

Abstract
Tumor-associated macrophages (TAMs) promote triple-negative breast cancer (TNBC) progression. Here, we report BRCA1-IRIS overexpressing (IRISOE) TNBC cells secrete high levels of GM-CSF in a HIF-1α- and a NF-κB-dependent manner to recruit macrophages to IRISOE cells and polarize them to pro-tumor M2 TAMs. GM-CSF triggered TGF-β1 expression by M2 TAMs by activating STAT5, NF-κB and/or ERK signaling. Despite expressing high levels of TGF-β1 receptors on their surface, IRISOE TNBC cells channeled TGF-β1/TβRI/II signaling towards AKT, not SMAD, which activated stemness/EMT-phenotypes. In orthotopic and syngeneic mouse models, silencing or inactivating IRIS in TNBC cells lowered the levels of circulating GM-CSF, suppressed TAM recruitment, and decreased the levels of circulating TGF-β1. Co-injecting macrophages with IRISOE TNBC cells induced earlier metastasis in athymic mice accompanied by high levels of circulating GM-CSF and TGF-β1. IRISOE TNBC cells expressed low levels of calreticulin (the “eat me” signal for macrophages) and high levels CD47 (the “don’t eat me” signal for macrophages) and PD-L1 (a T-cell inactivator) on their surface. Accordingly, IRISOE TNBC tumors had significantly few CD8+/PD-1+ cytotoxic T-cells and more CD25+/FOXP3+-regulatory T cells. These data show that the bi-directional interaction between IRISOE cells and macrophages triggers an immunosuppressive microenvironment within TNBC tumors that is favorable for the generation of immune-evading/stem-like/IRISOE TNBC metastatic precursors. Inhibiting this interaction may inhibit disease progression and enhance patients’ overall survival.
Keywords: BRCA1-IRIS, triple-negative breast cancer, metastasis, tumor-associated macrophages, GM-CSF, TGF-β1, SMAD, PD-L1, CD47, Calreticulin
Introduction
Evading immune system destruction is an effective strategy for cancer progression. This tolerogenic environment induced by factors released from tumor cells expands pro-tumor myeloid cells, such as macrophages (1). Tumor-associated macrophages (TAMs) can be polarized to anti-tumor, M1 macrophages that express metabolic enzymes; e.g., iNOS, cytokines; e.g., IL-12, chemokines; e.g., CXCL10, and transcription factors; e.g., IRF5 (2), or pro-tumor, M2 macrophages that express metabolic enzymes; e.g., ARG1, cytokines; e.g., IL-10, chemokines; e.g., CCL17, and transcription factors; e.g., IRF4 (1,2).
Granulocyte-macrophage colony-stimulating factor (GM-CSF) stimulates proliferation and survival of macrophages, neutrophils, eosinophils, dendritic cells, and microglia (3). GM-CSF also affects mature hemopoietic cells through cytokines production, antigen presentation, and phagocytosis (4,5). In mice and human, increased circulated GM-CSF level indicates late-stage tumors and/or tumor progression (6). At low-level, GM-CSF exerts an anti-tumor effect by activating dendritic cells (DCs) within the tumor, while at high-level, GM-CSF activity is mainly immunosuppressive, enriching tumor microenvironment of M2 TAMs (7).
Increased expression of the CD47 on cancer cell transduces a “don’t eat me” inhibitory signal through activating signal regulatory protein alpha (SIRPα) on macrophages (8). Preclinical models showed anti-CD47 therapies stimulate phagocytosis of cancer cells, in vitro, and anti-tumor immune responses, in vivo (8). In contrast, calreticulin expression at the cancer cell surface promotes phagocytosis of cancer cells by engaging the LRP1 receptor on macrophages (9).
The immunoinhibitory receptor, PD-1, is predominantly expressed on activated effector T-cells (10), or antigen-specific T-cells chronically exposed to the antigen (11). PD-1 ligands; PD-L1 is expressed on hematopoietic and cancer cells, and PD-L2 is expressed on macrophages and dendritic cells (12). PD-L1 on aggressive tumor cells engagement of PD-1 on T-cells leads to T-cell exhaustion (aka. anergy); including inhibition of proliferation and cytokines; e.g., IL-2 and IFN-γ production, and apoptosis resulting in tumor cells evasion of recognition/destruction by the immune system (13,14). Anti-PD-1 or anti-PD-L1 are now successful cancer immunotherapy that stimulates the adaptive immune system to attack cancer cells (14).
BRCA1-IRIS (aka IRIS, for In-frame Reading of Intron 11 Splice variant) is an oncogene produced by the alternative usage of the BRCA1 locus (15). IRIS expression is high in breast cancers, especially TNBCs (16). Deliberate IRIS overexpression (IRISOE) in normal human mammary epithelial (HME) cells or luminal A/ER+ cells converts them into genuine TNBC cells expressing basal, epithelial-to-mesenchymal (EMT) and stemness markers, and lacking ERα and BRCA1 proteins expression, in vitro and in vivo (17,18). HME/IRISOE cells formed genuine TNBCs in SCID mice (16,19), including the presence of a necrotic/hypoxic/inflamed cores within them (16,20). We recently named this core “the aggressiveness niche” (20), and showed experimental evidence that IRISOE TNBC metastatic precursors develop within this aggressiveness niche (19,21,22).
Here, we show that GM-CSF secretion from IRISOE cells activates STAT5, NF-κB, and ERK signaling in TAMs to enhance their proliferation, recruitment, survival, M2-polarization, and expression/secretion of TGF-β1. Inhibiting GM-CSF signaling attenuated TAMs recruitment, M2-polarization, and decreased the immunosuppressive ability of IRISOE cells leading to significantly reduced aggressiveness and regression of IRISOE tumors through an activated adaptive immune response.
Materials and Methods
Cell culture.
Mammary cell lines used here were from ATCC and maintained as previously described (19). The doxycycline (Dox)-inducible IRISOE cell lines (IRISOE1-5) generation and maintenance were described earlier (15). Orthotopic injection of these cell lines in SCID mice and maintaining mice on Dox-supplemented drinking-water led to primary (1°) orthotopic IRISOE mammary tumors. Few of these tumors used to develop 1° orthotopic IRISOE breast cancer cell lines “IRIS291, IRIS292, and IRIS293” used in the experiments described herein (16,19). These cell lines are maintained in Dox-supplemented RPMI 1640 medium containing 10% fetal bovine serum (FBS). IRIS291 and IRIS293 clones expressing Red Fluorescent Protein developed by a viral infection of a mCherry-expressing cDNA and antibiotic selection. Human THP1s from ATCC maintained in PRMI/10% FBS in our laboratory. Authentication of all commercial and in-house cell lines done using STR profiling and also tested for mycoplasma contamination.
Antibodies, recombinant proteins and drugs.
Mouse monoclonal (mono) anti-human IRIS and rabbit polyclonal (poly) anti-mouse Iris antibodies developed in our laboratory. Mouse (m) anti-human (h) GM-CSF (R&D #3209), Rabbit (Rb) poly-anti-m GM-CSF (ab9741), m mono-anti-h HIF-1α (Novusbio, NB100-105), Rb poly-anti-h p-p65/NF-κB (Cell Signaling, #3033), Rb clonal-anti-h γ-Tubulin (abcam, ab11321), Rb mono-anti-h Actin (Cell signaling, 13E5), Rat mono-anti-h GM-CSF Rα (R&D, #698423), m mono-anti-h and -m TGF-β1 (R&D, #9016), Rb poly-anti-h TβRII (Y424, sc-17007-R), Rb poly-anti-h Cyclin D1 (Thermo, #RB-010-P0), Rb poly-anti-h p-SMAD2 (S465, S467, Thermo, 44-244G), Rb mono-anti-h p-SMAD3 (S423, S425, Cell Signaling, C25A9), m mono-anti-m and -h TGF-β1 (Bio-Cell, 704719J1), FITC conjugated Rat mono-anti-h- and m-CD11b and isotype control (Cell signaling, #24442, #56722, respectively), F4/80 Rat mono-anti-m F4/80 (abcam, ab6640), PE conjugated mono-m anti-h CD206 and isotype control (Invitrogen, 12-2069, P3.6.2.8.1, respectively), Rat mono-anti-m CD25 (abcam, ab210333), Rb mono-anti-h and -m FOXP3 (abcam, ab215206), Rat mono-anti-m CD8 (BioLegend, 100702), Rb poly- anti-h Calreticulin (R&D, #681233), Rb mono-anti-h PD-L1 (abcam, ab251611).
Human recombinant (r)GM-CSF from R&D (7954-GM), reconstituted in sterile PBS to a stock concentration of 100μg/ml and used at 50ng/ml. Human rEGF from Cell Signaling (#8916), reconstituted in sterile PBS to a stock solution of 100μg/ml, and used at 10ng/ml. Human rTGF-β1 from R&D (7754-BH), reconstituted in 4mM HCl to a stock concentration of 100μg/ml, and used at 5ng/ml. Vehicles for these recombinant proteins were PBS and/or 0.2μM HCl.
The NF-κB inhibitor “JSH-23” from Sigma (#J4455, 10μM working concentration), the STAT5 inhibitor “IQDMA” from Abcam (ab141192, 10μM working concentration), the NF-κB inhibitor “Bay 11-7082” from Sigma (B5556, 10μM working concentration), the ERK inhibitor “PD98059” from Calbiochem (#51300, 10μM working concentration), the PI3`K inhibitor “LY294002” from Calbiochem (#440202, 10μM working concentration), the SMAD3 inhibitor “SIS3 from Calbiochem (#566405, 10μM working concentration), the TβRI/II inhibitor “LY2109761 from Santa Cruz (700874-71-1, 5μM working concentration).
Cytokine array.
Conditioned media (CM) from equal numbers from IRISOE cell lines (IRIS1-IRIS5) grown in the absence (i.e., naïve HME) or the presence (i.e., HME/IRIS) of Dox plated in serum-free medium for 20h used to screen for cytokine, chemokine and growth factors levels on antibody arrays (RayBio, Norcross, GA, USA). Assay performed according to the manufacturer’s instructions and as (23) repeated on 3 different preparations (i.e., n=3 for each cell line in the presence or absence of Dox).
siRNA transfection.
Naïve HME, IRIS291, and IRIS293 cells seeded at a density of 3X105 cells/well in a 6-well plate for 16-18h transiently transfected with siLuc siRNA, siHIF1α (using two different siRNA), siGM-CSF, siTGF-β1, siSMAD5, sip65, siERK or siIRIS using Xfect™ Transfection reagent (Clonetech) according to the manufacturer’s instructions. After 48h, media was changed, and cells exposed to normoxia (20% O2) or hypoxia (1% O2, using Hypoxia Chamber [STEMCELL technologies, #27310]) for an additional 24h, at which time CM collected for ELISA analysis and total proteins for western blot. At least n=3 for each assay was performed.
