

Abstract
Pancreatic ductal adenocarcinoma (PDA) is an aggressive malignancy characterized by paucity of tumor-proximal CD8+ T cells and resistance to immunotherapeutic interventions. Cancer-associated mechanisms that elicit CD8+ T-cell exclusion and resistance to immunotherapy are not well known. Here, using a Kras- and p53-driven model of PDA, we describe a mechanism of action for the pro-tumorigenic cytokine IL35 through STAT3 activation in CD8+ T cells. Distinct from its action on CD4+ T cells, IL35 signaling in gp130+CD8+ T cells activated the transcription factor STAT3, which antagonized intratumoral infiltration and effector function of CD8+ T cells via suppression of CXCR3, CCR5, and IFNγ expression. Inhibition of STAT3 signaling in tumor-educated CD8+ T cells improved PDA growth control upon adoptive transfer to tumor-bearing mice. We showed that activation of STAT3 in CD8+ T cells was driven by B cell– but not Treg-specific production of IL35. We also demonstrated that B cell–specific deletion of IL35 facilitated CD8+ T-cell activation independently of effector or regulatory CD4+ T cells and was sufficient to phenocopy therapeutic anti-IL35 blockade in overcoming resistance to anti–PD-1 immunotherapy. Finally, we identified a circulating IL35+ B-cell subset in patients with PDA and demonstrated that presence of IL35+ cells predicted increased occurrence of phosphorylated (p)Stat3+CXCR3–CD8+ T cells in tumors and inversely correlated with a cytotoxic T-cell signature in patients. Together, these data identified B cell–mediated IL35/gp130/STAT3 signaling as an important direct link to CD8+ T-cell exclusion and immunotherapy resistance in PDA.
Keywords: pancreatic cancer, regulatory B cells, IL35, T-cell exclusion
INTRODUCTION
Infiltration of cytotoxic T cells into the tumor parenchyma correlates with better outcomes in a variety of tumor types, especially in the context of immunotherapy (1). Thus, better understanding of the mechanisms that modulate T-cell trafficking and function in tumors is necessary in order to overcome inefficient immune responses. This is particularly relevant in pancreatic ductal adenocarcinoma (PDA), an aggressive and deadly disease often characterized by lack of infiltration and/or dampened functionality of CD8+ T cells (2,3). In the setting of PDA, immunotherapy has been unsuccessful (4,5). The mechanisms that restrict tumor-directed CD8+ T-cell function in PDA are thought to be linked to immunosuppression (6–8). Significant research efforts have described a variety of tumor cell-intrinsic or -extrinsic mechanisms that may act to restrict CD8+ T-cell activity in the PDA tumor microenvironment (TME), among which are myeloid cell recruitment and polarization, expansion of regulatory and ɣδ T cells, as well as modulation of T-cell infiltration by tumor cells themselves (9–17). These studies suggest that reversing immune suppression in pancreatic cancer could improve endogenous T-cell activity. Although it is becoming clear that contribution from both tumor cell-intrinsic and -extrinsic mechanisms may dictate the type of immunosuppression present in the TME, the mechanisms that these various immunosuppressive arms utilize in order to directly control T-cell function in PDA remain poorly defined.
We previously demonstrated that the cytokine IL35 promotes pancreatic tumorigenesis (18, 19). IL35 is a member of the IL12 family of cytokines, forms via heterodimerization of p35 and Ebi3, and is thought to signal in naïve CD4+ T cells through the IL35 receptor (IL35R, consisting of IL12Rβ2 and gp130 subunits). This activates STAT1 and STAT4 signaling pathways (20). Elevated IL35 can be detected in lymphoma cells and lung cancer, and predicts poor outcome in cases of leukemia, colorectal, and pancreatic cancer (20). Regulatory T cells (Tregs), naïve CD4+ T cells (iTr35), dendritic cells (DCs), and B cells are known to produce IL35 (11,19,21–25), whose expression has been linked to its ability to modulate immune responses via diverse mechanisms (20). IL35 expression in Tregs promotes CD8+ T-cell exhaustion in melanoma and colorectal cancer (21) and has also been shown to facilitate tumor growth via accumulation of myeloid-derived suppressor cells (MDSCs) and induction of angiogenesis (26). IL35 expression in B cells also dampens T-cell activity independently of Tregs in experimental autoimmune encephalomyelitis by suppressing macrophages and inflammatory CD4+ T cells (24). Because of these effects, it seems likely IL35 may impact functionality of diverse immune cell populations (20). We previously showed that IL35 exerts immunosuppressive effects in PDA and is primarily expressed by two immune cell populations, regulatory T and B cells (19). However, the mechanisms underlying IL35 function in PDA remain unclear, and are addressed in the current study.
MATERIAL AND METHODS
Mice
All mouse protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the UNC-CH. Animals were maintained in a specific pathogen-free facility. Six- to seven week-old wild-type CD45.2+ (WT) C57Bl/6J mice were purchased from The Charles River Laboratories (#027). The KrasLSL-G12D/+;Trp53LSL-R172H/+ and p48Cre/+ (KPC) mice have been previously described (27). Six- to seven week-old B6 CD45.1+ (B6.SJLPtprca.Ptprcb/BoyJ) mice were purchased from Jackson laboratories (#002014). Ebi3Tom.L/L and Foxp3Cre-YFP;Ebi3Tom.L/L mice were obtained from D. Vignali (University of Pittsburgh)(21, 28–31). CD19CreEbi3L/L mice were generated by crossing CD19Cre mice (32) to Ebi3Tom.L/L mice in our colony for two generations to obtain homozygocity at Ebi3 locus. Resulting mice lacked expression of IL35 in either B cells (BEbi3–/–) or Tregs (TregEbi3–/–). Littermates were used as controls.
BWT and Bp35–/– mice were obtained by a mixed bone marrow chimera method using lethally irradiated (1000 cGy radiation delivered from cesium source) C57BL/6J mice as recipients (24)(Supplementary Table S1). Bp35−/− or control BWT mice were obtained by reconstituting recipient mice with a mixture of bone marrow cells from B cell–deficient μMT mice (Jackson labs, #002288) or WT C57BL/6J mice (80%), respectively, and p35–/– mice (20%; Jackson labs, #002692). 10×106 bone marrow cells were injected intravenously into the WT recipients irradiated at 1000 cGy. The chimeras were used after 8 weeks and specific deletion of p35 in B cells was confirmed by PCR. Reconstitution was confirmed using flow cytometry for major immune subtypes.
Validation of cell type–specific knockouts
To validate that the deletion of p35 or Ebi3 genes was specific to the B-cell lineage, splenic CD19+ B cells, CD11b+ myeloid cells, and CD4+ T cells were isolated by FACS from Bp35−/−, BWT, BEbi3+/− and BEbi3−/− mice (please see “Lymphocyte isolation” section). To verify specific deletion of Ebi3 gene from Tregs, Foxp3+ (YFP+) Tregs, Foxp3– (YFP–) conventional T cells (Tcon), and CD19+ B cells were purified from TregEbi3+/– and TregEbi3–/– mice by FACS. All sorted populations and remaining non-T, -B, or -myeloid cells were lysed and genomic DNA was extracted using the DNA easy kit (Qiagen). PCR was used to check the presence of wild type p35 or Ebi3 allele in the immune cells (Primer sequences are listed in Supplementary Table S3).
Cell lines
The murine PDA cell line KPC 4662 was derived from a primary pancreatic tumor of C57Bl/6J KPC mice by Dr. Vonderheide’s lab (33). GFP-labeled KPC cells were generated as previously described (18). Cells were maintained at 37°C and 5% CO2 in complete DMEM (#11995–065, Gibco, 10% FCS and 1X Penstrep #15140–122, Gibco) and were confirmed to be mycoplasma and endotoxin free. The cells were used at <16 passages. Cells were confirmed to contain Kras, Cre, and p53 mutant alleles/transgene by genotyping.
Tumor growth and antibody blocking experiments
For intrapancreatic injection of cancer cells, mice were anesthetized using a ketamine (100 mg/kg)/xylazine (10 mg/kg; Med-Vet International) cocktail. The depth of anesthesia was confirmed by verifying an absence of response to toe pinch. An incision in the left flank was made, and 75,000 KPC cells in ice-cold PBS mixed at 1:1 dilution with Matrigel (#354234, Corning) in a volume of 50 μL were injected using a 28-gauge needle into a tail of the pancreas. The wound was closed in two layers, with running 5–0 Vicryl RAPIDE sutures (Ethicon) for the body wall, and 5–0 PROLENE sutures (Ethicon) for the skin. All animals were given the pain reliever buprenorphine (0.1 mg/kg; Med-Vet International) once subcutaneously after orthotopic surgery. For therapeutic experiments, mice received antibody treatment using anti-IL35 (V1.4C4.22) at 200 μg/week for 3 weeks, anti-IL27 (MM27–7B1) at 200 μg/week for 3 weeks, and/or anti–PD-1 (RMP1–14, BioXcell) at 200 μg/injection on days 7, 9, and 11, or their respective IgG isotype controls once an orthotopic tumor reached 4–5 mm (day 7)(Supplementary Table S2). Tumor growth was monitored by ultrasound, as described below. Three doses of antibody were given in total, on days 7, 9, and 11 after injection of KPC cells.
Adoptive transfer of CD8+ T cells
Spleens were harvested from orthotopically injected CD45.2+ mice after 3 weeks post KPC 4662 cell injections. CD8+ T cells were sorted from the spleens after red blood cell lysis using a BD FACS-ARIA III sorter, and purity of CD8+ T cells was >98%. Sorted CD8+ T cells were treated with plate-bound anti-CD3 (1 μg/mL), soluble anti-CD28 (2 μg/mL) as control or plate-bound anti-CD3 (1 μg/mL), soluble anti-CD28 (2 μg/mL), and STA-21 (20 μM) to inactivate pSTAT3 in CD8+ T cells for 48 hours. After 48 hours, 10×106 STA-21– or control-treated CD8+ T cells were adoptively transferred via tail vein injection into CD45.1+ mice. One day after adoptive transfer, 75,000 KPC 4662 cells were orthotopically transplanted into the pancreas of CD45.1+ mice. The recipient mice were sacrificed 21 days post-tumor cell injections, tumor size and weight were measured, and spleens and tumors were collected for further processing and analysis.
Depletion of CD8+ T cells, CD4+ T cells, and Tregs in vivo
For CD4+ and CD8+ T-cell depletion studies, 200 μg of anti-CD4 (Bio X cell, BP0003–1, clone GK1.4) and 200 μg of anti-CD8 (Bio X Cell, BP0004–1, clone 53–6.7) or an IgG isotype control rat IgG2b, κ and rat IgG2a, κ (Bio X Cell), respectively, were administered intraperitoneally daily starting 3 days prior to tumor cell injection and twice a week after tumor cell injection. Tregs were depleted using 500 μg of anti-CD25 clone PC 61.5.3 (BioXcell, BE0012), injected i.p. on day 3 and day 1 before pancreatic tumor implantation and repeated every 5 days for the duration of the experiment. Control mice received rat IgG1λ (BioXcell) isotype control. Mice were sacrificed 21 days after tumor implantation, tumor size and weight were measured, and spleen and tumor samples were collected for further processing. Depletion of cells was confirmed by flow cytometry at the end of the experiment.
Ultrasound imaging
Monitoring of orthotopic pancreatic tumors was preformed weekly on the Vevo 2100 Imaging System (VisualSonics Inc.) using the MS550D transducer. Mice were anaesthetized using isoflurane (2%) throughout the procedure. Hair was removed from left flank of each mouse by electric razor and hair removal cream (Nair, Church & Dwight). Images were taken at 11 mm image depth, and measurements were calculated using the Vevo LAB software (VisualSonics Inc.).
