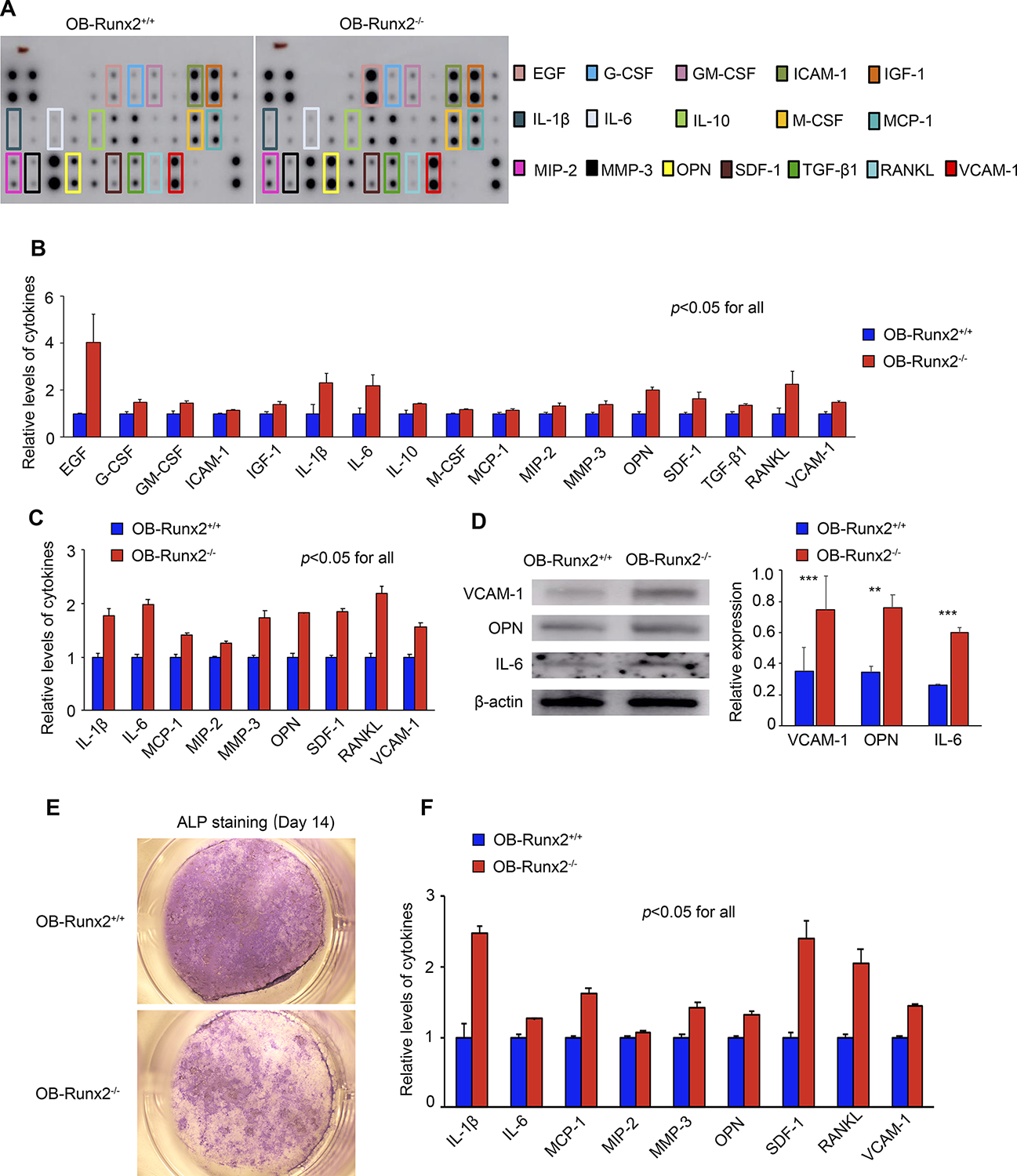
OB-Runx2 deficiency increases levels of protumorigenic and MDSC-stimulating cytokines in BM. A, Representative cytokine array of BM supernatants from OB-Runx2+/+ and OB-Runx2−/− mice before tumor cell injection. Specific cytokines of interest are identified by colored boxes. B, Cytokine levels were quantified by densitometric analysis and normalized to internal controls (n=3 mice/group, in duplicate). C, Cytokine array analysis of CM from Runx2+/+ and Runx2−/− OBs harvested from long bones. Cytokine levels were quantified by densitometric analysis and normalized to internal controls (n=3 mice/group, in duplicate). D, Representative Western blot showing VCAM-1, OPN, IL-6, and β-actin (loading control) expression in Runx2+/+ and Runx2−/− OBs harvested from mouse long bone (left panel). Quantification of VCAM-1, OPN, and IL-6 expression from three independent experiments was performed with Image J software (right panel). E, Primary pre-OBs harvested from the calvaria of newborn OB-Runx2+/+ and OB-Runx2−/− mice were cultured in osteogenic medium, with medium changed every 3 days. Representative images of ALP staining performed at day 14 for identification of differentiated, mature OBs (Leica KL300 LED stereomicroscope, 8X; Leica DFC310 FX digital color camera). F, Cytokine array analysis of CM (day 14) from calvarial OB cultures. Cytokine levels were quantified by densitometric analysis and normalized to internal controls (n=3 mice/group, in duplicate). Data are presented as mean ± SEM. **P < 0.01. ***P < 0.001.
To determine whether OBs lacking Runx2 are a source of these elevated cytokines, we isolated OBs from long bones of 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice (without tumor cell injection) and cultured these primary OBs in proliferation medium. After 3 days, the cytokine levels in the CM were measured by cytokine array. Compared with those from OB-Runx2+/+ mice, OBs from OB-Runx2−/− mice secreted significantly more pro-MM and MDSC-stimulating cytokines, notably including OPN, stromal cell-derived factor 1 (SDF-1), RANKL, vascular cell adhesion molecule 1 (VCAM-1), IL-1β, and IL-6 (22–24) (Fig. 2C). We also confirmed the elevated expression of VCAM-1, OPN, and IL-6 in the OBs of OB-Runx2−/− mice by Western blot (Fig. 2D). To determine whether OB expression and secretion of these pro-MM and MDSC-stimulating molecules is related to the differentiation state of OBs, we harvested calvarial pre-OBs from OB-Runx2+/+ and OB-Runx2−/− newborn mice and cultured these cells in osteogenic medium for 14 days. ALP staining (4,12) showed fewer mature OBs differentiated from Runx2−/− than from Runx2+/+ pre-OBs (Fig. 2E). Furthermore, levels of pro-MM and MDSC-stimulating cytokines were markedly higher in the CM of Runx2−/− pre-OB cultures than in CM of Runx2+/+ pre-OB cultures (Fig. 2F). In summary, these data demonstrate that OB-Runx2 deficiency hinders OB differentiation and maturation, causing OBs to remain in an immature state in which the production of pro-MM and MDSC-stimulating cytokines is enhanced.