Conditioned media transfer experiment.
Briefly, CM from normoxic or hypoxic (24h) naïve HME, IRIS291, IRIS292, or IRIS293 directly analyzed for secreted factors using ELISA or receptors expression using membrane preparations. The CM following certain siRNA transfection or hypoxia was added onto THP1 cells. Secretion from THP1 assessed using ELISA 24h later, and receptor expression on the surface determined using western blot of membrane extracts. At all steps, equal numbers of each cell type were seeded to avoid discrepancies due to cell number variations. At various steps in this protocol, specific NeuAb and/or drugs were added, as indicated in the Results section.
Cytokine ELISA.
Co-cultures CM or mice sera diluted in carbonate coating buffer (pH 9.6) was used to coat 96-well ELISA plates overnight at 4°C. Plates then washed three times with PBST (phosphate-buffered saline - 0.05% Tween-20), blocked with 2% bovine serum albumin for 1h at room temperature (RT) were incubated with primary antibody diluted in blocking solution for 2h at room temperature (RT). After washing primary antibodies, HRP-conjugated secondary antibodies were added for 1h at RT, and the reaction determined using Western Lightning Plus-ECL (PerkinElmer, Waltham, MA, USA) as a substrate. All experiments done in triplicates performed three separate times and shown as mean ± SD.
Co-culture experiment.
Boyden chambers (BD biosciences) of 8μm pore size (for migration) or 0.4μm-pore size (for secretome) analysis used to layer IRISOE cells in the lower chamber with or without neutralizing antibodies, and the THP1 cells in the transwell inserts. THP1s migration to the lower face of these inserts was counted and plotted 24-72h later. Co-cultures were performed under normoxic or hypoxic conditions, in triplicates 3 separate times.
Western blot.
Performed, as previously described (23). Protein lysates prepared from cell sonication (in PBS containing protease and phosphatase inhibitor, Thermo Scientific) or membrane fraction preparation were prepared. Protein concentrations estimated using Pierce™ BCA protein assay kit (Thermo Scientific) and 25μg of proteins denatured in NuPAGE LDS sample buffer (Thermo Scientific) were resolved on NuPAGE gels (Thermo Scientific). Proteins were electro-transferred to PVDF membranes that were blocked with 5% dry milk for 1h, washed five times (10 minutes/each) with PBS-T. Incubation with primary antibody was always done overnight at 4°C when membranes were again washed five times (10 min/each) with PBS-T and exposed to suitable HRP-conjugated secondary antibodies for 1h at RT. Washed membranes were developed using Western Lightning Plus-ECL as a substrate. Tubulin or actin was used as an internal loading control. All westerns were done at least three separate times.
Immunohistochemistry (IHC).
IHC analysis, as previously described (23), was done on 4μm-thick paraffin-embedded sections of tumor tissue excised from IrisOE syngeneic mammary tumors generated in BALB/c mice. After deparaffinization and rehydration, antigen retrieval for Iris was performed using pepsin (10μM) for 20 minutes at 37°C, whereas for all other antigens by boiling the slides in citrate buffer (pH 6.0) for 10 minutes in a microwave. Cooled slides were washed in PBS (3x15 minutes each), incubated in 3% hydrogen peroxide (H2O2) for 10 minutes to block endogenous peroxidase activity unless fluorescence analyses were performed. Following washing, slides were blocked with 10% normal goat serum for 1h at RT and probed with primary antibodies overnight at 4°C in a moist chamber. Slides were then incubated with suitable horseradish peroxidase (HRP), or Alexa Fluor conjugated secondary antibodies for 1h at RT (depending on the analysis). After washing, HRP-conjugated secondary antibody-treated slides were developed using Vector DAB substrate kit (Vector Laboratories) and counterstained with Meyer’s hematoxylin (Thermo Scientific) for two minutes, washed, dehydrated and mounted with Permount (Thermo Fisher Scientific). Alternatively, slides that were stained with Alexa Fluor conjugated secondary antibody were counterstained and mounted with VECTASHIELD mounting medium for fluorescence with DAPI (Vector Laboratories) and were imaged under the microscope. All experiments were done on at least 5 tumors from phenotypes or treatments.
RT/PCR and Real-time quantitative RT/PCR.
One hundred ng of total RNA was processed for quantitative RT-PCR (qRT-PCR) using iScript™ One-Step RT-PCR kit with SYBR Green (Bio-Rad) using EGFR mRNA, Forward: 5′-CCAGGACCCCCACAGCACTGCAGTGGGCAA-3′; Reverse: 5′-GTGGGTCTAAGAGCTAATGCGGGCATGGCT-3′, CK5 mRNA, Forward: 5′-GCGGTTCCTGGAGCAGCAGAACAAGGTTCT-3′, Reverse: 5′-CTGAGGTGTCAGAGACATGCGTCTGCATCT-3′, CK17 mRNA, Forward: 5′-CTGGCTGCTGATGACTTCCGCACCAAGTTT-3′; Reverse: 5′-CGCAGTAGCGGTTCTCTGTCTCCGCCAGGT-3′, CDH2 mRNA, Forward: 5′-ACAGTGGCCACCTACAAAGG-3′; Reverse: 5′-CCGAGATGGGGTTGATAATG-3′, Twist mRNA: Forward: 5′-GGAGTCCGCAGTCTTACGAG-3′; Reverse 5′-TCTGGAGGACCTGGTAGAGG-3′, SnailI mRNA, Forward 5′-CCTCCCTGTCAGATGAGGAC-3′; Reverse 5′-CCAGGCTGAGGTATTCCTTG-3′, Sox2 mRNA, Forward: 5′-TTCATCGACGAGGCTAAGCGGCTG-3′; Reverse: 5′-AGCTGCCGTTGCTCCAGCCGTTCA-3′, Oct4 mRNA, Forward: 5′-ACATGTGTAAGCTGCGGCC-3′; Reverse: 5′-GTTGTGCATAGTCGCTGCTTG-3′, Nanog mRNA, Forward: 5′-ATGCCTCACACGGAGACTGT-3′; Reverse: 5′-AGGGCTGTCCTGAATAAGCA-3′. The following primers for β-actin mRNA (internal control), Forward: 5′-ACAGAGCCTCGCCTTTGC-3′; Reverse: 5′-GCGGCGATATCATCATCC-3′ were used to normalize with expression and data are correlated to the calibrator sample.
The qPCR reactions were performed in a final volume of 25μl. The mean Ct was calculated for each sample and used to determine the ΔCt for this sample as follows: ΔCT = Ct for the gene of interest - Ct of the internal control gene (β-actin). Then the ΔΔCT was calculated as follows: ΔΔCT = [(Ct for the gene of interest - Ct of the internal control gene, β-actin) for sample A - (Ct for the gene of interest - Ct of the internal control gene (β-actin) for sample B], where sample B is the calibrator. For the statistical analysis, the ΔΔCT and not the raw Ct data were used (24). All were done in triplicates performed 3 separate times.
Fluorescence-activated cell sorting (FACS).
Single-cell suspensions from the in vivo treated tumors were processed for FACS analysis. One million cells were stained with specific fluorescently labeled antibodies on ice for 1h. Cells were then washed three times with FACS buffer (1% BSA in PBS) by centrifugation at 2000 rpm at 4°C for 10 minutes, and further incubated with FITC-conjugated secondary antibody for 30 minutes in ice. Cells were washed and then analyzed for surface staining on Gallios Flow Cytometer (Beckman Coulter, Pasadena, CA, USA). For sorting, Moflo XDP cell sorter (Beckman Coulter) was used. The data were analyzed with Kaluza Flow Cytometry Analysis Software v 1.2. Each FACS was performed 3 separate times in triplicates with identical results.
Orthotopic and Syngeneic mammary models.
All animal experiments were approved by the “Institutional Animal Care and Use Committee” (IACUC) of the University of Mississippi Medical Center and in accordance with the NIH guidelines. Athymic or BALB/c (6-8 weeks old) female mice (numbers are indicated in Results section) were injected in 2° left thoracic mammary fat pad with orthotopic 1° IRISOE mammary tumor cell lines; IRIS291 or IRIS293 (also RFP-expressing variants), or the human IRISOE/TNBC cell lines; MDA-MB-231 (hereafter, MDA231) or MDA-MB-468 (hereafter, MDA468) cell lines (17,20–22) expressing shCtrl or shIRIS, or the mouse TNBC cell line; 4T1 expressing shCtrl or shIris1 or shIris2, admixed or not with human THP1-macrophages (at 10:1 ratio). After IRISOE tumors reached a tumor volume indicated in figures and text, randomized mice were divided into groups that were injected intratumorally with vehicle or IRISpep four times every third day for two weeks. Tumor formation or death was monitored and recorded, as indicated in text and figures. A digital caliper was used to measure tumors every third day, and volume was calculated using the formula: volume = (length x width2)/2. Endpoints were death or sacrifice of mice and a collection of tumors and peripheral blood. Excised tumors were divided into several portions to flash-frozen, generate DNAs, RNAs, and proteins, prepare single cells as described in (25) for FACS analysis as indicated, or paraffin-embedding, sectioning (at 4μm) and IHC staining as described above.
Human samples staining and analysis.
Kaplan Meier Plotter was used to delineate the association between IRIS gene expression and disease-free survival (DFS) or overall survival (OS). Within each cohort, high expresser and low expresser patients were analyzed and compared for their OS and DFS, respectively. Breast tissue microarrays comprised of normal, DCIS, invasive, and metastatic samples were purchased from US Biomax, Inc. IHC protocols were described earlier (16). A semi-quantitative scoring system was used to identify the percentage of tumor cells showing positive staining (26). Scoring represents the overall stain intensity and percentage of cancer cells stained in 4 high magnification fields of each sample. Average overall staining intensity (27) was valued as percentage of cell stained/field: zero (<1% staining) was considered very weakly stained; 1 (1–10% staining) was considered weakly stained; 2 (10%–25% staining) was considered medium stained; 3 (25-50% staining) was considered strongly stained, and 4 (>50% staining) was considered very strongly stained. The positive staining scoring method is subjective, and artifacts such as high background or variable stain deposition can skew the results, and the scores for the two categories remain as separate functions and cannot be combined for analysis and comparison (28).