Lymphocyte isolation
Single-cell suspensions were prepared from tumors and spleens isolated from orthotopic and/or adoptive transfer models. Spleens were mechanically disrupted using the plunger end of a 5 mL syringe and resuspended in 1% FBS/PBS. Spleen samples were processed following RBC lysis (eBioscience; 00–4333-57). For isolation of tumor-infiltrating lymphocytes, tumor tissue was minced into 1–2 mm pieces and digested with collagenase IV (1.25 mg/mL; #LS004188, Worthington), 0.1% soybean trypsin inhibitor (#T9128, Sigma), hyaluronidase (1 mg/mL; #LS002592, Worthington), and DNase I (100 mg/mL; #LS002007, Worthington) in complete DMEM for 30 minutes at 37°C. Cell suspensions were passed through a 70-μm cell strainer (Falcon) and resuspended in RPMI media (Gibco). Lymphocytes were isolated from processed tumor tissues by OptiPrep (Sigma) density gradient centrifugation. MACS isolation of total CD45+ leukocytes (MACS Miltenyi #130–052-301) was performed on the leukocyte-enriched fraction according to Miltenyi’s protocol, and the purity was >90%. Cells were stained with fluorophore-labelled antibodies (Supplementary Table S2) for 30 minutes on ice in FACS buffer (PBS with 3% FCS and 0.05% sodium azide). After staining, cells were washed twice with FACS buffer and resuspended in sorting buffer (PBS with 1% FCS and 0.05% sodium azide). Cell sorting using a BD FACS ARIA III sorter was performed to isolate CD19+CD21hiCD5+CD1d+ regulatory B cells (Bregs), CD19+CD21loCD5-CD1d- conventional B cells (Bcon), CD4+YFP+ Tregs, and CD8+ T cells. Cells were collected in complete RPMI media containing 10% FCS with 1X Penstrep (#15140–122, Gibco) antibiotics. >97% purity was achieved.
B-cell and T-cell in vitro cultures
For in vitro CD8+ T-cell culture, splenic CD8+ T cells specific for the OVA257–264 (InvivoGen) antigen were sorted (>98% purity) from WT mice immunized with OVA257–264 for 1 week (10 μg/mouse). T cells were cultured with plate bound anti-CD3 (1 μg/mL, BioXCell) and soluble anti-CD28 (2 μg/mL, BioXCell), with the addition of recombinant IL35 (rIL35, 50 ng/mL)(Chimerigen Laboratories; CHI-MF-11135) and OVA257–264 (2 μg/mL) for 48 hours. For blocking STAT3 or STAT4 activity, cells were cultured with 50 μM fludarabine (Selleckchem), 20 μM STA-21, or 100 μM lisofylline (Santa Cruz Biotechnology) for 48 hours (Supplementary Table S2 for list of antibodies and reagents).
Mouse splenic Bregs (CD19+CD21hiCD5+CD1d+) were sorted by flow cytometry from spleens of tumor bearing WT, p35–/–, and Ebi3–/– mice (>97% purity), as described above. 100,000 Bregs or Bcon cells and 100,000 CD8+ T cells (1:1 ratio) were cocultured in the 96-well Transwell plates, with B cells occupying the top chamber and CD8+ T cells the bottom chamber (Corning; 3381) for 48 hours. B cells were activated by anti-CD40 (1 μg/mL, eBioscience) and LPS (2 μg/mL, Sigma) for 48 hours, and CD8+ T cells were activated by plate bound anti-CD3 (1 μg/mL) and soluble anti-CD28 (2 μg/mL) with OVA257–264 (2 μg/mL). Cytokine secretion of CD8+ T cells was evaluated by flow cytometry, as described below.
Intracellular cytokine and transcription factor staining
For ex vivo stimulation, sorted cells from tumors or spleens of orthotopic and/or adoptive transfer models (except for B cells, which were cultured in LPS and anti-CD40 prior to this step) were incubated with PMA (50 ng/mL; Sigma, #P8139) and ionomycin (200 ng/mL; Sigma, #I0634) in the presence of Golgistop Brefeldin A (1X, Biolegend) in complete RPMI medium for 5 hours at 37°C. Cells were washed and blocked with anti-CD16/CD32 (Fc Block, BD Biosciences, 0.1 mg per 100,000 cells) for 5 minutes on ice. Viability was assessed using the Live/Dead 7AAD (Biolegend; 420404) stain solution or Live/Dead Aqua cell stain kit (Life technologies). Cells were then washed and stained with labeled antibodies against surface markers on ice for 30 minutes in FACS buffer (PBS with 3% FCS and 0.05% sodium azide). After surface staining, cells were washed, fixed and permeabilized using cytofix/cytoperm buffer (BD, 554714) for 15 minutes at 4°C in the dark. Intracellular staining was performed using fluorophore-conjugated cytokine antibodies for 1 hour at 4°C in the dark. After intracellular staining, cells were washed and resuspended in FACS buffer for acquisition by flow cytometry. Intracellular staining for Foxp3 was performed using a Foxp3 staining kit (eBioscience, cat. No. 00–5523). For staining of phosphoproteins, cells were fixed with fixation buffer (Biolegend; 420801) at room temperature for 10 minutes and permeabilized with true-phos perm buffer (Biolegend; 425401) at –20°C overnight. Cells were then washed twice and resuspended in cell staining buffer (Biolegend; 420201). Fluorophore-conjugated phosphoprotein cocktail antibodies or isotype controls were added and incubated for 60 minutes at 4°C. After incubation, cells were washed, resuspended in FACS buffer, and samples were acquired on LSR II and LSRII Fortessa (BD Bioscience) and analyzed with FlowJo version 10.2 (TreeStar, Inc.). The human PBMC samples were processed as described above, but the blocking step was done with human BD Fc Block (BD Biosciences, 564219, 0.1 mg per 100,000 cells) for 5 minutes on ice. All antibodies and reagents listed in Supplementary Table S2.
Enzyme-linked immunosorbent assay (ELISA)
Peripheral blood samples were collected from mice using the retro-orbital method, and serum samples were prepared by centrifuging samples at 1000 rpm for 5 minutes at 4oC. All serum samples were stored at –80°C. 50μL of serum sample were used to detect circulating IL35. The concentration of serum IL35 was measured by using the Legend Max mouse IL35 heterodimer ELISA kit (Biolegend, USA, IL35 pre-coated ELISA kit, p35, EBI-3), and IL35 concentration was determined based on the recombinant IL35 standard that came with the kit. Absorbance were read at 450 nm within 15 minutes using Perkin Elmer Enspire multimode reader. All samples were assayed in triplicates.
Human samples
The study was carried out in accordance with the UNC Chapel Hill and Washington University School of Medicine guidelines and was approved by IRB ethics committees. Informed consent was obtained from the patients and healthy donors before blood donation. The study conducted in accordance to ethical standards such as Declaration of Helsinki.
PBMC isolation and B-cell enrichment:
Blood samples were collected from 30 treatment-naïve patients with pancreatic cancer and 30 healthy controls. PBMCs were obtained by separating the whole blood samples via density gradient centrifugation (Lymphoprep, Axis-shield). Briefly, blood samples were collected in yellow top ACD tubes (BD Vacutainer, 364606)(~20 mL each). Blood was diluted by addition of an equal volume of 0.9% NaCl. About 20 mL of diluted blood was layered on top of 10 mL Lymphoprep and centrifuged at 800 x g for 20 minutes at 18°C. The mononuclear cells were removed from the sample/medium interface using a Pasteur pipette. Cells were collected by centrifugation at 250 x g for 5 minutes and were washed three times with 1XRPMI-1640. The isolated PBMCs were then stained with anti-human CD19 (HIB19; Biolegend), CD24 (ML5; Biolegend), and CD38 (HB-7; Biolegend) in FACS buffer for 20 minutes on ice. CD19+CD24hiCD38+ Bregs and CD19+CD24loCD38– Bcon cells were sorted using a BD FACS ARIAIII, and cells were collected in complete RPMI media. >97% cell purity was achieved.
PDA patient samples
For the purposes of analyzing CD8+ T-cell and IL35+ immune cell infiltration, we examined 11 samples containing PDA lesions. Samples consisted of 5 μm sections that were cut from FFPE blocks provided by the Tissue Pathology Laboratory (TPL) of the UNC School of Medicine and were obtained after informed written consent. This study was conducted in accordance with the Declaration of Helsinki. All samples were anonymized prior to being transferred to the investigator’s laboratory and therefore met exempt human subject research criteria. Samples were selected on the basis of patient diagnosis with PDA.
Quantitative PCR (qPCR) analysis for gene expression
RNA was prepared from sorted CD19+CD24hiCD38+ Bregs and CD19+CD24loCD38- Bcon cells from human PBMCs using the RNeasy micro kit (Qiagen). CDNA was generated using high capacity cDNA-RT kit (Invitrogen). QPCR analysis (with 100 ng DNA template) were performed using the SSO advanced universal SYBR green super-mix reagent (Bio-Rad) and Applied Bio-System (ABS) platform. Results were normalized to the expression of β-actin, and each sample was run in triplicate. Gene expression was determined by the delta-delta Ct method (2–∆∆Ct). Primer sequences are listed in Supplementary Table S3.
Immunohistochemistry and immunofluorescence
Mouse splenic and tumor tissues for all models and treatments analyzed were fixed in 10% buffered formalin (Fisher Scientific) for 48 hours and embedded in paraffin. Six-micrometer sections were deparaffinized and rehydrated. A solution of 1% hydrogen peroxide (stock of 30% hydrogen peroxide, Sigma) in methanol at room temperature for 10 minutes was used to quench endogenous peroxidase activity. Antigen retrieval was done in a 10 mmol/L sodium citrate plus 0.05% Tween-20 solution (pH 6.1) for 15 minutes in a microwave oven. Blocking was performed for 1 hour at room temperature in a solution of 10% goat serum (Vector Laboratories), 10 mmol/L Tris–HCl, 0.1 mol/L magnesium chloride, 1% BSA, and 0.5% Tween-20. Sections were incubated with primary rat anti-CD8α (clone 53–6.7, BD Pharmingen) diluted in 2% BSA/PBS (final concentration of 2.5 mg/mL) overnight at 4°C. Secondary biotinylated goat anti-rat (Vector Laboratories) was diluted in 2% BSA/PBS (final concentration of 3.75 mg/mL) and incubated for 1 hour at room temperature. Tertiary ABC solution was prepared according to the manufacturer’s instructions (Vectastain ABC kit, Vector Laboratories) and incubated for 45 minutes at room temperature. Sections were developed using a 3,30-diaminobenzidine tetrahydrochloride kit (DAB peroxidase substrate kit, Vector Laboratories). Slides were then counterstained with Harris hematoxylin (Sigma), dehydrated, and mounted with DPX mounting media (Sigma). Images were acquired using Nikon Eclipse Ni-U microscope, with NIS-Elements software (Nikon).
For immunofluorescence, mouse pancreata were fixed, rehydrated, and subjected to antigen retrieval as described above. Blocking was done for 1 hour (10mM Tris-HCL, 0.1M MgCl2, 0.5% Tween-20, 1% BSA, 10% chicken serum; Vector Laboratories), primary antibodies were applied overnight at 4oC. Anti-mouse antibodies included phospho-Histone 3 (Millipore, 06–570) and CK8 (DSHB, Troma-I). Secondary antibodies were used at 1:200 for 1 hour at room temperature followed by incubation with 1 μg/ml DAPI for 10 minutes. Slides were washed and mounted with ProLong Gold anti-fade mounting medium (Invitrogen), and images were acquired with an Olympus BX61.
