

Abstract
Background/Aim:
Hepatic fat excess in non-alcoholic fatty liver disease (NAFLD) reflects an imbalance between fat accumulation and disposal. Conflicting data exist for the role of fatty acid oxidation (FAO), one of the disposal pathways, and have mostly come from studies delivering fatty acids (FA) intravenously. Whether FAO of orally-provided FA is affected in NAFLD is unknown.
Methods:
We performed a breath-test study to measure FAO in subjects with NAFLD and healthy controls. Subjects ingested [1-13C] palmitic acid (PA, 10 mg/kg) in a liquid meal and the rate of 13CO2 appearance in expired air was measured over 6 hours by a BreathID device (Exalenz) to obtain the cumulative percent dose recovered (CPDR), the total amount of ingested 13C recovered. CPDR was corrected by the results of a [1-13C] acetate breath test, performed 1–4 weeks later, to calculate the rate of PA β-oxidation.
Results:
PA oxidation was 27% lower in 43 subjects with NAFLD compared to 11 controls (CPDR 9.5±2.4% vs. 13.1±3.7%, p=0.0001) and this persisted after correcting for acetate (29.3±10.5 vs. 36.6 ±13.9, p=0.03). The decrease in FAO was not due to delayed transit as the time to peak 13C detection did not differ between groups (4.9±1.2 vs. 4.7±0.8 hours, p=0.7). Rates of PA oxidation were not correlated with obesity, hepatic or adipose insulin resistance, ALT, liver fat content and NAFLD histology.
Conclusion:
FAO of orally-delivered FA is decreased in NAFLD compared to healthy controls, likely reflecting decreased β-oxidation. The use of a breath test offers noninvasive dynamic assessment of FAO.
Keywords: β-oxidation, stable isotope, palmitic acid, acetate
Lay summary:
We used a breath test to study the metabolism of dietary fatty acids, the building blocks of fat, in individuals with fatty liver and compared them to normal weight individuals. We found that individuals with fatty liver break down less dietary fat, which can contribute to fat storage in the liver.
Introduction
Non-Alcoholic Fatty Liver Disease (NAFLD) is estimated to affect 30 % of the US population1; it commonly co-exists with features of the metabolic syndrome (obesity, hypertension, diabetes mellitus and hypertriglyceridemia) and its rising prevalence seems to accompany the increasing rates of obesity2.
Hepatocyte triglyceride content reflects a balance between lipid acquisition (fatty acid (FA) uptake or de-novo lipogenesis and esterification) and removal (β oxidation or metabolism, and export as a component of VLDL particles). In NAFLD, insulin resistance is associated with increased de-novo lipogenesis and adipose tissue lipolysis, both driving an increase of FA availability to hepatocytes2. Fabbrini et al3 found that the increase in very low density lipoprotein (VLDL) triglyceride secretion rate in subjects with NAFLD is not able to adequately compensate for the increased rate of intrahepatic triglyceride production. Whether FA oxidation (FAO) is also dysregulated in patients with NAFLD is unclear. We aim to determine if FAO of an oral fat load is different between subjects with NAFLD and healthy normal-weight controls.
Materials and methods
Subjects and study design
Forty-three subjects with NAFLD and 11 healthy normal-weight controls were enrolled in this prospective study (clinicaltrials.gov ). The study was approved by the NIDDK/NIAMS Institutional Review Board and all subjects gave written informed consent. All authors had access to study data and approved the final manuscript.
NAFLD group
Included were adults (>=21 years) of both genders with NAFLD defined as having imaging of the liver within 6 months of enrollment consistent with excess liver fat and at least one of:
elevated transaminases (ALT>31 U/L for men or >19 U/L for women, or AST >30 U/L) or
metabolic syndrome or diabetes.
Laboratory tests were obtained within 12 weeks of the breath test. Subjects were excluded if they had a history of excessive alcohol consumption (> 30 g/d for men or > 20 g/d for women), other causes of liver disease (e.g. viral hepatitis, autoimmune or genetic liver disease), decompensated cirrhosis, advanced lung or heart disease, insulin-dependent diabetes, allergy to milk or soy, pregnancy or nursing, and history of prior gastrointestinal resection.