The study also involves a cohort of 96 breast cancer patients with primary invasive ductal carcinomas recently diagnosed and treated at the National Cancer Institute (NCI), Cairo University, (Cairo, Egypt) between September 2009 and October 2012 were included in the study as recently reported by us (29). None of the patients showed metastasis at the time of the initial diagnosis. Expression of ER, PR, and HER-2/neu was assessed in all tumor samples. Based on this analysis 43 of the patients were negative for all three markers and thus considered TNBCs [mean age of 51.91 ± 12.34 SD, range: 30–78 years] and 53 showed expression of some/all of the markers and thus were considered non-TNBCs [mean age of 52.77 ± 12.13 SD, range: 27–81 years]. Twenty normal breast tissue samples obtained from reduction mammoplasty (mean age 35 ± 13.94 SD; range, 22–64 years) were included as controls in the study. WHO classification of breast tumors was used to grade the tumors, and American Joint Committee on Cancer’s Staging Manual (7th edition) was used to stage the tumors (30). All participants signed written informed consent before enrollment in the study that was approved by the Institutional Review Board (IRB) of the NCI, Cairo, Egypt, according to the 2011 Helsinki Declaration, CIOMS, Belmont Report, U.S. Common Rule. Patients enrolled in the study were ≥18 years old, and none of the patients were pregnant or breast-feeding, had an active second malignancy, or were involved in another clinical trial were excluded from the study. The median follow-up period was 33 months.
Statistical analysis.
Statistical analysis was performed using unpaired, two-tailed Student’s t-test. In all figures, data represents the mean from at least three separate biological repeats done in at least triplicates each +/− SD, *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
IRIS expression in breast cancers.
Immunohistochemical (IHC) analysis of paraffin-embedded breast cancer tissue microarrays (TMA, n=511, Biomax.us®) with a monoclonal anti-IRIS antibody (16) showed low expression in normal/near cancer tissues (n=66, Suppl. Fig. 1A1), increased in the DCIS tumors (n=180, Suppl. Fig. 1A2), increased further in invasive tumors (n=100, Suppl. Fig. 1A3) to reach the highest level in metastatic tumors (n=165, Suppl. Fig. 1A4).
IHC staining of another TMA (n=326, all subtypes, Hawaiian SEER collection) with anti-IRIS, -AKT1, -AKT2, or -p-AKT specific antibodies (16) showed 84% (274/326) of the tumors were IRISOE (i.e., express ≥2-fold over normal tissues). Among those, 64% (176/274, p-value 0.012), 65% (180/274, p=0.006), 68% (188/274, p=0.030) stained positive for AKT1, AKT2, and p-AKT, respectively (Suppl. Fig. 1B, left) (16). In a TNBC sub-cohort (n=72), 88% (63/72) were IRISOE tumors (16). Spearman correlation coefficient (r) analysis showed r=0.748 (p=0.00043) for IRIS and AKT1 or AKT2 overexpression, and r=0.834 (p=0.0038) for IRIS and p-AKT overexpression (Suppl. Fig. 1B, right).
We re-assessed an Egyptian breast cancer cohort (n=96) that contained 45% TNBC patients and 55% non-TNBC patients (29). Among the TNBC group, 65% (28/43), while 28% (15/53) among the non-TNBCs group were IRISOE patients (29). Only in the TNBC group, DFS (p=0.05) and OS (p=0.04) were significantly different between IRISOE and IRISlow patients (compare right to left, Suppl. Fig. 1C). Together, suggest that in TNBCs, the increases in IRIS and AKT1/2 expression/activation correlate with enhanced breast cancer aggressiveness, and reduced DFS and OS.
GM-CSF recruits TAMs to IRISOE cells.
Re-analysis of our recent antibody array screening for IRISOE cells secretome showed that compared to HME cells, CM of IRISOE cells contained >18-fold higher GM-CSF (CSF2, Fig. 1A), while same levels CSF3 (Fig. 1A). Recently, we proposed that the necrotic/hypoxic/inflamed core within an IRISOE TNBC tumor represents an “aggressiveness niche” (20), where IRISOE TNBC metastatic precursors are developed through bi-directional interactions between IRISOE TNBC cells and activated stromal cells, e.g., MSCs and TAMs (21,22).
Figure 1. GM-CSF from IRISOE cells recruits TAMs.

(A) Representative antibody array of indicated CM (n=3/each). (B) HIF-1α expression in indicated cells and conditions. (C) GM-CSF ELISA in CM of indicated cells conditions (n=3, in triplicates). (D) Level of p-p65/NF-κB in indicated cells and treatments. (E) GM-CSF ELISA of indicated cells and treatments (n=3, in triplicates. (F-L) Migration in Boyden chambers of indicated THP1s towards CM of indicated cells ± GM-CSF NeuAb (n=3, in triplicates). Scale bar=250μm (F), =50μm (G-L). (M) quantification of data in F-L. (N) Expression of CSF2R in indicated cells. (O) A schematic representation of the data presented above.
To evaluate the aggressiveness niche microenvironment role in GM-CSF production and secretion, siLuc- or siHIF-1α-transfected HME, IRIS291, and IRIS293 cells (48h) were exposed to normoxia (20% O2) or hypoxia (1% O2, for 24h). First, as we recently reported even under normoxic conditions, IRISOE cells maintain high-levels of HIF-1α (Fig. 1B). Secondly, ELISA analysis showed that normoxic siLuc-transfected HME cells secrete low-levels of GM-CSF (Fig. 1C). Hypoxia and HIF-1α silencing didn’t alter that (Fig. 1C). In contrast, normoxic siLuc-transfected IRIS291 or IRIS293 cells secrete ~5-fold higher GM-CSF that increased under hypoxia to 15-20-fold (Fig. 1C). HIF-1α silencing (Fig. 1B) in IRISOE cells decreased GM-CSF secretion in normoxic and hypoxic cells (Fig. 1C). It will be essential to assess the role of HIF-2α (31) in this process in the near future.
Inflammation activates NF-κB to promote the expression of cytokines (32). Compared to vehicle-treated HME, IRIS291, and IRIS293 cells, those treated with 10μM JSH-23 (inhibits NF-κB transcriptional activity) (33) showed significantly lower p-p65/NF-κB level (Fig. 1D). This led to significantly decreased GM-CSF secretion from IRIS291 and IRIS293 cells according to ELISA analysis (Fig. 1E).
Phorbol 12-myristate-13-acetate (PMA) treatment triggers differentiation of monocytic THP1 cells into none-polarized (M0) macrophages. We layered PMA-untreated or PMA-treated THP1s in 8μm inserts of Boyden chambers containing HME, IRIS291, or IRIS293 cells CM in the lower well (media changed daily). Compared to HME CM, IRIS291 or IRIS293 cells CM recruited 400-600% more PMA-untreated THP1s (72h later) that was blocked by GM-CSF neutralizing antibody (NeuAb, Fig. 1F). Moreover, HME CM recruited very low number of PMA-treated THP1s (24h later) that was not affected by GM-CSF NeuAb (compare Fig. 1G to 1H). In contrast, IRIS291 CM recruited significantly higher numbers of PMA-treated THP1 (compare Fig. 1I to 1G) that was blocked by GM-CSF NeuAb (compare Fig. 1J to 1I), Similarly, IRIS293 CM recruited significantly higher numbers of PMA-treated THP1 (compare Fig. 1K to 1G) that was also blocked by GM-CSF NeuAb (compare Fig. 1L to 1K). Quantitative analysis of these effects are presented in (Fig. 1M). Interestingly, compared to HME cells, IRISOE cells express ~3-fold higher of the GM-CSF receptor, CSF2Rα (Fig. 1N). Compared to M1 polarized THP1s, M2 polarized express >15-fold higher of CSF2Rα (Fig. 1N).
Taken together, these data suggest that: (1) in the aggressiveness niche, IRISOE stabilizes HIF-1α (even under normoxia) (22,34) and activates NF-κB (17,23) to trigger GM-CSF expression/secretion from TNBC cells; and (2) since CSF2Rα is the only high-affinity receptor, GM-CSF recruits M2 more efficiently than M1 polarized macrophages to IRISOE cells (Fig. 1O).
GM-CSF triggers TGF-β1 secretion from M2-TAMs.
ELISA analysis of CM from siLuc- or siGM-CSF-transfected HME, IRIS291, or IRIS293 cells (48h) exposed to normoxia or hypoxia (additional 24h) showed that untransfected and untreated THP1s or those exposed to normoxic siLuc- or siCSF2-transfected HME cells CM secrete low-levels of TGF-β1 (Fig. 2A). Hypoxic siLuc-transfected HME cells CM triggered a 2-fold higher of TGF-β1 secretion from THP1s that was blocked by GM-CSF silencing (Fig. 2A). In contrast, normoxic siLuc-transfected IRIS291 or IRIS293 cells CM induced ~5-fold higher TGF-β1 secretion from THP1s increased to >10-fold by hypoxic CM (Fig. 2A). In both situations, GM-CSF silencing blocked the ability of the CM to induce TGF-β1 secretion from THP1s (Fig. 2A).
Figure 2. TGF-β1 from macrophages enhances IRISOE cells aggressiveness.
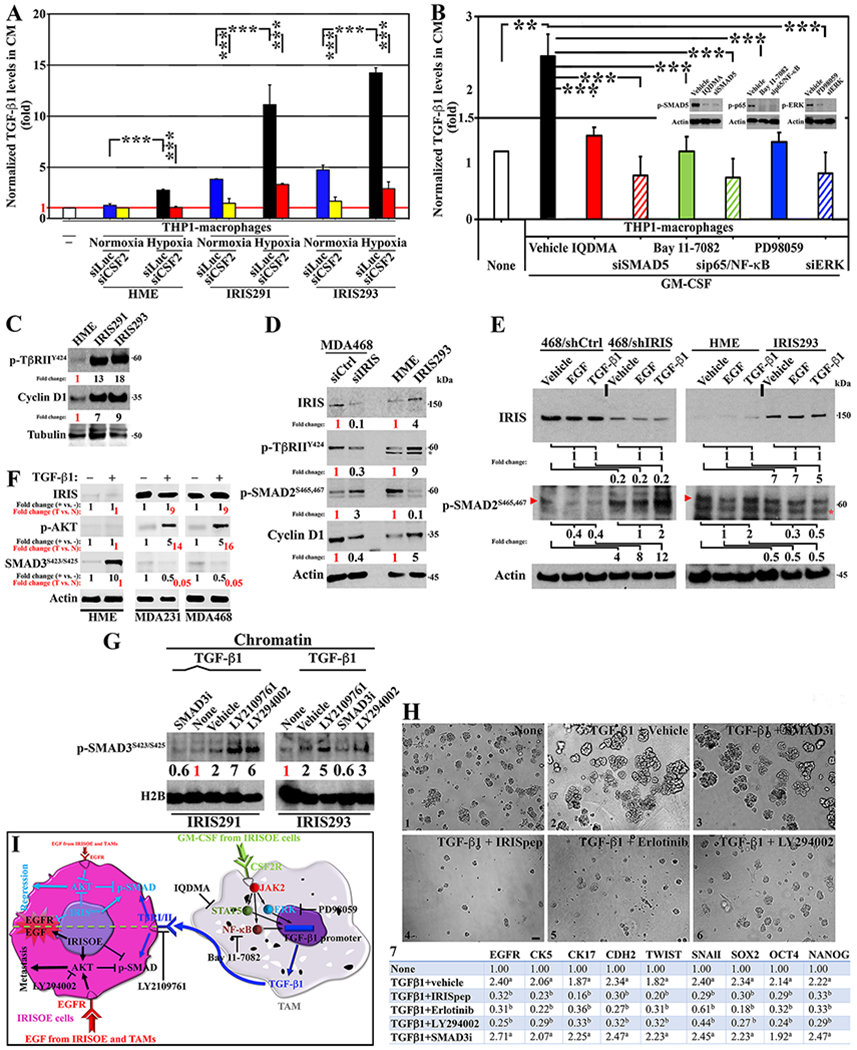
(A) ELISA for TGF-β1 in CM from indicated cells and treatments (n=3, in triplicates). (B) ELISA for TGF-β1 in CM from indicated cells and treatments (n=3, in triplicates). (C) Expression of p-TβRIIY422 and CyclinD1 in indicated cells (n=3). (D) Expression of IRIS, p-TβRIIY422, p-SMAD2S465,S467, and CyclinD1 in indicated cells (n=3). (E) Expression of IRIS and p-SMAD2S465,S467 in indicated cells and treatments (n=3). Arrowhead shows p-SMAD2S465,S467 position and asterisk shows non-specific band. (F) Expression of IRIS, p-AKT, and p-SMAD3S423,S425 in indicated cells and treatments (n=3). (G) Expression of p-SMAD3S423,S425 in indicated cells and treatments (n=3). (H) Representative images of acini of indicated cells in Matrigel supplemented with indicated factors/drugs (n=3, in triplicates). Scale bar =50μm. (H7) Normalized mRNA expression (fold) of the indicated factors in indicated cells and treatments. a is p=0.05, and b is p=0.001 (n=3, in triplicates). (I) A schematic representation of the data presented above.
