

Abstract
Epigenetic control is critical for regulation of gene transcription in mammalian cells. Among the most important epigenetic mechanisms are those associated with post-translational modifications of chromosomal histone proteins, which modulate chromatin structure and increased accessibility of promoter regulatory elements for competency to support transcription. A critical histone mark is trimethylation of histone H3 at lysine residue 27 (H3K27me3) that is mediated by Ezh2, the catalytic subunit of the Polycomb Group Complex PRC2 to repress transcription. Treatment of cells with the active vitamin D metabolite 1,25(OH)2D3, results in transcriptional activation of the CYP24A1 gene, which encodes a 24-hydroxylase enzyme that is essential for physiological control of vitamin D3 levels. We report that the Ezh2-mediated deposition of H3K27me3 at the CYP24A1 gene promoter is a requisite regulatory component during transcriptional silencing of this gene in osteoblastic cells in the absence of 1,25(OH)2D3. 1,25(OH)2D3 dependent transcriptional activation of the CYP24A1 gene is accompanied by a rapid release of Ezh2 from the promoter, together with binding of the H3K27me3-specific demethylase Utx/Kdm6a and thereby subsequent erasing of the H3K27me3 mark. Importantly, we find that these changes in H3K27me3 enrichment at the CYP24A1 gene promoter are highly dynamic, as this modification is rapidly reacquired following withdrawal of 1,25(OH)2D3.
Keywords: Vitamin D-mediated transcription, Epigenetic control of transcription, Osteoblast gene transcription
Introduction
Vitamin D3-dependent regulation of transcription is a principal mechanism for physiological control of gene expression in osteoblasts (Meyer et al., 2014; Meyer et al., 2010a; Meyer & Pike, 2013). The active ligand of vitamin D3, 1α,25-dihydroxy vitamin D3 (1,25(OH)2D3), binds to an intracellular specific Vitamin D Receptor (VDR) which trans-locates to the cell nucleus. VDR-ligand complex interacts with genomic regulatory elements (VDREs, for Vitamin D Responsive Element), particularly at target promoters and regulatory enhancers (Pike & Christakos, 2017; Pike et al., 2016; Pike et al., 2017). VDR can form high molecular weight complexes through specific protein-protein interactions with transcriptional co-activators and/or co-repressors in a ligand-dependent manner (Herdick & Carlberg, 2000; Meyer & Pike, 2013; Rachez & Freedman, 2000). Co-regulator factors include epigenetic modifiers that are recruited to VDR-targeted chromatin to alter the surrounding epigenetic landscape. There is consequential remodeling of chromatin organization that modifies specificity and activity of transcription (Carvallo et al., 2008; Kim et al., 2005; Montecino et al., 2007).
We and others have shown that changes in epigenetic histone marks accompany 1,25(OH)2D3-enhanced gene transcription in mammalian cells, including bone-derived osteoblasts (Carvallo et al., 2008; Montecino et al., 2007; Nurminen et al., 2018; Pike et al., 2016). These marks include histone H3 and H4 acetylation (H3ac and H4ac, respectively) mediated by co-activators that include p300/CBP and SRC-1 (Carvallo et al., 2008; Kim et al., 2005), and histone H3 and H4 methylation that include asymmetric di-methylation of histone H3 arginine residue 17 (H3R17me2a) and H4R3me2a mediated by the arginine methylases PRMT4 and PRMT1, respectively (Moena et al., in press).
The CYP24A1 gene has served as a paradigm for studying epigenetic mechanisms that regulate gene expression in cells exposed to 1,25(OH)2D3 (Kim et al., 2005; Meyer et al., 2010a; Pike et al., 2015). This gene encodes a vitamin D hydroxylase that catalyzes the transition of 1,25(OH)2D3 to the inactive metabolite 1,25,24(OH)3D3, thereby modulating long-term actions of the active ligand in nearly all tissues (Meyer & Pike, 2019). The CYP24A1 gene remains transcriptionally silent in cells grown in the absence of 1,25(OH)2D3 and is rapidly induced to express when the cells are exposed to 1,25(OH)2D3 (Dhawan & Christakos, 2010; Kim et al., 2005). Hence, CYP24A1 represents a gene model system that is transcriptionally activated in the presence of 1,25(OH)2D3 that is then inactivated by the enzymatic activity encoded by this gene.
Previous studies have shown that enrichment in the symmetric di-methylation of histone H4 arginine 3 residue (H4R3me2s) mark represents a signature of the transcriptionally-silent CYP24A1 gene promoter (Moena et al., in press; Seth-Vollenweider et al., 2014). This mark is deposited on the promoter by the arginine methylase PRMT5 in the absence of 1,25(OH)2D3 and can be erased upon ligand-dependent induction of CYP24A1 gene transcription. Recent results from our group demonstrated that knockdown of PRMT5 expression in osteoblastic cells and the concomitant reduction of H4R3me2s enrichment at the CYP24A1 promoter, is not accompanied by CYP24A1 gene transcription (Moena et al., in press). This result indicates that CY24A1 gene silencing in osteoblasts in the absence of 1,25(OH)2D3, occurs independent of the PRMT5-mediated deposition of the H4R3me2s mark. The possibility should thereby be considered that additional epigenetic mechanisms contribute to this transcriptional silencing.
Bioinformatics analyses using the database ENCODE Project Consortium (ENCODE Project Consortium (2012); Kent et al., 2002) website (accessed through the USCS Genome Browser: https://genome.uscs.edu) reveal that in several osteoblastic and non-osteoblastic cell types, the absence of 1,25(OH)2D3 results in a CYP24A1 gene locus that exhibits significant enrichment of H3K27me3. In general, this epigenetic mark is deposited by the Polycomb Group Complex PRC2, predominantly by the activity of its catalytic subunit Ezh2, and represses transcription (Kazanets et al., 2016; Raphael Margueron et al., 2008). Reflecting a related dimension of epigenetic control, H3K27me3 can be erased from chromatin through the activity of the Utx/Kdm6a and Jmjd3/Kdm6b demethylases, which are often associated with nuclear complexes that activate transcription (Agger et al., 2008; Shilatifard, 2012; Sze & Shilatifard, 2016). The expression of a large set of genes that are relevant for multiple physiological processes have been reported to be down-regulated by PRC2-Ezh2-dependent enrichment of H3K27me3 (Raphaël Margueron & Reinberg, 2011), further indicating that this epigenetic mark represents a critical component of the molecular mechanisms that control gene expression in eukaryotic cells.