For the purposes of analyzing status of pSTAT3 and CXCR3 expression on CD8+ T cells, 11 human PDA lesion samples were analyzed. Samples consisted of 5 μm sections that were cut from FFPE blocks provided by the Tissue Pathology Laboratory (TPL) of the UNC School of Medicine. All samples were anonymized prior to being processed. Triple IF (3-plex IF) stains were carried out in the Leica Bond-Rx fully automated staining platform (Leica Biosystems Inc., Norwell, MA). Slides were dewaxed in Bond™ Dewax solution (AR9222) and hydrated in Bond Wash solution (AR9590) (all from Leica Biosystems). Epitope retrieval for all targets was done for 20 minutes in Bond-epitope retrieval solution 1 pH6.0 (AR9661) or solution 2 pH9.0 (AR9640) (both from Leica Biosystems). The epitope retrieval was followed with 10 minutes of endogenous peroxidase blocking using Bond peroxide blocking solution (DS9800) (Leica Biosystems). Positive and negative controls (no primary antibody) and single-stain controls were done for 3-plex IF where one primary antibody was omitted to make sure that cross reactivity between the antibodies did not occur. The following antibodies were used: CD8 (4B11, Leica). CD8+ T cells were counted per 10× field-of-view (FOV), counting 3–6 FOV per tumor sample. Stained slides were counterstained with Hoechst 33258 (# H3569) and mounted with ProLong® Diamond Antifade Mountant (#P36961; Life Technologies (Carlsbad, CA). We previously estimated the average value of Ebi3+ cells/FOV across all tumor samples as 20 Ebi3+ cell/FOV. FOVs with number of Ebi3+ cells <20 were classified as Ebi3low and FOVs with number of Ebi3+ cells ≥20 were classified as Ebi3hi, as reported in Mirlekar, et al. (19).
Analysis of Ebi3, p35 and cytokeratin immunofluorescence in human PDA
Slides containing fluorescently labeled tissue sections were scanned in the Aperio ScanScope FL (Leica Biosystems) using the 20X objective and images were archived in TPL’s eSlide Manger database (Leica Biosystems). Images were manually annotated to demarcate tumor (all images) and lymphoid regions (present on three images) using Aperio ImageScope (v12.4.2.700). To reduce operator bias, annotations in lymphoid regions were made using the Hoechst nuclear counterstain layer (EBI3 and IL12a image layers were not visible). Annotated images were then analyzed using Tissue Studio software (Tissue Studio version 2.7 with Tissue Studio Library version 4.4.2; Definiens Inc., Carlsbad CA). For analysis of tumors, regions positive for cytokeratin were digitally separated from the surrounding stroma (Tissue Studio Composer software) followed by cellular analysis for co-expression of EBI3, IL12a and cytokeratin. In lymphoid areas, cellular analysis for co-expression of EBI3 and IL12a was performed on all cells within the annotation regions. Analysis data included the percentage of co-expressing cells and the average cellular fluorescent intensities for each antibody marker.
Generation and analysis of RNA-seq data from human B cells
An RNA-sequencing library preparation for human PBMC B-cell populations derived from three healthy volunteers and three treatment-naïve PDA patients was generated. PDA samples were pre-selected based on positivity for p35 and Ebi3 expression, as assessed by QPCR.Samples were sorted for human Bregs and Bcon markers (12 samples total), as described above, and template RNA was derived from freshly sorted single-cell suspensions using the Illumina TruSeq stranded library preparation protocol. Sequencing was done on the Illumina HiSeq4000 platform using 150bp paired-end chemistry and targeting 9×107 reads per sample. TCGA expression matrices were accessed at http://firebrowse.org. FASTQ files were aligned to the human reference genome using STAR v2.4.2. The BAM output files were then quantified using Salmon v0.8.2. FastQC v0.11.7 and MultiQC v1.5 were used to generate quality assurance reports. Statistical analyses were executed in R v3.3.3. Differential gene expression analysis was conducted on the resulting expression matrices using the DESeq2 R package. Genes that were found to be differentially upregulated in tumor-associated Breg subtypes compared to Bcon subtypes, with a Benjamin Hochberg corrected p-value of less than 0.1, were identified. A tumor-infiltrating Breg signature was calculated by taking the geometric mean of the expression values of the identified genes. The Breg signature was then calculated for TCGA pancreatic cancer tumors. Gene signatures for CD8+ T cells and cytotoxicity were calculated by taking the geometric mean of the CD8 T cell gene signature genes (as per (34)) and the genes PRF1 and GZMA as per Rooney et al (35), respectively. A scatterplot was generated from the tumor-infiltrating Breg signature and the ratio of the CD8+ T-cell to cytotoxicity signature using the ggplot2 R package. These gene signatures were calculated across tumor types in TCGA. Correlation coefficients were generated for each tumor type, and a volcano plot depicting the results was created using the ggplot2 R package. Gene Set Enrichment Analysis (GSEA v3.0) was used comparing different subtypes for genesets, C2 and C7, from the Molecular Signatures Database (MSigDB v6.2). B-cell RNA-sequencing data will be submitted to the NIH Gene Expression Omnibus (GEO) repository upon acceptance of the manuscript for publication, the link will be live at the time of publication.
Statistical analysis
At least 9–21 mice were used in each group, with a minimum of 6 mice in each group per experiment, and the experiments were repeated a minimum 3 times to validate reproducibility. Before analysis, data were examined for quality. Group means were compared with Student’s t tests. Significance in variations between two groups was determined by unpaired student t test (two-tailed). Statistical analysis was performed using GraphPad Prism software. Data are presented as mean±S.E.M. p<0.05 was considered statistically significant.
RESULTS
IL35 suppresses chemotactic receptor expression and effector function in CD8+ T cells
IL35-mediated tumor growth is associated with changes in T-effector and regulatory T-cell (Treg) subsets (19). To understand the cellular basis of IL35 immunosuppression, we depleted CD4+ T cells or CD25+ Tregs in p35–/– mice orthotopically injected with primary syngeneic KrasG12D;Trp53R172H;p48Cre/+ (KPC) mouse pancreatic cancer cells (Supplementary Fig. S1A-D)(19). Neither one of these depletion strategies rescued tumor growth, whereas, as previously shown, depletion of CD8+ T cells fully restored tumor growth, prompting us to ask whether IL35 may directly signal to CD8+ T cells (Supplementary Fig. S1A-D)(19). IL35 has been shown to signal through the STAT1/STAT4 heterodimer in CD4+ T cells. However, the mechanism of IL35 signaling in CD8+ T cells is not known (20). We found that recombinant IL35 elicited an increase in phosphorylation of STAT1, STAT3, and STAT4 in CD8+ T cells, raising the possibility that these pathways could regulate IL35-dependent CD8+ T-cell function (Fig. 1A). Because one of the features of IL35-loss is increased infiltration and activation of CD8+ T cells in the tumor parenchyma (19), we asked whether IL35 could directly affect the expression of chemokine receptors and/or IFNγ production by cytotoxic T lymphocytes by examining the expression of CXCR3 and CCR5, which have been implicated as important facilitators of intratumoral CD8+ T-cell infiltration (17,36). We found that incubation of activated OVA-specific CD8+ T cells with recombinant IL35 resulted in significantly attenuated expression of IFNγ and both CXCR3 and CCR5, consistent with the observation that p35 expression correlates with decreased CXCR3 in autoimmunity (Fig. 1B)(37).
Figure 1. IL35 mediates activation of STAT3, suppression of IFNɣ, and chemotactic receptors CXCR3 and CCR5 in gp130+CD8+ T cells.
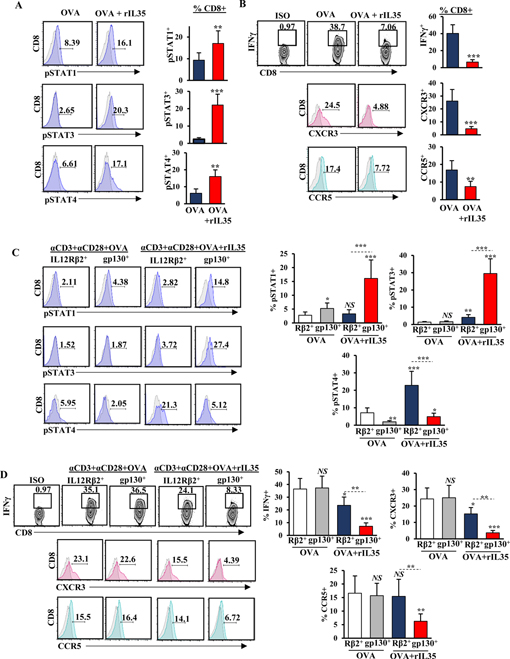
(A) Representative flow cytometry histograms (left) and quantification (right) of phospho(p)-STAT1, pSTAT3, and pSTAT4 in CD3+CD8+ T cells, activated with anti-CD3 (1 μg/mL) and anti-CD28 (2 μg/mL) in the presence of OVA (2 μg/mL) with and without recombinant (r)IL35 (50 ng/mL). Proportion of CD8+ T cells is indicated. (B) Representative flow cytometry plots and histograms (left) and quantification (right) of IFNɣ production and expression of CXCR3 and CCR5 in CD3+CD8+ T cells activated as in (A). (C) Representative flow cytometry histograms (left) and quantification (right) of pSTAT1, pSTAT3, and pSTAT in CD3+CD8+ T cells sorted for IL12Rβ2+ or gp130+ subsets (single-positive) activated as indicated in (A). Proportion of CD8+ T cells is shown. (D) Representative flow cytometry plots and histograms (left) and quantification (right) of IFNɣ production and expression of CXCR3 and CCR5 in CD3+CD8+ T cells sorted for IL12Rβ2+ or gp130+ subsets (single-positive) activated as indicated in (A). Proportion of CD8+ T cells is shown. Error bars indicate S.E.M.; p-values were calculated using Student’s t-test (unpaired, two-tailed); NS: not significant, *p<0.05; **p<0.01; ***p<0.001. Experiment were conducted using 6–8–week-old C57B6 mice with 6 mice per group in triplicate. Data represents 3 independent experiments.
In CD4+ T cells, IL35 mediates function via receptor chains IL12Rβ2 and gp130, which signal to activate STAT1 and STAT4 but not STAT3 (38). To understand how IL35R contributes to STAT3 activation in CD8+ T cells, we sorted T cells based on singular expression of either IL12Rβ2 or gp130 (Supplementary Fig. S2A). We found that when treated with IL35, IL12Rβ2+CD8+ T (subset-1) cells activated STAT4, and the gp130+ subset (subset-2) activated both STAT1 and STAT3 (Fig. 1C, Supplementary Fig. S2A). Consistent with this observation, treatment with rIL35 activated all three STATs in IL12Rβ2+gp130+CD8+ T cells (Supplementary Fig. S2B-C). Modulation in IFNγ, CXCR3, and CCR5 correlated with expression of the gp130+ subunit, although IL12Rβ2+CD8+ T cells somewhat downregulated IFNγ and CXCR3 but not CCR5 (Fig. 1D). Thus, IL35 signaling in gp130+CD8+ T cells impeded expression of CXCR3, CCR5, and IFNγ.