THP1 cells silenced from SMAD5, p65/NF-κB or ERK (48h) were exposed to 50ng/ml of recombinant (r)GM-CSF for 24h, or they were exposed to 50ng/ml of rGM-CSF in the presence of the vehicle, the STAT5 inhibitor; IQDMA, the NF-κB inhibitor; Bay 11-7082, or the ERK inhibitor; PD98059 (24h). ELISA analysis showed rGM-CSF induced >2-fold increase in TGF-β1 secretion by THP1s (vehicle, Fig. 2B). Decreasing expression or activity of SMAD5, p65/NF-κB, or ERK (Fig. 2B, inset) blockade rGM-CSF-induced TGF-β1 secretion from THP1s (Fig. 2B).
IRISOE channels TGF-β1 signaling towards the non-canonical pathway.
Compared to HME cells, IRIS291 and IRIS293 express 10-20-fold higher of TβRIIY424 (constitutively active by autophosphorylation on tumor cells (35)) correlated with a 5-10-fold higher Cyclin D1 expression (Fig. 2C). We transfected the TNBC cell line MDA-MB-468 that overexpress IRIS (17) (hereafter MDA468) with siLuc or siIRIS (72hr, Fig. 2D). In parallel, we grew HME and IRIS293 cells (24h). IRIS-silencing decreased TβRIIY424 expression >70%, and Cyclin D1 expression >60%, despite increasing p-SMAD2S465,S467 level by ~3-fold (Fig. 2D). Conversely, IRISOE triggered ~5-fold increase in TβRIIY424 and Cyclin D1 expression, despite decreasing p-SMAD2S465,S467 level by >90% (Fig. 2D).
MDA468/shCtrl, MDA468/shIRIS, HME or IRIS293 cells were all treated with vehicles, 10ng/ml rEGF or 5ng/ml rTGF-β1 (24h). Nuclear/chromatin extracts were probed for IRIS and p-SMAD2S465,S467 levels. rEGF treatment did not affect IRIS expression in any of the 4 cell lines (Fig. 2E), while decreased p-SMAD2S465,S467 level by ~60% in MDA468/shCtrl, and by ~70% in IRIS293 cells (Fig. 2E). Similarly, rTGF-β1 treatment had no effect on IRIS expression in any of the 4 cell lines (Fig. 2E), decreased p-SMAD2S465,S467 level by ~60% in MDA468/shCtrl cells and by ~50% in IRIS293 cells, and increased it by 2-fold in MDA468/shIRIS cells and HME cells (Fig. 2E). Importantly, IRIS-silencing increased p-SMAD2S465,S467 level ~4-fold, ~8-fold, and ~12-fold under vehicle, rEGF, or rTGF-β1 treatments, respectively in MDA468 cells (Fig. 2E), and overexpression decreased it by 50% under vehicle, rEGF or rTGF-β1 treatment in HME cells (Fig. 2E). Identical results were obtained using MDA231 and IRIS291 cells.
In total extracts, compared to vehicle, treatment with 5ng/ml of rTGF-β1 (48h) had no effect on IRIS or AKTT308/S473 levels, while induced 10-fold increase in p-SMAD3S423/S425 level (Fig. 2F, black) in HME cells. Compared to vehicle, treatment with 5ng/ml of rTGF-β1 (48h) had no effect on IRIS level, increased p-AKTT308/S473 level by 5-fold, while decreased p-SMAD3S423/S425 level by 50% in MDA231, and MDA468 cells (Fig. 2F, black). Moreover, compared to HME cells, the effect of rTGF-β1 (48h) translates to no effect on IRIS level, ~15-fold increase in p-AKTT308/S473 level, and ~95% decrease in p-SMAD3S423/S425 level in MDA231 and MDA468 cells (Fig. 2F, red).
IRIS291 and IRIS293 cells starved from serum (48hr) were exposed to 5ng/ml of rTGF-β1 plus 10μM of different inhibitors (Fig. 2G). In chromatin fraction, under basal condition, IRIS291 and IRIS293 showed low p-SMAD3S423,S425 level, and stimulation with rTGF-β1 increased that by ~2-fold in both cell lines (Fig. 2G). As expected SMAD3 inhibitor (SMAD3i) decreased rTGF-β1-induced p-SMAD3S423,S425 by >3-fold in both cell lines (from 2 to 0.6-fold, Fig. 2G). In contrast, inhibiting TβRI/II signaling using LY2109761 or PI3’K/AKT signaling using LY294002 increased p-SMAD3S423,S425 level by >5-fold and >3-fold in both cell lines, respectively (Fig. 2G).
TGF-β1/TβRI/II triggers EMT/stemness in IRISOE cells through AKT, not SMAD signaling.
We treated MDA468 cells in 2D cultures (24h) or in 3D-Matrigel (7days, media/factor/drugs changed every 3rd day) with none or 5ng/ml rTGF-β1 plus vehicle, or 2μM of IRIS inhibitory peptide (hereafter IRISpep (17,19,21–23)), 10μM of erlotinib, LY294002 or SMAD3i (24h). In the 3D-Matrigel cultures rTGF-β1 induced IRIS291 cells to form larger, unorganized and invasive acini (compare Fig. 2H1 and 2H2 (17). Interestingly, while SMAD3i had no effect (Fig. 2H3), IRISpep (Fig. 2H4), erlotinib (Fig. 2H5) and LY294002 (Fig. 2H6) all blocked these rTGF-β1-induced effects. This was also consistent with the real -time RT/qPCR on total RNAs from the 2D cultures that showed rTGF-β1 increased expression of basal; EGFR, CK5, CD17, EMT; CDH2, Twist, SnailI, and stemness; Sox2, Oct4 and Nanog markers (Fig. 2H7). IRISpep, erlotinib, and LY294002 blocked rTGF-β1-induced expression of these proteins, whereas SMAD3i had no effect (Fig. 2H7).
Taken together, these data suggest that: (1) GM-CSF secreted from IRISOE cells triggers TGF-β1 production and secretion from TAMs by activating CSF2Rα-induced SMDA5, p65/NF-κB and/or ERK signaling; and (2) in combination with (Suppl. Fig. 1B) and our previous studies (16,17,19,21–23), IRISOE-induced EGFR and TβRII expression allows EGF (secreted by IRISOE cells (29)) and TGF-β1 (secreted by TAMs (this study)) to re-direct canonical-SMAD signaling that promotes growth-suppression, apoptosis and genomic stability (35) into non-canonical-AKT signaling that promotes aggressiveness in IRISOE cells (cf. Fig. 2I).
In vivo evidence for TAM-induced IRISOE tumor aggressiveness.
To establish the role of TAMs in promoting IRISOE tumor aggressiveness, in vivo, we orthotopically injected 2x106 RFP-IRIS291 or RFP-IRIS293 cells alone or admixed with 2x105 THP1s (n=10/each, Fig. 3A). Compared to alone, THP1 co-injection increased IRIS291 tumor size from 0.9±0.2cm3 to 1.8±0.05cm3 (2-fold increase), and IRIS293 tumor size from 0.7±0.1cm3 to 1.7±0.08cm3 (~3-fold increase) within 10 weeks (Fig. 3B). Ex vivo analysis showed increase lung (Fig. 3C, upper arrowheads) and brain (Fig. 3C, lower arrowheads) metastases in mice co-injected with THP1s. THP1 co-injection also led to the death of 7 (IRIS291) and 6 (IRIS293) of the 10 mice (Fig. 3D).
Figure 3. Bidirectional interaction between IRISOE cells and M2-TAMs.