BM cytokines upregulated by OB-Runx2 deficiency attract MM cells
To determine whether the elevated cytokines/chemokines detected in the BM of OB-Runx2−/− mice can attract MM cells, we performed transwell migration assays with BM supernatants (BMS) serving as the chemoattractant (15,16). Compared with BMS from OB-Runx2+/+ mice, BMS from OB-Runx2−/− mice markedly stimulated the chemotaxis of 5TGM1 MM cells (Fig. 3A). Similarly, the CM harvested from Runx2−/− OB cultures significantly promoted the migration of these MM cells (Fig. 3B).
Figure 3.

OB-Runx2 deficiency attracts MM cells via increased cytokines in BM. A-C, Migration assays were conducted with 2×105 5TGM1 MM cells seeded on 8-μm pore inserts and, as a chemoattractant in the bottom of the well, either BMS from OB-Runx2+/+ or OB-Runx2−/− mice (n=3 mice/group) (A); CM obtained from primary OBs harvested from the long bones of OB-Runx2+/+ and OB-Runx2−/− mice and cultured in proliferating medium for 3 days (n=3 mice/group) (B), or BMS from OB-Runx2−/− mice pre-incubated for 30 minutes with neutralizing antibodies against VCAM-1 (10 μg/ml) or OPN (10 μg/ml) or with isotype IgG (control) (n=3 mice/group) (C). The number of MM cells migrated into the bottom of each well was counted after 24 hours. Each sample was analyzed in triplicate and each assay was performed at least two times. Data are presented as mean ± SEM. ***P < 0.001.
VCAM-1 and OPN have been reported as important soluble cytokines for MM bone metastasis (25,26), and the levels of both molecules were significantly increased in BMS of OB-Runx2−/− mice and CM of Runx2−/− OBs (Figure 2). Pretreatment of OB-Runx2−/− BMS with neutralizing antibodies specific for VCAM-1 or OPN significantly attenuated MM cell migration (Fig. 3C). These data demonstrate that the OB-Runx2 deficiency-induced increase in specific cytokines within the BM can attract MM cells and may thus directly contribute to MM dissemination to new bone sites.
OB-Runx2 deficiency enhances MDSC expansion and activation in BM
Immature myeloid cells are prevalent in the BM, representing 30% of all cells therein (27). These cells normally differentiate into mature myeloid cells such as granulocytes and macrophages (28,29). However, under certain pathological conditions, including cancer, immature myeloid cells maintain an undifferentiated state, rapidly expand, and acquire potent immunosuppressive potential, becoming activated MDSCs (22,29). Because cytokine arrays showed significant upregulation of multiple MDSC-stimulating cytokines, particularly IL-1β and IL-6, in the BM of OB-Runx2−/− mice and in the CM of cultured Runx2−/− OBs, we investigated the effects of OB-Runx2 deficiency on MDSC expansion and activation in vivo. BM cells harvested from 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice (without tumor cell injection) were stained with antibodies against markers of MDSCs and of MDSC activation (30–32) and analyzed by FACS. Compared with BM of OB-Runx2+/+ mice, BM of OB-Runx2−/− mice had significantly more MDSCs (Fig. 4A), and these MDSCs expressed higher levels of the MDSC activation markers arginase 1 (ARG1), inducible nitric oxide synthase (iNOS), and IL-10 (Fig. 4B) (30–32). These changes were specific to the BM, as no differences were found in the spleen or blood.
Figure 4.

OB-Runx2 deficiency promotes MDSC expansion and activation in BM via IL-1β and IL-6. The number and activation state of MDSCs isolated from BM of OB-Runx2+/+ and OB-Runx2−/− mice were determined by FACS. A, Representative FACS plot (left) and quantification (right) of MDSCs (Gr1+ CD11bhi) (n=6 mice/group). B, Expression of ARG1, iNOS, and IL-10 in MDSCs from OB-Runx2−/− mice relative to that in OB-Runx2+/+ mice, as determined by FACS (n=6 mice/group). C, MDSCs isolated from BM of OB-Runx2+/+ mice were cultured in medium mixed in a 1:1 ratio with PBS, BMS from OB-Runx2+/+ or OB-Runx2−/− mice, or BMS from OB-Runx2−/− mice pretreated for 30 minutes with neutralizing antibodies against IL-1β (20 μg/ml) or IL-6 (20 μg/ml) or isotype IgG control. After 48 hours in culture, MDSCs were counted in triplicate by a Z1 Dual Threshold Coulter Counter (Beckman Coulter) (n=3/culture group). D, Expression of ARG1, iNOS, and IL-10 in MDSCs from each group in (C) relative to that in the cells cultured with PBS, as determined by FACS (n=3/culture group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
To confirm that increased levels of MDSC-stimulating cytokines (e.g., IL-6 and IL-1β) caused the increase and activation of MDSCs in the BM of OB-Runx2−/− mice, MDSCs sorted from the BM of wild-type OB-Runx2+/+ mice were cultured in medium mixed in a 1:1 ratio with PBS (control) or BMS from OB-Runx2+/+ or OB-Runx2−/− mice (without tumor cell injection). Interestingly, compared with PBS, BMS from either mouse group stimulated MDSC expansion and expression of activation markers, but the increases were significantly greater with BMS from OB-Runx2−/− mice (Fig. 4C and D). Moreover, the addition of IL-1β or IL-6 neutralizing antibodies dramatically inhibited the MDSC expansion and activation by BMS from OB-Runx2−/− mice (Fig. 4C and D). Collectively, these findings indicate that OB-Runx2 deficiency promotes MDSC expansion and activation in BM by increasing the levels of MDSC-stimulating cytokines, namely IL-1β and IL-6, in the BM.
Accumulated MDSCs induce an immunosuppressive microenvironment in “pre-metastatic” bones of OB-Runx2−/− mice
T cell activation is required for tumor elimination by the immune system. Activated MDSCs can directly inhibit the proliferation and antitumor response of cytotoxic (tumor killing) CD8+ T cells by producing high levels of ARG1 and iNOS, and can indirectly suppress CD8+ T cell function via IL-10-mediated upregulation of regulatory T cells (Tregs) and regulatory B cells (Bregs) (30–32). Therefore, we examined the numbers of T cell subsets and Bregs in BM of 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice (without tumor cell injection) by FACS, considering the bones of OB-Runx2−/− mice to represent pre-metastatic bone sites. Compared with the BM of OB-Runx2+/+ mice, the BM of OB-Runx2−/− mice had significantly more Tregs and Bregs (Fig. 5A and B) and significantly fewer total T cells and cytotoxic CD8+ T cells (Fig. 5C-left and middle). In addition, compared with CD8+ T cells from the BM of OB-Runx2+/+ mice, those from the BM of OB-Runx2−/− mice expressed higher levels of programmed death 1 (PD-1) and T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) (Fig. 5C-right), two exhaustion markers that disable the CD8+ T cell response required to eliminate cancer cells (33,34).
Figure 5.