Healthy controls
Included were adults (>= 21years) of both genders with BMI <= 25 kg/m2 and no history of excessive alcohol consumption, medication use (except oral contraceptives), diabetes or liver disease and with normal liver transaminases and fasting glucose on screening laboratory tests.
Palmitate Breath test (PBT)
After an overnight fast, subjects were given 10mg/kg of [1-13C] palmitic acid (PA, Cambridge Isotope Laboratories, Tewksbury, MA) in a standardized heated liquid meal (Ensure®, Abbott Laboratories), to be consumed within 5 minutes. The appearance of 13C in expired CO2 was assayed by the BreathID (Exalenz Bioscience, Modiin, Israel) molecular correlation spectrometer device4,5. Subjects wore a nasal cannula connected to the device and the expired 13CO2/12CO2 ratio was measured continuously. After measurement of baseline levels, subjects ingested the test meal and measurements continued for 6 hours after that. Real-time graphical representation of the results was displayed during the test and the final results were downloaded as a computer file for off-line analysis.
During the test, subjects remained sedentary in a comfortable chair. Physical activity and talking were minimized. A 15-minute bathroom break was allowed after the first 3 hours of the test.
Acetate Correction
The rates of label appearance in expired CO2 after ingestion of labeled PA reflect the total rate of its metabolism. Although the labeled carbon can be metabolized to CO2 directly through β-oxidation and the tricarboxylic acid (TCA) cycle, there is also a possibility of label fixation during the exchange reactions of the TCA cycle and label retention in the bicarbonate pool. To control for this, all subjects also underwent an acetate breath test (ABT) to provide a correction factor for use in oxidation calculations6. ABT was performed in a similar manner to PBT with the test meal containing 2mg/kg 1[1-13C] acetate (AA, Cambridge Isotope Laboratories, Tewksbury, MA). PBT and ABT were performed 1–4 weeks apart to prevent tracer carry-over.
Dietary Assessment
To control for dietary habits, a 24-hour dietary recall was obtained by nutrition staff on the day of the study. Participants were asked to provide information on intake during the 24 hours before their visit, using a multi-pass approach to limit the extent of under reporting.
Measurement of insulin resistance
Insulin resistance was estimated by the homeostatic model assessment (HOMA-IR) calculated as plasma glucose (mg/dL) * plasma insulin (μU/mL)/4057. Adipo-IR, a measure of adipose tissue insulin resistance, was calculated as plasma free fatty acids (mmol/L) * plasma insulin (μU/mL)8. All values were collected in the fasting state.
Additional Data
Liver fat quantitation by 1H-magnetic resonance spectroscopy (MRS), liver stiffness measurement (LSM), and liver biopsies were not included in the study, but results were available for a subset of subjects who participated in other clinical trials. MRS was performed as previously described9. LSM was measured using vibration controlled transient elastography (VCTE, Fibroscan) with the M- or XL-probe as appropriate. The median result of 10 valid measurements is reported. Liver biopsies were scored according to the NASH-CRN scoring system10.
Patients were defined as having cirrhosis based on histology, LSM >= 10.3 or based on clinical and radiological findings.
Analysis and Statistical Methods
Breath test results are reported as the percent dose recovered (PDR) and cumulative percent dose recovered (CPDR). PDR reflects the rate of appearance of 13C in the expired air at any given time point, normalized to weight, height and dose. CPDR at any time point reflects the total percentage of 13C dose that was collected up to that time. The PA CPDR was divided by the AA CPDR for the same subject, to obtain the corrected PA CPDR.
Descriptive statistics are shown as frequencies or described with appropriate measures of central tendency. The Mann-Whitney test or Chi Square test were used to determine significance of the differences between the two subject groups where appropriate. Spearman rank test for correlation was used for secondary analyses of the association of the breath test results with baseline variables within the NAFLD groups. All p-values reported are two-sided with a level of significance of 0.05.
Primary Analysis
The study primary end point was the 6-hour PA CPDR. The primary analysis was the comparison of the primary end point between NAFLD subjects and controls.
Secondary Analyses
The timing of the peak PDR was compared between NAFLD subjects and controls.
Secondary post hoc analysis was done to assess the impact of other variables including age, BMI, lab results (ALT, AST, glucose, insulin, HOMA-IR, adipo-IR, triglycerides) on PA oxidation within the NAFLD group.