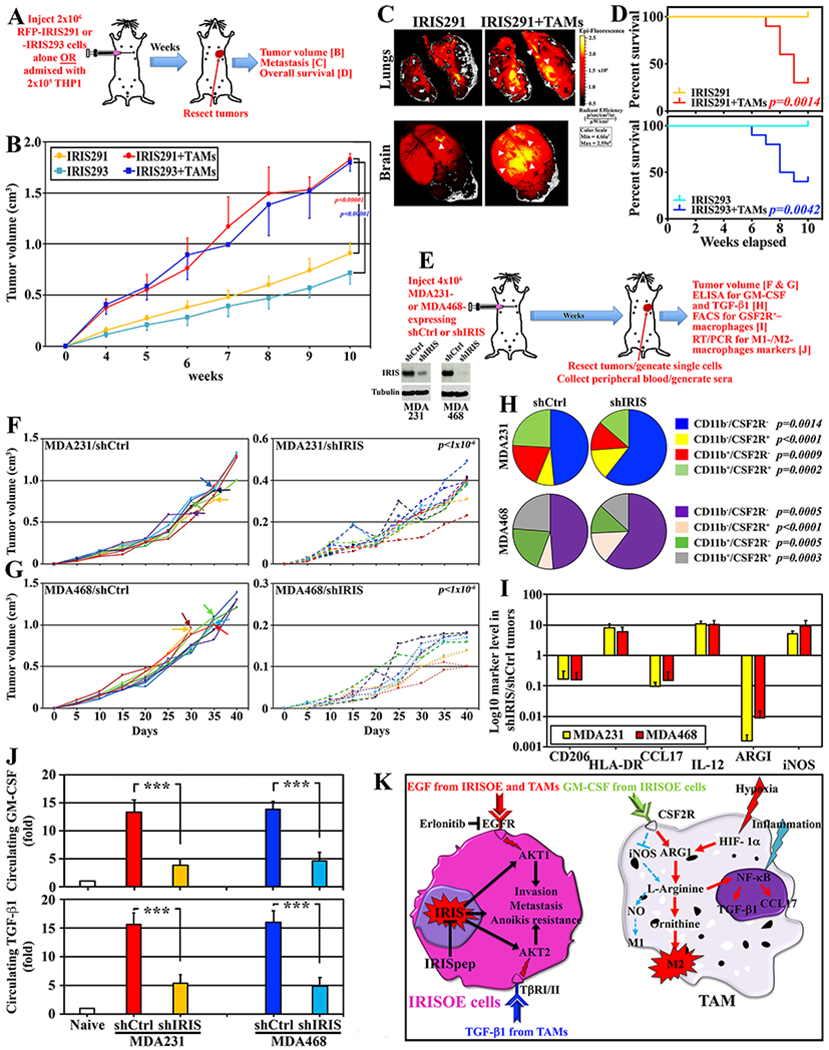
(A) Schematic description of the assays below. (B) Volume of tumors in athymic mice injected with 2x106 of RFP-IRIS291 or -IRIS293 alone (n=10/each) or admixed with 1x105 THP1s (n=10/each). (C) IRIS291 lung (upper) and brains (lower) metastasis in mice in (A). Identical results were obtained with IRIS293 cells. (D) OS in indicated mice injected with indicated cells combinations. (E) Schematic description of the assays below (insets show IRIS expression). (F and G) Volume of tumors developed in athymic mice injected indicated cells (arrows indicate the death of the mice). (H) Percentage of indicated fractions in indicated tumors. (I) Log10 of indicated markers in CD11b+ cells isolated from indicated tumors. (J) Level of GM-CSF and TGF-β1 in sera from mice bearing indicated tumors. (K) Schematic representation of the data presented above.
To further establish that, 4x106 MDA231 or MDA468 expressing shCtrl or shIRIS (Fig. 3E, inset) cells were injected in female athymic mice (see Fig. 3E). Forty days later, tumors and peripheral blood (PB) were collected from all mice (Fig. 3E). At day 40, shIRIS-expressing tumors were ~70% (MDA231, 0.38±0.07cm3 vs. 1.2±0.15cm3, Fig. 3F) and ~90% (MDA468, 0.15±0.03cm3 vs. 1.3±0.07cm3, Fig. 3G) smaller than shCtrl-expressing tumors. Additionally, only MDA231/shCtrl and MDA468/shCtrl tumors led to the death of 50% of the mice within the 40 days (Fig. 3G, left arrows).
Moreover, unpermeabilized single cell preparations generated from these tumors were co-labeled with anti-mouse CD11b (monocytes/macrophage marker) and CSF2R antibodies. We Gated FSC and SSC to remove debris and conjugates and normalized to isotype controls (in all subsequent experiments as well). This analysis revealed that compared to controls, CD11b− cells increased in MDA231/shIRIS tumors (73.8±3.7% vs. 56.2±4.7%, p=0.0002) and MDA468/shIRIS tumors (74.3±4.5% vs. 55.6±2.7%, p>0.00005), while CD11b+ cells decreased in MDA231/shIRIS tumors (26.2±3.7% vs. 43.8±4.7%, p=0.0002) and MDA468/shIRIS tumors (25.7±4.5% vs. 44.4±2.7%, p>0.00005, Fig. 3H). Importantly, compared to control tumors, CD11b−/CSF2R+ cells increased in MDA231/shIRIS tumors (13.3±1.1% vs. 7.8±0.6%, p<0.000001) and MDA468/shIRIS tumors (13.9±1.7% vs. 6.9±0.6%, p<0.000001) and CD11b+/CSF2R+ cells decreased in MDA231/shIRIS tumors (13.6±2.8% vs. 24.1±2.4%, p=0.0002) and MDA468/shIRIS tumors (13.2±2.9% vs. 23.8±2.4%, p>0.0003, Fig. 3H).
Real-time RT/qPCR analysis performed on total RNA isolated from these tumors showed that compared to MDA231/shCtrl tumors, MDA231/shIRIS tumors contained 8-, 11-, and 5-fold higher HLA-DR, IL-12, and iNOS (M1 markers), respectively, while >80% reduced expression of CD206, CCL17, and Arg1 (M2 markers, Fig. 3I, yellow). Similarly, compared to MDA468/shCtrl tumors, MDA468/shIRIS tumors contained 6-, 10- and 9-fold higher of HLA-DR, IL-12, and iNOS, respectively, while >90% reduction in CD206, CCL17, and ARG1 levels (Fig. 3I, red). Finally, ELISA analysis showed that compared to naïve mice (n=40, Fig. 3J, upper/white), MDA231/shCtrl or MDA468/shCtrl tumors-bearing mice (n=10/each) showed 12-15-fold higher, while MDA231/shIRIS or MDA468/shIRIS tumors-bearing mice (n=10/each) only 3-5-fold higher circulating GM-CSF and TGF-β1 (>60% decrease, Fig. 3J).
Taken together, these data suggest that: (1) the bidirectional interaction with TAMs enhances IRISOE cells’ aggressiveness, in vivo; and (2) IRIS silencing decreases TNBC tumors size, circulating GM-CSF and TGF-β1 levels, and the number of CSF2R+-macrophages recruited into these tumors (Fig. 3K).
Syngeneic model for the proposed bidirectional interaction.
The BALB/c TNBC tumor cell line; 4T1 (36) overexpress Iris (mouse IRIS homolog (37)) when compared to normal mouse HC11 cell line. We developed 2 shRNAs; shIris1 and shIris2 that target Iris intron 11 part and decrease Iris expression in 4T1 cells by >95% (Fig. 4A).
Figure 4. Syngeneic model for the bidirectional interaction of IrisOE cells and TAMs.
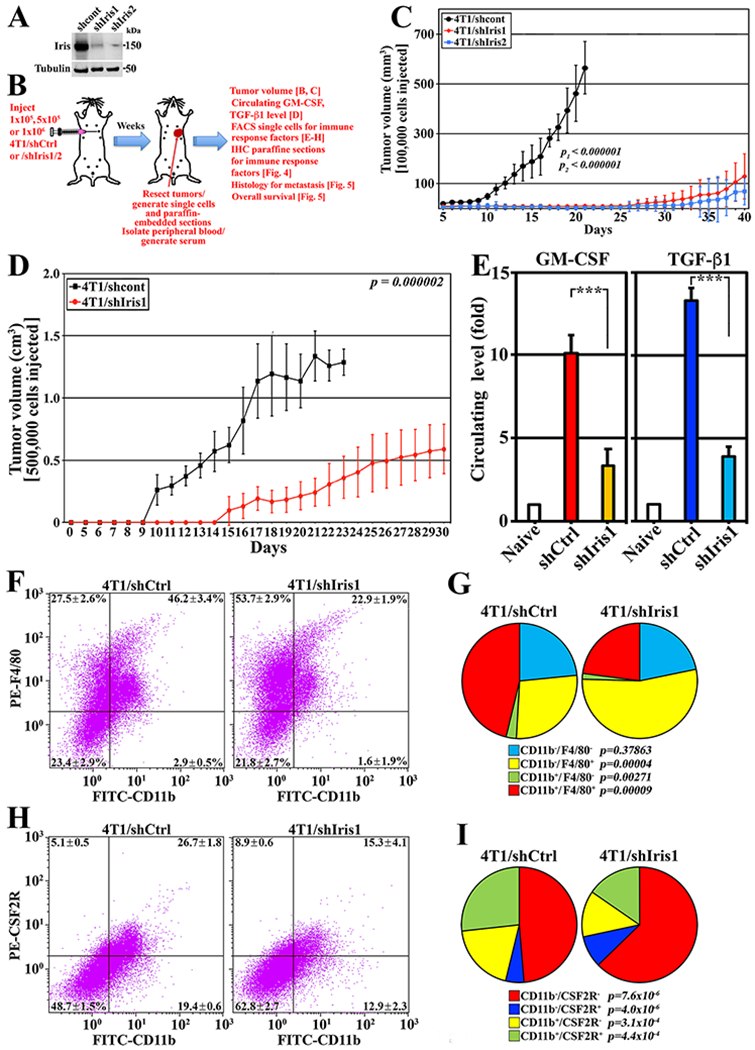
(A) Schematic description of the assays below. (B) Iris expression in 4T1/shCtrl, 4T1/shIris1, or 4T1/shIris2. (C and D) Volume of tumors developed in BALB/c mice injected with indicated cells. (E) Levels of GM-CSF and TGF-β1 in the circulation of indicated mice. (F and G) Percentage of indicated cell fractions in indicated tumors. (H and I) Percentage of indicated fractions in indicated tumors.
We injected 1x105 4T1/shCtrl, 4T1/shIris1 or 4T1/shIris2 cells (n=8/each) in female BALB/c mice (cf. Fig. 4B). Upper limits were ~0.5cm3 tumor volume or 40 days. The 4T1/shCtrl cells formed 0.58±0.1cm3 tumors by day 20, while 4T1/shIris1 cells 0.13±0.09cm3 (>75% reduction, p<0.000001), and 4T1/shIris2 cells 0.07±0.06cm3 (>85% reduction, p<0.000001) tumors by day 40 (Fig. 4C).
We injected 5x105 4T1/shCtrl cells or 4T1/shIris1 cells (n=10/each) in female BALB/c mice (cf. Fig. 4B). Upper limits were ~1.0cm3 tumor volume or 30 days. The 4T1/shCtrl cells formed 1.3±0.1cm3 tumors by day 20, while 4T1/shIris1 cells 0.59±0.2cm3 tumors (>55% reduction, p=0.000002) by day 30 (Fig. 4D). ELISA analysis on sera isolated from these mice showed that compared to naïve mice, 4T1/shCtrl tumor-bearing mice circulation contained 10.1±1.1 and 13.3±0.8-fold higher GM-CSF and TGF-β1, respectively, while 3.3±1.0 and 3.9±0.6-fold, respectively (>60% decrease, Fig. 4E) in 4T1/shIris1 tumor-bearing mice.