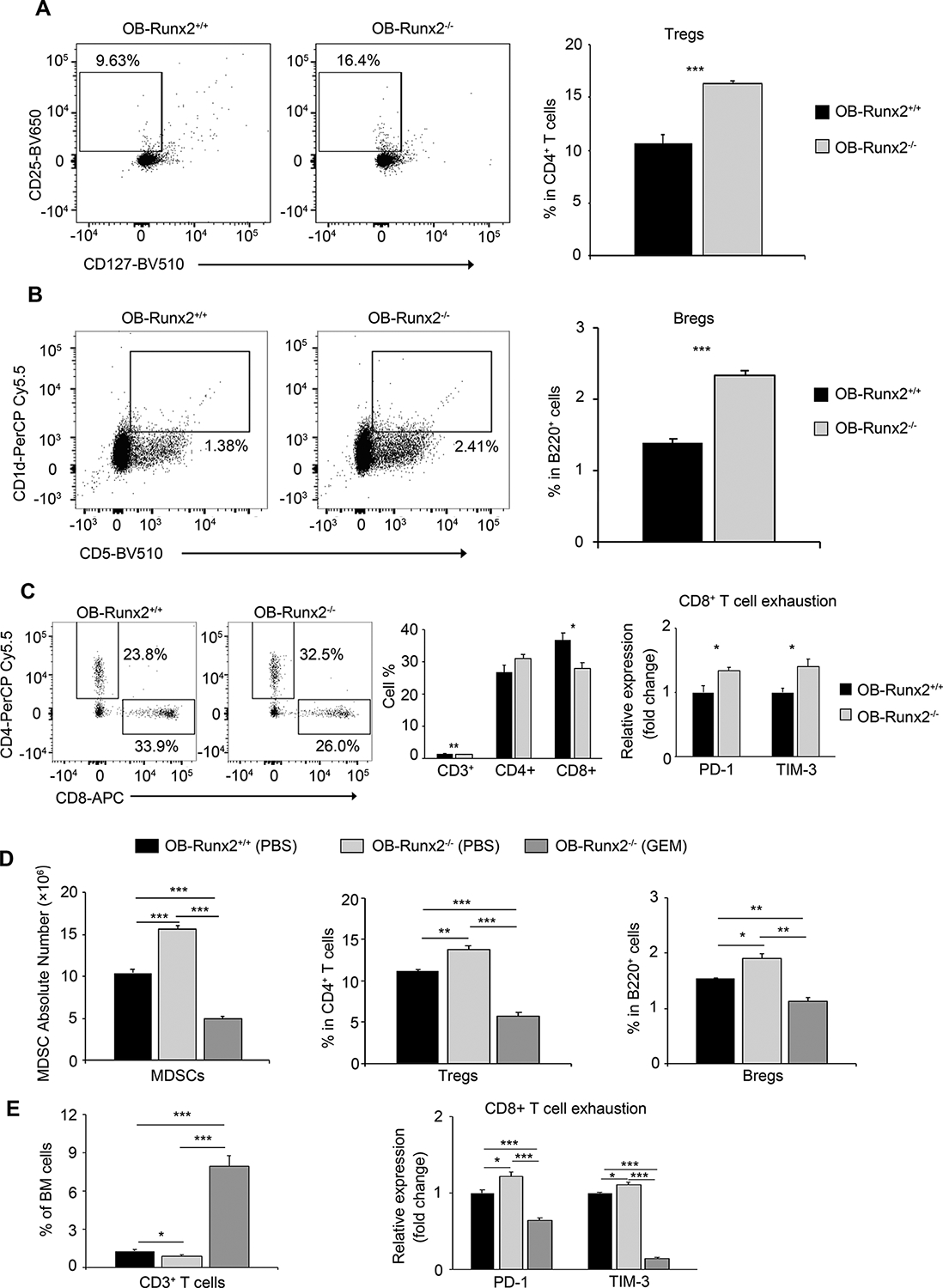
Accumulated MDSCs drive an immunosuppressive microenvironment in OB-Runx2-deficient mice. Total BM cells in non-tumor-bearing 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice were analyzed by FACS. Numbers shown in each plot represent the relative percentage of cells within the indicated gate (n=6 mice/group). A, Representative FACS plots (left) and the percentage of Tregs (CD25+CD127−) in the CD4+ T cell gate (right). B, Representative FACS plots (left) and the percentage of Bregs (CD5+CD1d+) in the B220+B cell gate (right). C, Representative FACS plots showing the percentage of CD4+ and CD8+ T cells in the CD3+ T cell gate (left), the percentage of total (CD3+) T cells among BM cells and the percentage of CD4+ and CD8+ T cells among total T cells (middle), and the expression of PD-1 and TIM-3 in CD8+ T cells (right) of OB-Runx2−/− mice relative to that in OB-Runx2+/+ mice. D-E, Immune cells in BM of non-tumor-bearing 5-week-old OB-Runx2+/+ and OB-Runx2−/− mice were profiled by FACS after weekly i.p. injection of PBS or GEM (60 mg/kg body weight/week) for 4 weeks (n=6 mice/group). D, Total number of MDSCs (Gr1+ CD11bhi) among BM cells (left), the percentage of Tregs among CD4+ T cells (middle), and the percentage of Bregs among B220+ cells (right). E, The percentage of total T cells among all BM cells (left) and the expression of PD-1 and TIM-3 in CD8+ T cells of OB-Runx2−/− mice relative to that in OB-Runx2+/+ mice (right). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
To confirm the central role of MDSCs in OB-Runx2 deficiency-induced immune suppression in BM, we depleted MDSCs in the BM of 5-week-old OB-Runx2−/− mice by 4-week treatment with GEM (Note: these mice were not injected with tumors, and GEM does not directly affect lymphocytes (17,18)). FACS analysis of BM cells confirmed a remarkable reduction in MDSC number after GEM treatment (Fig. 5D-left). Importantly, the depletion of MDSCs associated with a profound increase in the number of total T cells (Fig. 5E-left), a decrease in the number of Tregs and Bregs (Fig. 5D-middle and right), and decreased expression of PD-1 and TIM-3 in CD8+ T cells (Fig. 5E-right). Depletion of MDSCs in the BM of OB-Runx2−/− mice by 4-week treatment with 5-FU, another MDSC depletion agent (20,21), led to similar results. Specifically, FACS analysis of BM cells showed a significant decrease in the number of BM MDSCs, Tregs, and Bregs and decreased expression of PD-1 and TIM-3 in CD8+ T cells (Supplementary Fig. S3).
These results demonstrate that OB-Runx2 deficiency induces an immunosuppressive microenvironment in “pre-metastatic” new bone sites and MDSCs have a central role in this pathological process.