Results
Study participants
Forty-three NALFD subjects, with steatosis demonstrated by ultrasound, were enrolled in the study between March 2015 and May 2018. Baseline characteristics and metabolic features consistent with NAFLD shown in (Table 1). Although liver fat quantitation by 1H-magnetic resonance spectroscopy (MRS) and LSM were not included in the study, results were available for 14 (33%) and 35 (81%) of the NAFLD subjects, respectively (Table 1). Similarly, liver biopsies were not included in the study, but results were available for 11 (26%) of the NAFLD subjects, of whom 6 had evidence of steatohepatitis. Overall, 11(%) patients had compensated cirrhotic.
Table 1:
Baseline Demographic and Clinical Characteristics
| NAFLD (n=43) | Healthy (n=11) | P value | |
|---|---|---|---|
| Age (years) | 54 (42–59) | 33 (25–40) | <0.0001 |
| Male Sex (n, %) | 22 (55%) | 5 (45%) | 0.5 |
| Race (%) | 0.72 | ||
| Non-white | 26 (60%) | 6 (55%) | |
| White | 17 (40%) | 5 (45%) | |
| Hispanic ethnicity | 19 (44%) | 0 | |
| BMI (kg/m2) | 32.2 (28.5–35.3) | 21.8 (21–22.8) | <0.0001 |
| ALT (U/L) | 27 (28–64) | 16 (17–23) | <0.0001 |
| AST (U/L) | 28 (21–49) | 19 (13–19) | 0.001 |
| Platelets (K/uL) | 238 (206–250) | 221 (206–234) | 0.29 |
| Insulin (μU/mL) [n=42] | 23.8 (16.4–30.5) | 9 (5.1–10.1) | <0.0001 |
| Glucose (mg/dL) | 102 (83–162) | 88 (83–94) | 0.0003 |
| HOMA-IR [n=42] | 5.85 (4.2–7.9) | 1.97 (1.06–2.32) | <0.0001 |
| Triglyceride (mg/dL) | 142 (109–186) | 54 (44–88) | <0.0001 |
| Diabetes (n, %) | 11 (26%) | 0 | |
| Liver fat content by 1H-MRS (%) [n=14] | 16.6 (28–68.5) | NA | |
| Liver Stiffness Measurement (kPa) [n=35] | 3.5 (4.5–7.9) | NA | |
| Liver biopsy [n=11] | |||
| Steatohepatitis | 6 (55%) | NA | |
| NAFLD activity score (NAS) | 3 (2–5) | NA |
Results are expressed as median (IQR) or number (percentage). NA – not applicable
Healthy controls had normal BMI, ALT, glucose and insulin, and absence of clear metabolic abnormalities (Table 1).
Rates of oral palmitic acid oxidation
The rate of appearance of 13C in expired breath in NAFLD subjects within 6 hours after ingestion of labeled PA was 27% less in NAFLD subjects compared to healthy controls (CPDR 9.5±2.4% vs 13.1±3.7%, p=0.0001, Figure 1a–b), consistent with a lower rate of FAO.
Figure 1. Rates of PA and AA oxidation.

(A) Subjects with NAFLD have decreased recovery of 13C label from PA compared to healthy controls. (B) Cumulative 6-hour recovery of 13C from PA oxidation is significantly decreased in subjects with NAFLD. (C) Recovery of 13C label from AA is similar in both groups. (D) Cumulative 6-hour AA oxidation is similar between NAFLD and healthy controls. (E) After acetate correction, subjects with NAFLD still have significantly lower PA oxidation than healthy controls. PA – palmitic acid, AA – acetate, PDR – percent dose recovered, CPDR – cumulative percent dose recovered. Results presented as median with IQR
ABT results did not differ between groups (34.4±7.5% vs 38.1±9.7%, p=0.2, Figure 1c–d). After controlling for AA, the corrected PA 6-hour CPDR was also significantly lower in subjects with NAFLD compared to controls (29.3±10.5 vs. 36.6 ±13.9, p=0.03, Figure 1e).
Absorption of substrates
To ensure that the difference between the groups was not due to delayed absorption of PA in NAFLD subjects, time to the peak PA PDR was compared, and did not differ between groups (4.9±1.2 hours in NAFLD vs. 4.7±0.8 in controls, p=0.7, Figure 2a). AA was absorbed and metabolized more rapidly than PA and the timing of the peak of AA PDR was also similar between the groups (1.03±0.5 vs. 1.07±0.3 hours, p=0.9, Figure 2b).