Here, we report that Ezh2-mediated deposition of H3K27me3 is an important component of the CYP24A1 gene repression in osteoblastic cells in the absence of 1,25(OH)2D3. Release of Ezh2 from the CYP24A1 gene promoter, binding of Utx/Kdm6a and subsequent erasing of the H3K27me3 mark accompany CYP24A1 gene activation in response to 1,25(OH)2D3. Mechanistically, these changes at the CYP24A1 gene promoter are highly dynamic and persist in the presence of the ligand.
Methods
Cell culture and vitamin D3 treatment.
Rat osteosarcoma-derived ROS 17/2.8 cells (Majeska et al., 1980) were maintained in F-12 medium supplemented with 5% fetal bovine serum (FBS, Hyclone), 1.176 g/l NaHCO3, 0.118 g/l CaCl2 ×2H2O, and 6.670 g/l HEPES. Prior to the treatments with 1α,25 dihydroxy vitamin D3 (1,25(OH)2D3), cells were incubated for 24 h in media containing Charcoal/Dextran-treated fetal serum (Hyclone). Then, 10−8 M 1,25(OH)2D3 or vehicle, were added in fresh media for different periods of time (see below each figure).
Lentivirus production and infection of ROS17/2.8 cells.
HEK293FT cells (Life Technologies) were grown in 60 mm culture plates to 80–90% confluence; Lipofectamine 2000 reagent (Life Technologies) was used to transfect cells (following the manufacturer’s instructions) with the pCMVVSVg, pCMV-dR8.91 and pLKO.1-shRNA plasmids (at a 1:2:3 ratio, respectively, to a maximum of 10 μg total DNA per plate). The pLKO.1 empty vector was used to generate control non-coding viral particles. After 16–18 h, the culture medium was replaced, and cells maintained at 32 °C for additional 48 h. Supernatants containing pseudo-typed particles were collected and filtered through a PVDF filter (0.45 μm pore size). Aliquots of supernatants were immediately stored at −80 °C. ROS17/2.8 cells were plated in 6-well culture plates and infected for 48 h with 300 μl lentiviral particles carrying shRNA-Ezh2 (TRCN0000040075), shRNA-UTX (TRCN0000096240) or empty plasmids. All short hairpin-containing plasmids were acquired at Open Biosystems (GE Healthcare Dharmacon, UK).
Chromatin Immunoprecipitation (ChIP).
ChIP studies were performed as described previously (Carvallo et al., 2008; Soutoglou & Talianidis, 2002) with modifications. Cross-linked chromatin samples (200–300 bp) were obtained from ROS17/2.8 cells, grown in the presence or absence of 10−8 M 1,25(OH)2D3 or vehicle, that were incubated for 10 min with 1% formaldehyde and gentle agitation. Cross-linking was stopped by the addition of 0.125 M glycine and the cells washed with 10 ml of PBS, scraped off in 5 ml of PBS, and collected by centrifugation at 1,000g for 5 min. The cell pellet was resuspended in 1 ml of lysis buffer (50 mM Hepes pH 7.8, 20 mM KCl, 3 mM MgCl2, 0.1% NP-40, and a cocktail of proteinase inhibitors) and incubated for 10 min on ice. The cell extract was then collected by centrifugation at 1,000g for 5 min, resuspended in 0.3 ml of sonication buffer (50 mM Hepes pH 7.9, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% deoxycholate acid, 0.1% SDS, and a cocktail of proteinase inhibitors). Chromatin was sheared in a water bath sonicator Bioruptor (Diagenode, NJ, USA) to obtain fragments of 200–500 bp. Extracts were sonicated at high power for four pulses of 5 min each, 30 sec on, 30 sec off, and centrifuged at 16,000 × g for 15 min at 4 °C. Supernatant was collected, aliquoted, frozen in liquid nitrogen, and stored at − 80 °C; one aliquot was used for determining concentration by A260 measurements and chromatin size by electrophoretic analysis. Cross-linked extracts (500 A260 units) were re-suspended in sonication buffer to a final volume of 500 μL. Samples were pre-cleared by incubating with 2–4 μg of normal IgG and 50 μL of protein A/G-agarose beads (Santa Cruz Biotechnology, CA, USA) for 2 h, at 4 °C with agitation. Chromatin was centrifuged at 4000 × g for 5 min, the supernatant was collected and immunoprecipitated with either anti-EZH2 (07–689, Merck Millipore, MA, USA), anti-UTX (ab36938, Abcam), anti-H3K27me3 Millipore (07–449, Merck Millipore, MA, USA) and anti-H3K27Ac (ab4729, Abcam) for 12–16 h at 4 °C. The immunocomplexes were recovered with addition of 50 μL of protein A or G-agarose beads, followed by incubation for 1 h, at 4 °C with gentle agitation. Immunoprecipitated complexes were washed once with sonication buffer, twice with LiCl buffer (100 mM Tris-HCl pH 8.0, 500 mM LiCl, 1.0% NP-40, and 1.0% deoxycholic acid), and once with Tris–EDTA (TE) buffer pH 8.0 (2 mM EDTA and 50 mM Tris–HCl, pH 8.0), each time for 5 min at 4 °C; this was followed by centrifugation at 4000 × g for 5 min. Protein–DNA complexes were eluted by incubation with 100 μL of elution buffer (50 mM NaHCO3 and 1% SDS) for 15 min at 65 °C. Extracts were centrifuged at 16,000 × g for 30 s, and the supernatant was collected and incubated for 12–16 h at 65 °C, to revert the cross-linking. Proteins were digested with 25 μg of proteinase K (Merck Millipore) for 2 h at 50 °C, and the DNA was recovered by phenol/chloroform extraction and ethanol precipitation using glycogen (20 μg/mL) as a precipitation carrier. The PCR primers used to evaluate by qPCR the rat CYP24A1 gene promoter (−423/−199) were: 5’- TATTGGAAGGCGGACACTCT-3’ (forward) and 5’- GACTCCACCCCGGAGATAAC-3’ (reverse) and for the rat Runx2-p57 P1 gene promoter (−118/+29) were: 5’-GTGGTAGGCAGTCCCACTTT −3’ (forward) and 5’-TGTTTGTGAGGCGAATGAAG-3’ (reverse).
qPCR analyses.