OB-Runx2 deficiency impairs the antitumor immune response to MM cells in vivo
Activated CD8+ T cells release the proteins perforin and granzyme to induce tumor cell apoptosis (35). Thus, we next investigated the effect of OB-Runx2 deficiency on this immune response in tumor-bearing mice. 5TGM1-Luc MM cells were injected via tail vein into OB-Runx2+/+ and OB-Runx2−/− mice. BM cells were harvested and analyzed by FACS on days 1, 7, and 28 after tumor injection. The number of cytotoxic (granzyme B/perforin-expressing) CD8+ T cells in the BM of OB-Runx2+/+ and OB-Runx2−/− mice increased substantially between day 1 and day 7 after tumor cell injection but returned to low levels in the late stage (day 28) of tumor development. At each time point, however, the number of granzyme B+/perforin+ CD8+ T cells was significantly lower in OB-Runx2−/− than in OB-Runx2+/+ BM (Fig. 6A). In addition, the expression levels of the exhaustion markers PD-1 and TIM-3 in CD8+ T cells were significantly higher in OB-Runx2−/− BM than in OB-Runx2+/+ BM at each of these days during tumor development (Fig. 6B).
Figure 6.
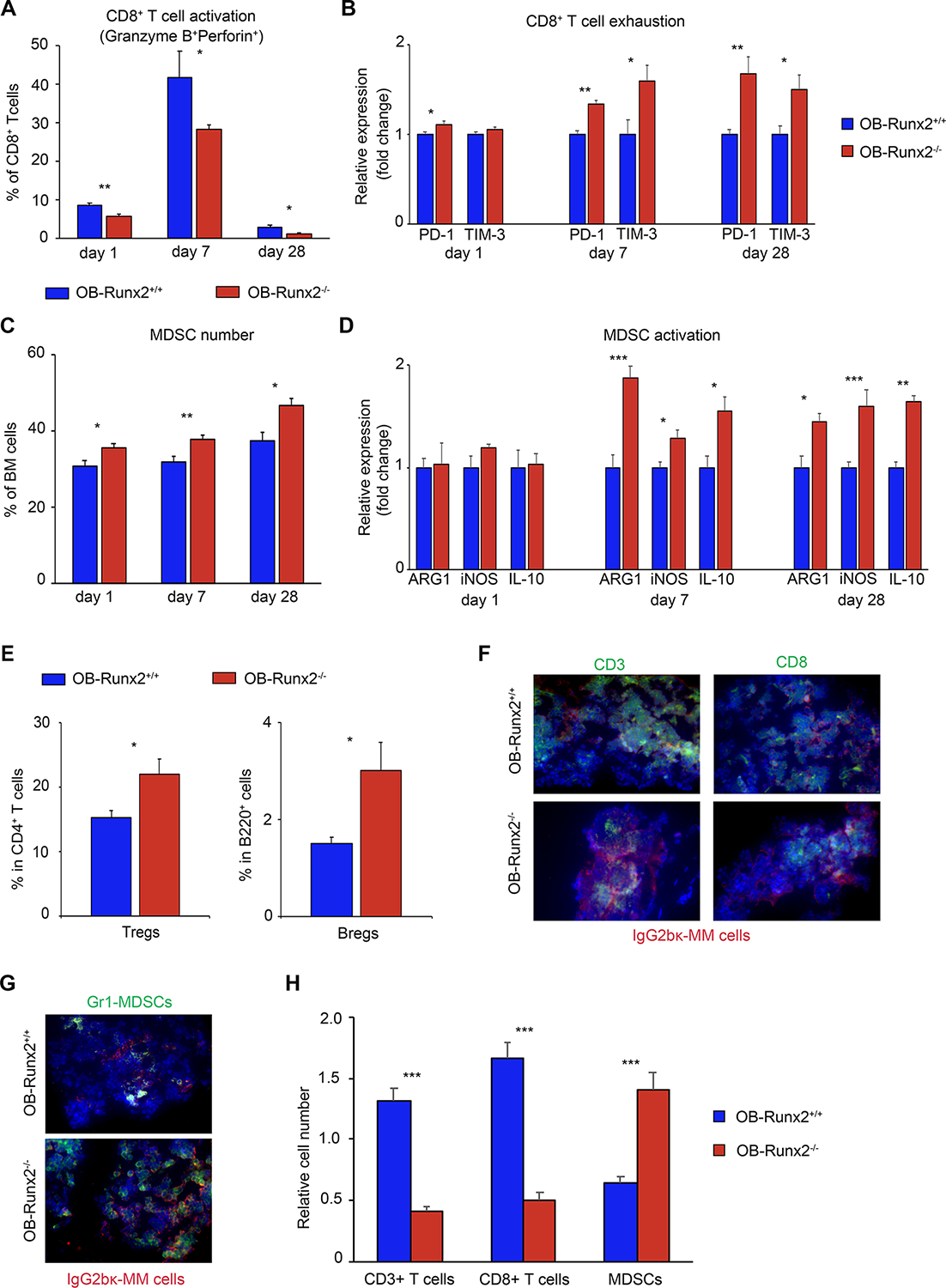
OB-Runx2 deficiency impairs the antitumor immune response to MM cells. OB-Runx2+/+ or OB-Runx2−/− mice were i.v. injected with 5TGM1-Luc MM cells (106 cells per mouse) (n=24 mice/group). BM cells were harvested on days 1, 7, and 28 after tumor cell injection for FACS analysis (n=6 mice/group/time point). A, Percentage of activated (granzyme B+ perforin+) CD8+ T cells in all CD8+ T cells. B, Expression of PD-1 and TIM-3 in CD8+ T cells of OB-Runx2−/− mice relative to that in OB-Runx2+/+ mice. C, Percentage of MDSCs among all BM cells. D, Expression of ARG1, iNOS, and IL-10 in MDSCs of OB-Runx2−/− mice relative to that in OB-Runx2+/+ mice. E, Percentage of Tregs among CD4+ T cells and of Bregs among B220+ cells on day 28. F-G, Representative images of immunofluorescence staining of bone sections harvested from mice 28 days after tumor cell injection showing localization of CD3+ T cells and CD8+ T cells (F) and MDSCs (G) within the tumors (400×, EVOS fluorescence microscope, Thermo Fisher). H, Quantification of the CD3+ T cells, CD8+ T cells, and MDSCs within tumors (n=6 mice/group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Because T cell activation and function can be inhibited by immunosuppressive cells such as MDSCs, Tregs, and Bregs (36), we also characterized immunosuppressive cells in the BM after tumor challenge. FACS analyses revealed significantly more MDSCs and greater expression of MDSC activation markers (Fig. 6C–D), as well as more Tregs and Bregs (Fig. 6E), in the tumor-bearing BM of OB-Runx2−/− mice than in the tumor-bearing BM of OB-Runx2+/+ mice.
Direct interaction of cytotoxic CD8+ T cells with tumor cells is required for tumor killing (37,38). In a parallel study, we harvested femurs and tibia from OB-Runx2+/+ and OB-Runx2−/− mice 28 days after the injection of MM cells. To evaluate immune cell infiltration within tumors, bones in which tumor cells were detected upon bioluminescence imaging were sectioned and stained for total T cells (CD3+), cytotoxic T cells (CD8+), immunosuppressive MDSCs (Gr-1+), and MM cells (IgG2bκ+). Tumors from OB-Runx2−/− mice harbored fewer CD3+ and CD8+ T cells and more MDSCs (Fig. 6F–H) than tumors from OB-Runx2+/+ mice did. Collectively, these discoveries demonstrate that OB-Runx2 deficiency suppresses CD8+ T cell function and tumor infiltration while increasing the function and tumor infiltration of immunosuppressive cells.