STAT3 inhibition in tumor-educated CD8+ T cells reduces pancreatic tumor growth
To understand which STAT signaling pathway was responsible for regulating IFNγ and/or chemokine receptors on CD8+ T cells, we used inhibitors of pSTAT1, pSTAT3, and pSTAT4 (fludarabine (Flu), STA-21, or lisofylline (Liso), respectively)(39–41) in vitro on antigen-directed CD8+ T cells in the presence or absence of rIL35 (Fig. 2A, Supplementary Fig. S3A-D). Phospho-STAT3 was not affected in antigen-stimulated CD8+ T cells, and treatment with STA-21 did not affect IFNγ production or CXCR3/CCR5 expression (Fig. 2A). However, reduction of IFNγ, as well as CXCR3 and CCR5 caused by treatment with OVA+rIL35 was rescued by addition of STA-21 (Fig. 2A-B). STAT1 and STAT4 activation was needed for sustaining IFNγ expression following antigenic stimulation of CD8+ T cells, likely reflecting signaling by antigen-presenting cells via IL12 (41)(Supplementary Fig. S3A-D). However, neither STAT1 nor STAT4 inhibition in activated CD8+ T cells treated with recombinant IL35 was sufficient to rescue IFNγ production and chemokine receptor expression (Supplementary Fig. S3A-D). Overall, these data suggest that IL35-mediated activation of STAT3 in CD8+ T cells impeded multiple axes required for their functionality.
Figure 2. STAT3 activation in CD8+ T cells is suppressive in vitro and in vivo.

(A) Representative flow cytometry plots and histograms of IFNɣ production and expression of CXCR3, CCR5, phospho(p)-STAT1, pSTAT3, and pSTAT4 in CD3+CD8+ T cells stimulated with anti-CD3 (1 μg/mL) and anti-CD28 (2 μg/mL) in the presence of OVA (2 μg/mL), and rIL35 (50 ng/mL) and/or STA-21 (20μM) inhibitor. Proportion of CD8+ T cells is indicated. (B) Quantification of IFNɣ production and expression of CXCR3, CCR5, pSTAT1, pSTAT3, and pSTAT4 in CD3+CD8+ T cells from (A). (C) Experimental schema used to test the role of Stat3 activation in CD8+ T cells. (D) Quantification of tumor weight from CD45.1+ mice 3 weeks post-orthotopic adoptive transfer of pre-treated with anti-CD3/CD28 with and without STA-21 CD45.2+CD8+ T cells and injection with KPC cells (n=6 mice/group). (E) Numbers (no.) of adoptively transferred CD45.2+ (left) and endogenous CD45.1+ (right) tumor-infiltrating CD3+CD8+ T cells 3 weeks post-orthotopic adoptive transfer to the CD45.1+ recipients indicated in (D). (F) Representative flow cytometry histograms (left) and quantification (right) of pSTAT1, pSTAT3, and pSTAT4 in intratumoral CD45.2+CD8+ T cells 3 weeks post-orthotopic adoptive transfer to CD45.1+ recipients indicated in (D). (G) Representative flow cytometry plots and histograms (left) and quantification (right) of IFNɣ production and expression of CXCR3 and CCR5 in intratumoral CD45.2+CD8+ T cells 3 weeks post-orthotopic adoptive transfer to CD45.1+ recipients indicated in (D). Error bars indicate S.E.M.; p-values were calculated using Student’s t-test (unpaired, two-tailed); NS: not significant, **p<0.01; ***p<0.001. Data represents 3 independent experiments.
We reasoned that if IL35-driven activation of STAT3 in CD8+ T cells was an important factor driving CD8+ T cell exclusion and inactivity in PDA, then inhibition of STAT3 activation in tumor-educated immunosuppressed CD8+ T cells could confer antitumor responses. To test this, we isolated splenic CD8+ T cells from WT CD45.2+ mice harboring KrasG12D;Trp53R172H;p48Cre/+ (KPC) orthotopic tumors, and treated T cells with either control or STA-21 ex vivo (Fig. 2C). Control or inhibitor-treated T cells were then adoptively transferred to WT CD45.1+ mice, followed by orthotropic injection of KPC tumor cells. We observed significantly reduced tumor growth in mice that received STA-21–pre-treated CD8+ T cells (Fig. 2D). This was accompanied by increased infiltration of CD45.2+CXCR3+CCR5+CD8+ T cells and significant increases in IFNγ production but no differences in infiltration by endogenous CD45.1+CD8+ T cells (Fig. 2E-G). Lethally irradiated mice adoptively infused with STA-21–treated CD8+ T cells were also able to better control tumor growth (Supplementary Fig. S4). These experiments suggested that activation of STAT3 in CD8+ T cells may regulate their infiltration and antitumor activity in vivo.
IL35 production by Tregs is dispensable for pancreatic tumor growth
Both Bregs and Tregs can produce IL35 in a variety of disease settings, including PDA (19, 20). IL35 production by Foxp3+ Tregs is important for tumor growth in models of melanoma and colorectal cancer (21). To determine whether Treg-derived IL35 confers immunosuppression in PDA, we used mice with Treg-specific Foxp3Cre-YFP-driven deficiency in Ebi3 (TregEbi3−/−)(Supplementary Fig. S5A-B)(21,28–31). We found that Treg-specific deficiency in IL35 did not alter orthotropic tumor growth, as KPC cells injected into either TregEbi3+/– or TregEbi3–/– littermates grew to a comparable tumor size, even though Ebi3 expression in CD4+ T cells was reduced to background levels (Supplementary Fig. S5C-D). Intratumoral and splenic frequency of total Foxp3+ Tregs, as well as cytokine production by B cells, was not altered in mice with Treg-specific deletion of Ebi3, suggesting that endogenous production of IL35 by Tregs in PDA does not control Treg or IL35+ B-cell expansion at the tumor site (Supplementary Fig. S5E-H). The frequency and activity of CD4+ and CD8+ T effector cells and frequencies of myeloid subsets were similar between TregEbi3+/– or TregEbi3–/– animals (Supplementary Fig. S5I-R). These observations highlight intrinsic differences in how different tumors types co-op IL35 and suggest that in the presence of IL35+ B cells, reduction of IL35 expression by Treg cells is not sufficient to alter PDA growth.
IL35+ B cells establish immunosuppression in PDA
To determine whether B cell–derived IL35 confers immunosuppression in PDA, we first generated mice with B-cell specific deficiency in the p35 (Bp35–/–) subunit of IL35 using a mixed bone marrow chimera approach (Fig. 3A, Supplementary Fig. S6A). Orthotopic injections of KPC cells resulted in attenuated PDA growth compared with controls (Fig. 3B). Analysis of IL35 expression in B cells confirmed near complete reduction in p35 protein in both splenic and intratumoral Bregs (Fig. 3C, Supplementary Fig. S6B-C). Previously, we observed that B cell–specific loss of IL35 resulted in decreased proliferation of cells transformed by KrasG12D alone (18). In contrast, KPC cells did not require B cell–derived IL35 for proliferation in vivo (Supplementary Fig. S6D). These findings, together with our observations suggesting that IL35 affected antitumor immune responses, prompted investigation into the immune functionality following B cell–specific deletion of IL35 (19).
Figure 3. B cell–derived IL35 promotes tumor growth and mediates suppression of IFNɣ and chemotactic receptors CXCR3 and CCR5 in CD8+ T cells.

(A) Experimental schema used to generate tumor-bearing mixed bone marrow chimeras containing B cell–specific deletion of p35 (Bp35–/–) and corresponding control BWT mice. (B) Quantification of tumor weight from BWT and Bp35–/– mice 3 weeks post-orthotopic injection with KPC4662 cells (n=6 mice/group). (C) Representative flow cytometry histograms (left) and quantification (right) of intracellular p35 expression in intratumoral and intrasplenic CD19+CD21hiCD5+CD1dhi Bregs from mice in (B). Proportion of p35+ Bregs is indicated. (D) Representative flow cytometry histograms (left) and quantification (right) of intratumoral CD45+CD3+CD4+CD25– effector T cells in BWT and Bp35–/– mice from (B). (E) Representative flow cytometry plots (left) and quantification (right) of intracellular IFNɣ (Isotype; Rat IgG1, κ) and TNFα (Isotype; Rat IgG1, κ) expression by CD3+CD4+ intratumoral T cells from mice in (B). Proportion of CD4+ T cells is indicated. (F) Representative flow cytometry histograms (left) and quantification (right) of intratumoral CD3+CD4+Foxp3+ Tregs from mice in (B). Proportion of CD4+ T cells is indicated. (G) Representative flow cytometry histograms (left) and quantification (right) of intracellular p35 (Isotype; Rat IgG2a, ĸ) and IL10 (Isotype; Rat IgG2b, κ) expression by intratumoral CD3+CD4+ T cells from mice in (B). (H) Quantification of frequency of tumor-infiltrating CD45+CD3+CD8+ T cells from BWT and Bp35–/– mice from (B) determined by flow cytometry. (I) Representative flow cytometry plots (left) and quantification (right) of IFNγ (Isotype; Rat IgG1, κ) in intratumoral CD45+CD3+CD8+ T cells from mice in (B). (J) Ratio of mean CD3+CD4+CD25– effector T cells (Teff) to Tregs (left) and ratio of mean CD3+CD8+ cytotoxic T cells to Tregs (right) were calculated based on the percent-positive lymphocyte population determined by flow cytometry. Error bars indicate S.E.M.; p-values were calculated using Student’s t-test (unpaired, two-tailed); NS: not significant, **p<0.01; ***p<0.001. Data represents 3 independent experiments.
Intratumoral immune profiling revealed elevated frequency of IFNɣ+ and TNFα+CD4+ effector T cells, as well as decreased in intratumoral but not splenic FoxP3+ Tregs, in Bp35−/− and BEbi3−/− mice compared to BWT mice, suggesting that B cell–derived IL35 may directly or indirectly regulate Treg frequency locally but not systemically (Fig. 3D-F, Supplementary Fig. S6E). Because it has been proposed that exogenous IL35 may promote expansion of suppressive Tregs by inducing its own expression in CD4+ T cells (20), we examined the ability of Tregs to produce IL35 in the context of B cell-specific deletion of this cytokine. Although the overall frequency of intratumoral Tregs was reduced, the ability of Tregs to produce IL35 remained intact (Fig. 3G, Supplementary Fig. S6E). Overall, this suggested that although B cell–directed IL35 signaling may regulate numbers of CD4+ T-cell subsets in PDA, IL35 expression by Tregs could be maintained in a cell autonomous fashion. The changes in the T-cell landscape were accompanied by increased infiltration and IFNɣ expression by effector CD8+ T cells, and overall, there was a significant increase in the ratio of CD4+ and CD8+ effector T cells to Tregs (Fig. 3H-J, Supplementary Fig. S6F-H). Concordant with our findings using global genetic loss of IL35, the frequency of myeloid cell subsets was not affected by B-cell loss of IL35 (Supplementary Fig. S6I-L). These results suggested that cancer-associated IL35 expression in B cells was sufficient to dampen productive T cell–mediated immune responses in PDA.