Quantitative PCR (qPCR) was performed using the Brilliant II SYBR Green QPCR master mix in a MX3000P spectrofluorometric thermal cycler (Stratagene-Agilent, CA, USA) according to the manufacturer’s recommendations. Efficiencies for each primer pair were adjusted to nearly 100%, modifying the primer concentration in the amplification mix.
Nuclear extracts and protein expression analyses.
Nuclear extracts from ROS17/2.8 cells, grown in the presence or absence of 10−8 M 1,25(OH)2D3 or vehicle, were prepared as previously described (Paredes et al., 2004). Protein levels were quantified using Bio-Rad Protein Assay reagent according to manufacturer’s instructions and using bovine serum albumin as standard (Bio-Rad, CA, USA). For western blot analyses, 10 μg of total protein were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and then transferred to a nitrocellulose membrane. Immunoblot was performed using the following antibodies: anti-TFIIB C-18 (sc-225, Santa Cruz Biotechnology, TX), anti-EZH2 (39901, Active Motif, CA) and anti-UTX/KDM6A (ab91231, Abcam). Immunoblotting was carried out using secondary antibodies conjugated to horseradish peroxide (Santa Cruz Biotechnology) and substrates for enhanced chemiluminescence (Thermo Scientific, IL, USA).
RT-PCR.
Total RNA was extracted with TRIzol (Life Technologies), according to the manufacturer’s protocol. Equal amounts of each sample (2 μg) were used for reverse transcription. qPCR was performed using Brilliant II SYBR® Green qPCR Master Mix (Agilent Technologies, CA). Expression of CYP24A1, EZH2, UTX and GAPDH was determined, using the following primers; rat CYP24A1: 5’- GCATGGATGAGCTGTGCGA-3’ (forward) and 5’- AATGGTGTCCCAAGCCAGC-3’ (reverse); EZH2: 5’- GCCAGACTGGGAAGAAATCTG −3’ (forward) and 5’- TCACTGGTGACTGAACACTCC −3’ (reverse); UTX 5’- ACAGTAATACGTGGCCTTGCTGGA-3’ (forward) and 5’- TTCATCTGCTGGTTGTAACAACTG −3’ (reverse); and GAPDH: 5’-CATGGCCTTCCGTGTTCCTA-3’ (forward) and 5’-CCTGCTTCACCACCTTCTTGAT-3’ (reverse).
Statistical analyses.
For ChIP assays, we used a one-way ANOVA analysis followed by the Dunnett post-test to compare significant changes with respect to control. For mRNA expression analysis, we used the Student’s t-test. In all the figures, error bars represent the mean ± S.E.M.; *P < 0.05, **P < 0.01, ***P < 0.001.
Statistical analyses
For ChIP assays, we used a one-way ANOVA analysis followed by the Dunnett post-test to compare significant changes with respect to control. For mRNA expression analysis by qPCR, we used the Student’s t-test. In all figures, error bars represent the mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
Results
PRC2-Ezh2 binds to the transcriptionally silent CYP24A1 promoter and is released following exposure to 1,25(OH)2D3.
Several groups have reported1,25(OH)2D3 mediated induction of the CYP24A1 gene in mammal cells (Dhawan & Christakos, 2010; Fetahu et al., 2014; Kim et al., 2005). We demonstrated that rat osteoblastic cells ROS 17/2.8 respond to 10−8 M 1,25(OH)2D3 by increasing expression of CYP24A1 mRNA in a time-dependent manner (Moena et al., in press), exhibiting a significant enhancement in response to the ligand within 2 h (Fig 1A). ChIP analyses using specific antibodies against VDR confirm that upregulation of CYP24A1 is accompanied by VDR binding to the CYP24A1 promoter (−423 to −199) in response to 1,25(OH)2D3 for two h (Fig 1B).
Figure 1. 1,25(OH)2D3 dependent expression of the CYP24A1 gene involves release of EZH2 and a reduction of H3K27me3 enrichment at the CYP24A1 promoter.
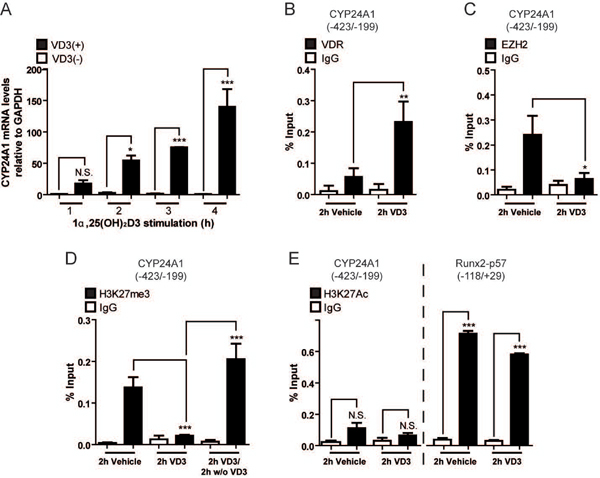
(A) ROS17/2.8 osteoblasts were treated with 10−8 M 1,25(OH)2D3 or vehicle for 1 to 4 h periods and CYP24A1 mRNA levels were determined by qRT-PCR using specific primers. Values were normalized against GAPDH mRNA. (B–C) Binding of VDR (B) and EZH2 (C) proteins to the CYP24A1 promoter in ROS17/2.8 cells exposed to 10−8 M 1,25(OH)2D3 or vehicle for 2 h, was determined by ChIP. (D) Enrichment of the H3K27me3 mark at the CYP24A1 promoter was determined by ChIP in samples from ROS17/2.8 cells exposed during 2 h to 10−8 M 1,25(OH)2D3 or vehicle, or alternatively, for 2h to 10−8 M 1,25(OH)2D3 followed for additional 2 h only to vehicle. (E) Enrichment of H3K27Ac mark at the CYP24A1 (left) and RUNX2 (right) promoters in ROS17/2.8 cells exposed to 10−8 M 1,25(OH)2D3 or vehicle for 2 h. ChIP values are expressed as % input ± SEM. Normal IgG was used as specificity control. Statistical analyses were carried out with respect to vehicle. *, p 0.05; **, p 0.01***, p 0.001; N.S. Statistically not significant differences.