MDSC depletion enhances antitumor immunity and decreases tumor burden in OB-Runx2−/− mice
Our studies in mice without MM tumor cells demonstrated that MDSCs have a central role in OB-Runx2 deficiency-induced immune suppression in BM. Specifically, the depletion of MDSCs profoundly increased in the number of total T cells and decreases the number of Tregs and Bregs and the expression of exhaustion markers in CD8+ T cells (Fig. 5 and Supplementary Fig. S3). To further confirm the central role of MDSCs in the impaired antitumor immunity induced by OB-Runx2 deficiency and to determine whether MDSC depletion reverses these immunosuppressive effects in OB-Runx2−/− mice, we depleted MDSCs in tumor-bearing OB-Runx2−/− mice as well as OB-Runx2+/+ counterparts. Starting 5 days after i.v. injection of 5TGM1-Luc MM cells, we i.p. injected GEM or PBS into the mice once weekly for 4 weeks. After 4 weeks of PBS treatment, bioluminescence imaging revealed substantially larger tumors in the BM of OB-Runx2−/− mice than in the BM of OB-Runx2+/+ mice. Although 4 weeks of GEM treatment inhibited tumor growth in both OB-Runx2+/+ and OB-Runx2−/− mice (Fig. 7A), the tumor reduction rate was much higher in GEM-treated OB-Runx2−/− mice than in OB-Runx2+/+ counterparts (8-fold vs. 2-fold difference in mean bioluminescence activity, respectively, compared with respective PBS-treated groups) (Fig. 7B). Serum IgG2bκ ELISA values confirmed these results (Fig. 7C).
Figure 7.
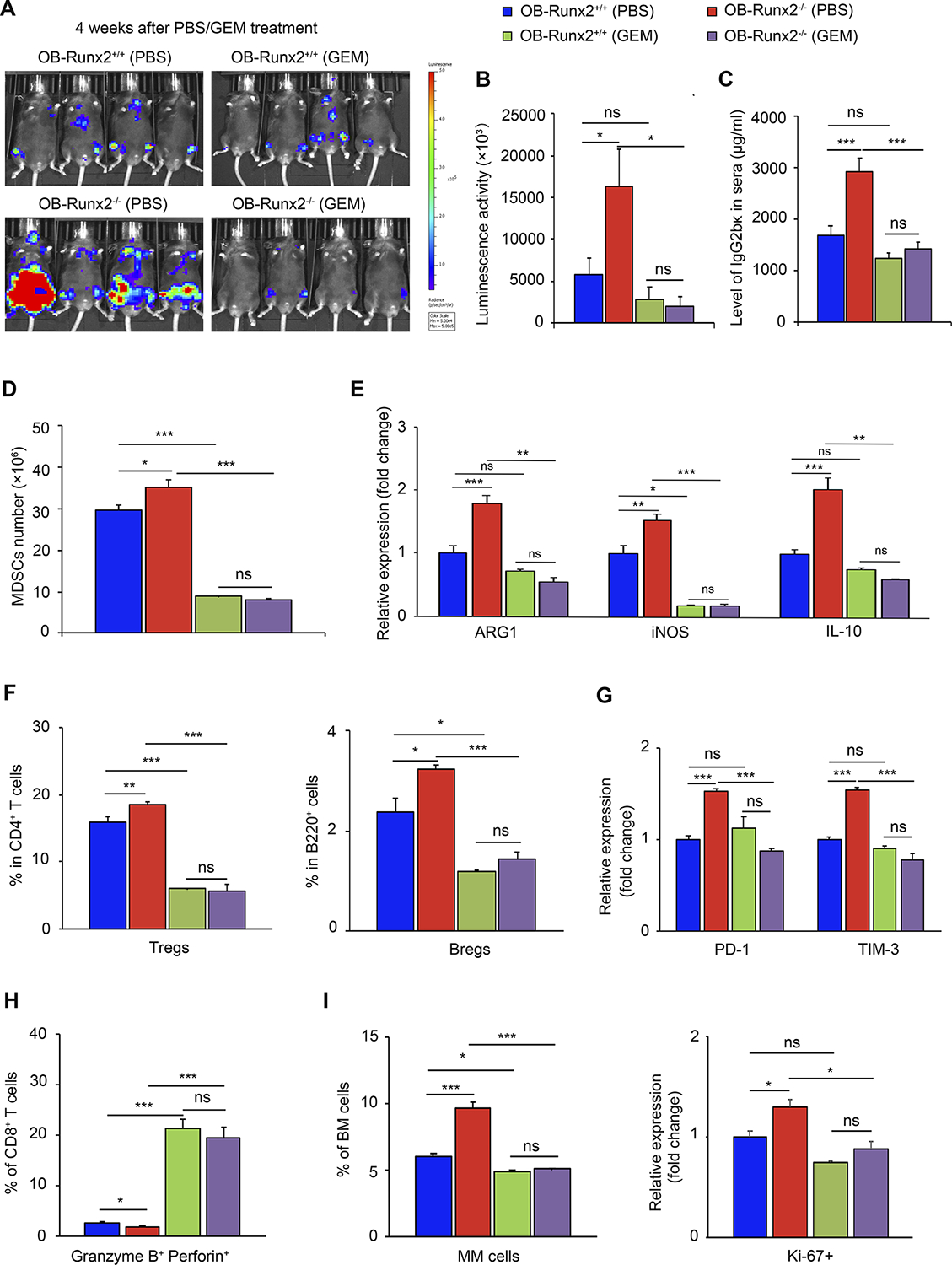
MDSC depletion enhances antitumor immunity and reduces tumor progression in OB-Runx2−/− mice. OB-Runx2+/+ and OB-Runx2−/− mice were i.v. injected with 5TGM1-Luc MM cells (106 per mouse). Five days after tumor cell injection mice began weekly treatment with PBS or GEM (60 mg/kg/week) (n=8 mice/group). A, Representative bioluminescence imaging and B, Mean bioluminescence activity in each group after 4 weeks of PBS or GEM treatment. C, Total tumor burden of each treatment group, measured by serum IgG2bκ ELISA after 4 weeks of PBS or GEM treatment. D-I, FACS analysis of BM cells from tumor-bearing bones after 4 weeks of PBS or GEM treatment. D, Total number of MDSCs (Gr1+ CD11bhi) among BM cells. E, Expression of ARG1, iNOS, and IL-10 in MDSCs of each group relative to that in PBS-treated OB-Runx2+/+ mice, as determined by FACS. F, Percentage of Tregs among CD4+ T cells (left) and of Bregs among B220+ cells (right). G, Expression of PD-1 and TIM-3 in CD8+ T cells of each group relative to that in CD8+ T cells of PBS-treated OB-Runx2+/+ mice, as determined by FACS. H, Percentage of activated (granzyme B+perforin+) cells among CD8+ T cells. I, Percentage of MM cells among BM cells (left) and expression of Ki-67 among all MM cells of each group relative to that in MM cells of PBS-treated OB-Runx2+/+ mice (right). Data are expressed as mean ± SEM. ns, not significant, *P < 0.05 **P < 0.01, ***P < 0.001.