Depletion of IL35 from B cells sensitizes PDA to immunotherapy
Anti–PD-1 checkpoint blockade does not impede murine PDA growth, and the response rate in human PDA has been poor (42). We have reported that complete genetic deficiency in IL35 confers increased PDA sensitivity to anti–PD-1 treatment, presumably through increased numbers of infiltrating CD8+ T cells. However, the cell type–specific contribution to the establishment of IL-35-driven immunosuppression and resistance to anti–PD-1 remains unclear (19). To develop a tractable model of B cell–specific loss of IL35, we generated a model where B cells lacked expression of Ebi3 (BEbi3–/–) subunit (Fig. 4A, Supplementary Fig. S7A) by crossing a mouse with a conditional deletion of floxed Ebi3 allele with a mouse coding for B cell–specific Cre recombinase (CD19Cre)(21,32). To understand if B cell–specific deficiency in IL35 was sufficient to confer sensitivity to anti–PD-1, we treated BEbi3+/– and BEbi3–/– mice with anti–PD-1 or isotype control (Fig. 4A). Orthotopic injections of KPC cells phenocopied our results with Bp35–/– mice and resulted in decreased PDA growth (Fig. 4B). Combination of anti–PD-1 with B cell–specific loss of IL35 conferred additional significant reduction in tumor growth compared to BEbi3+/– mice treated with either isotype or anti–PD-1 or BEbi3–/– mice treated with isotype control (Fig. 4B-C). Reduced Ebi3 protein was seen in splenic and intratumoral Bregs (Supplementary Fig. S7B-C). We also found that KPC cells were not dependent on B cell–derived Ebi3 for proliferation in vivo (Supplementary Fig. S7D). Loss of B cell–derived Ebi3 increased CD4+ effector T cells and decreased intratumoral Treg frequency (Supplementary Fig. S7E-G) and increased infiltration and IFNɣ expression by effector CD8+ T cells (Fig. 4D-F). Anti–PD-1–treated BEbi3–/– mice exhibited slightly upward trending changes in effector CD4+ T cells, comparable with those in IgG-treated BEbi3–/– mice. The ratio of CD4+ and CD8+ effector T cells to Tregs was significantly higher in BEbi3–/– mice treated with anti–PD-1 compared to the BEbi3–/– IgG cohort, and may contribute to better tumor control, but the frequency of myeloid cell subsets was not affected by B-cell loss of Ebi3 (Supplementary Fig. S7I-J, Supplementary Fig. S8A-E). Overall, these data suggested that elimination of IL35 expression from B cells alone may benefit anti–PD-1 efficacy by potentiating effector T-cell responses.
Figure 4. Production of IL35 by B cells impedes anti-PD1 therapy.

(A) Experimental schema used to generate tumor-bearing mice that contain B cell–specific deletion of Ebi3 (BEbi3–/–) and corresponding control BEbi3+/– mice. (B) Quantification of tumor weight from anti–PD-1–treated (200 μg) and IgG-treated (200 μg) BEbi3+/– and BEbi3–/– mice 3 weeks post-orthotopic injection with KPC cells (n=6 mice/group). (C) Quantification of tumor growth by ultrasound from BEbi3+/− and BEbi3−/− mice from (B). (D) Quantification of frequency of tumor-infiltrating CD45+CD3+CD8+ T cells from BEbi3+/− and BEbi3−/− mice from (B) determined by flow cytometry. (E) Representative flow cytometry plots of intratumoral CD45+CD3+CD8+ T cells stained for IFNγ (Isotype; Rat IgG1, κ) from mice in (B). (F) Quantification of intratumoral CD8+IFNγ+ T cells from all mice in (B). (G) Representative flow cytometry histograms (left) and quantification (right) of expression of CXCR3, CCR5, phospho(p)-STAT1, pSTAT3, and pSTAT4 in intratumoral CD3+CD8+ T cells isolated from BEbi3+/– and BEbi3–/– mice from (B). (H) Representative flow cytometry plots and histograms (left) and quantification (right) of IFNɣ production and expression of CXCR3, CCR5, pSTAT1, pSTAT3, and pSTAT4 in CD3+CD8+ T cells 48 hours after coculture with Breg cells in 1:1 ratio derived from the indicated mice. Experiment were done in triplicate with 6 mice each group. Error bars indicate S.E.M.; p-values were calculated using Student’s t-test (unpaired, two-tailed); NS: not significant, *p<0.05; **p<0.01; ***p<0.001. Data represents 3 independent experiments.
To determine which T-cell subset was responsible for reduced tumor growth when IL35 was deleted from B cells, we depleted either CD4+ T cells, CD25+ Treg, or CD8+ T cells (Supplementary Fig. S9A-I). Tumor growth was rescued only under conditions of CD8+ T-cell depletion, suggesting that B cells may utilize IL35 to directly control CD8+ T-cell functionality (Supplementary Fig. S9A-I). We then analyzed the role of B cell–derived IL35 in suppression of CD8+ T-cell function in vivo. In addition to altered expression of IFNγ (Fig. 3I, Fig. 4E-F), B cell–specific deletion of IL35 correlated with increased expression of both CXCR3 and CCR5 and a concordant decrease of pSTAT1, pSTAT3, and pSTAT4 in CD8+ T cells (Fig. 4G, Supplementary Fig. S10A-B). These results suggested that B cell–derived IL35 in PDA acted to restrain infiltration and effector function of cytotoxic CD8+ T cells.
To understand if IL35 production by B cells was sufficient to suppress CD8+ T-cell function, we established coculture assays where cytokine production in splenic B cells derived from tumor-bearing animals was stimulated by addition of LPS and anti-CD40. Activated B cells were seeded in Transwells and cocultured with activated OVA-directed CD8+ T cells. Activated B cells inhibited IFNγ production and chemokine receptor expression by CD8+ T cells in an IL35-dependent manner (Fig. 4H). This functional suppression correlated with increased phosphorylation of STAT1, STAT3, and STAT4 in CD8+ T cells (Fig. 4H).
Pharmacological blockade of IL35 reduces pancreatic tumor growth
Our published data indicates that global genetic deficiency in IL35 increases intratumoral cytotoxic CD8+ T cells and suggests that targeting IL35 genetically may be an effective strategy to convert PDA from an immunologically ‘cold’ to a ‘hot’ tumor (19). Treatments that increase tumor infiltration by CD8+ T cells represent a promising strategy for use in combination with checkpoint blockade. To determine how pharmacological blockade of IL35 affected PDA tumor growth and responsiveness to anti–PD-1, WT mice were orthotopically injected with KPC cells and treated with monotherapies or combination therapy (Fig. 5A). Blockade of IL35 resulted in significant reduction of serum IL35 and a concordant reduction of tumor growth (Fig. 5B, Supplementary S11A). Treatment with combination anti–PD-1 and IL35 blockade further improved tumor growth control (Fig. 5B). Because anti-IL35 targets the Ebi3 subunit, which is also a subunit of IL27, we tested whether depletion of IL27 affected PDA growth. Anti-IL27 (against the p28 subunit) did not elicit reduced tumor growth, suggesting that the action of anti-IL35 was on target (Supplementary Fig. S11B). Measurements of tumor growth during treatment with anti-IL35 revealed that both single-agent and combination with anti–PD-1 had an effect on tumor growth approximately 14 days post-treatment initiation (Fig. 5C). Survival of orthotopically injected mice treated with combination anti-IL35 and anti–PD-1 was also significantly improved (Fig. 5D). Treatment of animals bearing spontaneous KrasG12D;Trp53R172H;p48Cre/+ (KPC)-driven pancreatic cancer with either anti-IL35 alone or combination with anti–PD-1 also significantly reduced tumor growth (Fig. 5E). Reduced tumor growth in the orthotopic setting was accompanied by increased infiltration and activation of effector T cells, as well as a reduction in Treg frequency, corroborating our observations in models of complete or B cell–specific IL35 deficiency (Fig. 5F-H, Supplementary Fig. S11C-E). Mechanistically, administration of anti-IL35 significantly reduced the frequency of IL35-producing CD4+ T cells and Bregs, suggesting that IL35 may regulate its own expression in suppressive cell types (Supplementary Fig. S11F-G). We also found that the frequency of PD-1+CD8+ T cells increased in treated animals, suggesting that anti-IL35 blockade in PDA resulted in increased effector function of CD8+ T cells (Supplementary Fig. S11H). We confirmed that expression of CXCR3 and CCR5 was increased on CD8+ T cells derived from anti-IL35–treated animals, concordant with decreases in pSTAT3 and pSTAT4 activation, although no additional changes were observed upon addition of anti–PD-1 (Fig. 5I, Supplementary Fig. S11I).
Figure 5. IL35 blockade relieves immunosuppression of CD8+ T cells and synergizes with anti–PD-1.

(A) Schematic of the antibody treatment regimen. Anti-IL35 (200 μg first dose followed by 100 μg per week) or control IgG antibody was administered in therapeutic schedule (1 week after tumor cell injection on days 7, 11, and 15). Administration of anti–PD-1 (200 μg) was initiated on day 7 after tumors reached approximately 4–5 mm in diameter. Two more doses of anti–PD-1 were administered on days 9 and 11. Mice were sacrificed 3 weeks post-tumor cell injection or assessed for survival. (B) Quantification of tumor weight from WT mice treated with therapeutic anti-IL35, anti–PD-1, or combination (as in (A)) 3 weeks post-orthotopic injection with KPC cells (n=6 mice/group). (C) Quantification of tumor growth by ultrasound from WT mice in (B). (D) Survival plot of orthotopically injected WT mice from (B) treated with the indicated therapeutic antibodies. (E) Quantification of tumor growth by ultrasound from spontaneous KPC mice treated with therapeutic anti-IL35, anti–PD-1, or combination as in (A). Treatment was initiated when tumor measuring ~5mm was visualized by ultrasound. (n=6 mice/group). Data represents 3 independent experiments. (F) Quantification of frequency of orthotopic tumor-infiltrating CD45+CD3+CD8+ T cells from mice in (B). (G) Representative flow cytometry plots of intracellular IFNγ in intratumoral CD45+CD3+CD8+ T cells from the mice in (B). Proportion of CD8+ T cells is indicated. (H) Quantification of intratumoral CD8+IFNγ+ T cells from the mice in (B). (I) Quantification of CXCR3, CCR5, pSTAT3, and pSTAT4 expression in intratumoral CD3+CD8+ T cells from mice in (B). Error bars indicate S.E.M.; p-values were calculated using Student’s t-test (unpaired, two-tailed); NS: not significant, **p<0.01; ***p<0.001. Data represents 3 independent experiments.
Immature B cells produce IL35 and IL10 in human PDA
To understand which human B-cell subset may correspond to murine IL35+IL10+ B cells, we analyzed peripheral blood (PB) of healthy donors and treatment-naïve patients with PDA for presence of previously reported human Breg markers (Supplementary Fig. S12A)(43–47). CD19+ B cells from PB were sorted and lysed to assess their ability to express cytokines. QPCR revealed that the CD19+CD24hiCD27+ B10 subset produced IL10, whereas the CD19+CD24hiCD38hi immature B-cell subset was most similar to the murine Bregs and produced both IL10 and IL35 (Supplementary Fig. S12B). Analysis of the major immune cell subtypes from PB of PDA patients by flow cytometry was concordant with data from mouse models (19) and indicated that IL35 (p35+Ebi3+) was primarily expressed by B cells and CD4+ T cells, with a larger contribution from B cells (Supplementary Fig. S12C-D). Neither murine plasma cells nor human plasmablasts produced IL10 nor IL35, indicating that the cytokine-producing B cells in PDA did not develop from plasma cells, as has been suggested in prostate cancer (24,48)(Supplementary Fig. S13A-D).
Overall, the frequency of CD19+CD24hiCD38hi immature B cells in the PB of PDA patients was significantly higher than in healthy controls (Fig. 6A). We found that CD19+CD24hiCD38hi peripheral B cells from PDA patients expressed significantly higher IL35 and IL10 compared to healthy controls and Bcon cells (Fig. 6B). This observation is consistent with the notion that IL35 is induced in the context of inflammation (20). There was heterogeneity among patients with regards to frequency of PB immature IL35+ B cells, suggesting heterogeneity of Breg responses in PDA patient population (Fig. 6A-C). Thus, at least some PDA patients exhibit B cell–driven immunosuppressive mechanisms, as evidenced by increased Breg activity.