The CYP24A1 promoter exhibits significant enrichment of the repressive H3K27me3 epigenetic mark in both mouse osteoblasts and human breast cancer cells (ENCODE Database, accessed through the USCS Genome Browser: https://genome.uscs.edu) in the absence of 1,25(OH)2D3, when the CYP24A1 gene is transcriptionally silent (Saramäki et al., 2006; Zhang et al., 2011). These results indicated that the Polycomb Group Complex PRC2 binds to the CYP24A1 promoter to mediate H3K27me3 enrichment. As shown in Fig 1C, ChIP assays using antibodies against Ezh2, the catalytic subunit of PRC2, demonstrates that the H3K27 methyltransferase is enriched at the CYP24A1 promoter in cells grown in the presence of vehicle. Treatment with 1,25(OH)2D3 rapidly, within 2 h, results in dissociation of Ezh2 from the CYP24A1 promoter (Fig 1C), concomitant with interaction of the VDR (Fig 1B) and transcriptional upregulation of the CYP24A1 gene (Fig 1A). This release of Ezh2 from the CYP24A1 promoter is not due to a decreased expression of Ezh2 (data not shown).
ChIP analysis using antibodies against H3K27me3 confirm that this repressive epigenetic mark is enriched at the CYP24A1 promoter in the absence of 1,25(OH)2D3. The H3K27me3 mark is relinquished from this genomic sequence in cells exposed to 1,25(OH)2D3 for 2 h (Fig 2D). This 1,25(OH)2D3 dependent decrease in H3K27me3 is rapidly reversed in osteoblastic cells by transient exposure to ligand for 2 h and additional 2 h only with the vehicle (Fig 1D). Restoration of the H3K27me3 at the CYP24A1 promoter is comparable to the levels observed in the absence of 1,25(OH)2D3 (Fig 1D).
Figure 2. Enrichment of H3K27me3 at the CYP24A1 gene promoter is mediated by EZH2 binding.
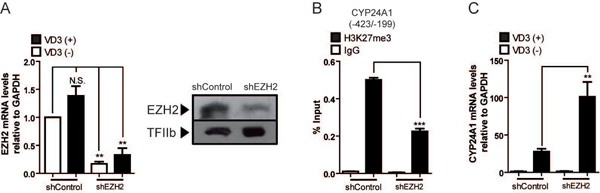
ROS17/2.8 osteoblasts were transduced for 48 h with lentiviruses encoding shRNAs against EZH2 or shRNA control, and then exposed to 10−8 M 1,25(OH)2D3 or vehicle for 2 h. (A) EZH2 knockdown was confirmed by measuring EZH2 mRNA (left) and protein (right) levels by qRT‐PCR and western blot analyses, respectively. GAPDH mRNA was determined as mRNA expression control. Detection of TFIIb was used to control for equal protein loading. (B) Effect of EZH2 knockdown on H3K27me3 enrichment at the CYP24A1 promoter. (C) Downregulation of EZH2 expression increases CYP24A1 gene responsiveness to 1,25(OH)2D3. CYP24A1 mRNA levels were measured by qRT-PCR, as described in figure legend 1. ChIP values are expressed as % input ± SEM. Normal IgG was used as specificity control. Statistical analyses were performed with respect to vehicle. **, p 0.01***; p 0.001; N.S. Statistically not significant differences.
The reduction in H3K27me3 (and of Ezh2) at the CYP24A1 promoter that accompanies 1,25(OH)2D3-dependent up-regulation of CYP24A1 transcription is not paralleled with a rapid enrichment of the active mark H3K27ac (Fig 1E, left panel), that accompanies active transcription of several mammalian genes and, particularly, active enhancers (Holmqvist & Mannervik, 2013; Nurminen et al., 2018; Sepulveda et al., 2017). As a control, using the same anti H3K27ac antibody we confirmed that this mark is found enriched at the Runx2 P1 promoter (Fig 1E, right panel), a bone-related gene that is strongly active in these cells and is transcriptionally independent of the presence of 1,25(OH)2D3.
Ezh2 is required for enrichment of H3K27me3 at the transcriptionally silent CYP24A1 gene promoter
We next determined whether Ezh2 indeed mediates deposition of H3K27me3 at the CYP24A1 promoter in osteoblastic cells. Knockdown of Ezh2 expression was carried out by infecting these cells with lentiviruses coding specific shRNAs against Ezh2 mRNA (Fig 2A). Reduced expression of Ezh2 results in a significant decrease of H3K27me3 enrichment (50 % or more) at the CYP24A1 gene promoter in cells grown with vehicle (Fig 2B). However, this downregulation of Ezh2 expression does not result in significant expression of CYP24A1 mRNA in the absence of 1,25(OH)2D3 (Fig 2C). Interestingly, the shRNA-mediated reduction of Ezh2 expression and the concomitant decreased enrichment of H3K27me3 at this promoter is accompanied by enhanced transcriptional responsiveness of the CYP24A1 gene to the 1,25(OH)2D3 ligand (Fig 2C). Thus, CYP24A1 mRNA expression following a 2 h treatment with the hormone increases about three-fold in the ROS 17/2.8 osteoblastic cells where Ezh2 had been previously depleted. Together, these results indicate that Ezh2 is necessary to mediate deposition of the H3K27me3 epigenetic mark at the CYP24A1 promoter in osteoblastic cells where this gene is transcriptionally silent. Our results also show that a reduction of the H3K27me3 mark is necessary but not sufficient to activate transcription of the CY24A1 gene in the absence of the 1,25(OH)2D3 ligand.