To evaluate the tumor-infiltrating myeloid compartment within these tumors, unpermeablized single cell preparations labeled with anti-mouse FITC-CD11b and PE-F4/80 or PE-CSF2R antibodies were FACS analyzed. The 4T1/shCtrl tumors contained ~50% CD11b− and ~50% CD11b+ fractions (Fig. 4F), increased to ~75% and ~25%, respectively in 4T1/shIris1 tumors (Fig. 4F). Compared to 4T1/shCtrl tumors, 4T1/shIris1 tumors showed increase in M1 population (CD11b−/F4/80+ cells, 27.5±2.6% vs. 53.7±2.9%, p=0.00004), and decrease in M2 population (CD11b+/F4/80+ cells, 46.2±3.4% vs. 22.9±1.9%, p=0.00009), and MDSC-population (CD11b+/F4/80− cell (38), 2.9±0.5 vs. 1.6±1.9%, p=0.00271, Fig. 4F and G).
Additionally, compared to 4T1/shCtrl tumors, in 4T1/shIris1 tumors the CD11b−/CSF2R− (48.7±1.5% vs. 62.8±2.7%, p=7.6x10−6), and CD11b−/CSF2R+ (5.1±0.5% vs. 8.9±0.6%, p= 4.0x10−6) populations increased (Fig. 4H and I). In contrast, the CD11b+/CSF2R− (19.4±0.6% vs. 12.9±2.3%, p=0.00031), and CD11b+/CSF2R+ (26.7±1.8% vs. 15.3±4.1%, p=0.00044) populations decreased (Fig. 4H and I).
Indeed, paraffin-embedded sections from these tumors IHC stained for myeloid cells’ markers (cf. Fig. 4B) confirmed the lack of CSF2R-expressing M2 CD206+-TAMs within 4T1/shIris tumors (compare Fig. 5A to B). In fact, these analyses also showed that 4T1/shCtrl tumors contained 30.8±7.3 F4/80+ cells/high power field (HPF) and 7.5±3.1 CD206+ cells/HPF, while 4T1/shIris1 tumors contained 18.7±4.9 F4/80+ cells/HPF (>40% decrease, p<0.000001) and 1.1±1.1 CD206+ cells/HPF (>80% decrease, p<0.000001, Fig. 5C and 5D). Moreover, 4T1/shCtrl tumors contained 25.5±5.5 CD25+ cells/HPF and 31.3±4.6 FOXP3+ cells/HPF, while 4T1/shIris1 tumors contained 9.8±3.5 CD25+ cells/HPF (>60% decrease p<0.000001) and 15.3±2.9 FOXP3+ cells/HPF (>50% decrease, p<0.000001, Fig. 5E and 5F). Finally, according to IHC (Fig. 5G) and FACS analysis performed on single-cell preparations from these tumors (Fig. 5H), 4T1/shCtrl tumors contained 25.7±2.7% CD8+ and showed almost complete absence of PD-1+-cells, while 4T1/shIris1 tumors 38.1±4.2 CD8+ cells (~50% increase, p=0.0001) and massive number of PD-1+ cells (Fig. 5G and 5H).
Figure 5. Immunosuppressive microenvironment in IrisOE TNBC syngeneic tumors.
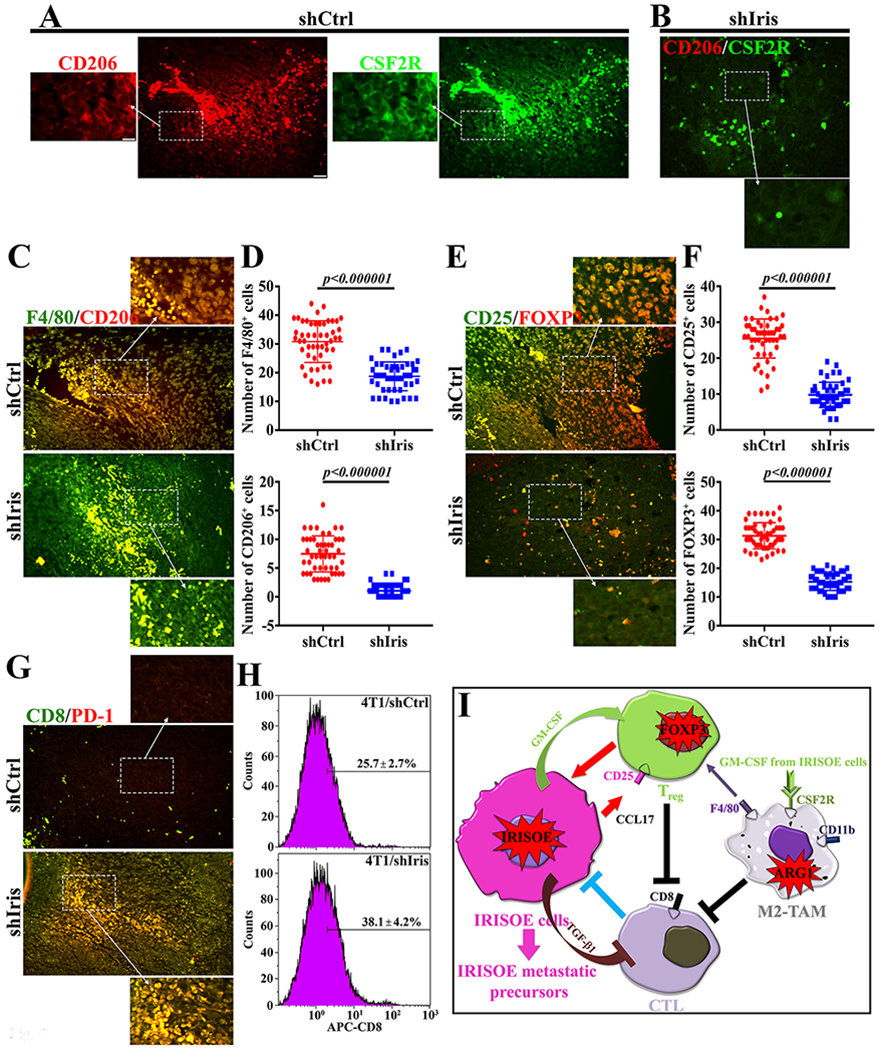
(A and B) Co-expression of M2 biomarker; CD206 and CSF2R in indicated tumors. (C) Co-expression of CD206 and general macrophage marker; F4/80 in indicated tumors. (D) Quantitative analysis of data in (C). (E) Co-expression of the Treg markers; CD25 (membranous) and FOXP3 (nuclear) in indicated tumors. (F) Quantitative analysis of data in (E). (G) Co-expression of the CTL markers; CD8 and PD-1 in indicated tumors. (H) FACS analysis of CD8+ cells in indicated tumors. (I) Schematic representation of the data presented above. Scale bar=100, insets=25μm.
Taken together, these data suggest that: (1) like IRIS, depleting Iris also reduces TNBC cells ability to form tumors; and (2) the bi-directional interaction between IrisOE cells and TAMs through GM-CSF and TGF-β1 triggers immunosuppressive microenvironment within TNBC tumors, including high levels of CSF2R-expressing M2 F4/80+/CD206+-TAMs and CD25+/FOXP3+-regulatory T-cells (Treg), while lower levels of CD8+/PD-1+-cytotoxic T cells (CTLs, Fig. 5I).
IRISOE/IrisOE triggers tumor metastasis.
We injected 1x106 of 4T1/shCtrl or 4T1/shIris2 cells (n=10/cells) in female BALB/c mice (cf. Fig. 4B). The limit was death from the disease within 40d. Tumors, adrenal glands, brains, hearts, kidneys, livers, lungs, and spleens were collected from each anesthetized mouse, paraffine-embedded on a single block, sectioned at 4μm and H&E stained (cf. Fig. 4B). As expected, 4T1/shCtrl tumors were larger. They showed evidence of aggressive TNBC cell morphology, including poorly differentiated epithelial cells with large hyperchromatic nuclei and relatively small cytoplasm (Fig. 6A, arrowheads), mesenchymal cells (Fig. 6A, arrows), and a large number of mitotic cells (Fig. 6A, asterisks). These mice should relatively low area of internal necrosis throughout the tumor (Fig 6B two-heads arrows). In contrast, 4T1/shIris2 tumors were small in size, contained differentiated epithelial cells only (Fig. 6C, arrowheads), and lacked almost completely mitotic cells (Fig. 6C). Moreover, compared to 4T1/shCtrl tumors, 4T1/shIris tumors exhibited extensive necrosis throughout the tumor (Fig 6B and 6D, two-heads arrows) accompanied by an increase in leukocytes infiltration (Fig. 6D, arrows).
Figure 6. Iris silencing prevents TNBCs metastasis and enhances mice OS.

(A-D) Representative images of indicated tumors, showing morphologically normal (arrowheads), mesenchymal-like cells (arrows), mitotic (asterisks) cells, necrosis (double-headed arrows) and infiltrated lymphocytes (arrows, D). (E-H) Lung metastasis, (I-L) liver metastasis, and (M-P) Kidney metastasis in mice injected with indicated cells. Scale bar=100μm (A, C, F, H, L, and N), 250μm (J, k Na M), and 500μm (B, D, E, G, I, O, and P). (Q) Overall survival in mice injected with 4T1/shCtrl cells (black) or 4T1/shIris2 cells (red).
Additionally, compared to lungs from 4T1/shCtrl cells injected mice, the lung of 4T1/shIris cells injected mice were free from metastasis (compare Fig. 6E and 6F to 6G and 6H). Similarly, compared to livers from 4T1/shCtrl cells injected mice, the livers of 4T1/shIris cells injected mice were free from metastasis (compare Fig. 6I and 6J to 6K and 6L). Finally, compared to kidneys from 4T1/shCtrl cells injected mice, the kidneys of 4T1/shIris cells injected mice were free from metastasis (compare Fig. 6M and 6N to 6O and 6P). Interestingly, in the lungs (Fig. 6F) and kidneys (Fig. 6M), metastasis was found within or close to afferent vessels and in most cases, appeared infiltrative, whereas in the liver were more localized, often appearing spherical (Fig. 6I and 6J). More importantly, all mice injected with the 1x106 4T1/shCtrl cells (n=10) died within 25 days (Fig. 6Q), while only 3 (n=10) mice injected with 1x106 4T1/shIris2 cells died by day 40 (Fig. 6Q, p=0.0001).
Taken together, these data suggest that IRISOE/IrisOE generates immunosuppressive microenvironment within TNBC tumors that is favorable for the generation of deadly metastatic precursors.
IRIS-inactivation triggers immune surveillance within TNBC tumors.