Figure 6. Identification of IL35-producing B cells in patients with PDA.

(A) Quantification of CD19+CD24hiCD38hi B cells in peripheral blood of healthy volunteers (n=30) and treatment-naïve PDA patients (n=30). Proportion of CD19+ cells is indicated. (B) Fold-change in Il10, p35, and Ebi3 from sorted CD19+CD24hiCD38hi Bregs or CD19+CD24loCD38lo Bcon cells from healthy volunteers or PDA patients (n=5 samples/B-cell group) determined by qPCR. Fold-change determined by comparing to healthy Bcon cells. (C) Fold-change in expression of Il10, p35, and Ebi3 from sorted CD19+CD24hiCD38hi Bregs in healthy donors or PDA patients determined by qPCR. (D) Representative immunofluorescence staining for CD8, CXCR3, and pSTAT3 in samples of human PDA. Arrow indicates pSTAT3+CD8+ T cells, arrowhead indicates pSTAT3–CD8+ T cells, and asterisks indicate pSTAT3+CD8– cells. Scale bars, 25μm. (E) Proportion of Ebi3-high and -low tumor regions as a function of immune aggregates (IA). Data was derived from counting 3–6 field-of-view (FOV)/tumor sample (n=11 tumor samples). (F) Proportion of pSTAT3+CD8+ T cells as function of low vs. high numbers of Ebi3+ immune cells. Each data point is the percent of pSTAT3+CD8+ T cells out of all CD8+ T cells/20x FOV. Data was derived from counting 3–6 FOV/tumor sample (n=11 tumor samples). (G) Proportion of CXCR3+CD8+ T cells as function of low vs. high numbers of Ebi3+ immune cells. Each data point is the percent of CXCR3+CD8+ T cells out of all CD8+ T cells/20x FOV. Data was derived from counting 3–6 FOV/tumor sample (n=11 tumor samples). (H) Proportion of CXCR3+CD8+ T cells as function of pSTAT3+ vs pSTAT3– in tumor regions high for Ebi3+ immune cells. Each data point is the percent of CXCR3+CD8+ T cells out of all CD8+ T cells/20x FOV. Data was derived from counting 3–6 FOV/tumor sample (n=11 tumor samples). (I) Correlation of the cancer Breg signature with the cytotoxic CD8+ T-cell index in PAAD from TCGA. (J) Correlation of the cancer Breg signature with the cytotoxic CD8+ T-cell index across TCGA subtypes. Error bars indicate S.E.M., p-values were calculated using Student’s t-test (unpaired, two-tailed); NS: not significant, **p<0.01; ***p<0.001. Data represents 3 independent experiments.
IL35+ immune cells correlate with pSTAT3+CXCR3–CD8+ T cells in human PDA
We previously documented the presence of intratumoral IL35+ B cells and T cells in human PDA (19). Consistent with our hypothesis that IL35 may suppress CD8+ T-cell infiltration, we reported that IL35+ immune cells negatively correlated with CD8+ T-cell infiltration into human tumor cell nests (19). To interrogate the relationship among IL35 expression, STAT3 activation, and CD8+ T-cell infiltration, we used multiplexed immunofluorescence to analyze primary human PDA tissues for CD8+ T cell–specific expression of pSTAT3 and CXCR3 in IL35 high (Ebi3hi) or IL35 low (Ebi3lo) regions (Fig. 6D) (19).We found that the majority of IL35+ cells resided at the interface between immune aggregates (IA) and tumor parenchyma, suggesting that IAs might serve as sites of active immune suppression in PDA (Fig. 6E). Consistent with the idea that IL35 may induce STAT3 activation in CD8+ T cells, we found that the presence of IL35+ immune cells positively correlated with pSTAT3 activation in CD8+ T cells, and there was very little STAT3 activation in the regions that contained few or no IL35+ cells (Fig. 6F). Overall, the percentage of pSTAT3+CD8+ cells was not high, but specifically correlated with presence of IL35+ immune cells. On the contrary, the presence of CXCR3+CD8+ T cells, overall, negatively correlated with IL35 expression (Fig. 6G). Analysis of IL35+ regions revealed a significant negative association between pSTAT3 activation and CXCR3 expression in CD8+ T cells (Fig. 6H). We also examined co-expression of Ebi3 and p35 proteins on archived human PDA samples and found that, consistent with reports, Ebi3 and p35 were co-expressed in cytokeratin+ cells at frequencies ranging from 0.37–89.26% of all cytokeratin+ cells, with a median value of 18.27% (Supplementary Fig. S14A-B)(49). Although the consequences of epithelial-specific IL35 expression in PDA on immune function are not yet clear, our initial analysis did not find a correlation between the frequency of IL35+ cancer cells and CD8+ T-cell infiltration into the tumor parenchyma (Supplementary Fig. S14C). These results suggested that IL35+ immune cells in the human PDA TME may restrict CD8+ T-cell infiltration into the tumor parenchyma by suppressing expression of CXCR3 via STAT3 activation.
To understand how human IL35+ B cells correlated with overall cytotoxic responses in human PDA, we performed bulk RNA-sequencing on sorted peripheral Bregs and Bcon cells from patients with PDA or healthy volunteers. A tumor-associated Breg signature was calculated as described in the Methods. This gene signature correlated with the ratio of a CD8 signature and a previously published cytotoxicity score (50). Specifically, the cancer-derived Breg signature inversely correlated with the ratio of the T-cell cytotoxicity score to the CD8 score in the TCGA PAAD pancreatic cohort (Figure 6I). We also found that several additional cancer types in TCGA presented with an upregulated Breg signature that negatively correlated with antitumor T-cell activity, suggesting that the role of IL35+ B cells in regulating T-cell responses in cancer was broader than previously appreciated (Figure 6J).
DISCUSSION
T cell–based immunotherapy is at the frontlines of clinical care for several types of cancer. Unfortunately, PDA thus far has failed to respond to checkpoint blockade therapy (42). Effective responses to immunotherapeutics has been shown to correlate with pre-existing intratumoral effector T-cell infiltration, which is lacking in a large proportion of PDA cases (1). Establishment of diverse immunosuppressive networks and physical barriers, such as dense desmoplasia, has been implicated in restricting endogenous antitumor responses and immunotherapy efficacy in PDA (6). Considerable effort has been put into delineating the contribution of some major immune infiltrating cells, such as myeloid cells and Tregs. Recruitment and pro-tumor polarization of macrophages and MDSCs have been shown to constrain CD8+ T-cell responses by engaging GM-CSF and CSFR1 axes (11,51). Tolerogenic CD206+MHCIIlow tumor-associated macrophages can also promote Treg expansion (52). Tregs, which are well-known drivers of suppression of tumor-directed CD8+ T-cell responses, are shown to also act on DCs and prevent productive antigen presentation (14). Investigations into rare subsets of immune cells present in PDA TME, such as ɣδ T cells, have yielded important insights into contributions of these cells to biology of PDA development (16).
A number of cell types have been shown to produce immunosuppressive IL35 in cancer, including Treg and B cells, as well as DCs (20). A study has suggested that a subset of patients with PDA have upregulated IL35 in tumor cells, although it is not clear what effect this has on immune function (49). Murine KPC cells used in our study do not produce IL35 and, therefore, recapitulate tumor biology where IL35 is sourced from the stromal microenvironment. Here, we showed that B cell–specific production of IL35 suppressed endogenous T-cell responses in PDA. Genetic or pharmacological targeting of IL35 resulted in decreased tumor growth associated with increased effector T-cell infiltration and IFNɣ production. We uncovered that sensitivity to anti–PD-1 was evident in the context of targeting IL35. We found that B cell–derived IL35 directly suppressed infiltration and activation of CD8+ T cells by downregulating chemotactic receptors CXCR3 and CCR5 and effector cytokine IFNγ, and this was dependent on IL35-induced phosphorylation of STAT3 in CD8+ T cells. Ex vivo inhibition of pSTAT3 activated CD8+ T cells from mice bearing PDA tumors and conferred antitumor activity upon adoptive transfer of inhibitor-treated T cells. These data point to a previously unappreciated role of B cells in directly suppressing infiltration and activity of tumor-directed CD8+ T cells in PDA.
Although B cells can account for up to 25% of immune infiltrate in some solid tumors and up to 15% in human PDA (53,54), the precise mechanisms of B-cell contribution to tumor growth are not well established. In concert with the idea that B lymphocytes play versatile roles in solid cancer biology, we and others have demonstrated that B cells promote PDA growth in coordination with hypoxia via immunoglobulin deposition or by production of IL35 (18, 54, 55). These findings have led to initiation of several clinical trials in PDA patients using combination chemotherapy and ibrutinib (an inhibitor of Btk found in both activated B cells and macrophages), marking an important foray into testing the contribution of B-cell and macrophage activation in human PDA (NCT02436668). However, given the diverse repertoire of B-cell function in cancer, it is important to better understand the consequences of disrupting the functionality of individual B-cell subsets in PDA.
Our data suggested that IL35+ B cells directly regulate infiltration and function of CD8+ T cells in PDA in a STAT3-dependent manner. CXCR3 and CCR5 are chemokine receptors expressed on effector T cells, can be upregulated by ligands CXCL9, CXCL10, and CXCL11, and have been shown to be important for T-cell infiltration into the tumor bed (36). Studies in melanoma and lung cancer models have found that genetic lack of STAT3 in T cells results in increased migration and effector function (56,57). These observations, together with our data and studies showing that STAT3 regulates proliferation, survival, cytotoxic gene expression, and memory function in CD8+ T cells that respond to viral infections, suggest that STAT3 could be a possible therapeutic target for enhancing antitumor CD8+ T cell–mediated responses (58,59). Further work needs to be done to establish if IL35 blockade and/or inhibition of STAT3 in effector T cells generates persistent memory responses and may provide further insight into strategies for combination immunotherapy. STAT inhibitor studies performed by us at this point were done ex vivo and are limited to proof-of-concept because all three inhibitors used (fludarabine, STA-21, and lysofylline) may have off-target effects (60–62).
Concordant with previous studies suggesting that B cells may influence Treg numbers in cancer, we found that B-cell production of IL35 was important for localized expansion of Foxp3+ Tregs (63). Specific triggers for IL35 production by Tregs in cancer have not been well-defined, although prior reports suggest that recombinant IL35 can induce IL35 in Tregs and naïve CD4+CD25–Foxp3– T cells (20). Our current data showed that IL35 in B cells does not affect production of IL35 by Tregs, so we can rule out B cells as being an initiating source of IL35 for T-cell lineages. It is possible that Treg-specific production of IL35 is sufficient to keep its expression going in a cell autonomous manner. It is also currently not known which cancer-associated inflammatory mediators induce IL35 production in B cells, and additional research dissecting the mechanisms of IL35 induction in immunosuppressive cells may provide novel targets for therapy.
With regards to immune-cell specific contribution of IL35 to tumor growth, our findings contrast data in mouse models of melanoma and colon cancer, which demonstrate that Treg-specific expression of IL35 is important in tumor growth control (21). It is possible that the difference in overall levels of IL35 produced by B cells and Tregs in distinct tumor models may account for this effect. We observed that in PDA, approximately 20% of Bregs and roughly 8% of CD4+ T cells produced IL35. This is in contrast with murine melanoma, where up to 40% of tumor-infiltrating Tregs are reported to produce IL35 (21).