Utx/Kdm6a demethylase is necessary to erase of H3K27me3 at the CYP24A1 promoter during 1,25(OH)2D3 dependent transcriptional activation of CYP24A1
Utx/Kdm6a demethylase catalyzes “erasing” of the H3K27me3 mark at target genome sequences and therefore to contribute to activate gene transcription (Agger et al., 2008; Shilatifard, 2012; Sze & Shilatifard, 2016). By ChIP analyses we determined that Utx is not bound to the CYP24A1 promoter in osteoblastic cells grown with vehicle (Fig 3A). Utx is rapidly enriched at this promoter sequence following treatment of osteoblastic cells with 1,25(OH)2D3 for 2 h (Fig 3A). This interaction pattern of Utx is inversely correlated with the enrichment levels of the H3K27me3 mark at the CYP24A1 promoter, indicating that Utx may be mediating the decrease in H3K27me3 that accompanies 1,25(OH)2D3-dependent CYP24A1 gene induction. To address this possibility, we down-regulated the expression of Utx in these cells by infecting with lentiviruses coding for a shRNA against the Utx mRNA (Fig 3B). As shown in Fig 3C, this Utx knockdown condition inhibits the reduction of H3K27me3 enrichment that accompanies 1,25(OH)2D3-dependent stimulation of CYP24A1 gene expression. This result further indicates that Utx mediates “erasing” of this repressive epigenetic mark from the CYP24A1 gene promoter in osteoblastic cells exposed to 1,25(OH)2D3. Importantly, downregulation of Utx expression in these same cells results in a significant inhibition (about 50%) in the ability of the CYP24A1 gene promoter to respond transcriptionally to 1,25(OH)2D3 (Fig 3D), further confirming the role of this demethylase during transcriptional activity of the CY24A1 gene. Other H3K27me3 demethylases (e.g. Jmjd3/Kdm6b) were not found to interact with the CYP24A1 promoter in these osteoblastic cells (not shown).
Figure 3. 1,25(OH)2D3 dependent binding of UTX to the CYP24A1 promoter is required for decreased H3K27me3 enrichment and transcriptional enhancement in response to 1,25(OH)2D3.
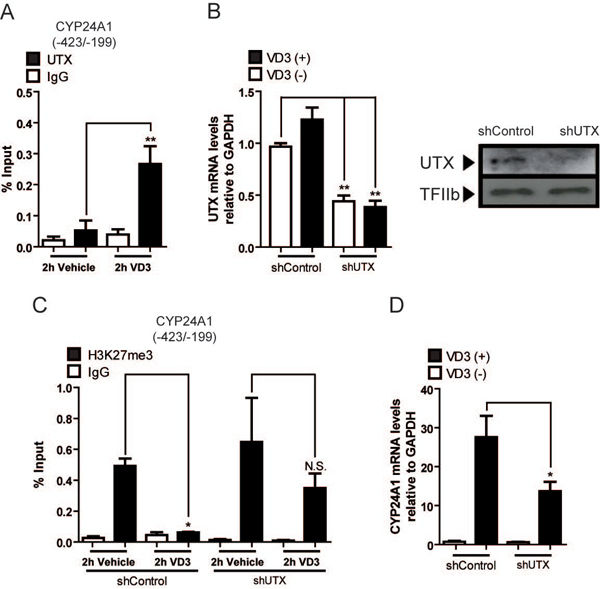
ROS17/2.8 osteoblastic cells were incubated with 10−8 M 1,25(OH)2D3 or vehicle for 2 h. (A) ChIP assays using antibodies against the UTX protein. (B-C) Downregulation of UTX expression in ROS17/2.8 cells infected for 48 h with lentiviruses encoding shRNAs against UTX or shRNA control, and subsequently exposed to 10−8 M 1,25(OH)2D3 or vehicle for 2 h. UTX knockdown was confirmed at mRNA (left) and protein (right) expression levels as described in figure legend 2. GAPDH mRNA is shown as mRNA expression control. Detection of TFIIb was used to control for equal protein loading. (C) Effect of UTX knockdown on H3K27me3 enrichment at the CYP24A1 promoter. (D) Knockdown of UTX decreases 1,25(OH)2D3 dependent CYP24A1 expression. ChIP values are expressed as % input ± SEM. Normal IgG was used as specificity control. Statistical analyses were performed with respect to vehicle. *, p 0.05; **, p 0.01; N.S. Statistically not significant differences.
Discussion
We report that Ezh2-dependent H3K27me3 enrichment at the CYP24A1 gene promoter is associated with epigenetic silencing of this gene in ROS 17/2.8 rat osteoblastic cells. We find that erasing of this H3K27me3 repressive mark by the Utx demethylase is necessary for full 1,25(OH)2D3-dependent transcription activation of the CYP24A1 gene in osteoblasts. Moreover, maintenance of CYP24A1 expression and of the concomitant H3K27me3 reduction at the CYP24A1 promoter, requires the continuous exposure of the cells to 1,25(OH)2D3. Together these results support a model (see Fig 4) where this epigenetic mark is dynamically controlled during 1,25(OH)2D3-dependent activation of the CYP24A1 gene is osteoblastic cells.
Figure 4. Proposed model describing control of histone H3K27me3 enrichment at the CYP24A1 gene promoter in osteoblastic cells.

Schematic representation of the CYP24A1 gene promoter showing the control of the H3K27me3 levels by EZH2 and UTX chromatin modifiers in ROS 17/2.8 osteoblasts grown in the presence or absence of 1,25(OH)2D3, respectively. The arrowhead represents the transcription start site (TSS) and the open ellipses, indicate nucleosomes localized along the proximal promoter region of the CYP24A1 gene. The sizes of modifications and modifiers reflect their respective levels of enrichment.