The effect of MDSC depletion on immune cell responses to MM invasion was determined by isolation of BM cells from bones in which tumor cells were detected. FACS analysis of these cells confirmed a significant decline in the number and activation of tumor-infiltrating MDSCs after GEM treatment (Fig. 7D–E). This MDSC depletion associated with a substantial decrease in the number of Tregs and Bregs (Fig. 7F), reduced expression of the CD8+ T cell exhaustion markers (Fig. 7G), and increased production of granzyme B and perforin in CD8+ T cells (Fig. 7H). Finally, GEM treatment of OB-Runx2−/− mice led to a significant decrease in MM cell number and proliferation (Ki-67 expression) in the BM (Fig. 7I).
To validate the effects of GEM-induced MDSC depletion, we treated tumor-bearing OB-Runx2+/+ and OB-Runx2−/− mice with 5-FU. Starting 5 days after i.v. injection of 5TGM1-Luc MM cells, we i.p. injected 5-FU or PBS into the mice once every 3 days for 4 weeks. FACS analysis of BM cells showed results similar to those obtained with GEM treatment (Supplementary Fig. S4A–F). Among BM MDSCs, the monocytic MDSC subset was the subset most increased by OB-Runx2 deficiency and the subset most depleted by 5-FU treatment (Supplementary Fig. S4A–B). We also assessed the effect of prior 5-FU-mediated MDSC depletion on the homing of MM cells to the BM. OB-Runx2+/+ and OB-Runx2−/− mice were treated with 5-FU or PBS for 2 weeks, then i.v. injected with 5TGM1-GFP MM cells. BM cells were isolated 1, 2, and 6 hours later and GFP+ MM cells in the BM were counted by FACS. Again, we found more tumor cells homing to bone in PBS-treated OB-Runx2−/− mice than in PBS-treated OB-Runx2+/+ mice. Furthermore, the reduction of MM cell bone-homing induced by prior 5-FU-mediated depletion of MDSCs was more pronounced in the OB-Runx2−/− mice (Supplementary Fig. S4G).
Together, our results demonstrate that MDSCs have a central role in OB-Runx2 deficiency-induced immune dysfunction, and targeting MDSCs can restore antitumor immunity and thus inhibit MM tumor growth in the OB-Runx2-deficient condition.
Discussion
MM cells release soluble factors that suppress Runx2 expression in OBs at new bone sites (4), but whether this effect feeds back to promote MM dissemination to these areas is unknown. Our results demonstrate that OB-Runx2 deficiency in new bone sites attracts MM cells, supports subsequent growth of arriving MM cells, and ultimately results in increased mortality of tumor-bearing mice. This MM promotion appears to be directly regulated by signaling of Runx2-deficient OBs and not related to bone formation/bone resorption, since we observed no change in the number of OBs, osteoclasts, or osteocytes in 5-week-old OB-Runx2−/− mice, which were used in this study. These findings point to OB-Runx2 deficiency as a novel mechanism for MM dissemination and progression in new bone sites.
Many cytokines, chemokines, and growth factors found in the BM have a chemoattractive effect on MM cells (39,40). In contrast, surveillance by immune cells within the BM is critical in preventing MM cell invasion (41). Here, we found that OB-Runx2 deficiency significantly increases the BM levels of multiple chemoattractants known to be involved in bone-homing of MM cells (22–24). Furthermore, Runx2 deficiency blocks pre-OBs and immature OBs from differentiating into mature OBs, and these undifferentiated OBs produce high levels of chemoattractants and immunosuppressive cytokines. Our findings are in line with those of a study by Kassen et al., which revealed a reduction in the number and activity of mature OBs across the entire disease course, along with the expansion of immature OBs beginning at the early stage of MM (7).
Importantly, the MM cell migration assays we conducted suggested that immature OBs from OB-Runx2−/− mice secrete factors that induce MM cell migration, including VCAM-1 and OPN. Overall, our results demonstrate that the increased production of chemoattractants induced by OB-Runx2 deficiency is a major cause of MM cell dissemination to new bone sites, and immature OBs lacking Runx2 are an important source of these chemoattractants in BM. In addition to soluble factors that attract MM cells, we found upregulated levels of cytokines necessary for the expansion and activation of immunosuppressive MDSCs in the BM of OB-Runx2−/− mice. Intriguingly, the MDSC-stimulating cytokines most substantially increased by OB-Runx2 deficiency are not the classical myeloid-specific growth factors (GM-CSF, G-CSF, M-CSF), but are the non-classical MDSC-stimulating cytokines IL-1β and IL-6, and we demonstrated that immature OBs lacking Runx2 are a source of these cytokines as well. We further demonstrated that increased levels of IL-1β and IL-6 in BM of OB-Runx2−/− mice are indeed responsible for the enhanced MDSC expansion and activation induced by OB-Runx2 deficiency. Although we focused on defining the effect of increased IL-1β and IL-6 on MDSC expansion and activation in the study, we are aware that these two molecules, especially IL-6, are also important for promoting MM cell proliferation (3,23) and may very likely contribute directly to the enhanced MM growth promoted by OB-Runx2 deficiency.
Cytotoxic CD8+ T cells are the primary cells involved in tumor elimination (30,42), but the antitumor function of these cells can be inhibited by immunosuppressive cells such as MDSCs, Tregs, and Bregs (30–32). Indeed, the immune cell profile in the BM of OB-Runx2−/− mice showed a significant increase in immunosuppressive MDSCs, Tregs, and Bregs and a decrease in the number of CD3+ and CD8+ T cells. Furthermore, the production of cytotoxins was attenuated and the expression of T cell exhaustion markers (PD-1 and TIM-3) was upregulated in CD8+ T cells from the BM of these mice, further suggesting that OB-Runx2 deficiency creates an immune suppressive microenvironment that favors MM dissemination (36,43).
MM challenge in OB-Runx2+/+ and OB-Runx2−/− mice confirmed these findings and demonstrated that this immune suppressive BM microenvironment induced by OB-Runx2 deficiency indeed promotes MM bone-homing and tumor growth. Immune dysfunction in the BM of OB-Runx2−/− mice occurred early after tumor inoculation and continued during tumor progression. In addition to the functional changes observed in the BM T cell population, the tumor infiltration of CD3+CD8+ T cells was attenuated in OB-Runx2−/− mice. At the same time, tumor infiltration by immunosuppressive MDSCs, Tregs, and Bregs dramatically increased, further indicating the involvement of these cells in diminishing antitumor immune activity in response to OB-Runx2 deficiency.