We injected 4x106 IRIS291 (n=20) and IRIS293 (n=20) cells in female athymic mice (cf. Fig. 3A). By day 20, all mice developed ~0.5cm3 tumors (Fig. 7A). Each group was randomized and intratumorally injected with scrambled peptide (Fig. 7A, black arrows, n=10/cell line) or the inhibitory IRIS peptide (IRISpep (17,23), Fig. 7A, red arrows, n=10/cell line) every 3rd day, a total of 4 times. By day 30 scrambled-injected IRIS291 2° orthotopic mammary tumors were 1.46±0.11cm3, and IRIS293 tumors were 1.49±0.11cm3, while IRISpep-injected IRIS291 tumors were 0.31±0.08cm3 (>75% regression, p,0.0002) and IRIS293 tumors were 0.34±0.14cm3 (>75% regression, p,0.004, Fig. 7A).
Figure 7. IRIS inactivation triggers immunity in TNBC tumors.
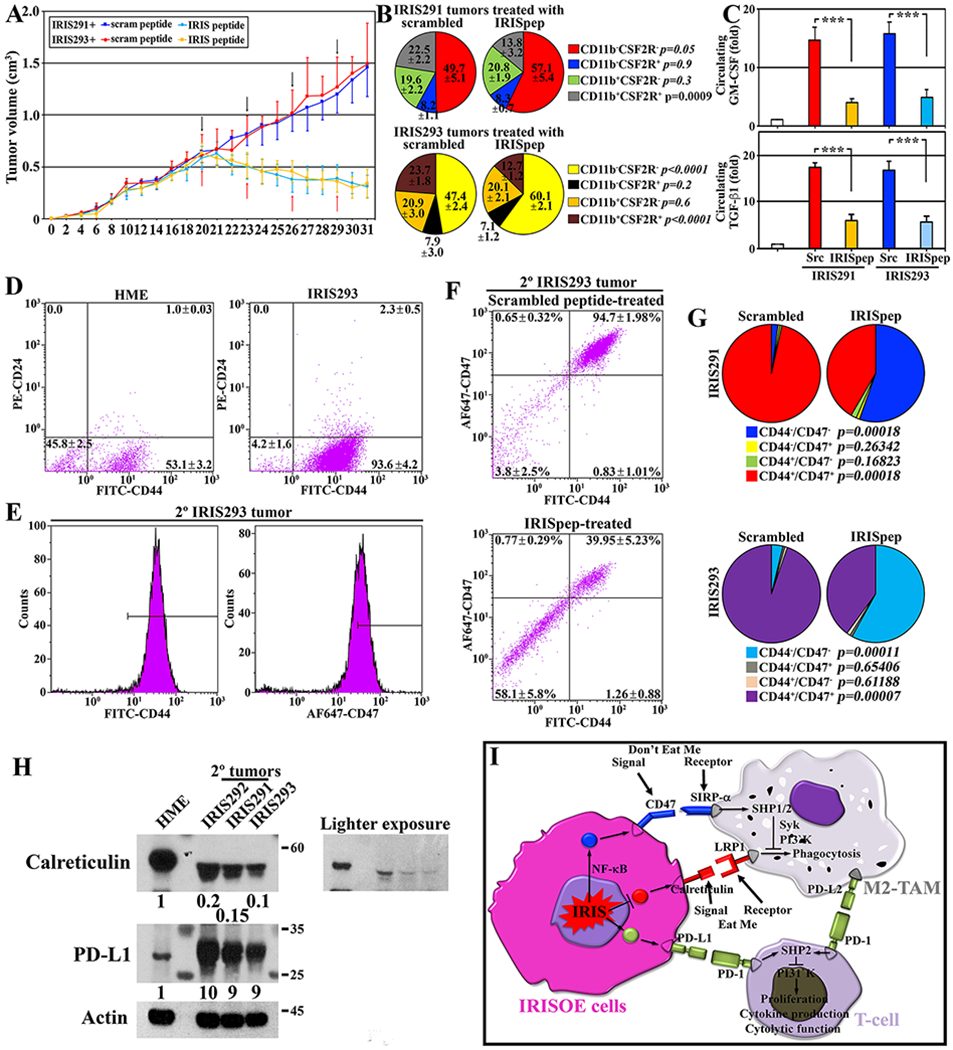
(A) Volume of established tumors in athymic mice intratumorally injected with scrambled (black arrows) or IRISpep (red arrows). (B) Percentage of indicated fractions in indicated tumors. (C) GM-CSF and TGF-β1 ELISA on sera from indicated tumor-bearing mice. (D) FACS for CD44 and CD24 staining in indicated cells. (E) FACS for CD44 (left) and CD47 (right) in 2° IRIS293 tumors. The figures are representative of 10 tumors. (F) FACS for CD44/CD47 on indicated tumors. (G) Summary of tumors IRIS291 and IRIS293 data shown in (F). P values are shown. (H) Expression of calreticulin, PD-L1 in HME, 1° IRIS292, or 2° IRIS291 or 2° IRIS293 tumors. The data is representative of five tumors/cell line. (I) Schematic representation of the data presented above.
Unpermeabilized single-cell preparations from each tumor were labeled with FITC anti-CD11b and PE anti-CSF2R antibodies. FACS analysis showed scrambled-injected IRIS291 tumors contained 49.7±5.1%, and IRIS293 tumors 47.4±2.1% of CD11b−/CSF2R− cells (Fig. 7B), while IRISpep-injected IRIS291 tumors 57.1±5.4% (~20% increased, p=0.05), and IRIS293 tumors 60.2±2.1% (>25% increase, p<0.0001, Fig. 7B). Correspondingly, scrambled-injected IRIS291 tumors contained 22.5±2.2%, and IRIS293 tumors 23.7±1.8% of CD11b+/CSF2R+ cells (Fig. 7B), while IRISpep-injected IRIS291 tumors 13.8±3.2% (40% decrease, p=0.0009), and IRIS293 tumors 12.7±1.2% (50% decreased, p<0.0001, Fig. 7B).
Additionally, ELISA analysis on sera isolated from these mice showed that compared to naïve mice, inactivating IRIS reduced the increase in circulating levels of GM-CSF by 74% and TGF-β1 by 65% in IRIS291 tumor-bearing mice, and by 70% and 65%, respectively in IRIS293 tumor-bearing mice (Fig. 7C).
FACS analysis showed parental HME cell line contained ~50% stem-like/CD44+CD24− cells, while IRISOE 1° tumors >95% (Fig. 7D) (19), the majority of which are also CD47+ (Fig. 7E). Within this population in scrambled-injected 2° IRIS293 tumors 3.8±2.5% were CD44−/CD47− cells, and 94.7±2.0% CD44+/CD47+ cells, while in IRISpep-injected tumors 58.1±5.8% were CD44−/CD47− cells (>15-fold increase, p=0.00011), and 40.0±5.2% CD44+/CD47+ cells (~60% decrease, p=0.00007, Fig. 7F and 7G). Similarly, in scrambled-injected 2° IRIS291 tumors 2.2±0.5% were CD44−/CD47− cells, and 96.6±1.4% CD44+/CD47+ cells (Fig. 7G), while in IRISpep-injected tumors 56.8±7.1% were CD44−/CD47− cells (>25-fold increase, p=0.00018), and 42.6±6.9% were CD44+/CD47+ cells (>55% decrease, p=0.00018, Fig. 7G).
Finally, these stem-like/CD44+CD24− cells isolated from the 2° IRIS291 and IRIS293 tumors and 1° IRIS292 tumors expressed 80-90% lower calreticulin and 9-10-fold higher PD-L1 on their surface compared to stem-like/CD44+CD24− isolated from HME cell line (Fig. 7H).
Taken together, these data suggest that: (1) IRISOE increases CD47 (don’t-eat-me initiator); (2) decreases calreticulin (eat-me initiator); and (3) increases PD-L1 expression on TNBC tumor cells, which promotes immunosuppression and TNBC deadly metastasis.
Discussion.
Tumor microenvironment (TME) plays a crucial role in promoting cancer metastasis. In the current study, we showed that within the aggressiveness niche (20) in TNBC tumors, IRISOE cells secrete high-levels of GM-CSF. At a low level, GM-CSF exerts an anti-tumor function by marking cancer cells for elimination by the immune system, or monocyte/macrophage phagocytosis (39). In contrast, high levels of GM-CSF expression exert a pro-tumor function, for example, by enhancing MMPs expression (39). An increased levels of GM-CSF in serum indicates poor prognosis in many cancers. In breast cancer, GM-CSF has a crucial role in the establishment of the pre-metastatic niche in the lungs (7,40). Increase serum GM-CSF levels in breast cancer-bearing mice promote differentiation of bone marrow-derived cells into neutrophils (TANs) that accumulate in the lungs to promote the metastatic niche (40). Interestingly, our syngeneic model showed that an increase in lung metastasis correlated with high-levels of circulating GM-CSF. It remains to be established whether this is accompanied by an increased in the number of TANs in the lungs of these mice.
GM-CSF silencing in IRISOE completely abolished TGF-β1 secretion by THP1s, suggesting bi-directional interaction with macrophages in the form of GM-CSF/TGF-β1. Like GM-CSF, TGF-β1 inhibition or overexpression results in carcinogenesis (41). TGF-β1 binding to TβRII recruits TβRI and activates it. During canonical signaling, activated TβRI/II phosphorylates SMAD2/3 that binds to SMAD4, translocates into the nucleus, and activates or repressors transcription. TGF-β1/TβRI/II can also signal through non-canonical pathways, including AKT (42). The fact that IRISOE blocked SMAD2/3 phosphorylation by TGF-β1 treatment in TNBC cells suggests that IRISOE blocks TGF-β1 canonical signaling and channels it towards non-canonical AKT signaling, perhaps to enhance stemness/EMT/aggressiveness in TNBC cells. Although we do not yet know the mechanism involved, IRISOE may upregulate one or more of the SMAD2/3 antagonistic pathways (43). We are evaluating this possibility. However, our data present another possible scenario. IRISOE upregulation of an autocrine EGF/EGFR signaling in TNBC cells (17,23) could block TGF-β1/TβRI/II canonical signaling (44) and enhances non-canonical signaling to increase TNBC cells aggressiveness (cf. Fig. 2I).
By inhibiting the “eat-me” signal and activating the “don’t eat-me” signal in macrophages, IRISOE effectively renders macrophages within the tumors into non-phagocytic/cytokine-producing factories (cf. Fig. 7I). How does IRISOE promote CD47 and suppress calreticulin expression in TNBC cells, remains to be seen? However, HIF-1α, NF-κB/p65, and AKT are all known inducer of CD47 and at the same time, are upregulated in IRISOE in TNBC cells (16,17,19–23). Calreticulin is an endoplasmic reticulum resident that facilitates the folding of major histocompatibility complex (MHC) class I molecules. Suppressing its expression would also influence antigen presentation to cytotoxic T cells within the tumor.