In the last few years, several groups have examined the presence and function of epigenetic post-translational modifications in chromosomal histone proteins that are associated with the CYP24A1 gene promoter (Fetahu et al., 2014; Meyer et al., 2010b; Moena et al., in press; Seth-Vollenweider et al., 2014). Our team has reported that specific transitions in enrichments of epigenetic histone marks accompany transcriptional upregulation of the CYP24A1 gene in osteoblastic cells exposed to 1,25(OH)2D3 (Moena et al., in press). These transitions involve passing from a H4R3me2s-enriched and H3ac/H4ac/H3R17me2a/H4Rme2a-poor CYP24A1 promoter chromatin in osteoblastic cells in the absence of 1,25(OH)2D3 to a H4R3me2s-poor and H3ac/H4ac/H3R17me2a/H4Rme2a-enriched condition at this promoter in osteoblastic cells exposed to the ligand. Whether H3K27me3 and H4R3me2s together are sufficient to maintain the CYP24A1 gene repressed in the absence of the hormone needs to be demonstrated. Our results showing that independent downregulation of the H3K27me3 and H4R3me2s marks from the CYP24A1 promoter does not result in transcriptional activation of this gene in the absence of ligand (Moena et al., in press; and this work), provides initial support for proposing a cooperative function of both histone marks during maintenance of CYP24A1 repression. Nevertheless, a potential contribution of additional, yet undetermined, epigenetic regulatory components to CYP24A1 silencing cannot be, at this point, discarded.
It is also necessary to consider that there is a requirement for concomitant deposition of active epigenetic histone marks to activate CYP24A1 transcription. Hence, a potential two-step process where erasing and then writing of epigenetic signatures is necessary during transcriptional activation of this gene. Among the necessary components for epigenetic writing we can identify the critical role of the VDR/SRC-1/p300 complex, which is formed in osteoblastic cells at target promoters in response to 1,25(OH)2D3 (Carvallo et al., 2008; Kim et al., 2005; Paredes et al., 2004; Sierra et al., 2003). This VDR/SRC-1/p300 complex brings into place the intrinsic histone acetyl transferase (HAT) activity of both SRC-1 and p300 coactivators (Carvallo et al., 2007; Kim et al., 2005), which can then mediate the increased histone acetylation that has been shown as critical during 1,25(OH)2D3 dependent CYP24A1 transcription activation (Moena et al., in press). Additionally, VDR-containing complexes can activate transcription by directly interacting with the RNA polymerase II holoenzyme at target gene promoters (Arriagada et al., 2010; Chiba et al., 2000; Sutton & MacDonald, 2003), including CYP24A1 (Carvallo et al., 2008; Montecino et al., 2007).
We recently established that histone acetylation at the CYP24A1 promoter in osteoblastic cells is hierarchically dominant over other active histone marks during induction of CYP24A1 gene transcription in response to 1,25(OH)2D3 (Moena et al., in press). Surprisingly, here we find that 1,25(OH)2D3-dependent decrease in H3K27me3 at the CYP24A1 promoter is not accompanied by a significant enrichment in H3K27ac, an epigenetic mark found present at transcriptionally active gene promoters in both osteoblastic and non-osteoblastic cells (Bustos et al., 2017; Rojas et al., 2015; Sepulveda et al., 2017). H3K27me3 and H3K27ac have been found to be mutually exclusive at regulatory genomic regions, although most studies indicate that H3K27ac is preferentially enriched at active enhancer and super-enhancer elements (Weiner et al., 2016). Therefore, together these observations suggest that the decrease in H3K27me3 at some promoters like CYP24A1 (perhaps changing to H3K27me2 or H3K27me1) in response to 1,25(OH)2D3 is sufficient to provide, in association with other active epigenetic histone marks, a proper epigenetic context to support transcription. Whether the absence of a H3K27me3 to H3K27ac transition during 1,25(OH)2D3 dependent CYP24A1 gene activation explains the subsequent rapid return to the H3K27me3 repressive condition at the CYP24A1 promoter after removal of 1,25(OH)2D3 from the media, represents a tempting working hypothesis. It will be important to determine in future investigations whether this paradigm also applies to other active genes in osteoblastic cells.
Presently, the specific mechanisms mediating the recruitment of both PRC2-Ezh2- and Utx-containing complexes to the CYP24A1 gene promoter in the absence and presence of 1,25(OH)2D3, respectively, are not fully understood. Extensive research in the last decade indicates that regulatory components that include non-coding RNAs, transcription factors and specific epigenetic readers, contribute to mediate binding of PRC2-Ezh2 complexes to target promoters during gene silencing (Kazanets et al., 2016; Margueron et al., 2008). On the other hand, the H3K27me3 demethylase Utx has been shown associated with complexes involved in transcription activation in mammals, including COMPASS (Agger et al., 2008; Shilatifard, 2012; Sze & Shilatifard, 2016). This complex catalyzes the deposition of the active histone mark H3K4me3 at target promoters, in close proximity to the TSS. Interestingly, H3K4me3 has been shown to accompany CYP24A1 gene transcription in response to 1,25(OH)2D3 (Nurminen et al., 2018; Singh et al., 2019), suggesting that Utx is bound to the CYP24A1 gene promoter as part of a COMPASS-like complex that is recruited in response to the ligand.
Here, our results shed light on PRC2 mediated epigenetic control of CYP24A1 gene repression in osteoblastic cells and demonstrates that during transcription activation of this gene in response to 1,25(OH)2D3, the PRC2 complex is released and replaced at the CYP24A1 promoter, by a Utx-containing complex. Future studies will need to address whether all of the reported epigenetic histone marks, as well as their writers, readers, and erasers, operate in a tightly coordinated manner to modulate chromatin structure and transcription activity using gene models like CYP24A1.
Acknowledgements
This work has been supported by FONDECYT 1170878 (To MM), FONDAP 15090007 (to MM) and R01 AR039588 (to GSS and JBL). DM was supported by a Doctoral fellowship from CONICYT.
Funding information
Grant/Award Number: 1500007; FONDAP, Grant/Award Number: 1170878; FONDECYT, Grant/Award Numbers: AR039588, R01.
Data availability: The data that support the findings of this study are available from the corresponding author upon reasonable request.
Footnotes
Conflict of interests
The authors declare that they have no conflict of interests.
Web site references
http://genome.ucsc.edu; The UCSC Human Genome Browser.