Our study supports the idea that B cell–derived IL35 promotes tumorigenesis via inhibition of CD8+ T cell infiltration and effector cytokine production, and we observed an increase in CD8+ T-cell PD-1 expression following anti-IL35 treatment, in concordance with an idea that newly activated T cells upregulate PD-1 expression overtime (27). It is also possible that physical location of immunosuppressive cells, with respect to tumor-reactive CD8+ T cells, may influence the nature of immunosuppression. Although our preliminary analysis did not indicate alterations in myeloid populations in either B cell– or Treg-specific deletion of IL35, it is still possible that IL35 may act on other immune cells apart from B cells and T cells.
Validation that IL35+ B cells have a role in inducing immunosuppression in human PDA was performed. We analyzed human B cells from patients with PDA using markers CD24 and CD38, previously used to identify IL10-producing B cells in patients with autoimmunity, and established that the CD24+CD38+ subset produced IL35 and was present at higher frequencies in PDA patients, suggesting disease-instigated expansion of this cell subtype (64). We also demonstrated that presence of IL35+ B cells inversely correlated with antitumor cytotoxic T-cell activity and expansion of Bregs across distinct cancer types, suggesting that Bregs are major drivers of immunosuppression in a variety of cancers.
Supplementary Material
Acknowledgements:
We thank B. Savoldo and G. Dotti for discussions and help with manuscript preparation.
Funding: This work was supported in part by R37 CA230786 (Y. P.-G.), University Cancer Research Fund at the University of North Carolina at Chapel Hill, United States (Y.P.-G.); AACR–PanCAN Pathway to Leadership Grant 13–70-25-PYLA (Y.P.-G.), V Foundation for Cancer Research grant V2016–016 (Y. P.-G.), Concern Foundation Conquer Cancer Now Award (Y.P.-G), the WUSTL SPORE in Pancreatic Cancer (R.C.F and W.G.H; P50CA196510), the WUSTL SPORE Career Enhancement Award grant 1P50CA196510–01A1 from the NCI (Y.P.-G), Cancer Cell Biology Training Program (CCBTP) grant 2T32CA071341–21 (D. M.) and R01 CA203689 (D.A.A.V). The UNC Flow Cytometry Core Facility, the UNC Translational Pathology Laboratory, the Animal Histopathology & Laboratory Medicine Core and the UNC Lineberger Animal Studies Core are supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC LCCC.
Footnotes
Conflict of Interest: D.A.A.V. declares competing financial interests. He has submitted patents that are pending or granted that cover IL35 and is entitled to a share in net income generated from licensing of these patent rights for commercial development. He also consults for several biopharmaceutical companies.
REFERENCES
- 1.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3. Epub 2014/05/16. doi: 10.1186/2051-1426-2-3. PubMed PMID: 24829760; PMCID: PMC4019906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–27. Epub 2007/10/03. doi: 10.1158/0008-5472.CAN-07-0175. PubMed PMID: 17909062. [DOI] [PubMed] [Google Scholar]
- 3.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, Idrisoglu S, Manning S, Nourbakhsh E, Wani S, Fink L, Holmes O, Chin V, Anderson MJ, Kazakoff S, Leonard C, Newell F, Waddell N, Wood S, Xu Q, Wilson PJ, Cloonan N, Kassahn KS, Taylor D, Quek K, Robertson A, Pantano L, Mincarelli L, Sanchez LN, Evers L, Wu J, Pinese M, Cowley MJ, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chantrill LA, Mawson A, Humphris J, Chou A, Pajic M, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Lovell JA, Merrett ND, Toon CW, Epari K, Nguyen NQ, Barbour A, Zeps N, Moran-Jones K, Jamieson NB, Graham JS, Duthie F, Oien K, Hair J, Grutzmann R, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Rusev B, Capelli P, Salvia R, Tortora G, Mukhopadhyay D, Petersen GM, Australian Pancreatic Cancer Genome I, Munzy DM, Fisher WE, Karim SA, Eshleman JR, Hruban RH, Pilarsky C, Morton JP, Sansom OJ, Scarpa A, Musgrove EA, Bailey UM, Hofmann O, Sutherland RL, Wheeler DA, Gill AJ, Gibbs RA, Pearson JV, Waddell N, Biankin AV, Grimmond SM. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52. Epub 2016/02/26. doi: 10.1038/nature16965. PubMed PMID: 26909576. [DOI] [PubMed] [Google Scholar]
- 4.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, Rosenberg SA. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–33. Epub 2010/09/16. doi: 10.1097/CJI.0b013e3181eec14c. PubMed PMID: 20842054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65. Epub 2012/06/05. doi: 10.1056/NEJMoa1200694. PubMed PMID: 22658128; PMCID: PMC3563263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol. 2013;25(2):200–5. Epub 2013/02/21. doi: 10.1016/j.coi.2013.01.006. PubMed PMID: 23422836; PMCID: PMC3647365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–76. Epub 2012/08/17. doi: 10.1158/1078-0432.CCR-11-3114. PubMed PMID: 22896693; PMCID: PMC3442232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017;24(3):100–5. Epub 2016/12/03. doi: 10.1038/cgt.2016.65. PubMed PMID: 27910857. [DOI] [PubMed] [Google Scholar]
- 9.Chao T, Furth EE, Vonderheide RH. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res. 2016;4(11):968–82. Epub 2016/11/03. doi: 10.1158/2326-6066.CIR-16-0188. PubMed PMID: 27737879; PMCID: PMC5110270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6. Epub 2011/03/26. doi: 10.1126/science.1198443. PubMed PMID: 21436454; PMCID: PMC3406187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–47. Epub 2012/06/16. doi: 10.1016/j.ccr.2012.04.024. PubMed PMID: 22698407; PMCID: PMC3721510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, Vonderheide RH. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res. 2015;3(4):399–411. Epub 2015/02/14. doi: 10.1158/2326-6066.CIR-14-0215. PubMed PMID: 25678581; PMCID: PMC4390506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA, Janowitz T, Jodrell DI, Tuveson DA, Fearon DT. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–7. Epub 2013/1½8. doi: 10.1073/pnas.1320318110. PubMed PMID: 24277834; PMCID: PMC3864274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017;20(3):558–71. Epub 2017/07/21. doi: 10.1016/j.celrep.2017.06.062. PubMed PMID: 28723561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Marinis JM, Beal AM, Savadkar S, Wu Y, Khan M, Taunk PS, Wu N, Su W, Wu J, Ahsan A, Kurz E, Chen T, Yaboh I, Li F, Gutierrez J, Diskin B, Hundeyin M, Reilly M, Lich JD, Harris PA, Mahajan MK, Thorpe JH, Nassau P, Mosley JE, Leinwand J, Kochen Rossi JA, Mishra A, Aykut B, Glacken M, Ochi A, Verma N, Kim JI, Vasudevaraja V, Adeegbe D, Almonte C, Bagdatlioglu E, Cohen DJ, Wong KK, Bertin J, Miller G. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell. 2018;34(5):757–74 e7. Epub 2018/11/14. doi: 10.1016/j.ccell.2018.10.006. PubMed PMID: 30423296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, Barilla R, Torres-Hernandez A, Hundeyin M, Mani VRK, Avanzi A, Tippens D, Narayanan R, Jang JE, Newman E, Pillarisetty VG, Dustin ML, Bar-Sagi D, Hajdu C, Miller G. gammadelta T Cells Support Pancreatic Oncogenesis by Restraining alphabeta T Cell Activation. Cell. 2016;166(6):1485–99 e15. Epub 2016/08/30. doi: 10.1016/j.cell.2016.07.046. PubMed PMID: 27569912; PMCID: PMC5017923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, Balli D, Hay CA, Sela Y, Merrell AJ, Liudahl SM, Gordon N, Norgard RJ, Yuan S, Yu S, Chao T, Ye S, Eisinger-Mathason TSK, Faryabi RB, Tobias JW, Lowe SW, Coussens LM, Wherry EJ, Vonderheide RH, Stanger BZ. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. 2018;49(1):178–93 e7. Epub 2018/07/01. doi: 10.1016/j.immuni.2018.06.006. PubMed PMID: 29958801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, Bar-Sagi D. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 2016;6(3):247–55. Epub 2015/12/31. doi: 10.1158/2159-8290.CD-15-0843. PubMed PMID: 26715643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mirlekar B, Michaud D, Searcy R, Greene K, Pylayeva-Gupta Y. IL35 Hinders Endogenous Antitumor T-cell Immunity and Responsiveness to Immunotherapy in Pancreatic Cancer. Cancer Immunol Res. 2018;6(9):1014–24. Epub 2018/07/08. doi: 10.1158/2326-6066.CIR-17-0710. PubMed PMID: 29980536; PMCID: PMC6125203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pylayeva-Gupta Y. Molecular Pathways: Interleukin-35 in Autoimmunity and Cancer. Clin Cancer Res. 2016;22(20):4973–8. Epub 2016/09/02. doi: 10.1158/1078-0432.CCR-16-0743. PubMed PMID: 27582486. [DOI] [PubMed] [Google Scholar]
- 21.Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, Beres AJ, Vogel P, Workman CJ, Vignali DA. Interleukin-35 Limits Anti-Tumor Immunity. Immunity. 2016;44(2):316–29. Epub 2016/02/14. doi: 10.1016/j.immuni.2016.01.013. PubMed PMID: 26872697; PMCID: PMC4758699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA. Cutting edge: Human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J Immunol. 2011;186(12):6661–6. Epub 2011/05/18. doi: 10.4049/jimmunol.1100315. PubMed PMID: 21576509; PMCID: PMC3110563. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Dixon KO, van der Kooij SW, Vignali DA, van Kooten C. Human tolerogenic dendritic cells produce IL-35 in the absence of other IL-12 family members. Eur J Immunol. 2015;45(6):1736–47. Epub 2015/03/31. doi: 10.1002/eji.201445217. PubMed PMID: 25820702; PMCID: PMC4617619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C, Li R, Jouneau L, Boudinot P, Wilantri S, Sakwa I, Miyazaki Y, Leech MD, McPherson RC, Wirtz S, Neurath M, Hoehlig K, Meinl E, Grutzkau A, Grun JR, Horn K, Kuhl AA, Dorner T, Bar-Or A, Kaufmann SHE, Anderton SM, Fillatreau S. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507(7492):366–70. Epub 2014/02/28. doi: 10.1038/nature12979. PubMed PMID: 24572363; PMCID: PMC4260166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, Wingfield PT, Kim SH, Egwuagu CE. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. 2014;20(6):633–41. Epub 2014/04/20. doi: 10.1038/nm.3554. PubMed PMID: 24743305; PMCID: PMC4048323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z, Liu JQ, Liu Z, Shen R, Zhang G, Xu J, Basu S, Feng Y, Bai XF. Tumor-derived IL-35 promotes tumor growth by enhancing myeloid cell accumulation and angiogenesis. J Immunol. 2013;190(5):2415–23. Epub 2013/0½5. doi: 10.4049/jimmunol.1202535. PubMed PMID: 23345334; PMCID: PMC3578001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, Cuevas C, Dotson AM, Tan X, Hotes JL, Greenberg PD, Hingorani SR. T Cells Engineered against a Native Antigen Can Surmount Immunologic and Physical Barriers to Treat Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2015;28(5):638–52. Epub 2015/11/04. doi: 10.1016/j.ccell.2015.09.022. PubMed PMID: 26525103; PMCID: PMC4724422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8(2):191–7. Epub 2006/12/01. doi: 10.1038/ni1428. PubMed PMID: 17136045. [DOI] [PubMed] [Google Scholar]