Figure 2. Time to peak 13C recovery.

(A) Time to peak PA PDR is similar between subjects with NAFLD and healthy controls. (B) Time to peak AA PDR is also similar between the 2 groups. PA – Palmitic acid, AA-Acetate, PDR – percent dose recovered.
Effect of Dietary Intake
Twenty-four-hour dietary recall data showed that total energy intake in the preceding day as well as the relative macronutrient contribution to the caloric intake did not differ between NAFLD and healthy controls (Table 2). The PA 6-hour CPDR did not correlate with dietary fat intake or other macronutrients (Table 3).
Table 2:
Dietary intake and macronutrient composition in NAFLD and healthy controls
| Macronutrient | NAFLD (n=35) | Healthy controls (n=11) | P value |
|---|---|---|---|
| Total energy (kcal) | 1722 (1234–2163) | 1677 (1315–2389) | 0.9 |
| Calories from Fat (%) | 34 (29–41.8) | 35.5 (32.2–41.6) | 0.39 |
| Calories from Carbohydrate (%) | 45.5 (33.5–50.8) | 45.3 (41.2–49.2) | 0.99 |
| Calories from Protein (%) | 19 (15–23) | 15.4 (13.6–19.2) | 0.08 |
Data shown as median (IQR)
Table 3:
Correlation of dietary total energy and macronutrient intake with PA oxidation
| Macronutrient | NAFLD | Healthy Controls | ||
|---|---|---|---|---|
| Energy (kcal) | R −0.10 | P 0.56 | R −0.37 | P 0.26 |
| Fat (grams) | R −0.07 | P 0.70 | R −0.24 | P 0.46 |
| Carbohydrates (grams) | R −0.06 | P 0.73 | R −0.5 | P 0.12 |
| Protein (grams) | R 0.05 | P 0.77 | R −0.15 | P 0.65 |
Data shown are Spearman correlation coefficients between PA 6-hr CPDR and energy and macronutrient intake
Effect of metabolic syndrome on PA oxidation
There was no correlation of PA CPDR with BMI or with indices of insulin resistance - HOMA-IR and Adipo-IR. PA CPDR also did not corelate with glucose, or TG levels (Figure 3).
Figure 3. Correlation of PA oxidation with metabolic syndrome features and liver fat.

The 6-hour palmitic acid CPDR in NAFLD subjects is not correlated with (A) HOMA-IR, (B) BMI, (C) Adipo-IR, (D) Glucose, (E) Triglycerides, (F) Liver fat percent by 1H-MRS or (G) ALT, (H) LSM.