A significant obstacle towards an effective TNBC tumor vaccine is the inefficient stimulation of immune cells present within the tumor (45). Reversing this immunosuppression requires activation of CTLs, and suppression of any immune inhibitory elements within the tumor, e.g., M2 macrophages (46). In fact, in TNBC tumors, chemotherapies induce the coordinated transcriptional induction of CD47 and PD-L1 mRNAs and proteins expression, leading to T-cell anergy or death (47). Interestingly, we recently showed that chemotherapies upregulate IRIS expression (17,23). Moreover, during cancer progression, the continuous antigenic stimulation results in T-cell exhaustion (48). PD-1 expression is a common feature of exhausted CD8 T-cells, and blockade of PD-1 restores T-cell function in vivo (49). Besides, the PD-L1 blockade could lead to a pro-inflammatory macrophage phenotype, e.g., production of higher-levels of TNFα and IL-12 (50). Our data combined implicate inhibiting IRIS expression or activity (with a specific drug, ongoing) could eliminate the IRISOE/CD47+/PD-L1+/stem-like cell populations within TNBCs involved in the tumors immune-evasion, chemo-resistance, and metastases. Alternatively, blocking PD-1 or PD-L1 signaling in IRISOE TNBC tumors could rescue exhausted CTLs, and increase the size, proliferation, and survival of macrophages within the tumor by upregulating expression of the MHCII and the costimulatory molecule CD86 (50) and elicit an endogenous and synergistic innate and adaptive immune response against these IRISOE TNBC tumors.
Supplementary Material
Significance:
The BRCA1-IRIS oncogene promotes breast cancer aggressiveness by recruiting macrophages and promoting their M2 polarization.
Acknowledgments:
Wael is Dr. Lawrence & Mrs. Bo Hing Chan Tseu, American Cancer Society Research Scholar. This research is supported by a National Cancer Institute grant R01 CA194447. The funding body had no role in the design of the study and collection, analysis, interpretation of data, and writing the manuscript.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References.
- 1.Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. The Journal of clinical investigation 2015;125:3365–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014;41:14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burgess AW, Camakaris J, Metcalf D. Purification and properties of colony-stimulating factor from mouse lung-conditioned medium. The Journal of biological chemistry 1977;252:1998–2003 [PubMed] [Google Scholar]
- 4.Cook AD, Braine EL, Hamilton JA. Stimulus-dependent requirement for granulocyte-macrophage colony-stimulating factor in inflammation. Journal of immunology (Baltimore, Md : 1950) 2004;173:4643–51 [DOI] [PubMed] [Google Scholar]
- 5.Morrissey PJ, Bressler L, Park LS, Alpert A, Gillis S. Granulocyte-macrophage colony-stimulating factor augments the primary antibody response by enhancing the function of antigen-presenting cells. Journal of immunology (Baltimore, Md : 1950) 1987;139:1113–9 [PubMed] [Google Scholar]
- 6.Su S, Liu Q, Chen J, Chen J, Chen F, He C, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer cell 2014;25:605–20 [DOI] [PubMed] [Google Scholar]
- 7.Reggiani F, Bertolini F. GM-CSF promotes a supportive adipose and lung microenvironment in metastatic breast cancer. Oncoscience 2017;4:126–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiskopf K Cancer immunotherapy targeting the CD47/SIRPalpha axis. European journal of cancer (Oxford, England : 1990) 2017;76:100–9 [DOI] [PubMed] [Google Scholar]
- 9.Feng M, Chen JY, Weissman-Tsukamoto R, Volkmer JP, Ho PY, McKenna KM, et al. Macrophages eat cancer cells using their own calreticulin as a guide: roles of TLR and Btk. Proceedings of the National Academy of Sciences of the United States of America 2015;112:2145–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. International immunology 1996;8:765–72 [DOI] [PubMed] [Google Scholar]
- 11.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006;443:350–4 [DOI] [PubMed] [Google Scholar]
- 12.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. Journal of immunology (Baltimore, Md : 1950) 2002;169:5538–45 [DOI] [PubMed] [Google Scholar]
- 13.Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity 2004;20:327–36 [DOI] [PubMed] [Google Scholar]
- 14.Ghebeh H, Mohammed S, Al-Omair A, Qattan A, Lehe C, Al-Qudaihi G, et al. The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressed in breast cancer patients with infiltrating ductal carcinoma: correlation with important high-risk prognostic factors. Neoplasia (New York, NY) 2006;8:190–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ElShamy WM, Livingston DM. Identification of BRCA1-IRIS, a BRCA1 locus product. Nature cell biology 2004;6:954–67 [DOI] [PubMed] [Google Scholar]
- 16.Shimizu Y, Luk H, Horio D, Miron P, Griswold M, Iglehart D, et al. BRCA1-IRIS overexpression promotes formation of aggressive breast cancers. PloS one 2012;7:e34102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanchard Z, Paul BT, Craft B, ElShamy WM. BRCA1-IRIS inactivation overcomes paclitaxel resistance in triple negative breast cancers. Breast cancer research : BCR 2015;17:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinha A, Paul B, Sullivan L, Sims H, El Bastawisy A, Yousef H, et al. BRCA1-IRIS overexpression promotes and maintains the tumor initiating phenotype: implications for triple negative breast cancer early lesions. Oncotarget 2016; In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinha A, Paul BT, Sullivan LM, Sims H, El Bastawisy A, Yousef HF, et al. BRCA1-IRIS overexpression promotes and maintains the tumor initiating phenotype: implications for triple negative breast cancer early lesions. Oncotarget 2017;8:10114–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.ElShamy WM, Sinha A, Said N. Aggressiveness Niche: Can It Be the Foster Ground for Cancer Metastasis Precursors? Stem cells international 2016;2016:4829106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan D, Paul BT, Koziol J, ElShamy WM. The pro- and anti-tumor roles of mesenchymal stem cells toward BRCA1-IRIS-overexpressing TNBC cells. Breast cancer research : BCR 2019;21:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryan D, Sinha A, Bogan D, Davies J, Koziol J, ElShamy WM. A niche that triggers aggressiveness within BRCA1-IRIS overexpressing triple negative tumors is supported by reciprocal interactions with the microenvironment. Oncotarget 2017;8:113294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paul BT, Blanchard Z, Ridgway M, ElShamy WM. BRCA1-IRIS inactivation sensitizes ovarian tumors to cisplatin. Oncogene 2015;34:3036–52 [DOI] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 2001;25:402–8 [DOI] [PubMed] [Google Scholar]
- 25.Ensley JF, Maciorowski Z, Pietraszkiewicz H, Klemic G, KuKuruga M, Sapareto S, et al. Solid tumor preparation for flow cytometry using a standard murine model. Cytometry 1987;8:479–87 [DOI] [PubMed] [Google Scholar]
- 26.Grayson W, Cooper K. Application of immunohistochemistry in the evaluation of neoplastic epithelial lesions of the uterine cervix and endometrium. Curr Diag Path 2003;9:19–25. [Google Scholar]
- 27.Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society 1981;29:577–80 [DOI] [PubMed] [Google Scholar]
- 28.Choudhury KR, Yagle KJ, Swanson PE, Krohn KA, Rajendran JG. A robust automated measure of average antibody staining in immunohistochemistry images. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society 2010;58:95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bogan D, Meile L, El Bastawisy A, Yousef HF, Zekri AN, Bahnassy AA, et al. The role of BRCA1-IRIS in the development and progression of triple negative breast cancers in Egypt: possible link to disease early lesion. BMC cancer 2017;17:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edge S, Compton C. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010;17:1471–4 [DOI] [PubMed] [Google Scholar]
- 31.Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. The Journal of clinical investigation 2010;120:2699–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Archiv : an international journal of pathology 2005;446:475–82 [DOI] [PubMed] [Google Scholar]
- 33.Ozkok A, Ravichandran K, Wang Q, Ljubanovic D, Edelstein CL. NF-kappaB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicology letters 2016;240:105–13 [DOI] [PubMed] [Google Scholar]
- 34.Li AG, Murphy EC, Culhane AC, Powell E, Wang H, Bronson RT, et al. BRCA1-IRIS promotes human tumor progression through PTEN blockade and HIF-1alpha activation. Proceedings of the National Academy of Sciences of the United States of America 2018;115:E9600–e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang F, Chen YG. Regulation of TGF-beta receptor activity. Cell & bioscience 2012;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan X, Bryniarski MA, Morris ME. In Vitro and In Vivo Efficacy of the Monocarboxylate Transporter 1 Inhibitor AR-C155858 in the Murine 4T1 Breast Cancer Tumor Model. The AAPS journal 2018;21:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pettigrew CA, French JD, Saunus JM, Edwards SL, Sauer AV, Smart CE, et al. Identification and functional analysis of novel BRCA1 transcripts, including mouse Brca1-Iris and human pseudo-BRCA1. Breast cancer research and treatment 2010;119:239–47 [DOI] [PubMed] [Google Scholar]
- 38.Fang Z, Wen C, Chen X, Yin R, Zhang C, Wang X, et al. Myeloid-derived suppressor cell and macrophage exert distinct angiogenic and immunosuppressive effects in breast cancer. Oncotarget 2017;8:54173–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong IS. Stimulatory versus suppressive effects of GM-CSF on tumor progression in multiple cancer types. Experimental & molecular medicine 2016;48:e242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quail DF, Olson OC, Bhardwaj P, Walsh LA, Akkari L, Quick ML, et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nature cell biology 2017;19:974–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang JJ, Blobe GC. Dichotomous roles of TGF-beta in human cancer. Biochemical Society transactions 2016;44:1441–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mu Y, Gudey SK, Landstrom M. Non-Smad signaling pathways. Cell and tissue research 2012;347:11–20 [DOI] [PubMed] [Google Scholar]
- 43.Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997;89:1165–73 [DOI] [PubMed] [Google Scholar]
- 44.Semlali A, Jacques E, Plante S, Biardel S, Milot J, Laviolette M, et al. TGF-beta suppresses EGF-induced MAPK signaling and proliferation in asthmatic epithelial cells. American journal of respiratory cell and molecular biology 2008;38:202–8 [DOI] [PubMed] [Google Scholar]
- 45.Tian H, Shi G, Wang Q, Li Y, Yang Q, Li C, et al. A novel cancer vaccine with the ability to simultaneously produce anti-PD-1 antibody and GM-CSF in cancer cells and enhance Th1-biased antitumor immunity. Signal transduction and targeted therapy 2016;1:16025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006;439:682–7 [DOI] [PubMed] [Google Scholar]
- 47.Samanta D, Park Y, Ni X, Li H, Zahnow CA, Gabrielson E, et al. Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America 2018;115:E1239–e48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jadhav RR, Im SJ, Hu B, Hashimoto M, Li P, Lin JX, et al. Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proceedings of the National Academy of Sciences of the United States of America 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends in immunology 2015;36:265–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer immunology research 2018;6:1260–73 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.