- 29.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6. Epub 2003/03/04. doi: 10.1038/ni904. PubMed PMID: 12612578. [DOI] [PubMed] [Google Scholar]
- 30.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr., Muller W, Rudensky AY. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28(4):546–58. Epub 2008/04/05. doi: 10.1016/j.immuni.2008.02.017. PubMed PMID: 18387831. [DOI] [PubMed] [Google Scholar]
- 31.Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability of the regulatory T cell lineage in vivo. Science. 2010;329(5999):1667–71. Epub 2010/10/12. doi: 10.1126/science.1191996. PubMed PMID: 20929851; PMCID: PMC4262151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25(6):1317–8. Epub 1997/03/15. PubMed PMID: 9092650; PMCID: PMC146582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–35. Epub 2012/06/16. doi: 10.1016/j.ccr.2012.04.025. PubMed PMID: 22698406; PMCID: PMC3575028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, Bruneval P, Fridman WH, Becker C, Pages F, Speicher MR, Trajanoski Z, Galon J. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–95. Epub 2013/10/22. doi: 10.1016/j.immuni.2013.10.003. PubMed PMID: 24138885. [DOI] [PubMed] [Google Scholar]
- 35.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. Epub 2015/01/17. doi: 10.1016/j.cell.2014.12.033. PubMed PMID: 25594174; PMCID: PMC4856474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, Luster AD, Evans SS. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. 2015;6:7458. Epub 2015/06/26. doi: 10.1038/ncomms8458. PubMed PMID: 26109379; PMCID: PMC4605273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dambuza IM, He C, Choi JK, Yu CR, Wang R, Mattapallil MJ, Wingfield PT, Caspi RR, Egwuagu CE. IL-12p35 induces expansion of IL-10 and IL-35-expressing regulatory B cells and ameliorates autoimmune disease. Nat Commun. 2017;8(1):719. Epub 2017/09/30. doi: 10.1038/s41467-017-00838-4. PubMed PMID: 28959012; PMCID: PMC5620058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, Satoskar AR, Garcia KC, Hunter CA, Drake CG, Murray PJ, Vignali DA. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol. 2012;13(3):290–9. Epub 2012/02/07. doi: 10.1038/ni.2227. PubMed PMID: 22306691; PMCID: PMC3529151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank DA, Mahajan S, Ritz J. Fludarabine-induced immunosuppression is associated with inhibition of STAT1 signaling. Nat Med. 1999;5(4):444–7. Epub 1999/04/15. doi: 10.1038/7445. PubMed PMID: 10202937. [DOI] [PubMed] [Google Scholar]
- 40.Chalmin F, Mignot G, Bruchard M, Chevriaux A, Vegran F, Hichami A, Ladoire S, Derangere V, Vincent J, Masson D, Robson SC, Eberl G, Pallandre JR, Borg C, Ryffel B, Apetoh L, Rebe C, Ghiringhelli F. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity. 2012;36(3):362–73. Epub 2012/03/13. doi: 10.1016/j.immuni.2011.12.019. PubMed PMID: 22406269. [DOI] [PubMed] [Google Scholar]
- 41.Bright JJ, Du C, Coon M, Sriram S, Klaus SJ. Prevention of experimental allergic encephalomyelitis via inhibition of IL-12 signaling and IL-12-mediated Th1 differentiation: an effect of the novel anti-inflammatory drug lisofylline. J Immunol. 1998;161(12):7015–22. Epub 1998/12/23. PubMed PMID: 9862738. [PubMed] [Google Scholar]
- 42.Johansson H, Andersson R, Bauden M, Hammes S, Holdenrieder S, Ansari D. Immune checkpoint therapy for pancreatic cancer. World J Gastroenterol. 2016;22(43):9457–76. Epub 2016/12/07. doi: 10.3748/wjg.v22.i43.9457. PubMed PMID: 27920468; PMCID: PMC5116591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42(4):607–12. Epub 2015/04/23. doi: 10.1016/j.immuni.2015.04.005. PubMed PMID: 25902480. [DOI] [PubMed] [Google Scholar]
- 44.Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol. 2015;27(10):479–86. Epub 2015/06/14. doi: 10.1093/intimm/dxv038. PubMed PMID: 26071023; PMCID: PMC4587489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Masson A, Bouaziz JD, Le Buanec H, Robin M, O’Meara A, Parquet N, Rybojad M, Hau E, Monfort JB, Branchtein M, Michonneau D, Dessirier V, Sicre de Fontbrune F, Bergeron A, Itzykson R, Dhedin N, Bengoufa D, Peffault de Latour R, Xhaard A, Bagot M, Bensussan A, Socie G. CD24(hi)CD27(+) and plasmablast-like regulatory B cells in human chronic graft-versus-host disease. Blood. 2015;125(11):1830–9. Epub 2015/0½2. doi: 10.1182/blood-2014-09-599159. PubMed PMID: 25605369. [DOI] [PubMed] [Google Scholar]
- 46.Menon M, Rosser EC, Mauri C. Identification and Isolation of Regulatory B Cells in Mouse and Human. Methods Mol Biol. 2019;1899:55–66. Epub 2019/01/17. doi: 10.1007/978-1-4939-8938-6_5. PubMed PMID: 30649765. [DOI] [PubMed] [Google Scholar]
- 47.Banko Z, Pozsgay J, Szili D, Toth M, Gati T, Nagy G, Rojkovich B, Sarmay G. Induction and Differentiation of IL-10-Producing Regulatory B Cells from Healthy Blood Donors and Rheumatoid Arthritis Patients. J Immunol. 2017;198(4):1512–20. Epub 2017/01/15. doi: 10.4049/jimmunol.1600218. PubMed PMID: 28087671. [DOI] [PubMed] [Google Scholar]
- 48.Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, Kallies A, Nutt SL, Sakaguchi S, Takeda K, Kurosaki T, Baba Y. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity. 2014;41(6):1040–51. Epub 2014/12/09. doi: 10.1016/j.immuni.2014.10.016. PubMed PMID: 25484301. [DOI] [PubMed] [Google Scholar]
- 49.Huang C, Li N, Li Z, Chang A, Chen Y, Zhao T, Li Y, Wang X, Zhang W, Wang Z, Luo L, Shi J, Yang S, Ren H, Hao J. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat Commun. 2017;8:14035. Epub 2017/0½0. doi: 10.1038/ncomms14035. PubMed PMID: 28102193; PMCID: PMC5253665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, Ziv E, Culhane AC, Paull EO, Sivakumar IKA, Gentles AJ, Malhotra R, Farshidfar F, Colaprico A, Parker JS, Mose LE, Vo NS, Liu J, Liu Y, Rader J, Dhankani V, Reynolds SM, Bowlby R, Califano A, Cherniack AD, Anastassiou D, Bedognetti D, Rao A, Chen K, Krasnitz A, Hu H, Malta TM, Noushmehr H, Pedamallu CS, Bullman S, Ojesina AI, Lamb A, Zhou W, Shen H, Choueiri TK, Weinstein JN, Guinney J, Saltz J, Holt RA, Rabkin CE, Cancer Genome Atlas Research N, Lazar AJ, Serody JS, Demicco EG, Disis ML, Vincent BG, Shmulevich L. The Immune Landscape of Cancer. Immunity. 2018;48(4):812–30 e14. Epub 2018/04/10. doi: 10.1016/j.immuni.2018.03.023. PubMed PMID: 29628290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, Yokoyama WM, Hawkins WG, Fields RC, Randolph GJ, DeNardo DG. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity. 2017;47(3):597. Epub 2017/09/21. doi: 10.1016/j.immuni.2017.08.018. PubMed PMID: 28930665; PMCID: PMC5664180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui R, Yue W, Lattime EC, Stein MN, Xu Q, Tan XL. Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget. 2016;7(31):50735–54. Epub 2016/05/19. doi: 10.18632/oncotarget.9383. PubMed PMID: 27191744; PMCID: PMC5226617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coronella-Wood JA, Hersh EM. Naturally occurring B-cell responses to breast cancer. Cancer Immunol Immunother. 2003;52(12):715–38. Epub 2003/08/16. doi: 10.1007/s00262-003-0409-4. PubMed PMID: 12920480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, Gorjestani S, Liudahl SM, Truitt M, Olson P, Kim G, Hanahan D, Tempero MA, Sheppard B, Irving B, Chang BY, Varner JA, Coussens LM. Bruton Tyrosine Kinase-Dependent Immune Cell Crosstalk Drives Pancreas Cancer. Cancer Discov. 2016;6(3):270–85. Epub 2015/12/31. doi: 10.1158/2159-8290.CD-15-0827. PubMed PMID: 26715645; PMCID: PMC4783268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D, Vonderheide RH, Simon MC. Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia. Cancer Discov. 2016;6(3):256–69. Epub 2015/12/31. doi: 10.1158/2159-8290.CD-15-0822. PubMed PMID: 26715642; PMCID: PMC4783189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kujawski M, Zhang C, Herrmann A, Reckamp K, Scuto A, Jensen M, Deng J, Forman S, Figlin R, Yu H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 2010;70(23):9599–610. Epub 2010/12/02. doi: 10.1158/0008-5472.CAN-10-1293. PubMed PMID: 21118964; PMCID: PMC3017475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yue C, Shen S, Deng J, Priceman SJ, Li W, Huang A, Yu H. STAT3 in CD8+ T Cells Inhibits Their Tumor Accumulation by Downregulating CXCR3/CXCL10 Axis. Cancer Immunol Res. 2015;3(8):864–70. Epub 2015/05/31. doi: 10.1158/2326-6066.CIR-15-0014. PubMed PMID: 26025380; PMCID: PMC4527884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu CR, Dambuza IM, Lee YJ, Frank GM, Egwuagu CE. STAT3 regulates proliferation and survival of CD8+ T cells: enhances effector responses to HSV-1 infection, and inhibits IL-10+ regulatory CD8+ T cells in autoimmune uveitis. Mediators Inflamm. 2013;2013:359674. Epub 2013/11/10. doi: 10.1155/2013/359674. PubMed PMID: 24204098; PMCID: PMC3800609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ciucci T, Vacchio MS, Bosselut R. A STAT3-dependent transcriptional circuitry inhibits cytotoxic gene expression in T cells. Proc Natl Acad Sci U S A. 2017;114(50):13236–41. Epub 2017/1½9. doi: 10.1073/pnas.1711160114. PubMed PMID: 29180433; PMCID: PMC5740647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lukenbill J, Kalaycio M. Fludarabine: a review of the clear benefits and potential harms. Leuk Res. 2013;37(9):986–94. Epub 2013/06/22. doi: 10.1016/j.leukres.2013.05.004. PubMed PMID: 23787174. [DOI] [PubMed] [Google Scholar]
- 61.Munoz J, Dhillon N, Janku F, Watowich SS, Hong DS. STAT3 inhibitors: finding a home in lymphoma and leukemia. Oncologist. 2014;19(5):536–44. Epub 2014/04/08. doi: 10.1634/theoncologist.2013-0407. PubMed PMID: 24705981; PMCID: PMC4012967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen M, Yang Z, Wu R, Nadler JL. Lisofylline, a novel antiinflammatory agent, protects pancreatic beta-cells from proinflammatory cytokine damage by promoting mitochondrial metabolism. Endocrinology. 2002;143(6):2341–8. Epub 2002/05/22. doi: 10.1210/endo.143.6.8841. PubMed PMID: 12021199. [DOI] [PubMed] [Google Scholar]
- 63.Yuen GJ, Demissie E, Pillai S. B lymphocytes and cancer: a love-hate relationship. Trends Cancer. 2016;2(12):747–57. Epub 2017/06/20. doi: 10.1016/j.trecan.2016.10.010. PubMed PMID: 28626801; PMCID: PMC5472356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, Mauri C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010;32(1):129–40. Epub 2010/01/19. doi: 10.1016/j.immuni.2009.11.009. PubMed PMID: 20079667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.