Effect of features of liver disease on PA oxidation
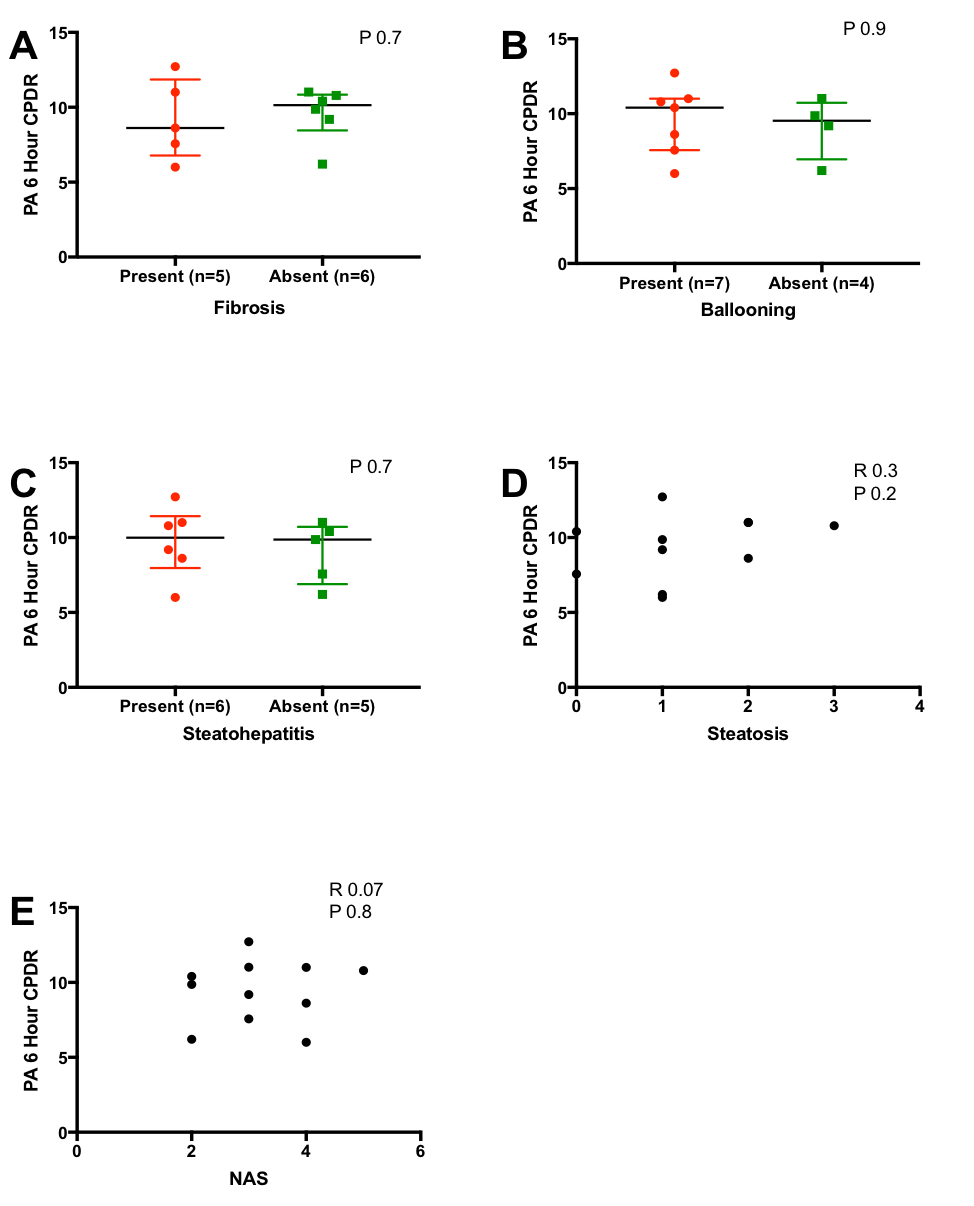
PA CPDR was not associated with ALT levels (Figure 3). There was no correlation of PA CPDR with liver fat content assessed by 1H-MRS in the 14 subjects that had available data. In the subset of subjects with available histology, PA CPDR was not associated with histological scores or with the presence of steatohepatitis, although we are underpowered in this subgroup analysis to detect a statistically-significant difference (Supplementary Figure). PA CPDR was not associated with LSM assessed by VCTE (Figure 3).
Patients with cirrhosis had PA CPDR that was not different from non-cirrhotic (6.2 vs. 9.6, p=0.2)
Effect of race and age on PA oxidation
Beyond metabolic parameters, the NAFLD cohort differed from the controls in age and Hispanic ethnicity. However, PA CPDR did not differ between Hispanic and non-Hispanic NAFLD subjects (8.8±2.5vs. 9.5±2.3, p=0.38) and did not correlate with age (R=0.2, P=0.19), ruling them out as confounders.
Discussion
In this study we aimed to determine if oxidation of an orally-delivered FA differs between NAFLD and healthy normal weight controls, utilizing an oral PBT. We found that subjects with NAFLD had a marked reduction of PA oxidation compared to healthy controls and this difference persisted after acetate correction. Reduced PA oxidation was observed irrespective of NAFLD severity.
The β-oxidation pathway is a cyclic process in which acyl-CoAs are shortened, and the two carboxy-terminal carbon atoms are released as acetyl-CoA each time a cycle is fully completed11. Acetyl-CoA can later be converted into ketone bodies (β-hydroxybutyrate or acetoacetate) or can be incorporated into the TCA for full oxidation12. Since acetate enters the TCA cycle directly as acetyl-CoA, the results of the ABT can serve to control for label fixation in the exchange reactions of the TCA and for retention in the bicarbonate pool6. By correcting the PA oxidation by the acetate correction factor, we are measuring predominantly the rates of β-oxidation.