Finally, we determined how the increase in MDSCs identified in OB-Runx2-deficient BM inhibits the anti-MM activity of CD8+ T cells. Specifically, we found that MDSCs in the BM of OB-Runx2−/− mice produce more ARG1 and iNOS, which directly inhibit CD8+ T cells, and more IL-10, which can indirectly suppress CD8+ T cells by upregulating Tregs and Bregs (30–32). The increase in MDSC number and activation was detected before and after MM inoculation in OB-Runx2−/− mice. Importantly, we found that depletion of MDSCs in the BM significantly enhanced BM immune function in both OB-Runx2+/+ and OB-Runx2−/− mice before MM inoculation and substantially reversed all of the immunosuppressive effects induced by OB-Runx2 deficiency. These results confirm the central role of MDSCs in creating an immunosuppressive BM microenvironment in pre-metastatic bone regions and demonstrate that OB-Runx2 deficiency-induced BM immunosuppression can be reversed by depletion of MDSCs.
Of clinical relevance, MDSC depletion after tumor inoculation not only reversed all the immunosuppressive effects induced by OB-Runx2 deficiency but also significantly prevented MM cell homing to bone and inhibited MM cell proliferation and progression in bone. These discoveries suggest that targeting MDSCs can inhibit MM dissemination and progression in bone by restoring antitumor immunity. GEM and 5-FU are FDA-approved anti-cancer agents used in the treatment of a variety of solid tumors (44) (45), and the effects of these drugs as MDSC inhibitors in the treatment of solid cancers have been reported (18–21). However, GEM and 5-FU are not commonly used as anti-MM drugs in patients, although they are occasionally used in relapsed or refractory MM patients (46–48). Because we have demonstrated that GEM and 5-FU can restore BM immunity by depleting MDSCs in the BM in both non-tumor-bearing and tumor-bearing OB-Runx2−/− mice, and because the doses of GEM and 5-FU used in our animal experiments were significantly lower than the doses used in cancer patients (seven times lower, 180 vs. 1250 mg/m2 body surface area/week) (49), we believe that the remarkable tumor inhibition by GEM and 5-FU treatment in OB-Runx2−/− mice is at least partially due to MDSC depletion and the consequently recovered antitumor function of cytotoxic CD8+ T cells. Our results shed light on the potential of GEM and 5-FU for use in patients with MM who have high levels of BM MDSCs induced by OB-Runx2 deficiency or by any other causes. Future studies should examine the therapeutic potential of combined administration of clinically used anti-MM drugs and low doses of GEM or 5-FU to simultaneously target MM cells and stimulate anti-MM immunity in BM. In addition, our previous study and studies from other groups have shown that MM cells inhibit Runx2 expression in pre-OBs and inhibit OB differentiation by secreting DKK1 or inducing recruitment of HDAC1 and EZH2 to the Runx2 promoter (4,9); targeting these molecules may be another approach to prevent MM progression promoted by OB-Runx2 suppression.
In conclusion, our findings demonstrate that the OB-Runx2 deficiency induced within pre-metastatic bone sites by soluble factors released by MM cells in primary sites can feed back to invite MM cell dissemination to and progression in these new bone areas. Mechanistic studies demonstrated that OB-Runx2 deficiency creates a highly chemoattractant and immunosuppressive BM microenvironment that is responsible for MM bone-homing and progression in new bone sites. Furthermore, Runx2-deficient OBs are one important direct source of the increased chemoattractants and immunosuppressive cytokines in BM, and the MDSCs that accumulate in response to these cytokines have a central role in suppressing antitumor immunity and promoting MM progression. Finally, our study demonstrates that targeting MDSCs by GEM or 5-FU restores antitumor immunity and reduces MM tumor burden in BM, providing a rationale for the use of these two agents in combination with commonly used anti-MM drugs for MM treatment in patients.
Supplementary Material
Acknowledgments
We thank Drs. Ralph Sanderson and Fenghuang Zhan for the generous gift of 5TGM1 cells. We thank the UAB Animal Imaging Core for assistance with mouse bioluminescence imaging, the UAB Histomorphometry and Molecular Analysis Core for tissue processing, and the UAB Flow Cytometry Core for aid in flow experiments (NIH P30 AR048311, NIH P30 AI27667). This work was supported by National Institutes of Health (NIH) grant R01CA151538 (YY), an International Myeloma Foundation Senior Award (YY), an American Society of Hematology (ASH) Bridge Grant Award (YY), UAB CCSG P30 CA013148 grant (YY), a Chinese Government Studying Abroad Scholarship (XX), and an International Program Fund for doctoral students from Sun Yat-sen University, China (CZ)
Footnotes
Conflicts of Interest Disclosures
The authors disclose no potential conflicts of interest.
This work was presented in part as an oral presentation at the 2017 ASH Annual Meeting in Atlanta, GA
References
- 1.Anderson KC, Carrasco RD. Pathogenesis of myeloma. Annual review of pathology 2011;6:249–74 [DOI] [PubMed] [Google Scholar]
- 2.Vande Broek I, Vanderkerken K, Van Camp B, Van Riet I. Extravasation and homing mechanisms in multiple myeloma. Clin Exp Metastasis 2008;25:325–34 [DOI] [PubMed] [Google Scholar]
- 3.Roodman GD. Genes associate with abnormal bone cell activity in bone metastasis. Cancer Metastasis Rev 2012;31:569–78 [DOI] [PubMed] [Google Scholar]
- 4.Ruan J, Trotter TN, Nan L, Luo R, Javed A, Sanderson RD, et al. Heparanase inhibits osteoblastogenesis and shifts bone marrow progenitor cell fate in myeloma bone disease. Bone 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y, Ren Y, Ramani VC, Nan L, Suva LJ, Sanderson RD. Heparanase enhances local and systemic osteolysis in multiple myeloma by upregulating the expression and secretion of RANKL. Cancer research 2010;70:8329–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roodman GD. Osteoblast function in myeloma. Bone 2011;48:135–40 [DOI] [PubMed] [Google Scholar]
- 7.Kassen D, Lath D, Lach A, Evans H, Chantry A, Rabin N, et al. Myeloma impairs mature osteoblast function but causes early expansion of osteo-progenitors: temporal changes in bone physiology and gene expression in the KMS12BM model. Br J Haematol 2016;172:64–79 [DOI] [PubMed] [Google Scholar]
- 8.Giuliani N, Colla S, Morandi F, Lazzaretti M, Sala R, Bonomini S, et al. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005;106:2472–83 [DOI] [PubMed] [Google Scholar]
- 9.Adamik J, Jin S, Sun Q, Zhang P, Weiss KR, Anderson JL, et al. EZH2 or HDAC1 Inhibition Reverses Multiple Myeloma-Induced Epigenetic Suppression of Osteoblast Differentiation. Mol Cancer Res 2017;15:405–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komori T. Signaling networks in RUNX2-dependent bone development. J Cell Biochem 2011;112:750–5 [DOI] [PubMed] [Google Scholar]