References
- Agger K, Christensen J, Cloos PA, & Helin K (2008). The emerging functions of histone demethylases. J Current opinion in genetics development, 18(2), 159–168. 10.1016/j.gde.2007.12.003 [DOI] [PubMed] [Google Scholar]
- Arriagada G, Henriquez B, Moena D, Merino P, Ruiz-Tagle C, Lian JB, Stein GS, Stein JL, & Montecino M (2010). Recruitment and subnuclear distribution of the regulatory machinery during 1α, 25-dihydroxy vitamin D3-mediated transcriptional upregulation in osteoblasts. J The Journal of steroid biochemistry molecular biology, 121(1–2), 156–158. 10.1016/j.jsbmb.2006.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos F, Sepúlveda H, Prieto CP, Carrasco M, Díaz L, Palma J, Lattus J, Montecino M, & Palma V (2017). Runt‐Related Transcription Factor 2 Induction During Differentiation of Wharton’s Jelly Mesenchymal Stem Cells to Osteoblasts Is Regulated by Jumonji AT‐Rich Interactive Domain 1B Histone Demethylase. J Stem Cells, 35(12), 2430–2441. 10.1002/stem.2704 [DOI] [PubMed] [Google Scholar]
- Carvallo L, Henriquez B, Olate J, Van Wijnen AJ, Lian JB, Stein GS, Onate S, Stein JL, & Montecino M (2007). The 1α, 25-dihydroxy Vitamin D3 receptor preferentially recruits the coactivator SRC-1 during up-regulation of the osteocalcin gene. The Journal of steroid biochemistry molecular biology, 103(3–5), 420–424. 10.1016/j.jsbmb.2006.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvallo L, Henríquez B, Paredes R, Olate J, Onate S, Van Wijnen AJ, Lian JB, Stein GS, Stein JL, & Montecino M (2008). 1α, 25‐dihydroxy vitamin D3‐enhanced expression of the osteocalcin gene involves increased promoter occupancy of basal transcription regulators and gradual recruitment of the 1α, 25‐dihydroxy vitamin D3 receptor‐SRC‐1 coactivator complex. J Journal of cellular physiology, 214(3), 740–749. 10.1002/jcp.21267 [DOI] [PubMed] [Google Scholar]
- Chiba N, Suldan Z, Freedman LP, & Parvin JD (2000). Binding of liganded vitamin D receptor to the vitamin D receptor interacting protein coactivator complex induces interaction with RNA polymerase II holoenzyme. J Journal of Biological Chemistry, 275(15), 10719–10722. 10.1074/jbc.275.15.10719 [DOI] [PubMed] [Google Scholar]
- Dhawan P, & Christakos S (2010). Novel regulation of 25‐hydroxyvitamin D3 24‐hydroxylase (24 (OH) ase) transcription by glucocorticoids: Cooperative effects of the glucocorticoid receptor, C/EBPβ, and the Vitamin D receptor in 24 (OH) ase transcription. J Journal of cellular biochemistry, 110(6), 1314–1323. 10.1002/jcb.22645 [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. J Nature biotechnology, 489(7414), 57 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetahu IS, Höbaus J, & Kállay E (2014). Vitamin D and the epigenome. J Frontiers in physiology, 5, 164 10.3389/fphys.2014.00164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdick M, & Carlberg C (2000). Agonist-triggered modulation of the activated and silent state of the vitamin D3 receptor by interaction with co-repressors and co-activators. J Journal of molecular biology, 304(5), 793–801. 10.1006/jmbi.2000.4267 [DOI] [PubMed] [Google Scholar]
- Holmqvist P-H, & Mannervik M (2013). Genomic occupancy of the transcriptional co-activators p300 and CBP. J Transcription, 4(1), 18–23. https://dx.doi.org/10.4161%2Ftrns.22601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanets A, Shorstova T, Hilmi K, Marques M, & Witcher M (2016). Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. J Biochimica et Biophysica Acta -Reviews on Cancer, 1865(2), 275–288. 10.1016/j.bbcan.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, & Haussler D (2002). The human genome browser at UCSC. J Genome research, 12(6), 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Shevde NK, & Pike JW (2005). 1, 25‐Dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA‐binding, co‐activator recruitment, and histone acetylation in intact osteoblasts. J Journal of Bone Mineral Research, 20(2), 305–317. 10.1359/JBMR.041112 [DOI] [PubMed] [Google Scholar]
- Majeska RJ, Rodan SB, & Rodan GA (1980). Parathyroid hormone-responsive clonal cell lines from rat osteosarcoma. J Endocrinology, 107(5), 1494–1503. 10.1210/endo-107-5-1494 [DOI] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, & Reinberg D (2008). Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Molecular cell, 32(4), 503–518. 10.1016/j.molcel.2008.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, & Reinberg D (2011). The Polycomb complex PRC2 and its mark in life. J Nature biotechnology, 469(7330), 343 10.1038/nature09784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MB, Benkusky NA, Lee C-H, & Pike JW (2014). Genomic determinants of gene regulation by 1, 25-dihydroxyvitamin D3 during osteoblast-lineage cell differentiation. J Journal of Biological Chemistry, 289(28), 19539–19554. 10.1074/jbc.M114.578104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MB, Goetsch PD, & Pike JW (2010a). A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1α, 25-dihydroxyvitamin D3. J Journal of Biological Chemistry, 285(20), 15599–15610. 10.1074/jbc.M110.119958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MB, Goetsch PD, & Pike JW (2010b). Genome-wide analysis of the VDR/RXR cistrome in osteoblast cells provides new mechanistic insight into the actions of the vitamin D hormone. J The Journal of steroid biochemistry molecular biology, 121(1–2), 136–141. 10.1016/j.jsbmb.2010.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MB, & Pike JW (2013). Corepressors (NCoR and SMRT) as well as coactivators are recruited to positively regulated 1α, 25-dihydroxyvitamin D3-responsive genes. J The Journal of steroid biochemistry molecular biology, 136, 120–124. 10.1016/j.jsbmb.2012.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MB, & Pike JW (2019). Mechanistic homeostasis of vitamin D metabolism in the kidney through reciprocal modulation of Cyp27b1 and Cyp24a1 expression. J The Journal of steroid biochemistry molecular biology, 105500. 10.1016/j.jsbmb.2019.105500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moena D, Merino P, Lian JB, Stein GS, Stein JL, & Montecino M (in press). Switches in histone modifications epigenetically control vitamin D3-dependent transcriptional upregulation of the CYP24A1 gene in osteoblastic cells. Journal of cellular physiology. 10.1002/jcp.29420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino M, Stein GS, Cruzat F, Marcellini S, Stein JL, Lian JB, Van Wijnen AJ, & Arriagada G (2007). An architectural perspective of vitamin D responsiveness. J Archives of biochemistry biophysics, 460(2), 293–299. 10.1016/j.abb.2006.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurminen V, Neme A, Seuter S, & Carlberg C (2018). The impact of the vitamin D-modulated epigenome on VDR target gene regulation. J Biochimica et Biophysica Acta -Gene Regulatory Mechanisms, 1861(8), 697–705. 10.1016/j.bbagrm.2018.05.006 [DOI] [PubMed] [Google Scholar]