NAFLD commonly occurs together with the metabolic syndrome13, and this accounts for many of the differences in baseline characteristics between our NAFLD subjects and healthy controls. It is thus unclear if the difference between the NAFLD and controls in PA oxidation is specific to NAFLD or due to the presence of coexisting metabolic syndrome. However, since the CPDR did not correlate with obesity (BMI), HOMA-IR, or adipose tissue insulin resistance, which are common features of the metabolic syndrome, it is likely to be specific to NAFLD. A study comparing FAO between subjects with metabolic syndrome with or without NAFLD would help answer that question and assess the impact of metabolic syndrome exclusive of NAFLD, but this would be difficult to perform. Studies have shown that the prevalence of NAFLD in subjects with metabolic syndrome can be 70–80%14, rendering it difficult to find subjects with metabolic syndrome who have absolutely no evidence of NAFLD.
To ensure that the difference in PA oxidation was not due to a delay in absorption in the NAFLD group, as subjects with insulin resistance and obesity can have impaired gastrointestinal motility, we compared the time to peak PDR between groups and found no difference that explains the decrease in FAO.
Previously Lambert et al15 described a “second meal effect” that refers to the impact of previous macronutrient consumption on the metabolism of meal triglyceride. To rule this out as a cause for our findings we evaluated meal composition in both groups and found that total caloric and macronutrient intake was similar in the 24 hours preceding the study. Furthermore, total energy and macronutrient composition did not correlate with PA oxidation.
Age and the proportion of Hispanic subjects were the only non-metabolic parameters that differed between the NAFLD and healthy control groups. The healthy controls were younger than the NAFLD group and as expected the NAFLD subjects were predominantly Hispanic. However, within the NAFLD group, age did not correlate with PA oxidation and there was no difference in FAO between Hispanics and non-Hispanics. Thus, the difference between the groups in rates of PA oxidation are not due to difference in baseline parameters.
The results of breath testing with the BreathID device are comparable to those of “classic” off-line breath testing using isotope ratio mass spectroscopy16 but allow for real-time assessment of test validity and results. The same device with a different substrate (¹³C-Urea) is approved by the FDA for the diagnosis of H. Pylori infection17.
Our study assessed total body FAO, but the actual site of oxidation that accounts for the difference between NAFLD and controls is unclear. Fatty acids can be oxidized in most tissues (except the brain and red blood cells) but three are quantitatively more important than others: adipose tissue, skeletal muscle and liver.
Given that our study hints at NAFLD as the cause for the impaired FAO, it is plausible that the difference is predominantly driven by reduction in hepatic FAO; however, this cannot be definitively proven with our study design.
There are currently no reliable methods for measuring hepatic FAO in vivo. Ketone bodies are produced mainly in liver mitochondria since few other tissues in the body are capable of ketogenesis and indirect measurement of hepatic FAO, assessed by plasma ketone body concentrations, suggests that hepatic FAO is either increased or normal in subjects with NAFLD3. However, this is an indirect measurement and could be affected by other parameters.
Mitochondrial dysfunction plays a key role in the pathogenesis of NAFLD. Data from patients with NASH show loss of hepatic mitochondrial cristae and a paracrystalline inclusion of unknown composition18–20. In vivo data in humans using various 13C-labeled substrates (methionine and ketoisocaproic acid) suggested a lower rate of mitochondrial oxidation in patients with NAFLD20–22. These results are in line with our findings.
Other studies investigating whole body and hepatic fat oxidation have reported conflicting results, which may be affected by the method of measurement. FAO has been studied using labeled fatty acids, indirect calorimetry or indirectly using measurement of plasma ketone bodies, as discussed above. In a study by Croci et al, obese/overweight patients with NAFLD showed reduced whole-body FAO compared with healthy controls when assessed with indirect calorimetry under basal, insulin stimulation (hyper-insulinemic-euglycemic clamp) and exercise23. These results are consistent with our findings. However, several studies using tracer methodology showed an increase in FAO in NAFLD subjects24,25. Most of these studies have been done using labeled FA given as intravenous infusions with frequent blood sampling and prolonged duration of testing to reach an isotopic steady state. The prolonged infusion of fatty acid to a peripheral vein at a constant rate does not reflect physiologically the fluctuating levels of fatty acids in the circulation and the complex hormonal and metabolic response to an oral source of caloric load. Furthermore, there may be inherent differences in the fate of FA derived from peripheral adipose tissue (mimicked by an IV infusion) and those derived from an oral load. Although PBT with oral loading has been used in the past to evaluate FAO in healthy subjects26, to the best of our knowledge our study is the first to utilize it to assess FAO in subjects with NAFLD and compare them to healthy controls.