- 11.Javed A, Chen H, Ghori FY. Genetic and transcriptional control of bone formation. Oral and maxillofacial surgery clinics of North America 2010;22:283–93, v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adhami MD, Rashid H, Chen H, Clarke JC, Yang Y, Javed A. Loss of Runx2 in committed osteoblasts impairs postnatal skeletogenesis. J Bone Miner Res 2015;30:71–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bakkus MH, Asosingh K, Vanderkerken K, Thielemans K, Hagemeijer A, De Raeve H, et al. Myeloma isotype-switch variants in the murine 5T myeloma model: evidence that myeloma IgM and IgA expressing subclones can originate from the IgG expressing tumour. Leukemia 2001;15:1127–32 [DOI] [PubMed] [Google Scholar]
- 14.van den Akker TW, Radl J, Franken-Postma E, Hagemeijer A. Cytogenetic findings in mouse multiple myeloma and Waldenstrom’s macroglobulinemia. Cancer Genet Cytogenet 1996;86:156–61 [DOI] [PubMed] [Google Scholar]
- 15.Trotter TN, Li M, Pan Q, Peker D, Rowan PD, Li J, et al. Myeloma cell-derived Runx2 promotes myeloma progression in bone. Blood 2015;125:3598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trotter TN, Gibson JT, Sherpa TL, Gowda PS, Peker D, Yang Y. Adipocyte-Lineage Cells Support Growth and Dissemination of Multiple Myeloma in Bone. Am J Pathol 2016;186:3054–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wesolowski R, Markowitz J, Carson WE 3rd. Myeloid derived suppressor cells - a new therapeutic target in the treatment of cancer. J Immunother Cancer 2013;1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sawant A, Schafer CC, Jin TH, Zmijewski J, Tse HM, Roth J, et al. Enhancement of antitumor immunity in lung cancer by targeting myeloid-derived suppressor cell pathways. Cancer Res 2013;73:6609–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol 2009;9:900–9 [DOI] [PubMed] [Google Scholar]
- 20.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 2010;70:3052–61 [DOI] [PubMed] [Google Scholar]
- 21.De Veirman K, Menu E, Maes K, De Beule N, De Smedt E, Maes A, et al. Myeloid-derived suppressor cells induce multiple myeloma cell survival by activating the AMPK pathway. Cancer Lett 2019;442:233–41 [DOI] [PubMed] [Google Scholar]
- 22.Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. Journal of leukocyte biology 2015;98:913–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manier S, Sacco A, Leleu X, Ghobrial IM, Roccaro AM. Bone marrow microenvironment in multiple myeloma progression. Journal of biomedicine & biotechnology 2012;2012:157496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anborgh PH, Mutrie JC, Tuck AB, Chambers AF. Role of the metastasis-promoting protein osteopontin in the tumour microenvironment. J Cell Mol Med 2010;14:2037–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okada T, Hawley RG, Kodaka M, Okuno H. Significance of VLA-4-VCAM-1 interaction and CD44 for transendothelial invasion in a bone marrow metastatic myeloma model. Clin Exp Metastasis 1999;17:623–9 [DOI] [PubMed] [Google Scholar]
- 26.Caers J, Gunthert U, De Raeve H, Van Valckenborgh E, Menu E, Van Riet I, et al. The involvement of osteopontin and its receptors in multiple myeloma cell survival, migration and invasion in the murine 5T33MM model. Br J Haematol 2006;132:469–77 [DOI] [PubMed] [Google Scholar]
- 27.Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun 2017;8:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katoh H, Watanabe M. Myeloid-Derived Suppressor Cells and Therapeutic Strategies in Cancer. Mediators of inflammation 2015;2015:159269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun 2017;8:14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faccio R. Immune regulation of the tumor/bone vicious cycle. Ann N Y Acad Sci 2011;1237:71–8 [DOI] [PubMed] [Google Scholar]
- 31.Zhao E, Wang L, Dai J, Kryczek I, Wei S, Vatan L, et al. Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. Oncoimmunology 2012;1:152–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balkwill F, Montfort A, Capasso M. B regulatory cells in cancer. Trends in immunology 2013;34:169–73 [DOI] [PubMed] [Google Scholar]
- 33.Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis 2015;6:e1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011;117:4501–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 2015;15:388–400 [DOI] [PubMed] [Google Scholar]
- 36.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009;9:162–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weigelin B, Krause M, Friedl P. Cytotoxic T lymphocyte migration and effector function in the tumor microenvironment. Immunol Lett 2011;138:19–21 [DOI] [PubMed] [Google Scholar]
- 38.Martinez-Lostao L, Anel A, Pardo J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin Cancer Res 2015;21:5047–56 [DOI] [PubMed] [Google Scholar]
- 39.Aggarwal R, Ghobrial IM, Roodman GD. Chemokines in multiple myeloma. Exp Hematol 2006;34:1289–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmadzadeh A, Kast RE, Ketabchi N, Shahrabi S, Shahjahani M, Jaseb K, et al. Regulatory effect of chemokines in bone marrow niche. Cell Tissue Res 2015;361:401–10 [DOI] [PubMed] [Google Scholar]
- 41.Lee SJ, Borrello I. Role of the Immune Response in Disease Progression and Therapy in Multiple Myeloma. Cancer Treat Res 2016;169:207–25 [DOI] [PubMed] [Google Scholar]
- 42.Candolfi M, Curtin JF, Yagiz K, Assi H, Wibowo MK, Alzadeh GE, et al. B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia 2011;13:947–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han S, Yang Y. Phenotypic and functional dissection of myeloid-derived suppressor cells. Applied Biological Chemistry 2016;59:367–71 [Google Scholar]
- 44.Ciccolini J, Serdjebi C, Peters GJ, Giovannetti E. Pharmacokinetics and pharmacogenetics of Gemcitabine as a mainstay in adult and pediatric oncology: an EORTC-PAMM perspective. Cancer Chemother Pharmacol 2016;78:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahlberg R, Lorenzen S, Thuss-Patience P, Heinemann V, Pfeiffer P, Mohler M. New Perspectives in the Treatment of Advanced Gastric Cancer: S-1 as a Novel Oral 5-FU Therapy in Combination with Cisplatin. Chemotherapy 2017;62:62–70 [DOI] [PubMed] [Google Scholar]
- 46.Offidani M, Mele A, Corvatta L, Marconi M, Malerba L, Olivieri A, et al. Gemcitabine alone or combined with cisplatin in relapsed or refractory multiple myeloma. Leuk Lymphoma 2002;43:1273–9 [DOI] [PubMed] [Google Scholar]
- 47.Zheng H, Yang F. Gemcitabine in Treating Patients with Refractory or Relapsed Multiple Myeloma. Asian Pacific Journal of Cancer Prevention 2014;15:9291–3 [DOI] [PubMed] [Google Scholar]
- 48.Valdez BC, Wang G, Murray D, Nieto Y, Li Y, Shah J, et al. Mechanistic studies on the synergistic cytotoxicity of the nucleoside analogs gemcitabine and clofarabine in multiple myeloma: relevance of p53 and its clinical implications. Exp Hematol 2013;41:719–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 2016;7:27–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.