- Paredes R, Arriagada G, Cruzat F, Villagra A, Olate J, Zaidi K, Van Wijnen A, Lian JB, Stein GS, & Stein JL (2004). Bone-specific transcription factor Runx2 interacts with the 1α, 25-dihydroxyvitamin D3 receptor to up-regulate rat osteocalcin gene expression in osteoblastic cells. J Molecular cellular biology, 24(20), 8847–8861. 10.1128/MCB.24.20.8847-8861.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike JW, & Christakos S (2017). Biology and mechanisms of action of the vitamin D hormone. J Endocrinology Metabolism Clinics, 46(4), 815–843. 10.1016/j.ecl.2017.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike JW, Meyer MB, Benkusky NA, Lee SM, John HS, Carlson A, Onal M, & Shamsuzzaman S (2016). Genomic determinants of vitamin D-regulated gene expression In Vitamins & Hormones (Vol. 100, pp. 21–44): Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike JW, Meyer MB, John HCS, & Benkusky NA (2015). Epigenetic histone modifications and master regulators as determinants of context dependent nuclear receptor activity in bone cells. J Bone, 81, 757–764. 10.1016/j.bone.2015.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike JW, Meyer MB, Lee S-M, Onal M, & Benkusky NA (2017). The vitamin D receptor: contemporary genomic approaches reveal new basic and translational insights. J The Journal of clinical investigation, 127(4), 1146–1154. 10.1172/JCI88887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachez C, & Freedman LP (2000). Mechanisms of gene regulation by vitamin D3 receptor: a network of coactivator interactions. J Gene, 246(1–2), 9–21. [DOI] [PubMed] [Google Scholar]
- Rojas A, Aguilar R, Henriquez B, Lian JB, Stein JL, Stein GS, Van Wijnen AJ, Van Zundert B, Allende ML, & Montecino M (2015). Epigenetic control of the bone-master Runx2 gene during osteoblast-lineage commitment by the histone demethylase JARID1B/KDM5B. J Journal of Biological Chemistry, 290(47), 28329–28342. 10.1074/jbc.M115.657825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saramäki A, Banwell CM, Campbell MJ, & Carlberg C (2006). Regulation of the human p21 (waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D 3 receptor. J Nucleic acids research, 34(2), 543–554. 10.1093/nar/gkj460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda H, Aguilar R, Prieto CP, Bustos F, Aedo S, Lattus J, Van Zundert B, Palma V, & Montecino M (2017). Epigenetic signatures at the RUNX2‐P1 and Sp7 gene promoters control osteogenic lineage commitment of umbilical cord‐derived mesenchymal stem cells. J Journal of cellular physiology, 232(9), 2519–2527. 10.1002/jcp.25627 [DOI] [PubMed] [Google Scholar]
- Seth-Vollenweider T, Joshi S, Dhawan P, Sif S, & Christakos S (2014). Novel mechanism of negative regulation of 1,25-dihydroxyvitamin D3-induced 25-hydroxyvitamin D3 24-hydroxylase (Cyp24a1) Transcription: epigenetic modification involving cross-talk between protein-arginine methyltransferase 5 and the SWI/SNF complex. J Journal of Biological Chemistry, 289(49), 33958–33970. 10.1074/jbc.M114.583302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A (2012). The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. J Annual review of biochemistry, 81, 65–95. 10.1146/annurev-biochem-051710-134100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra J, Villagra A, Paredes R, Cruzat F, Gutierrez S, Javed A, Arriagada G, Olate J, Imschenetzky M, & Van Wijnen AJ (2003). Regulation of the bone-specific osteocalcin gene by p300 requires Runx2/Cbfa1 and the vitamin D3 receptor but not p300 intrinsic histone acetyltransferase activity. J Molecular cellular biology, 23(9), 3339–3351. 10.1128/mcb.23.9.3339-3351.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Verma A, Sallin MA, Lang F, Sen R, & Sen JM (2019). Noncoding variations in Cyp24a1 gene are associated with Klotho‐mediated aging phenotypes in different strains of mice. J Aging cell, 18(3), e12949. https://dx.doi.org/10.1111%2Facel.12949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutoglou E, & Talianidis I (2002). Coordination of PIC assembly and chromatin remodeling during differentiation-induced gene activation. J Science, 295(5561), 1901–1904. 10.1126/science.1068356 [DOI] [PubMed] [Google Scholar]
- Sutton AL, & MacDonald PN (2003). Vitamin D: more than a “bone-a-fide” hormone. J Molecular Endocrinology, 17(5), 777–791. 10.1210/me.2002-0363 [DOI] [PubMed] [Google Scholar]
- Sze CC, & Shilatifard A (2016). MLL3/MLL4/COMPASS family on epigenetic regulation of enhancer function and cancer. J Cold Spring Harbor perspectives in medicine, 6(11), a026427. 10.1101/cshperspect.a026427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Lara-Astiaso D, Krupalnik V, Gafni O, David E, Winter DR, Hanna JH, & Amit I (2016). Co-ChIP enables genome-wide mapping of histone mark co-occurrence at single-molecule resolution. J Nature biotechnology, 34(9), 953 10.1038/nbt.3652 [DOI] [PubMed] [Google Scholar]
- Zhang C, Tang W, Li Y, Yang F, Dowd DR, & MacDonald PN (2011). Osteoblast-specific transcription factor Osterix increases vitamin D receptor gene expression in osteoblasts. J PloS one, 6(10), e26504. 10.1371/journal.pone.0026504 [DOI] [PMC free article] [PubMed] [Google Scholar]