The strengths of our study include firstly, the measurement of FAO in a physiologic manner by providing the substrate orally in a mixed meal. Secondly, we applied stringent selection of healthy controls by using strict ALT and BMI cutoffs to avoid inadvertent recruitment of NAFLD subjects in this group27. Thirdly, unlike isotopic ratio mass spectroscopy in which breath samples are collected intermittently, the BreathID device continuously senses exhaled breath in real-time, providing continuous data measurement throughout the 6-hour study.
An important limitation of our study is that not all subjects with NAFLD had liver biopsies or liver fat quantification by H1-MRS. This limits our ability to infer a correlation between liver fat content and FAO and to assess whether FAO differs between NAFL and non-alcoholic steatohepatitis. Future studies may address this.
Although the healthy controls were not required to have liver imaging to prove absence of steatosis, their likelihood of having it is very low given the strict exclusion criteria27. To validate this assumption, we examined available data from the NHANES III survey. We identified 539 participants with interpretable ultrasound who met our healthy control criteria by reporting excellent health, taking no medications, drinking <= 30 drinks/month, having normal BMI and ALT, and no evidence for viral hepatitis, diabetes, hypertension or stroke. Only 7% of these participants had significant steatosis. Furthermore, 7 (64%) of the healthy controls in our study had liver ultrasounds in another clinical trial, and all were negative for steatosis. Thus, we believe these subjects are adequate as healthy controls
Secondly, it is possible that full oxidation was not captured during the 6-hour study period. Additionally, we evaluated a single fatty acid, and results of PA oxidation might not be generalizable to oxidation of other FA or triglycerides. However, palmitic acid has been the standard tracer used in FAO studies. Lastly, our study is specifically underpowered for H1-MRS and liver biopsy data analysis as this was not part of the study design.
In conclusion, we found that subjects with NAFLD have decreased oxidation of orally-delivered palmitic acid, likely reflecting decreased β-oxidation. The use of a breath test offers a noninvasive dynamic tool for the assessment of fatty acid oxidation.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements:
We would like to acknowledge Jean-Pierre Raufman MD (NIDDK T32 training grant program director at University of Maryland), Sara Turner and Amber Courville (NIH department of clinical nutrition), Exalenz Bioscience (James McLoughlin and Avraham Hershkowitz M.Sc.) for technical support, Cythia Moylan MD and Greg Samsa PhD (Duke Master’s Program committee) for manuscript feedback.
Financial support: This study was supported by the Intramural Research Program of NIDDK. GN was supported by T32 DK067872.
List of Abbreviations:
- NAFLD
Non-alcoholic fatty liver disease
- FAO
fatty acid oxidation
- PA
palmitic acid
- CPDR
cumulative percent dose recovered
- AA
acetate
- FA
fatty acid
- PBT
palmitate breath test
- TCA
tricarboxylic acid cycle
- ABT
acetate breath test
- PDR
percent dose recovered
- LSM
liver stiffness measurement
- VCTE
vibration controlled transient elastography
Footnotes
Clinical Trial Number:
Conflicts of interest: All authors have no conflicts of interest to disclose
References
- 1.Argo CK, Caldwell SH. Epidemiology and natural history of non-alcoholic steatohepatitis. Clin Liver Dis. 2009;13(4):511–531. [DOI] [PubMed] [Google Scholar]
- 2.Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48(4):434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51(2):679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmilovitz-Weiss H, Niv Y, Pappo O, et al. The 13C-caffeine breath test detects significant fibrosis in patients with nonalcoholic steatohepatitis. J Clin Gastroenterol. 2008;42(4):408–412. [DOI] [PubMed] [Google Scholar]
- 5.Stravitz RT, Reuben A, Mizrahi M, et al. Use of the methacetin breath test to classify the risk of cirrhotic complications and mortality in patients evaluated/listed for liver transplantation. J Hepatol. 2015;63(6):1345–1351. [DOI] [PubMed] [Google Scholar]
- 6.Bergouignan A, Schoeller DA, Votruba S, Simon C, Blanc S. The acetate recovery factor to correct tracer-derived dietary fat oxidation in humans. Am J Physiol Endocrinol Metab. 2008;294(4):E645–653. [DOI] [PubMed] [Google Scholar]
- 7.Gutierrez-Buey G, Nunez-Cordoba JM, Llavero-Valero M, Gargallo J, Salvador J, Escalada J. Is HOMA-IR a potential screening test for non-alcoholic fatty liver disease in adults with type 2 diabetes? Eur J Intern Med. 2017;41:74–78. [DOI] [PubMed] [Google Scholar]
- 8.Sondergaard E, Espinosa De Ycaza AE, Morgan-Bathke M, Jensen MD. How to Measure Adipose Tissue Insulin Sensitivity. J Clin Endocrinol Metab. 2017;102(4):1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reeder SB, Cruite I, Hamilton G, Sirlin CB. Quantitative Assessment of Liver Fat with Magnetic Resonance Imaging and Spectroscopy. J Magn Reson Imaging. 2011;34(4):spcone. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander-Tetri BA, Network NCR. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology. 2011;53(3):810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33(5):469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25(1):42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lallukka S, Yki-Jarvinen H. Non-alcoholic fatty liver disease and risk of type 2 diabetes. Best Pract Res Clin Endocrinol Metab. 2016;30(3):385–395. [DOI] [PubMed] [Google Scholar]
- 14.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. [DOI] [PubMed] [Google Scholar]
- 15.Lambert JE, Parks EJ. Postprandial metabolism of meal triglyceride in humans. Biochim Biophys Acta. 2012;1821(5):721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goetze O, Selzner N, Fruehauf H, Fried M, Gerlach T, Mullhaupt B. 13C-methacetin breath test as a quantitative liver function test in patients with chronic hepatitis C infection: continuous automatic molecular correlation spectroscopy compared to isotopic ratio mass spectrometry. Aliment Pharmacol Ther. 2007;26(2):305–311. [DOI] [PubMed] [Google Scholar]
- 17.Shirin H, Kenet G, Shevah O, et al. Evaluation of a novel continuous real time (13)C urea breath analyser for Helicobacter pylori. Aliment Pharmacol Ther. 2001;15(3):389–394. [DOI] [PubMed] [Google Scholar]
- 18.Caldwell SH, Swerdlow RH, Khan EM, et al. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. 1999;31(3):430–434. [DOI] [PubMed] [Google Scholar]
- 19.Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183–1192. [DOI] [PubMed] [Google Scholar]
- 20.McCullough A, Previs S, Kasumov T. Stable isotope-based flux studies in nonalcoholic fatty liver disease. Pharmacol Ther. 2018;181:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banasch M, Ellrichmann M, Tannapfel A, Schmidt WE, Goetze O. The non-invasive (13)C-methionine breath test detects hepatic mitochondrial dysfunction as a marker of disease activity in non-alcoholic steatohepatitis. Eur J Med Res. 2011;16(6):258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Portincasa P, Grattagliano I, Lauterburg BH, Palmieri VO, Palasciano G, Stellaard F. Liver breath tests non-invasively predict higher stages of non-alcoholic steatohepatitis. Clin Sci (Lond). 2006;111(2):135–143. [DOI] [PubMed] [Google Scholar]
- 23.Croci I, Byrne NM, Choquette S, et al. Whole-body substrate metabolism is associated with disease severity in patients with non-alcoholic fatty liver disease. Gut. 2013;62(11):1625–1633. [DOI] [PubMed] [Google Scholar]
- 24.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14(6):804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iozzo P, Bucci M, Roivainen A, et al. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology. 2010;139(3):846–856, 856 e841–846. [DOI] [PubMed] [Google Scholar]
- 26.DeLany JP, Windhauser MM, Champagne CM, Bray GA. Differential oxidation of individual dietary fatty acids in humans. Am J Clin Nutr. 2000;72(4):905–911. [DOI] [PubMed] [Google Scholar]
- 27.Takyar V, Nath A, Beri A, Gharib AM, Rotman Y. How healthy are the “Healthy volunteers”? Penetrance of NAFLD in the biomedical research volunteer pool. Hepatology. 2017;66(3):825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.